

Abstract
Microfluidic technology overcomes many of the limitations to traditional analytical methods in microbiology. Unlike bulk-culture methods, it offers single-cell resolution and long observation times spanning hundreds of generations; unlike agarose pad-based microscopy, it has uniform growth conditions that can be tightly controlled. Because the continuous flow of growth medium isolates the cells in a microfluidic device from unpredictable variations in the local chemical environment caused by cell growth and metabolism, authentic changes in gene expression and cell growth in response to specific stimuli can be more confidently observed. Bacillus subtilis is used here as a model bacterial species to demonstrate a "mother machine"-type method for cellular analysis. We show how to construct and plumb a microfluidic device, load it with cells, initiate microscopic imaging, and expose cells to a stimulus by switching from one growth medium to another. A stress-responsive reporter is used as an example to reveal the type of data that may be obtained by this method. We also briefly discuss further applications of this method for other types of experiments, such as analysis of bacterial sporulation.
Keywords: Cellular Biology, Issue 131, Bacteria, microfluidics, PDMS, fluorescence microscopy, exponential growth, medium switching
Introduction
One of the most striking features of life on Earth is its great resilience and variety. A central goal of molecular biology is to understand the logic by which cells use genes and proteins to maximize their growth and fitness under a wide variety of environmental conditions. To achieve this goal, scientists must be able to confidently observe how individual cells grow, divide, and express their genes under a given set of conditions, noting how cells respond to subsequent changes in their environment. However, traditional analytical methods in microbiology have technical limitations that affect the types of questions that can be addressed. For example, bulk culture-based analyses have been very useful over the years, yet they offer only population-level data that can mask meaningful cell-to-cell variations or the behaviors of smaller sub-populations of cells in the total population. Single-cell analyses of living bacteria based on light microscopy reveal single-cell behavior but are also technically limited. Bacteria are typically immobilized on agarose pads containing growth medium, but cell growth and division crowds the microscopic view and depletes the available nutrients after just a few cell cycles, substantially limiting the observation time1,2. Moreover, the local depletion of nutrients and the concomitant buildup of metabolic byproducts due to cell growth are constantly changing the local cell growth environment in ways that are difficult to measure or predict. Such environmental changes using agarose pads pose a challenge to studies of steady-state behaviors or of cellular responses to specific changes in growth conditions3.
Microfluidic technology, in which a liquid medium is continuously flowed through microfabricated devices, offers a solution to classic experimental limitations. A microfluidic device can keep individual cells in position for live-cell microscopy while the flow of growth medium constantly provides cells with fresh nutrients and washes away metabolic byproducts and excess cells, thereby creating a highly uniform growth environment. Under constant growth conditions, cell behaviors can be observed in isolation from the influence of environmental factors, permitting an unimpeded view of the internal logic of cells. As the fluid flow prevents the microfluidic device from becoming crowded with cells, observation of single cell lineages for tens or hundreds of generations becomes possible4,5. Such long observation times permit the detection of otherwise undetectable long-term or rare cell behaviors. Finally, the composition of the medium that flows through the device can be altered at will, allowing cells to be observed as they respond to the onset of a stress or to the introduction or removal of a particular compound.
Microfluidics has already enjoyed a number of important applications. For instance, it has been used in tissue-, organ-, or body-on-a-chip devices, in which multiple human cell types are co-cultured to simulate an in vivo condition6; for the study of nematode movement in microstructured environments7; to examine interactions among bacterial biofilms (e.g., 8); and for the encapsulation and manipulation of tiny volumes of cells or chemicals (e.g., 9). Microfluidic devices have also become increasingly popular in the field of microbiology (for excellent reviews, see 10 and 11), especially as their physical and flow properties are well-matched to natural microbial niches12. For instance, microfluidics has been recently employed by microbiologists for such purposes as precisely measuring cell growth and division13,14,15, analyzing pathogen movement16, monitoring quorum sensing17 and physiological transitions18, and for protein counting19, among many other examples. The method presented here is specifically designed for the analysis of single bacterial cell lineages rather than combinations of strains or species. The microfluidic device demonstrated here utilizes one variation of the "mother machine" design4, in which cells are grown single-file within a microfluidic trench with one closed end and one open end; cell growth and division pushes progeny cells up and out of the open end into the fluid flow. Our analyses typically focus only on the "mother" cell that is confined at the closed end of the trench. We consider this method as an advancement over previous light microscopy-based single-cell analytical techniques, such as cell immobilization on agarose pads. While B. subtilis is used as a model here, the method is also applicable to other bacterial species (Escherichia coli is another common model; some species with different cell sizes or morphologies may require the fabrication of new devices with different dimensions). The use of fluorescent reporters to mark cells and to visualize changes in gene expression requires the use of genetically tractable species; however, analyses of cell growth and morphology are possible even without fluorescent markers.
