

Abstract
Microsatellite instability (MSI), a well-established driver pathway in colorectal carcinogenesis, can develop in both sporadic and hereditary conditions via different molecular alterations in the DNA mismatch repair (MMR) genes. MMR protein immunohistochemistry (IHC) is currently widely used for the detection of MMR deficiency in solid tumors. The IHC test, however, can show varied staining patterns, posing challenges in the interpretation of the staining results in some cases. Here we report a case of an 80-year-old female with a colonic adenocarcinoma that exhibited an unusual “null” IHC staining pattern with complete loss of all 4 MMR proteins (MLH1, MSH2, MSH6, and PMS2). This led to subsequent MLH1 methylation testing and next generation sequencing which demonstrated that the loss of all MMR proteins was associated with concurrent promoter hypermethylation of MLH1 and double somatic truncating mutations in MSH2. These molecular findings, in conjunction with the patient's age being 80 years and the fact that the patient had no personal or family cancer history, indicated that the MMR deficiency was highly likely sporadic in nature. Thus, the stringent Lynch syndrome type surveillance programs were not recommended to the patient and her family members. This case illustrates a rare but important scenario where a null IHC phenotype signifies complex underlying molecular alternations that bear clinical management implications, highlighting the need for recognition and awareness of such unusual IHC staining patterns.
Keywords: Lynch syndrome, microsatellite instability, MMR immunohistochemistry, MLH1 methylation, double somatic mutation, next generation sequencing
Introduction
Aberrant function of mismatch repair (MMR) genes leads to the microsatellite instability (MSI) phenotype, which is a well-established mechanism in the development of several different types of cancers, most notably colorectal and endometrial adenocarcinomas. Germline defects in the major MMR genes – MLH1, MSH2, MSH6, and PMS2 – are associated with Lynch syndrome (LS), predisposing patients to colorectal, endometrial, ovarian, urothelial, upper gastrointestinal, and pancreatic carcinomas.1 Although approximately 15-20% of colorectal cancers show aberrant MMR protein expression or MSI high (MSI-H) phenotype, only about 3% of patients have germline mutations diagnostic of LS;1–3 thus, the majority of MSI-H colorectal cancers (CRCs) are sporadic in nature. In these sporadic cases, the dominant underlying molecular mechanism is MLH1 promoter hypermethylation.1 In recent years, somatic biallelic MMR gene mutations/alterations have emerged as another mechanism underpinning MSI.3,4
The MMR proteins are routinely assessed by pathologists through the use of immunohistochemistry (IHC) as an initial test for detecting MMR deficiency.5 The MMR proteins function as dimers of MLH1/PMS2 (MutLα) and MSH2/MSH6 (MutSα) wherein the stability of PMS2 and MSH6 requires an intact MLH1 and MSH2, respectively, but the stability of MLH1 and MSH2 can be maintained even when PMS2 or MSH6 is lost due to the fact that the function of PMS2 or MSH6 can be compensated for by other MMR proteins. Consequently, specific patterns of protein loss as detectable by IHC can lead to accurate prediction of the affected MMR gene. For example, concurrent loss of MLH1 and PMS2 on IHC would indicate abnormality in MLH1 whereas isolated loss of PMS2 would suggest abnormality in PMS2.
IHC staining patterns, however, can vary, reflecting the complex biology underlying MMR deficiency; a heightened awareness of unusual IHC findings and their biological implications is thus crucial in ensuring the most effective use of this tool in the work-up of cases with MMR deficiency. Well documented unusual IHC patterns include 1) no MLH1 loss in the presence of a pathogenic MLH1 mutation, presumably reflecting retained antigenicity of the mutant MLH1 protein 6,7; 2) concurrent loss of MLH1/PMS2/MSH6 whereby defects in MLH1/PMS2 trigger a loss of MSH6 due to secondary instability of a coding microsatellite in MSH6 8; and 3) chemoradiation associated downregulation of MSH6 expression which results in near complete or complete MSH6 loss in treated tumors.2,5 Concurrent loss of all 4 MMR proteins represents yet another unusual IHC pattern that, to our knowledge, has only been reported once, in a LS patient.2 Here, we add a novel case of MMR protein ‘null’ colorectal carcinoma that is associated with sporadic aberrations in both MLH1 and MSH2.
