

Abstract
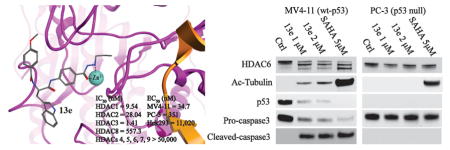
Previously, we designed and synthesized a series of o-aminobenzamide-based histone deacetylase (HDAC) inhibitors, among which the representative compound 11a exhibited potent inhibitory activity against class I HDACs. In this study, we report the development of more potent hydrazide-based class I selective HDAC inhibitors using 11a as a lead. Representative compound 13b showed a mixed, slow, and tight binding inhibition mechanism for HDAC1, 2, and 3. The most potent compound 13e exhibited low nanomolar IC50s toward HDAC1, 2, and 3 and could down-regulate HDAC6 in acute myeloid leukemia MV4-11 cells. The EC50 of 13e against MV4-11 cells was 34.7 nM, which is 26 times lower than its parent compound 11a. In vitro responses to 13e vary significantly and interestingly based on cell type: in p53 wild-type MV4-11 cells, 13e induced cell death via apoptosis and G1/S cell cycle arrest, which is likely mediated by a p53-dependent pathway, while in p53-null PC-3 cells, 13e caused G2/M arrest and inhibited cell proliferation without inducing caspase-3-dependent apoptosis.
Graphical Abstract
INTRODUCTION
Targeting histone modifying histone deacetylases (HDACs) to restore the expression of tumor suppressor genes has shown clinical benefits especially for the treatment of cancer.1 HDACs together with histone acetyltransferases (HATs) form two families of enzymes with reversible actions for modifying the acetylation status of chromosomal histones and nonhistone proteins. HDACs remove acetyl groups from the lysine residues, resulting in a “closed” configuration of chromatin blocking the access of the transcription machinery to DNA, and finally suppresses gene expression. HATs, however, perform antagonist actions that trigger an “open” state of chromatin and promote gene expression.2,3 Remarkably, a growing number of nonhistone proteins are also described as targets of HDACs and HATs, which include transcription factors, DNA binding nuclear receptors, signal mediators, transcriptional coregulators, and cytoskeletal proteins.4–7 Acetylation of nonhistone targets impacts protein stability, protein cellular localization, and protein–protein/protein–nucleotide interactions, which can finally influence cell proliferation, survival, and apoptosis.8,9 The tumor suppressor p53 was the first reported nonhistone target of HDACs and HATs.10 Acetylation of p53 activates its sequence-specific DNA binding activity and consequently increases activation of its target genes.11 Furthermore, acetylated p53 induces cell apoptosis and basic autophagy by transcriptionally upregulating tuberous sclerosis 2 (TSC2), AMP-activated protein kinase (AMPK), and damage-regulated autophagy modulator (DRAM), thereby suppressing the mammalian target of rapamycin (mTOR) and the unc-51-like autophagy activating kinase 1 (ULK1) complex further downstream in MES-SA cells.12 p53-family members p63 and p73, which can compensate for p53, could also mediate apoptosis.13,14
To date, a total of 18 HDAC isoforms have been identified in humans, 11 of which contain a zinc-binding site; these can be divided into classes I, II, and IV, while class III HDACs (SIRT1–7) require NAD+ for their activity.15 Class I HDACs, including HDAC 1, 2, 3, and 8, are homologous to yeast Rpd3 (reduced potassium dependency-3) protein. Class II comprises HDAC 4, 5, 6, 7, 9, and 10 and is structurally related to yeast Hda1 (histone deacetylases 1). Class III possesses only one member, HDAC11.16
So far, four HDAC inhibitors (HDACIs) have been approved by the FDA: vorinostat (SAHA),17 romidepsin (FK228),18 belinostat (PXD-101),19 and panobinostat (LBH589)20 for the treatment of cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), or multiple myeloma (MM) (Figure 1). Understanding the structural regions of HDACIs is essential to design potent and selective inhibitors. Pharmacophore models of most HDACIs contain three structural parts: a cap group, a linker, and a zinc-binding group (ZBG). The cap group interacts with the surface of the enzyme; the linker occupies the long hydrophobic tunnel leading to the zinc site; and the ZBG functions in the bottom catalytic site.21,22 The classification of HDACIs primarily depends on their ZBG chemical structures: hydroxamates, benzamides, aliphatic acids, electrophilic ketones, and so on. To some extent, modification of the ZBG can introduce a change in potency and in the selectivity profile.23–25 For example, most hydroxamates are pan-HDACIs, while the benzamides have increased class I selectivity. Although hydroxamic acid is the most commonly used ZBG, it suffers from susceptibility to hydrolysis leading to inactive carboxylic acid as well as glucuronidation-based inactivation.26 This structural instability contributes the main reason for poor pharmacokinetic profiles (e.g., short t1/2 and low Cmax) of clinical hydroxamate HDACIs.27,28
Figure 1.

FDA-approved HDACIs.
Selective HDACIs have attracted increased attention as they are hypothesized to elicit fewer side effects than the pan-HDACIs. Class I HDACs are the most relevant with regards to the initiation and development of cancer among all the Zn2+-dependent HDACs;29 therefore, discovery of potent class I selective HDACIs is of great interest for the potential treatment of cancer. Currently, most class I HDACIs, especially those in clinical research, are benzamide HDACIs bearing o-aminobenzamide as the ZBG. Additionally, modifications to the o-aminobenzamide moiety led to alterations in inhibitor activity. Recent studies demonstrated that aryl substituents on the o-aminobenzamide of benzamide HDACIs led to dramatic improvements in potency and selectivity for HDAC1 and 21,30,31 or for HDAC3.32,33
In our previous study, we designed and synthesized a series of HDAC class I selective benzamide HDACIs bearing a tryptophan cap group. The most potent compound 11a, (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)-methyl)-N-(2-aminophenyl)benzamide, exhibited potent inhibitory activity with IC50 against HDAC1, 2, and 3 of 20.6, 157, and 138 nM, respectively.34 However, like the ZBG, o-aminobenzamide reaches its limit in its inhibitory potency and is susceptible to glucuronidation-based inactivation.26 In 2015, Wang et al. reported new HDAC class I selective inhibitors with a benzoylhydrazide scaffold.35 Our group also generated a series of class I HDAC selective inhibitors that incorporated a hydrazide as the ZBG, as shown for representative compound 3b (Figure 2). Furthermore, we demonstrated that the hydrazide motif displayed allosteric inhibition kinetics and was impervious to glucuronidation.26 As part of our current study, we modified our lead compound 11a by changing the linker and introducing the hydrazide motif as ZBG (Figure 2). Herein, we discuss the structure–activity relationships (SARs), isotype selectivity, enzymatic inhibition kinetics, and p53 status-dependency mechanism for this new series of compounds.
Figure 2.

Modification of the lead compound 11a.
CHEMISTRY
Scheme 1 shows the synthesis of intermediates 4a and 4b. Methyl protection of 1a and 1b afforded 2a and 2b, respectively, which underwent amino acid condensation with compound 3 to achieve 4a and 4b.
Scheme 1. Synthesis of Intermediates 4a and 4ba.
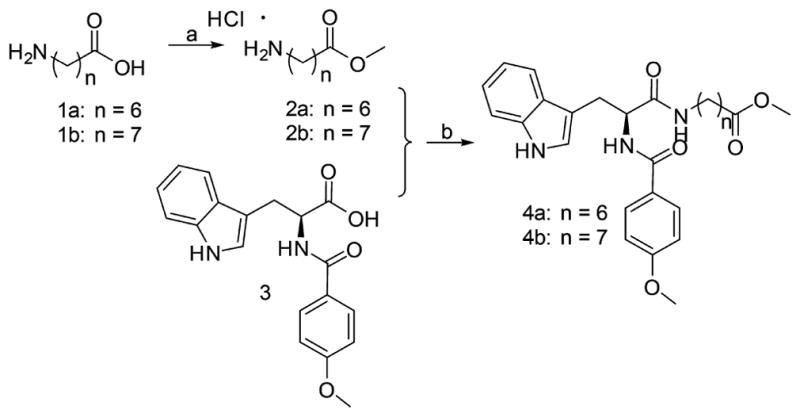
aReagents and conditions: (a) CH3COCl, CH3OH, reflux, yield 95%; (b) TBTU, TEA, DCM, yield 55%.
Compounds 4a, 4b, 5, 6, and 7 were treated with hydrazine monohydrate to achieve hydrazides 8, 9, 10, 11, and 12, respectively. The hydrazides were reacted with different aliphatic aldehydes, then reduced by NaBH3CN to give the end products, 13a–13h (Scheme 2). Intermediate 14 was reacted with cyclopentylhydrazine or phenylhydrazine by a TBTU-mediated amide formation to afford 15a and 15b, respectively (Scheme 3). Compound 16 (Scheme 4) was subjected to a similar reaction in order to produce 17, which was treated with propionaldehyde and then NaBH3CN to obtain end product 18.
Scheme 2. Synthesis of Target Compounds 13a–13ha.

aReagents and conditions: (a) hydrazine monohydrate, CH3OH, reflux, yield 95%; (b) different aliphatic aldehyde, MgSO4, CH3CH2OH, yield 98%; (c) NaBH3CN, CH3OH, concentrated hydrochloric acid, 60%.
Scheme 3. Synthesis of Target Compound 15a.

aReagents and conditions: (a) cyclopentylhydrazine, benzylhydrazine, or pyrazolidine, TBTU, TEA, DCM, yield 50%.
Scheme 4. Synthesis of Target Compound 18a.

aReagents and conditions: (a) hydrazine monohydrate, TBTU, TEA, DCM, yield 75%; (b) propionaldehyde, CH3CH2OH, MgSO4, yield 90%; (c) NaBH3CN, CH3OH, concentrated hydrochloric acid.