The present protocol excludes the process of fabricating the silicon master using photolithography, which has been extensively described elsewhere5; masters can also be easily outsourced from microfabrication facilities. It includes the molding of a PDMS device from a reinforced silicon master; bonding the device to a piece of cover glass; assembling the microfluidic inlet and outlet plumbing, including pinch valves to permit medium switching; passivating the device, preparing bacterial cells, and loading the device with bacterial cells; attaching the plumbing to the device and equilibrating the cells; and loading the device onto a fluorescence microscope for imaging. Because many different image acquisition and processing software tools can be used to visualize and analyze different data of interest4,5, example images are shown, but image-capture methods are not included in this protocol.
Protocol
1. PDMS Device Casting
In a dust-free environment, mix PDMS polymer and curing agent at a 10:1 ratio and degas under vacuum for at least 10 min.
Place a silicon master (or an epoxy or polyurethane replica thereof) on a piece of unbroken aluminum foil in a polystyrene Petri plate. Pour the degassed PDMS mixture over the master to a depth of approximately 5 mm. Degas the Petri plate for at least 10 min. Use a gentle stream of air to break surface bubbles. NOTE: The dimensions of the two-tiered device used are provided in the accompanying CAD file5 (see Supplementary File 1). A plastic replica master may partially float during degassing; if this occurs, push down the replica with a clean plastic applicator.
Cure PDMS for at least 1 h in an oven at 60 °C and cool to room temperature. Remove the aluminum foil containing the master and cured PDMS from the Petri plate. Carefully peel off the aluminum foil from the back of the master, then carefully peel the PDMS from the master. NOTE: Alternatively, PDMS may be cured overnight at room temperature. The relative fragility of a silicon-wafer master may be overcome by using epoxy adhesive to permanently bond the wafer to a 1/16 inch-thick aluminum disc of the same size as the wafer.
As a wafer typically includes several microfluidic devices with slightly different dimensions (see Supplementary File 1), cut a single device to size using a clean, sharp scalpel. NOTE: For routine B. subtilis work, use devices with 1.0-µm-wide cell trenches, which are marked with a C or F in the accompanying CAD file.
2. Device Punching and Bonding
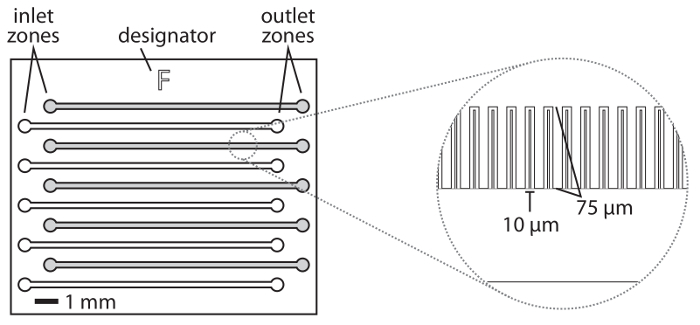
Place a piece of translucent office tape (see Table of Materials) over the patterned side of the device to enhance the visibility of the patterned features. The inlet and outlet ports are identified as circular areas at opposite ends of the visible fluidic channels (Figure 1). To improve their visibility when punching holes in the device for the inlet and outlet pins, mark the inlets and outlets with dots using a fine-tipped marker. NOTE: To ensure that the inlet and outlet pins are not crowded, every other channel is punched, beginning with the channel just beneath the alphabetical designator for the device.
Flip the PDMS device so that the patterned side is down, and punch through the device to the marked dots using an 0.75-mm biopsy punch; discard the punchings. NOTE: The devices are punched with the patterned side down because the hole generated by the biopsy punch is typically slightly flared where the punch enters the polymer. Punching the device in an inverted orientation ensures that the hole on the patterned side has a sharp border.
Using a stereomicroscope, ensure that the punched holes overlap with the inlet and outlet areas of the device. Use a fine-tipped tweezers to remove any extraneous fragments of PDMS from the punched holes.
Use adhesive tape to remove dust from both sides of the device and place into a clean polystyrene Petri plate. Prepare a 22 mm x 40 mm cover glass by wetting with 100% isopropyl alcohol and rubbing with a cleanroom wipe; dry with a stream of dust-free air or nitrogen.
- Place the punched PDMS device feature side up together with the cleaned cover glass into an oxygen plasma cleaner.
- Plasma-treat the device with the following settings: vacuum, 70 mTorr; O2 pressure, 200 mTorr; duration, 15 s; power, 30 W. Immediately after the plasma treatment period finishes, remove the device and cover glass from the plasma cleaner.