Case Report
Our case is that of an 80-year-old woman who was noted to have colonic thickening on a surveillance CT for an adrenal cortical adenoma. Her other medical history was non-contributory. She had no family history of colon cancer nor of Lynch syndrome. Colonoscopy revealed a large mass in the ascending colon, and a right hemicolectomy ensued which revealed a 9.4cm pT3N2b adenocarcinoma.
Routine H&E sections showed that the tumor was poorly differentiated, exhibiting a solid growth pattern with only minimal gland formation. By IHC, the tumor cells were positive for low molecular weight cytokeratin (clone CAM 5.2, Becton & Dickinson). IHC for MMR proteins was carried out as previously described with monoclonal antibodies to MLH1 (clone M1, Ventana), MSH2 (clone: G219-1129, Cell Marque, Rocklin CA, USA), MSH6 (clone 44, Ventana), and PMS2 (clone A16.4, BD Biosciences).8 Surprisingly, this revealed a null phenotype with loss of all four proteins (Figure 1). Internal controls (stromal cells) were positive, indicating the staining pattern was valid.
Figure 1.
H&E section of the colonic carcinoma showing that the tumor is poorly differentiated with a solid growth pattern (A). By immunohistochemistry, the tumor cells are positive for cytokeratin CAM5.2 (B); they show loss of staining for MLH1 (C), PMS2 (D), MSH2 (E), and MSH6 (F) with appropriate positive internal control for each of the 4 stains (all figures at 200× magnification; scale bar, 500 μm).
To confirm the MMR status and to explore the genomic mutational profile, we sequenced matched tumor-normal tissue using a custom hybridization capture-based next-generation sequencing (NGS) assay, MSK-IMPACT, to identify somatic mutations.9 Polymorphisms which are present in both the blood and tumor DNA concurrently are filtered out and not reported. In addition to detection of somatic mutations in a large panel of cancer-associated genes (410 genes using MSK-IMPACT v5), MSK-IMPACT also allows the detection of the microsatellite instability (MSI) status by applying the MSIsensor program and evaluating the length variations of the microsatellite regions attainable from the aligned sequencing data.10
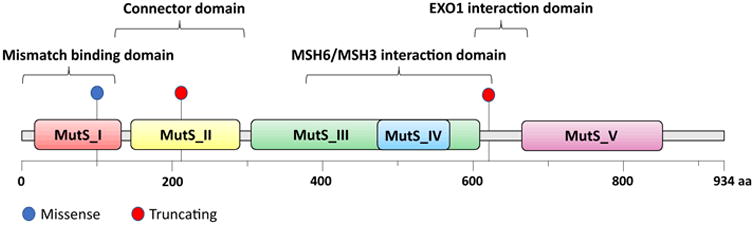
Indeed, our patient's tumor (50% tumor purity) showed a markedly elevated MSIsensor score (29.17) and was therefore deemed to be MSI-high. Not surprisingly, the tumor demonstrated a high number of mutations, specifically, 58, including a BRAF V600E mutation and 3 distinct somatic mutations of MSH2 (Figure 2). One of these was MSH2 c.1861 C>T (p.R621*) in exon 12, with a variant allele frequency (VAF) of 0.24. This is a known pathogenic truncating mutation in exon 12 found in LS families.11 The two other mutations, c.298G>A (p.V100I) (VAF 0.06) and c.633dupG(p.K212Efs*20) (VAF 0.18), did not appear in common publicly available databases (ClinVar, Clinvitae, COSMIC, MutDB, UMD Colon, InSIGHT colon cancer, MMR DB); however, the latter is a frameshift mutation that leads to a premature stop codon in exon 3. Given that MSH2 has 16 exons, this aberration is very likely pathogenic since it truncates the vast majority of the protein. None of these mutations are present in the blood DNA and thus all are somatic in nature. The complete loss of MSH2 staining on IHC suggested that these mutations represented biallelic aberrations.