RESULTS AND DISCUSSION
In our previous study, we designed and synthesized a series of o-aninobenzamide-based HDACIs, among which the representative compound 11a exhibited potent inhibitory activity against class I HDACs (IC50: HDAC1, 20.6 nM; HDAC2, 157 nM; HDAC3, 138 nM).34 Although the o-aminobenzamide motif is the most commonly used ZBG for selective class I HDACIs, it features several drawbacks: lower inhibitory activity, potential for heptatotoxicty due to the potential imine metabolites, and glucuronidation-based inactivation. To explore new HDACI motifs, Wang and colleagues demonstrated a novel hydrazide-based class I HDACI, based on which our lab generated a more potent series of class I HDAC selective inhibitors with hydrazide as a potential ZBG.26,35 We have shown that hydrazide compounds are not glucuronidated,26 and a preliminary pharmacokinetics study in mice showed the Cmax and AUC of our hydrazide compound LP-411 is at least 200 times higher than all FDA-approved HDACIs. Further, the t1/2 is much longer than approved hydroxamate HDACIs (data not shown), which indicates hydrazide motifs have better in vivo stability. The side effect of hydrazide-based HDACIs is still unknown because they have not been tested in clinic. However, compounds bearing hydrazine/hydrazide motif are used clinically as antidepressants and antituberculosis agents for decades with great safety profiles.36 Potential toxicity of the most recognizable hydrazide-containing agent isoniazid is hepatotoxicity, but the rate was as low as 0.5–2%,37 and rarely was the hepatotoxicity lethal (1/25000).38 Due to the improved pharmacokinetic profile and low toxicity of hydrazide, we optimize benzamide 11a by keeping its cap and linker unchanged and introducing different hydrazide motifs to achieve compounds 13a–13d, 15a, and 15b (Table 1). More specifically, 13a–13d possess linear alkyl chains two to five carbons in length on the hydrazine motif, while 15a and 15b bear cyclic hydrazide substituents. HDAC1, 2, 3, and 6 enzymatic inhibition experiments and antiproliferative assays based on the acute myeloid leukemia MV4-11 cells were conducted to efficiently screen the newly designed compounds (Table 1). Compounds 13a–13d with linear alkyl motifs on hydrazide exhibited nanomolar IC50s toward HDAC1, 2, and 3. Further, unlike the parent compound 11a, 13b–13d exhibited a degree of HDAC3 selective inhibition. Among the inhibitors tested, 13b bearing a propyl hydrazide substituent exhibited the greatest selectivity toward HDAC1, 2, and 3: 13b’s IC50 against HDAC3 was 5.63 nM, ~51-fold and ~11-fold lower than against HDAC2 and HDAC1, respectively. However, compound 15a with cyclopentyl and 15b with benzyl motifs showed no inhibition against HDAC1, 2, or 3 up to 5000 nM. The compounds did not inhibit HDAC6 with the exception of 13a, which has an ethyl hydrazide moiety. All of the hydrazide compounds displayed submicromolar EC50s against MV4-11 cells. The EC50 of the most potent compound (13b) was 138.7 nM, 6.6 times more potent than parent benzamide 11a.
Table 1.
HDAC1, 2, 3, and 6 Inhibitory Activity and in Vitro Antiproliferative Activity of 13a–13d, 15a, 15b, and 11aa
| ||||||
|---|---|---|---|---|---|---|
| Compounds No. | R | IC50 (nM)
|
EC50 (nM, MV4-11 cells) | |||
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | |||
| 13a |
|
95.46±5.78 | 88.77±10.37 | 34.90±6.08 | 392.7±29.25 | 658.2±78.3 |
| 13b |
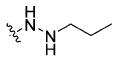
|
63.28 ±6.36 | 287.1±47.31 | 8.50±1.80 | >100000 | 138.7±15.5 |
| 13c |
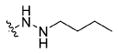
|
53.68 ±8.17 | 193.5±10.23 | 16.54±1.28 | >100000 | 642.1±93.2 |
| 13d |
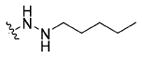
|
29.78 ±1.79 | 332.1±21.07 | 10.87±1.02 | >100000 | 557.8±48.3 |
| 15a |
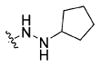
|
>5000 | >5000 | >5000 | >100000 | >20000 |
| 15b |
|
>5000 | >5000 | >5000 | >100000 | >20000 |
| 11a |
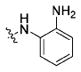
|
20.6±3.8 | 157.1±19.9 | 138.4±10.4 | >100000 | 911.8±100.7 |
IC50 and EC50 values are the mean of at least three experiments ± the standard error of the mean.
In order to explore the inhibition mechanism of hydrazide versus benzamide HDAC inhibitors, we conducted in vitro Vmax studies for hydrazide compound 13b and its parent benzamides 11a using recombinant HDACs 1, 2, and 3. Lineweaver–Burke analysis showed that 13b clearly demonstrates a convergence in the second quadrant for HDAC1, 2 and 3, indicative of mixed inhibition (Figure 3A–C). As indicated in our previous work, heterodimer HDACs appear to contain an allosteric binding pocket created by the interface of the two monomers in the heterodimer (Figure 4).26 Therefore, one way to interpret the results shown in Figure 3A–C is that, in addition to being able to bind to the same site as the enzymatic substrate (competitive), 13b may have an alternate binding site (i.e., a noncompetitive allosteric site). Parent inhibitor 11a displayed entirely different binding kinetics against HDAC1, HDAC2, and HDAC3. For HDAC1, convergence of Lineweaver–Burke plots for varied inhibitor concentrations directly on the y-axis indicates competitive inhibition (Figure 3D); however, the compound displayed mixed-type inhibition toward HDAC2 and HDAC3.
Figure 3.

Lineweaver–Burke plots of enzyme kinetics data in the presence of inhibitors. Y-axes units: (pmoles acetylated substrate cleaved/min)−1. X-axes units: (μmoles) −1. (A–C) Compound 13b for HDAC1, 2, and 3 respectively. Intersection in 2nd quadrant indicative of mixed inhibition. (D–F) Compound 11a for HDAC1, 2, and 3, respectively. Intersections on y-axis and in 2nd quadrant are indicative of mixed and competitive inhibition. Representative plots of n ≥ 3 experiments.
Figure 4.
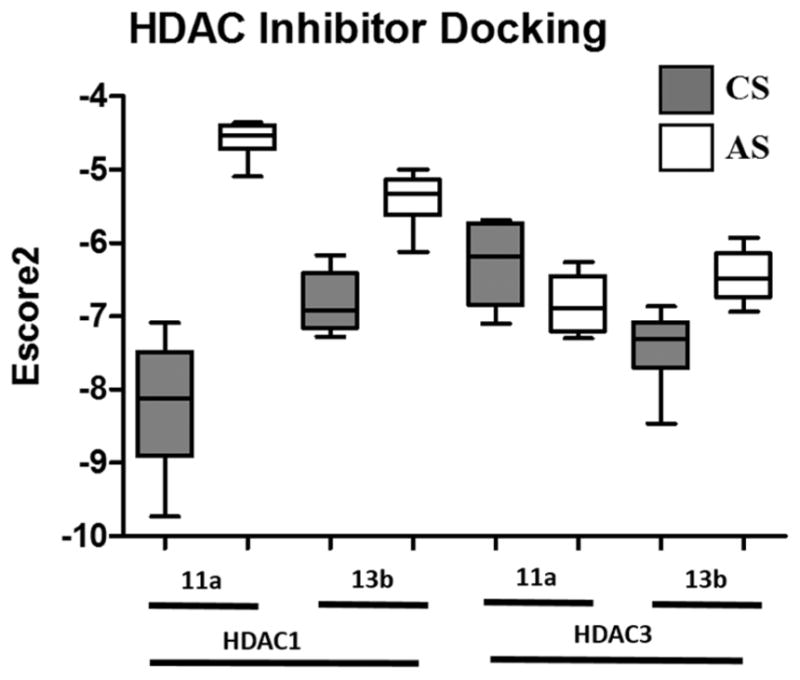
Molecular docking probe of the catalytic and allosteric binding pockets of HDAC1 (PDB: 5ICN) and HDAC3 (PDB: 4A69). Data is represented as a box and whisker plot using the top 10 docking poses for each condition. The CS was defined as the catalytic pocket that incorporated the Zn metal, and the AS was defined as the allosteric pocket created by the interface between the HDAC heterodimer.
To better understand the potential binding modes and patterns between the two protein heterodimers, we tested 11a and 13b in four conditions each by probing the competitive site (CS) and the allosteric site (AS) of HDAC1 and HDAC3 using computational docking. These results were particularly useful in determining how much variability was in potential binding modes for each of these eight total conditions (Figure 4), and the potential interaction maps of 11a and 13b with both sites of HDAC1 and HDAC3 (Supporting Information Figures S2 and S3). The overall sizes of CS and AS for each isoform were reasonably similar (Figure 5). As shown using scoring in the docking simulations (Figure 4), there was a very large discrimination between the high affinity CS for 11a in HDAC1 and the very low affinity AS for 11a in HDAC1. The interaction of 11a with the HDAC1 CS also indicated it could form hydrogen bonds with Cys100, Tyr303, His140, and Gly149 as well as a π–π interaction with Phe150, while interaction of 11a with HDAC1 AS was weaker and appeared to only partially bury the indole moiety (Supporting Information Figure S2). Therefore, 11a has a higher preference with HDAC1 CS, which might be the reason 11a exhibited competitive inhibition against HDAC1. For 13b interaction with HDAC1, we see a modest preference for the CS over the AS (Figure 4); furthermore, interaction of 13b with HDAC1 CS and AS also showed similar hydrogen bonding (Supporting Information Figure S3). Compounds 11a and 13b binding to HDAC3 appeared to be almost equal in preference for the two sites in the protein (Figure 4). More possible interactions were found with the HDAC3 AS than HDAC1 AS (Supporting Information Figures S2 and S3), which is in agreement with the kinetics data where both of these compounds displayed mixed inhibition toward HDAC3.
Figure 5.