- Invert the PDMS and place it on the center of the cover glass so that the patterned side contacts the glass; ensure that the feeding channels (running between the inlet and outlet areas) are aligned with the long axis of the cover glass. Press down very gently to ensure that the PDMS seals against the glass.
Bake the assembled device in an oven for at least 1 h at 60 °C. After baking, manually verify that the PDMS is bonded to the glass by pushing gently on one corner of the device. NOTE: Once the device is bonded, it should be used within 24 h, as the polymer will become increasingly hydrophobic with increasing time after plasma treatment.
3. Microfluidic Plumbing Preparation
Cut lengths of polymer tubing (inner diameter (ID) 0.02 inch, outer diameter (OD) 0.06 inch) to appropriate lengths to reach from the syringe pump to the switching apparatus (if used) and the device when mounted on the microscope. Cut as many lengths as there are microfluidic lanes.
Assemble Y junctions for pinch valves (if using) by cutting and attaching 2-cm lengths of flexible silicone tubing (0.03 inch ID, 0.065 inch OD) to the top barbs of the "Y" and 1-cm lengths of silicone tubing to the bottom barb of the "Y."
Cut 10-cm (or appropriate) lengths of polymer tubing; connect them at one end to the switch junction by inserting them into the 1-cm silicone tubing segments on the bottom barb of the "Y." Connect them at the other end to 21G needles that have been removed from their plastic syringe adaptors and bent approximately 90° approximately 9 mm from the needle end.
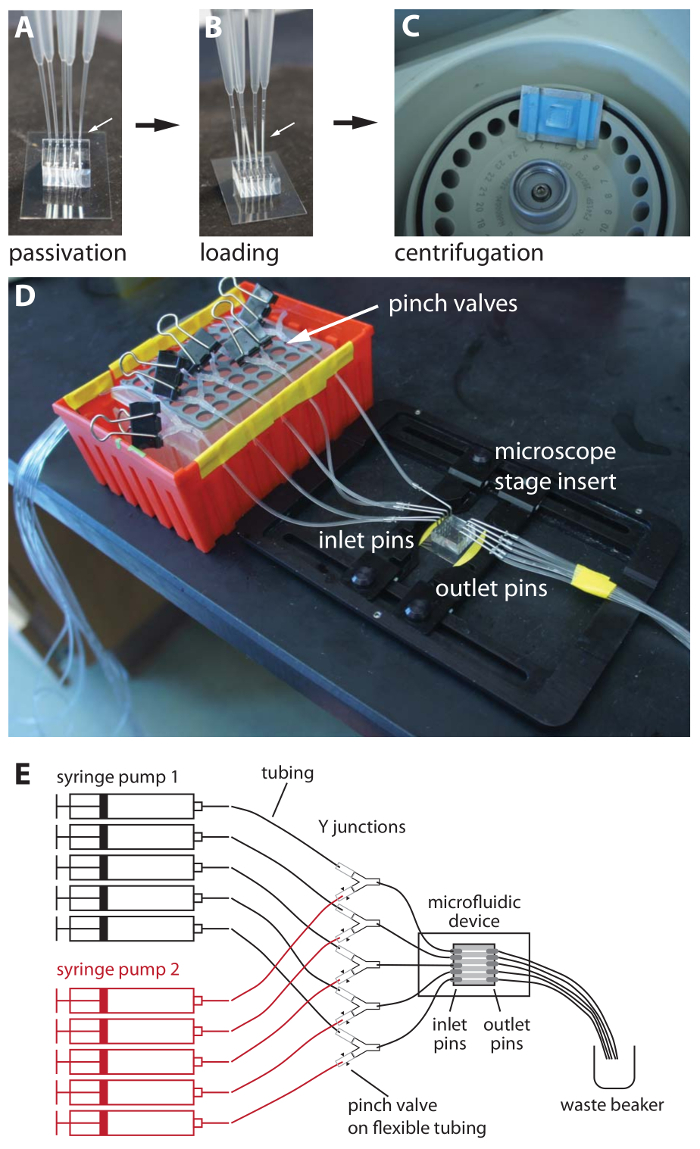
Connect 21G blunt needles to one end of the tubing segments that will run from the syringes to the switch apparatus by inserting the needles into the tubing. Insert the other ends of each length of tubing into the silicone tubing segments on the inlet barbs of the Y junctions. NOTE: Use some sort of apparatus to hold the pinch valves and junctions in place; this can easily be made from a micropipette tip box (Figure 2D).
Cut lengths of polymer tubing to carry waste from the outlet of the device to a waste beaker. Connect them at one end to bent 21G needles prepared as described above. Tape the other end of the tubes into a waste beaker.