Figure 2.
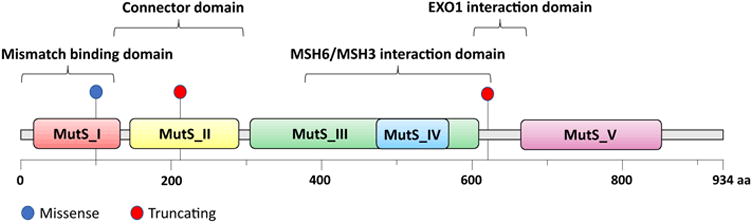
Schematic of the MSH2 protein domains and the location of the three MSH2 mutations found in our patient's tumour.15
To determine whether the IHC loss of MLH1 and PMS2 might be related to MLH1-promoter methylation, the tumor was tested for 5 CpG sites within the MLH1 promoter (in region -209 to -181 from the transcription start site). The PCR product was subjected to pyrosequencing on a Pyromark Q24 pyrosequencer. The degree of methylation for each site was calculated as follows: methylation % = peak height of methylated (C)/(peak height of methylated [C] + peak height nonmethylated [T]) × 100. The MLH1 hypermethylation levels were considered as present if all 5 CpG sites are methylated at >=10%. All 5 sites were methylated above 10% in our patient's tumor, thus confirming the presence of methylation.
The patient had normal recovery from the surgery and was discharged home in stable condition. However, a follow-up CT scan, performed 3 months after surgery, revealed new hepatic metastasis. The patient chose not to receive further treatment. The patient also denied germline mutation testing; nonetheless, given the molecular results above, the MMR deficiency was regarded as highly likely sporadic in nature, and no high risk surveillance protocols were recommended to the patient or her family members.
Discussion
In this report, we illustrate an unusual MMR IHC staining pattern whereby all 4 MMR proteins are lost in a colonic carcinoma. Our case is the first instance of a null IHC phenotype that is the result of co-existing sporadic MLH1 promoter methylation and MSH2 somatic mutation, the latter representing a newly emerged mechanism for MSI-H in CRC.3
In our case, the sporadic nature of MLH1 loss is well supported by the demonstration of both MLH1 promoter methylation and BRAF V600E mutation in the tumor. We speculate that the loss of MSH2 is also sporadic secondary to the somatic mutations detected in MSH2 gene. Of the 3 mutations detected, two are known or highly likely to be deleterious. Since the sequencing platform we used (MSK-IMPACT) does not report alterations of the matched germline controls, these mutations are only present in the tumor and thus are somatic. Although we do not have direct evidence that the mutations are in trans, their deleterious nature and relative positioning in the gene coupled with the complete loss of MSH2 on IHC, suggests that they affected both MSH2 alleles. This argument is strengthened by the lack of clinical evidence to suggest the alternative possibility of a hereditary condition. In fact, the patient's clinical profile, particularly the age of cancer diagnosis being 80 years and the lack of personal or family cancer history, strongly favors a sporadic condition, and serves as evidence (albeit indirect) to support that the loss of MSH2 (as well as MSH6) is most likely a consequence of the somatic mutations in the MSH2 gene. Unfortunately, at this point, we are not able to ascertain the patient's germline data to further corroborate this argument.