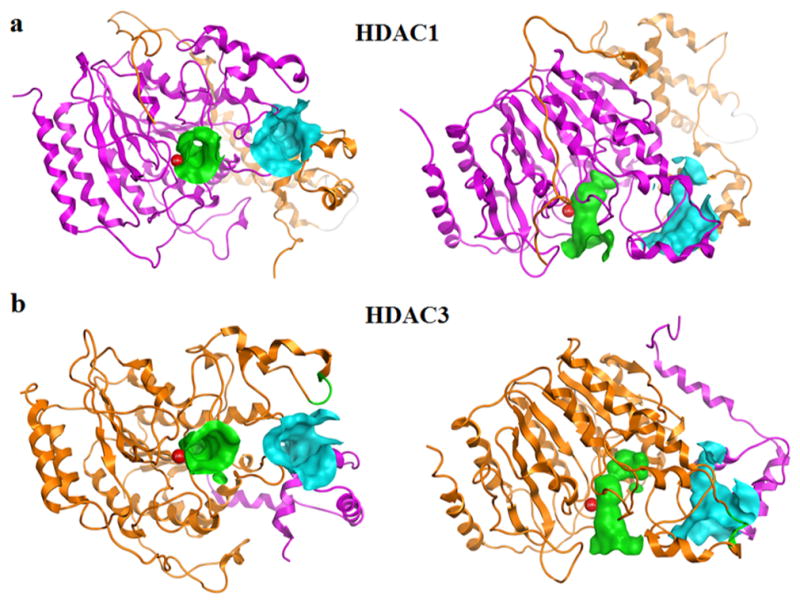
Molecular imaging and binding pocket identification for HDAC1 and HDAC3 heterodimers. HDAC1 (PDB: 5ICN) and HDAC3 (PDB: 4A69) are depicted as ribbon diagrams with each chain of the heterodimer a unique color. Zn is shown as a red space filling sphere. The catalytic site (CS) and allosteric site (AS) are depicted as surfaces using the ActiveLP color scheme with green and blue, respectively.
To further examine whether the binding of inhibitor to the enzymes was time dependent, we determined IC50 curves of 13b and 11a against HDAC1, 2, and 3 for several preincubation times (15, 30, 60, and 90 min; Figure 6). Dose–response curves and calculated IC50 values for 13b and 11a all varied with enzyme–inhibitor preincubation time. For example, after a 90 min preincubation, the IC50 of 13b for HDAC3 was 5.7 nM, a 3-fold decrease compared to the IC50 measured after 15 min preincubation; additionally, the IC50 did not reach a steady-state value for 90 min (Figure 6C). Similar results were also observed for 13b with HDAC1 and 2 and for 11a with HDAC1, 2, and 3 (Figure 6). These observations suggest that 13b and 11a are both slow, tight-binding inhibitors of class I HDACs.
Figure 6.

IC50 curves of 13b and 11a for HDAC1, 2, and 3. The red, blue, yellow, and green showed inhibitor incubated with enzyme for 15, 30, 60, and 90 min, respectively. Compounds 13b and 11a both showed time-dependent inhibition toward HDAC1, 2, and 3. Figures are representative of n ≥ 2 experiments.
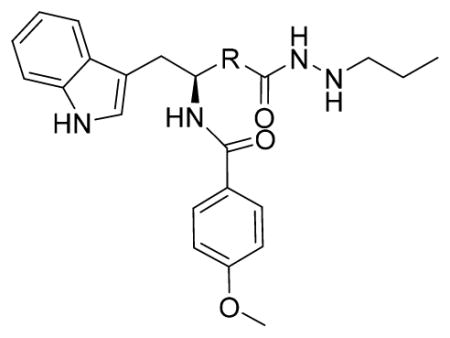
After optimizing the ZBG, we explored the effect of varying the linker on compounds’ activity and selectivity. For this purpose, we kept the cap group and ZBG of the most selective, potent compound (13b, Table 1) unchanged and introduced several different linker groups to obtain compounds 13e–13h and 18 (Table 2). The modified compounds maintained 13b’s selectivity pattern in subclass I HDACs and showed no inhibition against HDAC6 up to 100 μM. Surprisingly, 13e, with a linker simply truncated by a single methylene spacer compared to 13b, exhibited much higher activity against HDAC1, 2 and 3, and higher in vitro activity against MV4-11 cells as well. IC50s of 13e against HDAC1, 2, and 3 were 9.54, 28.04, and 1.41 nM, which were 6.6-fold, 10.2-fold, and 6.1-fold lower than those for 13b, respectively. What’s more, the antiproliferative activity of 13e (EC50: 34.7 nM) in MV4-11 cells was 4.1 times higher than 13b (EC50: 138.7 nM).
Table 2.
HDAC1, 2, 3, and 6 Inhibitory Activity and in Vitro Antiproliferative Activity of 13e–13h and 18a
| ||||||
|---|---|---|---|---|---|---|
| Compounds No. | R | IC50 (nM) | EC50 (nM, MV4-11 cells) | |||
|
| ||||||
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | |||
| 13b |
|
63.28±10.24 | 287.1±38.2 | 8.50±0.89 | >100000 | 138.7±23.5 |
| 13e |
|
9.54±1.34 | 28.04±4.12 | 1.41±0.57 | >100000 | 34.7±4.2 |
| 13f |
|
80.19±10.29 | 209.0±30.5 | 12.44±15.26 | >100000 | 1254±101 |
| 13g |
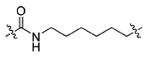
|
91.59±15.42 | 191.3±25.3 | 5.205±1.44 | >100000 | 652.7±89.3 |
| 13h |
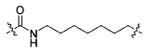
|
52.77±3.80 | 199.3±10.4 | 12.09±3.21 | >100000 | 1687±239 |
| 18 |
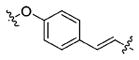
|
28.88±2.78 | 283.3±43.6 | 2.816±0.76 | >100000 | 243.0±48.2 |
IC50 and EC50 values are the mean of at least three experiments ± the standard error of the mean.
As 13e is the most potent compound, we then tested the IC50 of 13e for the other Zn2+-dependent HDACs (HDAC4, 5, 7, 8, and 9) in order to assess its subtype selectivity profile. Inhibitory activity toward HDAC10 and 11 could not be determined because there are no appropriate substrates.39 As expected, 13e did not show inhibition against any Class IIa HDACs (HDAC4, 5, 7, and 9) up to 50 μM (Table 3 and Supporting Information Figure S4). IC50 for 13e against Class I member HDAC8 was 626.12 nM, which was about 58.4-fold, 20.0-fold, and 395.2-fold higher than HDAC1, HDAC2, and HDAC3, respectively (Table 3 and Supporting Information Figure S5). Therefore, 13e is a HDAC1, 2, and 3 selective inhibitor, or HDAC class I selective inhibitor if we take the modest HDAC8 inhibition into consideration. To examine the specificity of 13e, we further tested the inhibitory activity of 13e against two different Zn2+ metalloenzymes, MMP-2/MMP-9 and APN/CD13, that have been suggested to play a role in cancer survival.40,41 Results indicated that 13e did not inhibit the two enzymes at concentrations up to 50 μM (Supporting Information Figures S6 and S7), suggesting that hydrazide HDAC inhibitors’ anticancer activity was not due to nonspecific metalloprotease inhibition. We also predicted a binding mode of 13e with CS and AS of HDAC1 and HDAC3. The hydrazide functional group could form a bidentate interaction with Zn2+, which is similar to hydroxamic acid in the CS (Supporting Information Figure S8).
Table 3.
Compound 13e Inhibitory Activity for HDAC1-11
| HDAC classes | subtype | IC50 (nM)a |
|---|---|---|
| class I | HDAC1 | 9.54 ± 1.34 |
| HDAC2 | 28.04 ± 4.12 | |
| HDAC3 | 1.41 ± 0.57 | |
| HDAC8 | 557.3 ± 97.58 | |
| class IIa | HDAC4 | >50000 |
| HDAC5 | >50000 | |
| HDAC7 | >50000 | |
| HDAC9 | >50000 | |
| class IIb | HDAC6 | >50000 |
IC50 and EC50 values are the mean of at least three experiments ± the
standard error of the mean.
To follow up on these experiments, we examined the HDAC inhibitory activity of our HDACIs in cell culture, selecting the most potent compound (13e) for analysis in MV4-11 cells with SAHA as a positive control. Results showed compound 13e can markedly increase the level of acetylated histone H3 and acetylated histone H4, which was consistent with the HDAC1, 2, and 3 inhibition activities (Figure 7). Unexpectedly, the level of acetyl-tubulin also increased, although 13e does not inhibit HDAC6, which controls the deacetylation of tubulin. We hypothesize that HDAC6 was down-regulated when treated with 13e; thus, we also characterized the level of HDAC6. As expected, HDAC6 was nearly absent following treatment with ≥0.5 μM 13e or with 5 μM SAHA for 24 h. Further studies showed that 13e-induced HDAC6 repression requires more than 12 h of treatment. This suggested HDAC6 down-regulation is likely due to transcriptional repression, and not through protein degradation (proteinase and lysosome, etc.), as the latter one would occur within hours. HDAC6 is required for tumor cells to be resistant to anoikis and then facilitate tumor cell invasion and metastasis.42 Another study showed oncogenic K-ras contributes to SAHA resistance by upregulating HDAC6 and c-myc expression.43 Therefore, down-regulation of HDAC6 may have benefits in overcoming HDAC inhibition resistance. To the best of our knowledge, ours is the first report of a hydrazide HDAC class I inhibitor that down-regulated HDAC6 instead of acting through direct inhibition on that HDAC isoform.
Figure 7.