Prepare appropriate volumes of media containing 0.1 mg/mL bovine serum albumin (BSA) and fill the desired size and number of syringes. Connect the BSA-loaded syringes to the prepared 21G needles attached to the inlet tubing and load syringes into syringe pumps. NOTE: For typical experiments 5 channels of the device are used, each with a 20-mL syringe. An experiment in which the medium is switched will require 2 banks of 5 syringes each. Here, the pump containing the first bank is referred to as the phase-1 pump, and the pump containing the second bank, with the post-switch medium, is referred to as the phase-2 pump.
Purge air from the inlet plumbing by running the syringe pumps at a high flow rate (>500 µL/min), beginning by orienting the syringe pumps vertically and tapping the syringes to bring air bubbles to the top. Once air has been purged from the syringes, place the syringe pump horizontally and progressively tap or flick the polymer lines from the syringes up through the switch apparatus to dislodge and purge air bubbles. Repeat this process for the second bank of syringes (if switching media).
After air bubbles are purged, set the phase-2 syringe pump (i.e., with the medium that will be used second, after the switch) to 1.5 µL/min, then pause the flow. Place small binder clips onto the segments of flexible tubing on the branch of the Y junctions corresponding to the second medium phase. Continue to run the phase-1 syringe pump at a modest flow rate (e.g., 10 µL/min) for at least 30 min to purge the second medium from the inlet tubing downstream of the Y junction.
4. Cell Culture Preparation
Grow a preculture of the strain of interest in the desired medium overnight, to stationary phase. The bacterial strain should contain a mutation that renders the cells immobile, so that the cells do not swim out of the channels of the device. NOTE: In this protocol, the bacterial strain is B. subtilis 3610 containing a hagA233V substitution (this mutation causes the flagellum to be straight rather than helical, preventing cell motility), a PrsbV-mNeonGreen reporter for cell stress, and a constitutive Phyperspank-mNeptune (red) reporter to highlight cells. LB Lennox medium is used in the example protocol. Typical strains for microfluidics experiments contain one or more fluorescent transcriptional reporters and a constitutively produced, cytoplasmic fluorescent protein to facilitate automated detection of cells. Growth defects were not observed when LB Lennox rich medium was used, but B. subtilis growth in minimal media may be inhibited under microfluidic conditions because secreted siderophores are washed away by the medium flow. Under such circumstances, growth may be restored by the addition of sodium citrate and ferric chloride to the medium, as reported for S750 medium11.
The morning of the experiment, dilute the B. subtilis cells 1:50 into a baffled 250-mL flask containing 25 mL of growth medium. Grow the cells for 5 - 6 h in shaking culture at 37 °C to an optical density of greater than 1. NOTE: A dense cell culture is preferable because the stationary-phase B. subtilis cells are smaller and less often found in cell chains, facilitating device loading. Different bacterial species may require other medium or growth conditions for optimal loading.
Using a syringe fitted with a 5-µm pore-size filter, filter approximately 15 mL of cell culture into a 15-mL conical tube to remove B. subtilis cell chains (it is unnecessary for E. coli and may not be necessary with other species). NOTE: Relatively few cells should be removed; the filtrate should be turbid.
Centrifuge the filtered culture for 10 min at 4,000 x g at room temperature. Pour off the supernatant and resuspend the cell pellet in the residual supernatant fluid remaining in the tube, adding approximately 500 µL of fresh medium if necessary to facilitate pipetting the cell suspension.
5. Device Loading
Gently place empty thin gel-loading micropipette tips (see Table of Materials) into the outlet holes of the microfluidic device.
Using thin gel-loading tips with a P200 micropipette, passivate the device by injecting medium containing 1 mg/mL BSA into the inlet holes, observing the tips in the outlet holes to monitor filling of the microfluidic channels (Figure 2A). Incubate the device at room temperature for approximately 5 min. NOTE: BSA is commonly used as a blocking or passivation agent that will bind to the hydrophobic surface of the PDMS polymer, increasing its hydrophilicity and reducing the binding of other proteins or of cells (via cell surface molecules).
Using thin gel-loading tips, load each channel with the resuspended cells (from Section 4), using the tips in the outlet channel to monitor the progress of the cells through the device (Figure 2B). NOTE: Loading is facilitated by setting the P200 micropipette to its maximum 200-µL volume and then aspirating a cushion of air before aspirating a small volume (approximately half the volume of the thin section of the pipette tip) of cells. The volume of resuspended cells need not be precise, as the volume of the PDMS device is small relative to that of the micropipette. We know of no upper limit to the density of the resuspended cells, as long as the cell suspension can be pipetted into the device. Cell clumps can clog the device and should be avoided by thorough pipetting and/or vortex mixing.
Gently remove the gel-loading tips from the outlet holes, working one at a time to prevent damage to the device.