Double somatic hits as an etiology for MSI-H was first reported by Sourrouille et al in 2013 and confirmed by subsequent studies.3,4 In some series, this mechanism explains the majority of previously unexplained MSI-H CRCs, i.e. CRCs that are MSI-H but with no detectable germline mutation and no MLH1 promoter methylation. The term “Lynch-like syndrome” has been used for these unexplained MSI-H cancers. This term, however, is not accurate as the condition is neither a distinct syndrome nor one that warrants automatic “Lynch-like” management. Notably, determination of double somatic hits as etiology of MSI-H requires careful considerations as illustrated by our case. In addition to the need to confirm the pathogenicity of the specific somatic mutations/alterations, it is also important to exclude the possibility of a hereditary condition before accepting the case as sporadic in nature. To this effect, it is important to bear in mind that certain LS type and non-LS type hereditary conditions can cause somatic inactivation of the MMR genes and result in MSI-H CRC. A notable example of LS type hereditary condition is via somatic mosaicism (whereby the causative somatic and germinal mosaicism in an MMR gene is not easily detectable in the germline DNA from the blood cells and yet can be transmitted to the next generation).3 Well documented examples of non-LS type hereditary conditions include germline biallelic MUTYH mutations (MUTYH-associated polyposis) and POLE germline variants.12,13 Thus, sufficient clinical and/or molecular evidence is needed before a definitive conclusion can be drawn. In the event that no alternative etiology is detected, the NCCN recommends that individuals with double somatic mutations/changes in the MMR genes be regarded as likely not having LS, and management should be based on personal/family history.14 In our patient, given the late age at diagnosis and the lack of personal and family cancer history, intensive “Lynch-like” follow up and family surveillance were not recommended.
In summary, our case illustrates an unusual MMR IHC null-phenotype associated with concurrent somatic inactivation of MLH1 and MSH2 in a colonic carcinoma, MLH1 inactivated via promoter hypermethylation and MSH2 loss of function associated with somatic mutations. Our case not only serves to increase awareness of unusual MMR IHC staining patterns in CRC, it also highlights the importance of MMR gene somatic alterations in the development of MSI in these tumors, and the versatility of NGS platforms in detecting both MSI and MMR gene mutation.
References
- 1.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagen CE, Lefferts J, Hornick JL, Srivastava A. “Null pattern” of immunoreactivity in a Lynch syndrome-associated colon cancer due to germline MSH2 mutation and somatic MLH1 hypermethylation. Am J Surg Pathol. 2011;35:1902–5. doi: 10.1097/PAS.0b013e318237c6ab. [DOI] [PubMed] [Google Scholar]
- 3.Sourrouille I, Coulet F, Lefevre JH, Colas C, Eyries M, Svrcek M, et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. 2013;12:27–33. doi: 10.1007/s10689-012-9568-9. [DOI] [PubMed] [Google Scholar]
- 4.Mensenkamp AR, Vogelaar IP, van Zelst-Stams WAG, Goossens M, Ouchene H, Hendriks-Cornelissen SJB, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146:643–646.e8. doi: 10.1053/j.gastro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Shia J. Evolving approach and clinical significance of detecting DNA mismatch repair deficiency in colorectal carcinoma. Semin Diagn Pathol. 2015;32:352–61. doi: 10.1053/j.semdp.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn JMD. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Airaud F, Küry S, Valo I, Maury I, Bonneau D, Ingster O, et al. A de novo germline MLH1 mutation in a Lynch syndrome patient with discordant immunohistochemical and molecular biology test results. World J Gastroenterol. 2012;18:5635–9. doi: 10.3748/wjg.v18.i39.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shia J, Zhang L, Shike M, Guo M, Stadler Z, Xiong X, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol Off J U S Can Acad Pathol Inc. 2013;26:131–8. doi: 10.1038/modpathol.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn JMD. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinforma Oxf Engl. 2014;30:1015–6. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dymerska D, Serrano-Fernández P, Suchy J, Pławski A, Słomski R, Kaklewski K, et al. Combined iPLEX and TaqMan assays to screen for 45 common mutations in Lynch syndrome and FAP patients. J Mol Diagn JMD. 2010;12:82–90. doi: 10.2353/jmoldx.2010.090063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet EJHG. 2014;22:1334–7. doi: 10.1038/ejhg.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elsayed FA, Kets CM, Ruano D, van den Akker B, Mensenkamp AR, Schrumpf M, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet EJHG. 2015;23:1080–4. doi: 10.1038/ejhg.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provenzale D, Gupta S, Ahnen DJ, Bray T, Cannon JA, Cooper G, et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw JNCCN. 2016;14:1010–30. doi: 10.6004/jnccn.2016.0108. [DOI] [PubMed] [Google Scholar]
- 15.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]