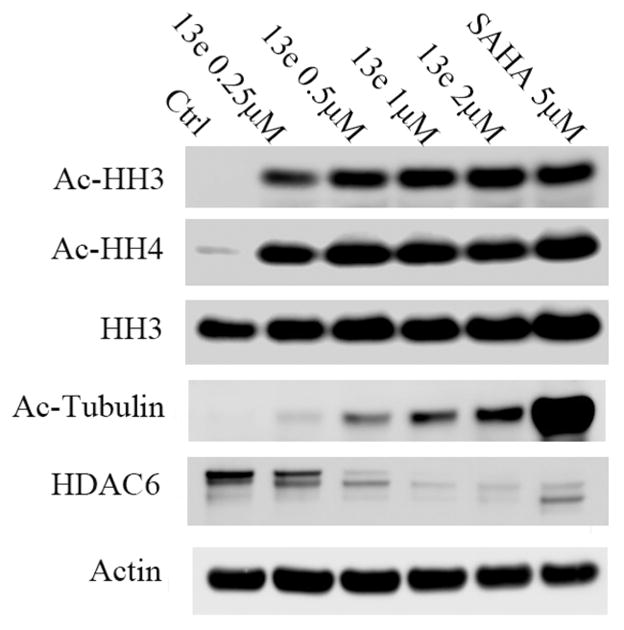
Western blot analysis of acetylated histone H3 (AcHH3), acetylated histone H4 (AcHH4), histone H3 (HH3), acetylated tubulin, and HDAC6 in MV4-11 cell lines after 24 h treatment with 13e at 0.25, 0.5, 1, and 2 μM, using 5 μM SAHA as positive control. Actin was used as a loading control. Figures are representative of n ≥ 2 experiments.
We tested our HDACIs against the acute myeloid leukemia cell line MV4-11 and carried out further evaluations of the antiproliferative activity of 13e against a prostate p53 null cancer cell line (PC-3) and human embryonic kidney cells, HEK293 (a nontumor cell line). The EC50 curves are shown in Figure 8. The EC50 value (11 020 nM) against the nontumor cell line HEK293 was more than 300-fold and 30-fold higher than against the MV4-11 and PC-3 cell lines, respectively, revealing our compounds’ cytotoxic selectivity for tumor cells over nontransformed cells. EC50 value of 13e toward PC-3 was 351.0 nM, which was more than 10 times higher than toward MV4-11 cells. Interestingly, after 48 h treatment of 13e at high concentration, almost all MV4-11 cells were dead, while ~20% of PC-3 remained viable compared to control; therefore, we speculated that 13e actually causes cell death in MV4-11 cells while merely preventing cell proliferation in PC-3 cells without killing the cells.
Figure 8.
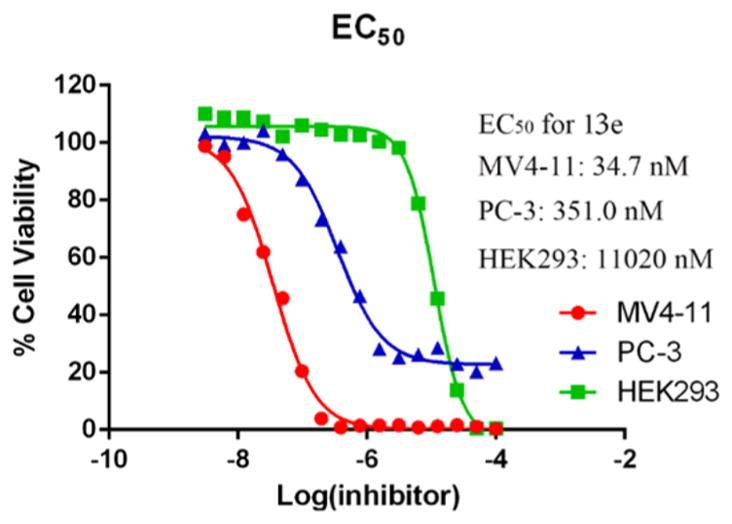
EC50 of 13e against MV4-11, PC-3, and HEK293 cells after 48 h treatment. Figures are representative of n ≥ 2 experiments.
In order to assess this hypothesis, we examined the MV4-11 and PC-3 cell numbers after treatment with 10 and 2 μM 13e for 24 and 48 h, respectively. As with the EC50 assay, the CellTiter-Blue (a buffered solution containing highly purified resazurin) was used as the assay reagent for determining cell viability. Functional mitochondria in viable cells convert the reagent to highly fluorescent resorufin.44,45 Fluorescence intensity of resorufin is therefore directly correlated to the number of viable cells in culture. In contrast to MV4-11 cells, for which the viable cell population decreased with time (Figure 9), the viable PC-3 cell population either slightly increased (2 μM) or remained unchanged (10 μM) following 13e treatment. We further examined the PC-3 cell line with a phase-contrast light microscope for changes in cell morphology in response to 48 h treatment with 10 and 2 μM 13e. Meeting our expectation, 13e did not obviously affect cell morphology at the concentration of 2 μM. Even at 10 μM, cells were still alive and attached to the culture flask (Figure 10). These observations were consistent with our hypothesis. Collectively, these data indicate that there are different, cell-type-dependent in vitro responses to our HDAC inhibitor. It is of interest to note that these two cell lines are distinct in their p53 genetic status: MV4-11 possesses wild-type p53, while PC-3 is p53 null. Activation and accumulation of p53 in the nucleus regulates the transcription of numerous target genes using specific DNA response elements, which occurs along with apoptosis, cell cycle arrest, DNA repair, and differentiation. As the first tumor suppressor gene linked to apoptosis, p53 can function as a “master regulator” of the apoptotic program not only by the activation of pro-apoptotic proteins such as Bax, Bak, and PUMA and repression of antiapoptotic Bcl-2 proteins but also by the activation of CD95 and TRAIL receptor 2 (TRAIL-R2/DR5), which can sensitize cells to death-receptor-mediated apoptosis.46,47 In human tumors, the p53 tumor suppressor gene is the most frequently mutated gene, and loss of wild type p53 function can both disable apoptosis and accelerate tumor development in transgenic mice.48 As p53 is a critical initiator of apoptosis, to further assess whether the different responses of MV4-11 cells and PC-3 cells to 13e was related to p53 status, we conducted Western blot analysis and determined the levels of pro-caspase3, cleaved caspase3, and cleaved PARP (poly ADP-ribose polymerase) in each cell line after 24 h of treatment with 1 and 2 μM of 13e (Figure 11). In MV4-11 cells, 13e caused the cleavage of inactive pro-caspase3, resulting in the subsequent increase in the levels of cleaved active caspase3 and cleaved PARP, indicating an activation of apoptosis. In PC-3 cells, 13e treatment resulted in no observed cleavage of pro-caspase3 or PARP. Also, in contrast to MV4-11 cells, 13e did not induce down-regulation of HDAC6 nor increase the level of acetylated tubulin in PC-3 cells. However, unlike HDAC6 down-regulation, in both cell lines 13e did not induce down-regulation of class I HDAC1. Other studies have demonstrated that introduction of exogenous wild-type p53 into cancer cells and its subsequent overexpression result in growth arrest, initially shown to occur at the G1/S cell cycle checkpoint.49–51 Thus, we examined the cell cycle of MV4-11 cells and PC-3 cells after treatment with 13e for 24 h. Consistent with previous reports, 13e induced G1/S and sub-G1 cell cycle arrest in MV4-11 cells (Figure 12). The population of G1 and sub-G1 cells increased after 24 h treatment with 0.5 μM 13e, and sub-G1 cells increased in a dose-dependent manner, which indicated significant cellular apoptosis (Figure 12 and Supporting Information Figure S10).52,53 While different from MV4-11 cells, 13e triggered G2/M arrest in PC-3 cells (Figure 12 and Supporting Information Figure S11). Based on these data, we hypothesized that the effects of 13e were mediated through a p53-dependent mechanism resulting in a G1/S and sub-G1 cell cycle arrest and cell death.
Figure 9.

Plots of resorufin fluorescence intensity over time for MV4-11 and PC-3 cells treated with 10, 2, or 0 μM 13e. Fluorescence intensity is directly proportional to the number of viable cells in culture. Figures are representative of n ≥ 2 experiments.
Figure 10.

PC-3 cell status after 48 h treatment with vehicle (DMSO), 2 μM 13e, and 10 μM 13e. Cells are less proliferative but remain alive after treatment with 13e. Figures are representative of n ≥ 2 experiments.
Figure 11.
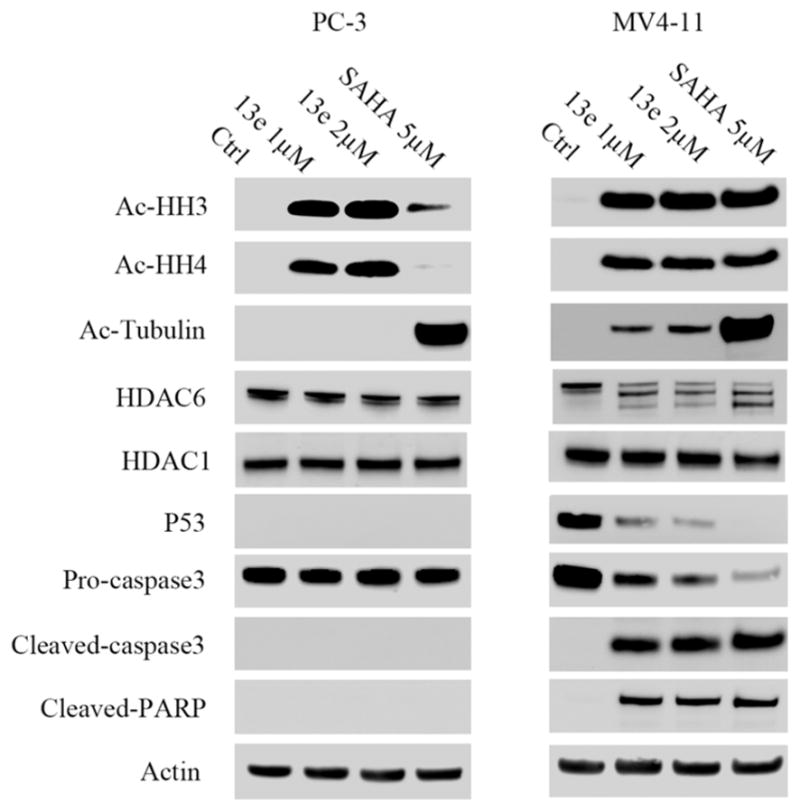
Western blot analysis of acetylated histone H3, acetylated histone H4, acetylated tubulin, HDAC6, p53, pro-caspase3, cleaved-caspase3, and cleaved-PARP in MV4-11 cells and PC-3 cells after 24 h treatment with 13e at 1 μM and 2 μM, using 5 μM SAHA as positive control. Actin was used as a loading control. Figures are representative of n ≥ 2 experiments.