Centrifuge the device in a bench-top microcentrifuge in an appropriate rotor adaptor at approximately 6,000 x g for 10 min (Figure 2C). NOTE: A custom-machined aluminum rotor adaptor that was designed to fit into a microcentrifuge rotor (Figure 2C) was used; different centrifuge models may be used with appropriate rotor adaptors. The important feature of the adaptor is that it provides lateral force in the direction of the closed ends of the cell trenches, thereby forcing cells into the side channels.
Verify successful loading under the microscope (Figure 3) but without affixing the device to the slide holder/stage insert. NOTE: In this protocol, a 60X, 1.4 NA Ph3 oil-immersion objective is used for both verification at this stage and for subsequent imaging.

6. Device Assembly, Equilibration, and Mounting
Carefully mount the loaded device onto a stage insert by taping the cover glass on either side of the PDMS to the bottom of the stage insert (i.e., invert the stage insert before attaching the device). Invert the stage insert-device assembly so that the PDMS is facing up and place on a soft surface, such as a dust-free wipe (Figure 2D).
Adjust the flow rate of the phase-1 syringe pump to 35 µL/min. Working with one lane at a time, insert the inlet needle and then the outlet needle (i.e., running to the waste beaker; Figure 2E).
Wipe away any excess medium with a clean dust-free wipe and visually inspect the device for leaks. Examine the outlet tubing for the appearance of excess cells (typically appearing as a turbid stripe) and the medium meniscus, which will slowly move toward the waste beaker.
Permit the device to run at 35 µL/min for approximately 15 - 30 min, until all the connected lanes are draining into the waste beaker.
Set the phase-1 syringe pump to 1.5 µL/min and pause the flow. Bring the entire pump and device apparatus to the microscope (this process is aided by placing it all on a rolling cart) and restart the phase-1 medium flow at 1.5 µL/min. Carefully mount the stage insert with the device onto an inverted fluorescence microscope, using tape as necessary to route the inlet and outlet tubing.
Locate desired positions on the device for imaging and begin imaging as desired. Note that cells typically require a few hours (approximately 10 generations) to reach steady-state exponential-phase growth, and the onset of image acquisition may be delayed as desired so that imaging begins after the equilibration period. NOTE: The steady-state growth of cells held in exponential phase should be verified during subsequent image analysis by tracking cell division times and ensuring that they have reached a constant minimum value.
7. Medium Switching
Between successive rounds of imaging, pause the flow of the phase-1 syringe pump. Carefully unclip the binder clips from the phase-2 silicone tubing, moving each to the silicone tubing on the phase-1 branch of the Y junction. Un-pause the flow of the phase-2 syringe pump (which was set to 1.5 µL/min in step 3.8) and continue imaging. NOTE: At 1.5 µm/min with a 10-cm length of polymer tubing downstream of the Y junctions, the second-phase medium reaches the device in approximately 50 min. The time to the device can be empirically tested using media that contain fluorescent tracer particles.
Representative Results
Successful initial cell loading, as assessed by phase-contrast microscopy before attaching the microfluidic plumbing to the device, would be considered as having all or nearly all of the microfluidic side channels containing one or more bacterial cells (Figure 3A). Optimal loading would show several cells in each channel, but channels will nonetheless fill with cells due to cell growth during the equilibration period (Figure 3B). Lanes with poor loading, in which relatively few (<50%) side channels are loaded with cells, may be used, but the resulting data density will be lower due to fewer cell lineages being imaged at each imaging position.
Once the device is initially loaded, it is equilibrated by attaching the microfluidic plumbing and flowing medium through the device at a relatively high flow rate of 35 µL/min. The equilibration step serves to remove air bubbles and excess cells in the feeding channel from the device, and to encourage the cells in the side channels to begin growing. The cells that are removed from the device upon medium flow can often be observed by the naked eye as a light-colored band moving through the outlet tubing towards the waste container; the movement of such bands serves as a visual indicator that fluid is flowing normally through the plumbing and the device. Microscopic inspection of an equilibrated device should reveal a few cells confined in single file in most of the side channels (Figure 3B, 0 min).
Once an equilibrated device is placed on the microscope under flow at 1.5 µL/min at 37 °C, the cells will resume uniform and constant exponential-phase growth over the course of approximately 2 h (Figure 3B). Constant exponential growth should be directly verified after image analysis by the appearance of a plateau in the generation time of the cells. At this point, the cells are ready for fluorescence and/or brightfield imaging as desired. A successfully loaded and equilibrated device can typically be reliably imaged for periods of at least 24 h (Figure 4A, C). Introducing a medium switch (Figure 4A-C) expands the range of experiments that can be conducted but also increases the frequency of catastrophic cell-death events (Figure 4D), presumably through the introduction of air bubbles that were not successfully purged from the tubing. Another event that may cause catastrophic cell death is that clusters of cells located at the inlet may become dislodged and pass through the feeding channel, as shown in Figure 4D. We do not at present understand why this causes catastrophic cell death in the device, and we have not yet devised a method to prevent such events from occurring.