Figure 12.
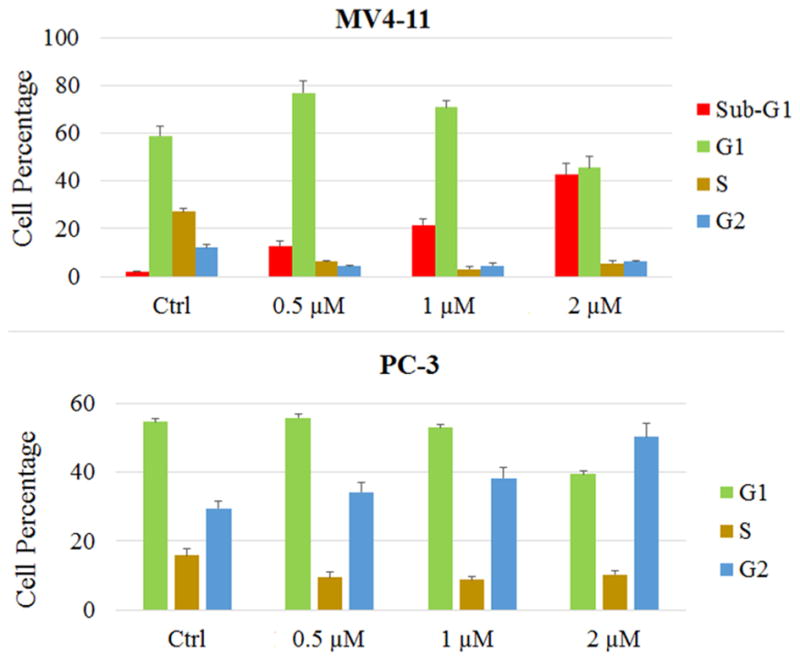
Cell cycle analysis of MV4-11 cells and PC-3 cells after treatment with 13e for 24 h. Figures are representative of n ≥ 2 experiments.
We also noticed that 13e can cause p53 degradation in MV4-11 cells (Figure 11), as degradation of p53 happened within 6 h with treatment of 0.5 μM 13e (Supporting Information Figure S9). p53 was degraded through the proteasome,54 and proteasome inhibitor bortezomib blocks this degradation upon hydrazide HDAC inhibitor treatment (Supporting Information Figure S12). Degradation of p53 is linked to concurrent depletion of antiapoptotic protein such as c-FLIP and XIAP, resulting in cellular apoptosis.55 In addition, inactivation of p53 by deletion, depletion, or inhibition in nontransformed or malignant human cell lines (such as HFFF2 fibroblasts, HCT116 colon cancer cells, SH-SY5Y neuroblastoma, and HeLa cervical cancer cells) can trigger autophagy;56,57 both apoptosis and autophagy can result in cell death.
CONCLUSION
A new series of hydrazide-based HDACIs were designed, synthesized, and had SAR interrogated. Their isotype selectivity, enzymatic binding kinetics, and in vitro anticancer activities were also characterized. Modifications to the hydrazide motifs as well as to the linker fragments of the compounds affected both HDAC inhibitory activity and efficacy against MV4-11 leukemia cells. Of the variations to the hydrazide motif, 13b with a three-carbon linear alkyl substituent exhibited the best selectivity and antiproliferative activity. Enzymatic inhibition kinetics studies showed that 13b was a slow and mixed-type inhibitor toward HDAC1, 2, and 3. Subsequent linker modifications of 13b led to the optimized compound 13e, which showed low nanomolar IC50 toward HDAC1, 2 and 3, submicromolar IC50 against HDAC8, and no inhibitory activity against class IIa (HDAC4, 5, 7, 9) and class IIb (HDAC6) up to 50 μM. The EC50 of 13e toward MV4-11 cells was 34.7 nM, which was about 26 times lower than that of the parent compound 11a. Compound 13e displayed no inhibition of HDAC6 up to 100 μM, though the compound did markedly increase the level of acetylated tubulin via a mechanism involving down-regulation of HDAC6. Cell based studies showed that 13e exhibited completely different mechanisms of antiproliferation activity against two different cancer cell lines, MV4-11 and PC-3. These cell lines have distinct p53 genetic statuses: MV4-11 is p53 wild-type, while PC-3 is p53 null. Our studies indicated that 13e-induced death of p53 wild type MV4-11 cells might be mediated by a p53-dependent apoptotic pathway resulting in G1/S and sub-G1 cell cycle arrest. In p53 null PC-3 cells, 13e could not induce apoptosis, instead producing cytostatic effects via promotion of only G2/M arrest.
EXPERIMENTAL SECTION
General Chemistry
All solvents, reagents, and compound precursors were purchased from Sigma-Aldrich or other chemical vendor and used as received, unless otherwise noted. 1H NMR and 13C NMR data were collected in deuterated solvent using a Bruker Nanobay 400 MHz instrument with TMS as an internal standard. Chemical shifts (δ) are given in parts per million, and coupling constants (J) are reported in hertz (Hz). Mass spectral data were gathered on a Thermo LCQ Fleet mass spectrometer using electrospray ionization. Purification was performed using a Teledyne Isco Combiflash 200 on prepacked C18-Aq columns. All target compounds were at least 95% pure as confirmed via UV detection of ESI-LCMS, performed on an Agilent 1100 HPLC instrument using an ODS HYPERSIL column (5 μm, 4.6 mm × 250 mm) using a gradient of water/methanol with 0.1% formic acid added.
(4-Methoxybenzoyl)-L-tryptophan (3), methyl (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)benzoate (5), methyl (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)-propanamido)benzoate (6), methyl (S)-6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)hexanoate (7), (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido) methyl)benzoic acid (1 4), and (S,E)-3-(4-(3-(1 H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)phenyl)acrylic acid (16) were synthesized as described previously.34
General Procedure for the Preparation of 2a and 2b
7-Methoxy-7-oxoheptan-1-aminium Chloride (2a)
7-Aminoheptanoic acid (1.45 g, 10 mmol) was dissolved in 200 mL of methanol. To this solution, acetyl chloride (2.4 g, 30 mmol) was added dropwise at 0 °C. The mixed solution was refluxed at 75 °C for 5 h. Volatiles were removed via vacuum, and the residue was recrystallized from ethyl acetate to afford the desired product 2a as a white powder, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (s, 2H), 3.58 (s, 3H), 2.78–2.68 (m, 2H), 2.31 (t, J = 7.4 Hz, 2H), 1.60–1.45 (m, 4H), 1.34–1.23 (m, 5H). ESI-MS m/z: 160.25 [M + H]+.
8-Methoxy-8-oxooctan-1-aminium Chloride (2b)
Using the synthetic method of 2a, compound 8-aminooctanoic acid gave 2b as a white powder, 93% yield.
General Procedure for the Preparation of 4a and 4b
Methyl (S)-7-(3-(1H-Indol-3-yl)-2-(4-methoxybenzamido)propanamido)-heptanoate (4a)
Compound 3 (1.02 g, 3 mmol) was dissolved in 50 mL of dichloromethane, and to this solution was added 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU, 1.05 g, 3.6 mmol) and trimethylamine (0.6 mL, 4.5 mmol). Thirty minutes later, 2a (0.52 g, 3.3 mmol) was added followed by 0.6 mL of additional triethylamine. The reaction was allowed to stir overnight, then washed with 1 N HCl (2 × 30 mL), saturated Na2CO3 (2 × 30 mL), and brine (2 × 30 mL), then dried over MgSO4. Volatiles were removed under vacuum to get the crude product 4a, which was used for the next step in the synthesis of 11 without further purification. ESI-MS m/z: 480.17 [M + H]+.
Methyl (S)-8-(3-(1H-Indol-3-yl)-2-(4-methoxybenzamido)-propanamido)octanoate (4b)
Using the synthetic method of 4a, compounds 3 and 2b gave 4b as a white solid, 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.00 (t, J = 5.6 Hz, 1H), 7.81 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.2 Hz, 1H), 7.05 (t, J = 7.0 Hz, 1H), 6.95–6.99 (M, 3H), 4.69–4.63 (m, 1H), 3.80 (s, 3H), 3.58 (s, 3H), 3.22–2.99 (m, 4H), 2.28 (t, J = 7.4 Hz, 2H), 1.54–1.48 (m, 2H), 1.42–1.29 (m, 2H), 1.23 (s, 6H). ESI-MS m/z: 494.17 [M + H]+.
General Procedure for the Preparation of 8, 9, 10, 11, and 12
(S)-N-(1-((4-(Hydrazinecarbonyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (8)
To a solution of 5 (2.4 g, 5 mmol) in methanol was added hydrazine monohydrate (1.6 g, 25 mmol). The mixture was refluxed at 75 °C for 48 h. Volatiles were then removed to afford compound 8, white solid, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 9.71 (s, 1H), 8.65 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 7.87–7.81 (m, 2H), 7.75 (d, J = 8.3 Hz, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.3 Hz, 2H), 7.21 (d, J = 2.2 Hz, 1H), 7.06 (dd, J = 11.1, 4.0 Hz, 1H), 6.98 (t, J = 8.0 Hz, 3H), 4.76 (td, J = 9.5, 5.0 Hz, 1H), 4.47 (s, 2H), 4.36 (d, J = 5.9 Hz, 2H), 3.80 (s, 3H), 3.22 (ddd, J = 24.2, 14.5, 7.3 Hz, 2H). ESI-MS m/z: 486.08 [M + H]+.
(S)-N-(1-((4-(Hydrazinecarbonyl)phenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (9)
Using the synthetic method of 8, compound 6 and hydrazine monohydrate gave 9 as a white solid, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.45 (s, 1H), 9.66 (s, 1H), 8.55 (d, J = 7.7 Hz, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.80 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 2.2 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 7.01–6.97 (m, 3H), 4.92–4.83 (m, 1H), 4.45 (s, 2H), 3.81 (s, 3H), 3.30–3.22 (m, 2H). ESI-MS m/z: 472.00 [M + H]+.
(S)-N-(1-((6-Hydrazinyl-6-oxohexyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (10)
Using the synthetic method of 8, compound 7 and hydrazine monohydrate gave 10 as a white solid, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.92 (s, 1H), 8.28 (d, J = 8.0 Hz, 1H), 8.02 (t, J = 5.3 Hz, 1H), 7.81 (d, J = 8.7 Hz, 2H), 7.68 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.05 (t, J = 7.4 Hz, 1H), 7.00–6.95 (m, 3H), 4.69–4.63 (m, 1H), 4.17 (s, 2H), 3.80 (s, 3H), 3.23–2.97 (m, 4H), 1.99 (t, J = 7.4 Hz, 2H), 1.52–1.44 (m, 2H), 1.39–1.34 (m, 2H), 1.30–1.14 (m, 2H). ESI-MS m/z: 466.17 [M + H]+.
(S)-N-(1-((7-Hydrazinyl-7-oxoheptyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (11)
Using the synthetic method of 8, compound 4a and hydrazine monohydrate gave 11 as a white solid, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.93 (s, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.01 (t, J = 5.6 Hz, 1H), 7.81 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 2.2 Hz, 1H), 7.05 (t, J = 7.0 Hz, 1H), 6.95–6.99 (m, 3H), 4.69–4.63 (m, 1H), 4.23 (s, 2H), 3.80 (s, 3H), 3.21–3.00 (m, 4H), 2.00 (t, J = 7.4 Hz, 2H), 1.53–1.42 (m, 2H), 1.38–1.35 (m, 2H), 1.21 (s, 4H). ESI-MS m/z: 480.25 [M + H]+.
(S)-N-(1-((8-hydrazinyl-8-oxooctyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (12)
Using the synthetic method of 8, compound 4b and hydrazine monohydrate gave 12 as a white solid, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.91 (s, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.00 (t, J = 5.6 Hz, 1H), 7.81 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.2 Hz, 1H), 7.05 (t, J = 7.0 Hz, 1H), 6.99–6.95 (m, 3H), 4.69–4.63 (m, 1H), 4.14 (s, 2H), 3.80 (s, 3H), 3.23–2.98 (m, 4H), 1.99 (t, J = 7.4 Hz, 2H), 1.51–1.45 (m, 2H), 1.41–1.31 (m, 2H), 1.22 (s, 6H). ESI-MS m/z: 494.25 [M + H]+.
General Procedure for the Preparation of 13a–13h
(S)-N-(1-((4-(2-Ethylhydrazine-1-carbonyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (13a)
Compound 8 (0.97 g, 2 mmol) was dissolved in 50 mL of ethanol, then acetaldehyde was added followed by 20 mmol of MgSO4. Upon completion of the reaction, MgSO4 was filtered off, and the volatiles were removed from the filtrate under vacuum. The residue was dissolved in 50 mL of methanol, followed by the addition of NaBH3CN and 2 drops of concentrated HCl. The mixture was allowed to stir overnight, volatiles were removed under vacuum, and the crude product was purified on combi-flash system C18 reverse phase columns eluted with acetonitrile and water to give 13a, a white powder, 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (d, J = 1.6 Hz, 1H), 9.98 (s, 1H), 8.67 (t, J = 6.0 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.77 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 2.2 Hz, 1H), 7.06 (t, J = 8.0 Hz, 1H), 7.01–6.97 (m, 3H), 5.10 (s, 1H), 4.83–4.72 (m, 1H), 4.37 (d, J = 5.9 Hz, 2H), 3.81 (d, J = 7.5 Hz, 3H), 3.31–3.15 (m, 2H), 2.82 (q, J = 7.2 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.57, 166.22, 165.65, 162.08, 143.33, 136.55, 132.08, 129.82, 127.71, 127.47, 127.24, 126.76, 124.20, 121.36, 118.99, 118.69, 113.79, 111.79, 111.02, 55.80, 54.90, 45.98, 42.33, 28.02, 13.57. ESI-MS m/z: 514.08 [M + H]+. (λ254) Purity 97.9%. Retention time: 7.74 min.
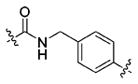
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((4-(2-propylhydrazine-1-carbonyl)benzyl)amino)propan-2-yl)-4-methoxybenzamide (13b)
Using the synthetic method of 13a, compound 8 and propionaldehyde gave 13b as a white solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.97 (s, 1H), 8.67 (t, J = 5.9 Hz, 1H), 8.42 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.32 (s, 1H), 7.29 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 1.8 Hz, 1H), 7.07 (t, J = 7.3 Hz, 1H), 7.01–6.97 (m, 3H), 5.10 (s, 1H), 4.80–4.75 (m, 1H), 4.37 (d, J = 5.8 Hz, 2H), 3.80 (s, 3H), 3.30–3.15 (m, 2H), 2.76 (t, J = 7.0 Hz, 2H), 1.55–1.40 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.57, 166.22, 165.61, 162.08, 143.31, 136.55, 132.09, 129.82, 127.72, 127.47, 127.24, 126.77, 124.20, 121.35, 118.99, 118.69, 113.79, 111.79, 111.02, 55.80, 54.90, 53.58, 42.33, 28.02, 21.33, 12.15. ESI-MS m/z: 528.17 [M + H]+. (λ254) Purity 97.0%. Retention time: 8.30 min.
(S)-N-(1-((4-(2-Butylhydrazine-1-carbonyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (13c)
Using the synthetic method of 13a, compound 8 and butyraldehyde gave 13c as a white solid, 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.97 (s, 1H), 8.67 (t, J = 6.0 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 7.89–7.82 (m, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 2.2 Hz, 1H), 7.10–7.03 (m, 1H), 7.02–6.94 (m, 3H), 5.08 (s, 1H), 4.82–4.70 (m, 1H), 4.37 (d, J = 5.9 Hz, 2H), 3.80 (s, 3H), 3.33–3.15 (m, 2H), 2.79 (t, J = 7.0 Hz, 2H), 1.51–1.30 (m, 4H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.56, 166.22, 165.60, 162.07, 143.31, 136.55, 132.08, 129.81, 127.71, 127.46, 127.23, 126.76, 124.19, 121.35, 118.98, 118.69, 113.79, 111.79, 111.02, 55.79, 54.89, 51.38, 42.33, 30.27, 28.01, 20.31, 14.40. ESI-MS m/z: 542.08 [M + H]+. (λ254) Purity 99.9%. Retention time: 8.71 min.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((4-(2-pentylhydrazine-1-carbonyl)benzyl)amino)propan-2-yl)-4-methoxybenzamide (13d)
Using the synthetic method of 13a, compound 8 and valeraldehyde gave 13d as a white solid, 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.98 (s, 1H), 8.68 (t, J = 5.8 Hz, 1H), 8.42 (d, J = 7.9 Hz, 1H), 7.84 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.1 Hz, 2H), 7.22 (s, 1H), 7.06 (t, J = 7.4 Hz, 1H), 7.01–6.97 (m, 3H), 5.08 (s, 1H), 4.76 (dd, J = 13.0, 9.3 Hz, 1H), 4.36 (d, J = 5.7 Hz, 2H), 3.80 (s, 3H), 3.36–3.14 (m, 2H), 2.77 (t, J = 7.0 Hz, 2H), 1.44–1.46 (m, 2H), 1.39–1.23 (m, 4H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) 172.58, 166.22, 165.59, 162.07, 143.31, 136.54, 132.06, 129.82, 127.69, 127.46, 127.23, 126.74, 124.21, 121.35, 118.99, 118.69, 113.78, 111.79, 111.01, 55.79, 54.89, 51.66, 42.31, 29.35, 27.98, 27.77, 22.53, 14.42. ESI-MS m/z: 556.25 [M + H]+. (λ254) Purity 98.5%. Retention time: 9.03 min.
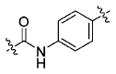
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((4-(2-propylhydrazine-1-carbonyl)phenyl)amino) propan-2-yl)-4-methoxybenzamide (13e)
Using the synthetic method of 13a, compound 9 and propionaldehyde gave 13e as a white solid, 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.50 (s, 1H), 9.92 (s, 1H), 8.59 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.80 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 2.1 Hz, 1H), 7.06 (t, J = 7.1 Hz, 1H), 7.01–6.97 (m, 3H), 5.06 (s, 1H), 4.91–4.85 (m, 1H), 3.81 (s, 3H), 3.27–3.25 (m, 2H), 2.75 (t, J = 7.1 Hz, 2H), 1.54–1.41 (m, 2H), 0.94–0.88 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.99, 166.37, 165.32, 162.15, 142.12, 136.49, 129.85, 128.30, 128.21, 127.68, 126.53, 124.32, 121.39, 119.08, 118.68, 113.85, 111.78, 110.66, 55.81, 53.63, 31.16, 21.34, 12.14. ESI-MS m/z: 514.08[M + H]+. (λ254) Purity 95.9%. Retention time: 8.36 min.