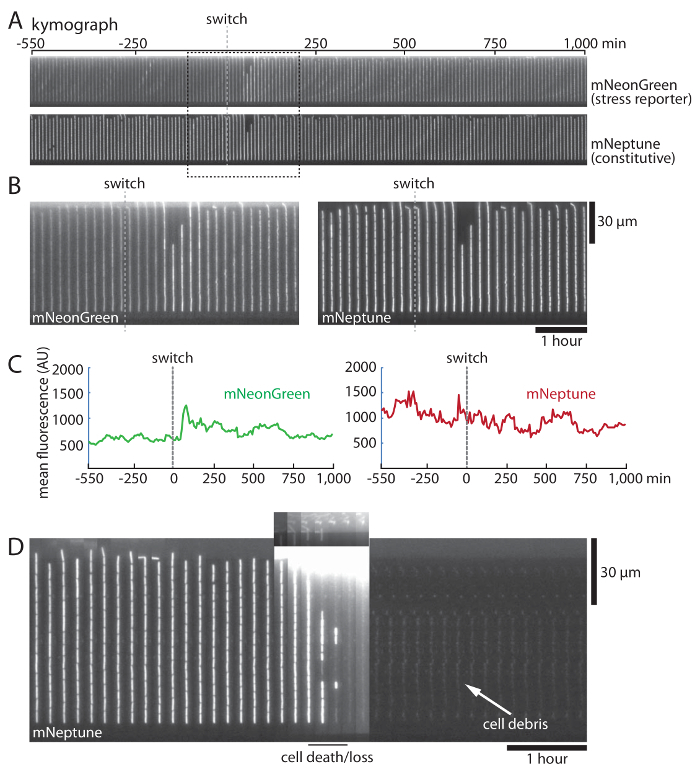
Figure 1: Schematic of the microfluidic device. A device schematic is shown approximately to scale. The alphabetic designator is labeled along with the inlet and outlet zones that are punched to connect the blunt-ended needles. Typically, half of the patterned channels are used (shaded gray). The cell trenches are oriented towards the designator. The accompanying CAD file (see Supplementary File 1) shows all of the features on the device. Please click here to view a larger version of this figure.
Figure 2: Microfluidic device passivation, cell loading, and equilibration. (A) A bonded device after passivation with BSA-containing medium. Gel-loading tips are kept in the outlet holes to monitor the passage of medium and cells through the device. The medium meniscus is visible in the thin portion of the gel-loading tips (arrow). (B) Device after cell loading. The cells are visible as a translucent white layer in the gel-loading tips (arrow). (C) The gel-loading tips are removed before centrifugation in a custom-fabricated centrifuge adaptor. Medical tape (blue) is placed on the aluminum parts to cushion the device. (D) Following centrifugation and verification of loading, the device is taped to a microscope stage insert and attached to the inlet and outlet pins. In the image shown, medium switching is made possible by pinch valves and Y-connectors that are arranged in an apparatus made from a micropipette tip box. (E) Schematic view of an entire assembled device, from the syringe pumps to the waste container. The outlet tubing runs to a small waste beaker. Please click here to view a larger version of this figure.
Figure 3: Cell loading and growth in the microfluidic device. (A) From left to right, phase-contrast micrographs of a device containing medium only before cell loading, the same device after cells have been added via pipetting but before centrifugation, and the same device after centrifugal loading. (B) Time course of cell adaptation and growth in the device (LB, 37 °C, flow 1.5 µL/min). At the beginning of adaptation, the stationary-phase cells are relatively short and narrow. As the cells adapt and return to exponential-phase growth (60 - 120 min), they adopt a longer and wider morphology. After 120 min, all cells in the device are resuming uniform exponential-phase growth, and the device is ready for imaging. Please click here to view a larger version of this figure.
Figure 4: Cell growth and medium switching in the microfluidic device. (A) Kymographs showing 550 min of growth before and 1,000 min of growth after a medium switch (grey dashed line) into 2% ethanol as a stressor. Images were captured at 10-min intervals. The top panel shows a transcriptional mNeonGreen reporter (green channel) for a stress-induced gene. The bottom panel shows a constitutive mNeptune reporter (red channel) that is used in this case for automated cell segmentation. (B) Close-up view of the portions of the kymographs in the dashed box in panel A, showing a transient response in the green channel following the medium switch. Note that growth continues through the switch, although the presence of the ethanol causes the cells to become shorter and grow at a slightly slower pace. Time scale is shown by the horizontal bar, and distance is shown by the vertical bar. (C) Example traces of analyzed fluorescence data of a single cell lineage from the experiment shown in panel A. The vertical dashed lines indicate the medium switch into 2% ethanol as a stressor. (D) Example of a failed medium switch. When the second medium reaches the cells, the cells in the channels are lysed. The switch is accompanied by a large number of cells that pass by in the main channel, causing a bright fluorescent signal (a re-scaled image corresponding to the washed-out region is shown just above it). After the switch, no further cells are observed, although dimly fluorescent cell debris remains in the channels. Time scale is shown by the horizontal bar, and distance is shown by the vertical bar. Please click here to view a larger version of this figure.