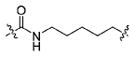
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((6-oxo-6-(2-propylhydrazinyl)-hexyl)amino)propan-2-yl)-4-methoxybenzamide (13f)
Using the synthetic method of 13a, compound 10 and propionaldehyde gave 13f as a white solid, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (d, J = 1.5 Hz, 1H), 9.22 (d, J = 5.7 Hz, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.03 (t, J = 5.6 Hz, 1H), 7.82 (d, J = 8.9 Hz, 2H), 7.68 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 2.2 Hz, 1H), 7.09–7.02 (m, 1H), 7.01–6.92 (m, 3H), 4.76 (d, J = 5.5 Hz, 1H), 4.70–4.64 (m, 1H), 3.80 (s, 3H), 3.23–2.99 (m, 4H), 2.68–2.57 (m, 2H), 2.00 (t, J = 7.4 Hz, 2H), 1.57–1.43 (m, 2H), 1.44–1.30 (m, 4H), 1.29–1.15 (m, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.12, 171.34, 166.00, 162.03, 136.50, 129.75, 127.76, 126.82, 124.08, 121.29, 119.01, 118.61, 113.77, 111.73, 111.09, 55.78, 54.70, 53.50, 39.01, 33.93, 29.25, 28.20, 26.44, 25.44, 21.22, 12.06. ESI-MS m/z: 508.25 [M + H]+. (λ254) Purity 95.9%. Retention time: 7.98 min.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((7-oxo-7-(2-propylhydrazinyl)-heptyl)amino)propan-2-yl)-4-methoxybenzamide (13g)
Using the synthetic method of 13a, compound 11 and propionaldehyde gave 13g as a white solid, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (d, J = 1.4 Hz, 1H), 9.22 (s, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.01 (t, J = 5.6 Hz, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 2.1 Hz, 1H), 7.05 (dd, J = 11.1, 3.9 Hz, 1H), 7.00–6.95 (m, 3H), 4.79 (s, 1H), 4.70–4.64 (m, 1H), 3.80 (s, 3H), 3.23–2.99 (m, 4H), 2.61 (t, J = 7.1 Hz, 2H), 2.00 (t, J = 7.4 Hz, 2H), 1.49–1.45 (m, 2H), 1.43–1.34 (m, 4H), 1.22 (s, 4H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.10, 171.39, 166.03, 162.03, 136.50, 129.75, 127.76, 126.81, 124.06, 121.29, 119.00, 118.61, 113.78, 111.73, 111.08, 55.78, 54.72, 53.50, 39.03, 33.91, 29.38, 28.73, 28.17, 26.50, 25.64, 21.22, 12.06. ESI-MS m/z: 522.25 [M + H]+. (λ254) Purity 98.0%. Retention time: 8.29 min.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((8-oxo-8-(2-propylhydrazinyl)-octyl)amino)propan-2-yl)-4-methoxybenzamide (13h)
Using the synthetic method of 13a, compound 12 and propionaldehyde gave 13h as a white solid, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (d, J = 1.7 Hz, 1H), 9.29 (s, 1H), 8.25 (d, J = 8.1 Hz, 1H), 8.00 (t, J = 5.6 Hz, 1H), 7.78 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 2.2 Hz, 1H), 7.08–7.02 (m, 1H), 7.01–6.92 (m, 3H), 4.76 (s, 1H), 4.69–4.63 (m, 1H), 3.78 (s, 3H), 3.20–2.98 (m, 4H), 2.59 (t, J = 7.0 Hz, 2H), 1.99 (t, J = 7.3 Hz, 2H), 1.50–1.44 (m, 2H), 1.42–1.29 (m, 4H), 1.19 (s, 6H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.23, 171.71, 166.24, 162.08, 136.48, 129.71, 127.68, 126.61, 124.03, 121.38, 118.93, 118.72, 113.84, 111.77, 110.89, 55.77, 54.77, 53.45, 33.89, 29.30, 28.84, 28.13, 26.60, 25.58, 21.10, 11.98. ESI-MS m/z: 536.33 [M + H]+. (λ254) Purity 99.0%. Retention time: 8.54 min.
General Procedure for the Preparation of 15a–15c and 17
(S)-N-(1-((4-(2-Cyclopentylhydrazine-1-carbonyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (15a)
Compound 14 (0.47 g, 1 mmol) was dissolved in 50 mL of dichloromethane and to this solution was added 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU, 0.35 g, 1.2 mmol) and trimethylamine (0.2 mL, 1.5 mmol). Thirty minutes later, cyclopentylhydrazine (0.12 g, 1.2 mmol) was added. The reaction was allowed to stir overnight, then washed with brine (2 × 30 mL) and dried over MgSO4. Volatiles were removed under vacuum and the crude residue purified on combi-flash system C18 reverse phase columns eluted with acetonitrile and water to yield the desired product. 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 10.86 (d, J = 1.7 Hz, 1H), 8.76 (t, J = 6.0 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H), 7.91–7.84 (m, 4H), 7.70 (d, J = 7.8 Hz, 1H), 7.37–7.31 (m, 3H), 7.22 (d, J = 2.1 Hz, 1H), 7.08–7.04 (m, 1H), 7.00–6.97 (m, 3H), 4.82–4.70 (m, 1H), 4.39 (d, J = 5.9 Hz, 2H), 3.96–3.85 (m, 1H), 3.80 (s, 3H), 3.33–3.14 (m, 3H), 1.97–1.64 (m, 6H), 1.62–1.48 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.65, 166.21, 165.79, 162.08, 145.32, 136.55, 129.84, 129.18, 128.38, 127.70, 127.48, 126.73, 124.20, 121.33, 118.97, 118.68, 113.79, 111.80, 110.96, 62.23, 55.81, 55.01, 42.33, 28.59, 28.00, 24.40. ESI-MS m/z: 554.17 [M + H]+. (λ254) Purity 98.4%. Retention time: 8.67 min.
(S)-N-(3-(1H-Indol-3-yl)-1-oxo-1-((4-(2-phenylhydrazine-1-carbonyl)benzyl)amino)propan-2-yl)-4-methoxybenzamide (15b)
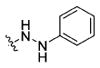
Using the synthetic method of 15a, compound 14 and phenylhydrazine gave 15b as a white solid, 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (d, J = 1.8 Hz, 1H), 10.33 (d, J = 3.0 Hz, 1H), 8.71 (t, J = 6.0 Hz, 1H), 8.44 (d, J = 7.9 Hz, 1H), 7.91 (d, J = 2.9 Hz, 1H), 7.85 (d, J = 8.6 Hz, 4H), 7.71 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.1 Hz, 3H), 7.23 (d, J = 2.2 Hz, 1H), 7.18–7.16 (m, 2H), 7.11–7.04 (m, 1H), 7.03–6.95 (m, 3H), 6.79 (d, J = 7.6 Hz, 2H), 6.73 (t, J = 7.3 Hz, 1H), 4.83–4.72 (m, 1H), 4.39 (d, J = 5.8 Hz, 2H), 3.80 (s, 3H), 3.37–3.16 (m 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.61, 166.65, 166.23, 162.08, 150.03, 143.81, 136.54, 131.86, 129.83, 129.22, 127.72, 127.38, 126.74, 124.22, 121.36, 119.04, 118.70, 113.79, 112.78, 111.80, 111.02, 55.80, 54.90, 42.36, 28.02. ESI-MS m/z: 562.17 [M + H]+. (λ254) Purity 96.8%. Retention time: 9.75 min.
(S,E)-N-(1-(4-(3-Hydrazinyl-3-oxoprop-1-en-1-yl)phenoxy)-3-(1H-indol-3-yl)propan-2-yl)-4-methoxybenzamide (17)
Using the synthetic method of 15a, compound 16 and hydrazine monohydrate gave 17 as a colorless oil, 55% yield. ESI-MS m/z: 485.17 [M + H]+.
General Procedure for the Preparation of 18
(S,E)-N-(1-(1H-Indol-3-yl)-3-(4-(3-oxo-3-(2-propylhydrazinyl)prop-1-en-1-yl) phenoxy)-propan-2-yl)-4-methoxybenzamide (18)
Using the synthetic method of 13a, compound 17 and propionaldehyde gave 18 as a white solid, 65% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.51 (s, 1H), 8.36 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.63 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 15.8 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.18 (d, J = 2.2 Hz, 1H), 7.06 (dd, J = 11.1, 3.9 Hz, 1H), 7.02–6.93 (m, 5H), 6.39 (d, J = 15.8 Hz, 1H), 5.04 (s, 1H), 4.65–4.51 (m, 1H), 4.19 (dd, J = 9.8, 6.4 Hz, 1H), 4.07 (dd, J = 9.9, 5.2 Hz, 1H), 3.81 (s, 3H), 3.11 (d, J = 7.0 Hz, 2H), 2.69 (t, J = 7.1 Hz, 2H), 1.48–1.37 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.24, 164.65, 162.00, 160.08, 138.65, 136.63, 129.63, 129.51, 128.02, 127.87, 127.15, 123.86, 121.39, 118.81, 118.24, 115.45, 113.85, 111.86, 111.30, 69.48, 55.80, 53.60, 50.18, 27.21, 21.25, 12.04. ESI-MS m/z: 527.25 [M + H]+. (λ254) Purity 97.9%. Retention time: 9.24 min.
In Vitro HDACs Inhibition Fluorescence Assay
All of the HDAC enzymes were bought from BPS Bioscience. In vitro HDAC inhibition assays were conducted as previously described. In brief, 20 μL of recombinant HDAC enzyme solution (HDAC1–9) was mixed with various concentrations of tested compound (20 μL). The mixture was incubated at 30 °C for 1 h (for the time-dependent assay, the mixture was incubated for 15, 30, 60, and 90 min, respectively), then 10 μL of fluorogenic substrate (Boc-Lys (acetyl)-AMC for HDAC1, 2, 3, and 6, Boc-Lys (trifluoroacetyl)-AMC for HDAC 4, 5, 7, 8, 9, and 11) was added. After incubation at 30 °C for 2 h, the catalysis was stopped by addition of 10 μL of developer containing trypsin and trichostatin A (TSA). Thirty minutes later, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 360 and 460 nm, respectively. The inhibition ratios were calculated from the fluorescence intensity readings of tested wells relative to those of control wells, and the IC50 curves and values were determined by GraphPad Prism 6.0, using the “log(inhibitor) vs. normalized response – variable slope” function.