Discussion
This microfluidics protocol is flexible in that many of the steps may be modified to optimize its use with a particular species or strain or for a specific purpose. Indeed, in this protocol we have made modifications to the original "mother machine" concept4 to optimize its use with B. subtilis. Often, the trenches in which the cells are confined constitute a single-layer feature, whereas in this protocol we use two-layer cell trenches, with a shallow channel surrounding the cells. The two-layer design was introduced as a way to maximize the flux of nutrients to and metabolites from B. subtilis cells, especially in long (>75 µm) trenches5; however, single-layer features often suffice for optimal cell growth and are commonly used for E. coli, especially when they are shorter (~25 µm)4. We typically use longer trenches, as they reduce the frequency with which B. subtilis cell chains, which arise stochastically, are pulled out of their trenches by the medium flow in the main feeding channel5.
Further modifications, such as the use of devices with different feature dimensions (e.g., channel widths) or characteristics (e.g., shape, number of open ends) may be substituted as appropriate. For instance, the use of cell trenches that are open on both ends (rather than at only one end, as in the present protocol) has been used in other studies1,20,21. Trenches that are open on both ends avoid cell aging because they are constantly being filled with newborn cells (as opposed to mother machines, where the oldest cell is tracked and newer cells are pushed out of the channel), although the fact that older cells are pushed out both ends typically limits their observational window to fewer than 10 generations1,21. We typically seek the longer observational windows (hundreds of generations) offered by a mother machine, which make it possible to measure even memoryless processes for which waiting times are tens or hundreds of generations long5. We have also used the mother machine design to observe cells that are not exponentially growing, as in our analyses of sporulation22. In such cases, additional cells throughout the growth trenches can be meaningfully analyzed22.
Many of the steps in the protocol are relatively forgiving in that they may be modified without substantially affecting the protocol outcome. For instance, an earlier version of the protocol called for cleaning the cover glass by sequential 10-min bath sonication steps in 10 M KOH and then pure water, but we found that good device bonding and operation was achieved using the simpler isopropanol-based cleaning step described here. If a bonding issue arose that cast suspicion on the cleanliness of the cover glass, reverting to a more-stringent cleaning protocol is recommended. Similarly, while we use a centrifugation step to maximize cell loading into the cell trenches, reasonably full loading can be achieved even without this step, provided that very concentrated cells are used for the loading step. This alternative strategy could be especially useful for researchers who do not have a readily available centrifuge adapter to spin the device. Again, different concentrations of passivating/lubricating agent (BSA in this protocol) may be used; we routinely use concentrations as high as 1 mg/mL. In cases where a strain may be sensitive to BSA, acceptable results may be achieved even in the absence of a passivating agent. Indeed, when we were using a microfluidic device to examine sporulation, which requires cell starvation, we were concerned that BSA might act as a potential nutrient source for the cells, and so we omitted it from the medium flow without observing any substantial adverse effects22.
Notably, the continuous flow of medium through the device can elicit slightly different phenotypes compared with cell growth in a closed system, such as a flask. For example, nutrient or compound depletion can be challenging, as cells can often scavenge trace compounds from the medium flow. Indeed, we have observed that inducing cell sporulation via starvation requires a longer time in a microfluidic device than in a flask. Similarly, we have observed that induction of energy stress using the oxidative phosphorylation decoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) requires several-fold higher concentrations in the microfluidic device than reported in flask culture23. Therefore, some optimization may be required to achieve satisfactory results with different medium additives.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This project was funded by the National Institutes of Health under GM018568. This protocol was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. CNS is part of Harvard University. Many thanks are due to Thomas Norman and Nathan Lord for their work in conceiving and fabricating the master template used for the devices shown here and in building the original version of the apparatus. We also thank Johan Paulsson for his valuable collaborative advice and thank members of his lab for their advice and continued improvements to bacterial microfluidic apparatuses.
References
- Moffitt JR, Lee JB, Cluzel P. The single-cell chemostat: an agarose-based, microfluidic device for high-throughput, single-cell studies of bacteria and bacterial communities. Lab Chip. 2012;12(8):1487–1494. doi: 10.1039/c2lc00009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, et al. Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat Protoc. 2011;7(1):80–88. doi: 10.1038/nprot.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusny C, et al. Technical bias of microcultivation environments on single-cell physiology. Lab Chip. 2015;15(8):1822–1834. doi: 10.1039/c4lc01270d. [DOI] [PubMed] [Google Scholar]
- Wang P, et al. Robust growth of Escherichia coli. Curr Biol. 2010;20(12):1099–1103. doi: 10.1016/j.cub.2010.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman TM, Lord ND, Paulsson J, Losick R. Memory and modularity in cell-fate decision making. Nature. 2013;503(7477):481–486. doi: 10.1038/nature12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perestrelo AR, Aguas AC, Rainer A, Forte G. Sensors. 12. Vol. 15. Basel: 2015. Microfluidic Organ/Body-on-a-Chip Devices at the Convergence of Biology and Microengineering; pp. 31142–31170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johari S, Nock V, Alkaisi MM, Wang W. On-chip analysis of C. elegans muscular forces and locomotion patterns in microstructured environments. Lab Chip. 2013;13(9):1699–1707. doi: 10.1039/c3lc41403e. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Coupling between distant biofilms and emergence of nutrient time-sharing. Science. 2017;356(6338):638–642. doi: 10.1126/science.aah4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, et al. One-step generation of cell-laden microgels using double emulsion drops with a sacrificial ultra-thin oil shell. Lab Chip. 2016;16(9):1549–1555. doi: 10.1039/c6lc00261g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel DB, Diluzio WR, Whitesides GM. Microfabrication meets microbiology. Nat Rev Microbiol. 2007;5(3):209–218. doi: 10.1038/nrmicro1616. [DOI] [PubMed] [Google Scholar]
- Taheri-Araghi S, Brown SD, Sauls JT, McIntosh DB, Jun S. Single-Cell Physiology. Annu Rev Biophys. 2015;44:123–142. doi: 10.1146/annurev-biophys-060414-034236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi A, Karig D, Kumar A, Ardekani AM. Interplay of physical mechanisms and biofilm processes: review of microfluidic methods. Lab Chip. 2015;15(1):23–42. doi: 10.1039/c4lc01095g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FB, et al. Long-term microfluidic tracking of coccoid cyanobacterial cells reveals robust control of division timing. BMC Biol. 2017;15(1):11. doi: 10.1186/s12915-016-0344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos M, et al. A constant size extension drives bacterial cell size homeostasis. Cell. 2014;159(6):1433–1446. doi: 10.1016/j.cell.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri-Araghi S, et al. Cell-size control and homeostasis in bacteria. Curr Biol. 2015;25(3):385–391. doi: 10.1016/j.cub.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siryaporn A, Kim MK, Shen Y, Stone HA, Gitai Z. Colonization, competition, and dispersal of pathogens in fluid flow networks. Curr Biol. 2015;25(9):1201–1207. doi: 10.1016/j.cub.2015.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JL, et al. Probing prokaryotic social behaviors with bacterial "lobster traps". MBio. 2010;1(4) doi: 10.1128/mBio.00202-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Z, et al. Measuring bacterial adaptation dynamics at the single-cell level using a microfluidic chemostat and time-lapse fluorescence microscopy. Analyst. 2014;139(20):5254–5262. doi: 10.1039/c4an00877d. [DOI] [PubMed] [Google Scholar]
- Okumus B, et al. Mechanical slowing-down of cytoplasmic diffusion allows in vivo counting of proteins in individual cells. Nat Commun. 2016;7:11641. doi: 10.1038/ncomms11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng SW, Mukherji S, Moffitt JR, de Buyl S, O'Shea EK. Robust circadian oscillations in growing cyanobacteria require transcriptional feedback. Science. 2013;340(6133):737–740. doi: 10.1126/science.1230996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JD, et al. Programmable, Pneumatically Actuated Microfluidic Device with an Integrated Nanochannel Array To Track Development of Individual Bacteria. Anal Chem. 2016;88(17):8476–8483. doi: 10.1021/acs.analchem.6b00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JR, Cabeen MT, Wiggins PA, Paulsson J, Losick R. Noise in a phosphorelay drives stochastic entry into sporulation in Bacillus subtilis. EMBO J. 2017;36(19):2856–2869. doi: 10.15252/embj.201796988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabeen MT, Russell JR, Paulsson J, Losick R. Use of a microfluidic platform to uncover basic features of energy and environmental stress responses in individual cells of Bacillus subtilis. PLoS Genet. 2017;13(7):e1006901. doi: 10.1371/journal.pgen.1006901. [DOI] [PMC free article] [PubMed] [Google Scholar]