Recombinant HDACs 1, 2, and 3 Vmax Studies
All of the HDAC enzymes were bought from BPS Bioscience. Forty microliters of recombinant human HDAC enzyme (HDAC1, 2, and 3) solution was added in 96-well format to black U-bottom plates. Forty microliters of inhibitor at various concentrations, diluted in HDAC buffer, was added. The mixture was allowed to incubate for 2 h at 30 °C before the addition of 20 μL of serially diluted fluorogenic substrate Boc-Lys(acetyl)-AMC in HDAC buffer. Two hours later, 20 μL of 5 mg/mL trypsin and 1 μM TSA solution was added to quench the reaction. Fluorescence was read at 360 nm (ex.)/460 nm (em.) using a Tecan M200 Pro. Vmax and Km were determined using GraphPad Prism’s Michaelis–Menten function. Corresponding Lineweaver–Burke double reciprocal plots were derived from these values and plotted accordingly.
MMP-2/MMP-9 Inhibitory Activity
InnoZyme Gelatinase (MMP-2/MMP-9) Activity Assay Kit was bought from EMD Chemicals, Inc. One hundred micromolar 13e (45 μL), 40 μM MMP inhibitor (45 μL), and activation buffer (45 μL) were added to designated wells. Then 45 μL of MMP-2/MMP-9 diluted in activation buffer was added to each well. After 10–15 min incubation, 10 μL of substrate working solution was added. The mixture was incubated at 37 °C for 2 h, and fluorescence was read at 320 nm (ex.)/405 nm (em.) using a Tecan M200 Pro.
CD13 Inhibition Assay
Forty microliters of various concentrations of compound solutions were added into a 96-well clear plate followed by the addition of CD13 enzyme (microsomal aminopeptidase from porcine kidney microsomes, Sigma) solution. Five min later, the substrate (L-leucine-p-nitroanilide) was added, and the mixture was incubated at 37 °C for 30 min. The absorbance was measured at 405 nm with a Microplate Reader.
Molecular Docking Against HDAC1 and HDAC3
Modeling and simulations were performed using MOE (Chemical Computing Group, Inc.). Structural PBD files used HDAC1 PDB code 5ICN58 and HDAC3 PDB code 4A69.59 Before simulations, proteins were protonated at T = 310 K, pH 7.3, salt at 200 mM, and using GB/VI electrostatics, but no energy minimization was performed. We employed two sites per protein docking. For each HDAC, two binding sites were defined using MOE Sitefinder to create docking dummies. The CS was defined as the catalytic pocket, which incorporated the Zn metal, and the AS allosteric site was defined by the pocket created by the interface between the HDAC heterodimer. Then each site was docked individually with the probe ligands 11a and 13b leading to eight conditions (two proteins, two sights, and two compounds). Simulations left the protein rigid and flexed the ligand. For each docking simulation, initial placement calculated 50 poses using triangle matching placement with London dG scoring, then the top 20 poses were refined using force field and Affinity dG scoring (Escore2). The top 10 poses from these simulations were analyzed.
MV4-11, PC-3, and HEK293 EC50 Analysis
All cell lines were maintained in RPMI1640 medium containing 10% FBS at 37 °C in a 5% CO2 humidified incubator. Specific numbers of cells (20000 MV4-11 cells, 10000 HEK293 cells, or 5000 PC-3 cells) were plated per well in 96-well clear plates and preincubated for 12 h, then serially diluted inhibitor was added. Cells were allowed to incubate with compounds for 48 h. CellTiter-Blue (resazurin cell viability assay reagent) was then added to each well at a final concentration of 0.125 mg/mL. The mixture was allowed to incubate for 2–4 h until sufficient color change occurred. Cell viability was measured as a function of resorufin fluorescence intensity using a Tecan M200 Pro spectrophotometer, 560 nm/590 nm (excitation/emission). Data were normalized to control wells, and background was removed. EC50s were determined using GraphPad Prism’s “log(inhibitor) vs. normalized response – variable slope” function.
Western Blot Analysis
MV4-11 cells (1,500,000 per well) or PC-3 cells (300,000 per well) was plated in flat-bottom six-well plates and allowed to grow for 12 h, and then treated with different concentrations of compounds. Twenty-four hours later, cells were harvested, washed by PBS, and lysed with RIPA buffer, which comprised 50 mM Tris Base, 150 mM NaCl, 5 mM EDTA, 0.1% (v/v) SDS, 0.5% (v/v) sodium deoxycholate, and 1% (v/v) Triton-x-100. After lysing, the suspension was ultrasonicated and centrifuged at 14000 rpm for 15 min at 4 °C. The mixture of 75 mL of supernatant and 25 mL of β-mercaptoethanol/LDS solution (15:85) was heated at 90 °C for 15 min and normalization according to BCA test carried out before loading. Lysates were run on Invitrogen NuPAGE 4–12% Bis-Tris 15 well gels at 170 V for approximately 60 min in MES buffer. Gels were transferred to methylcellulose and run at 30 V for 3 h. Membranes were incubated at 4 °C overnight with 1/1000 primary antibodies, which were diluted in 2.5% (w/v) milk or 5% (w/v) bovine serum albumin. The membrane was washed twice with TBST buffer before being incubated with secondary antibodies. Images were acquired using a GE ImageQuant LAS 4000.
Cell Cycle Analysis
MV4-11 cells (1,500,000 per well) or PC-3 cells (300,000 per well) was plated in flat-bottom six-well plates and allowed to grow for 12 h, and then treated with different concentrations of compounds. Twenty-four hours later, cells were harvested and washed with PBS; the supernatant was discarded. The cells were fixed in 2 mL of 75% cold ethanol at 4 °C overnight, then washed twice in cold PBS. Five hundred microliters of RNase A (20ug/mL) in PBS was added, and the mixture was incubated at 37 °C for 30 min, followed by the addition of 500 μL of propidium iodide (50 μg/mL in PBS). After 30 min at 4 °C, cells were analyzed by flow cytometry.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health/National Cancer Institute (CA163452 to C.J.C.), the National Institute of General Medical Sciences (P20GM103542) from the National Institutes of Health, and in part by pilot research funding from an American Cancer Society Institutional Research Grant awarded to the Hollings Cancer Center, Medical University of South Carolina.
ABBREVIATIONS USED
- MgSO4
magnesium sulfate
- HCl
hydrochloric acid
- Na2CO3
sodium carbonate
- NaBH3CN
sodium cyanoborohydride
- FBS
fetal calf serum
- PBS
phosphate buffered saline
- TSA
trichostatin A
- SDS
sodium dodecyl sulfate
- EDTA
ethyl-enediaminetetraacetic acid
- AS
allosteric site
- CS
catalytic site
Footnotes
Accession Codes
Structural PBD files used: HDAC1 (PDB code 5ICN) and HDAC3 (PDB code 4A69).
Author Contributions
The manuscript was written by X.L. Synthesis of compounds was performed by X.L. Design of the compounds was performed by X.L., C.J.C., and Y.Z. Assays and biological data collection were performed by X.L., E.S.I., J.L., X.K. R.A.H., Y.K.P., and X.L. performed the molecular modeling. All authors have given approval to the final version of the manuscript.
Notes
The authors declare the following competing financial interest(s): C.J.C. and R.A.H. are the co-founders of Lydex Pharmaceuticals. The other authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.8b00136.
Vmax plots of 13b and 11a toward recombinant HDACs 1 and 3; predicted binding mode and interaction of 11a, 13b, and 13e with the catalytic site (CS) and allosteric site (AS) binding modes of HDAC1 and HDAC3; 13e inhibition ratio for HDAC4, 5, 6, 7, 8, and 9 at the concentration of 50 and 10 μM; 13e inhibitory curve for HDAC1, 2, 3, and 8; 13e inhibition ratio for MMP-2/MMP-9 and APN/CD13 at the concentration of 10 and 50 μM; Western blot of HDAC6 and p53 status in MV4-11 cells after 6 and 12 h treatment by 13e; cell cycle analysis of 13e in MV4-11 cells and PC-3 cells after 24 h treatment; bortezomib blocks the degradation of p53 after 6 h treatment with hydrazide HDACi; 1H NMR spectral of intermediates 2a, 4b, 8, 10, 11, and 12 and all target compounds 13a–13h, 15a, 15b, and 18; 13C NMR of all target compounds 13a–13h, 15a, 15b, and 18 (PDF) Molecular formula strings of target compounds 13a–13h, 15a, 15b, and 18 (CSV)
References
- 1.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Fang H, Jiao J, Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr Med Chem. 2008;15:2840–2849. doi: 10.2174/092986708786242796. [DOI] [PubMed] [Google Scholar]
- 4.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther. 2010;10:935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Krämer OH, Baus D, Knauer SK, Stein S, Jäger E, Stauber RH, Grez M, Pfitzner E, Heinzel T. Acetylation of Stat1 modulates NF-κB activity. Genes Dev. 2006;20:473–485. doi: 10.1101/gad.364306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bilton R, Trottier E, Pouysségur J, Brahimi-Horn MC. ARDent about acetylation and deacetylation in hypoxia signalling. Trends Cell Biol. 2006;16:616–621. doi: 10.1016/j.tcb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Wu Q, Li W, Wang C, Fan P, Cao L, Wu Z, Wang F. Ultra-deep lysine crotonylome reveals the crotonylation enhancement on both histones and non-histone proteins by SAHA treatment. J Proteome Res. 2017;16:3664–3671. doi: 10.1021/acs.jproteome.7b00380. [DOI] [PubMed] [Google Scholar]
- 9.Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 11.Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 12.Mrakovcic M, Kleinheinz J, Fröhlich LF. Histone deacetylase inhibitor-induced autophagy in tumor cells: implications for p53. Int J Mol Sci. 2017;18:1883. doi: 10.3390/ijms18091883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Napoli M, Flores ER. The family that eats together stays together: new p53 family transcriptional targets in autophagy. Genes Dev. 2013;27:971–974. doi: 10.1101/gad.219147.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 15.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 16.Krieger V, Hamacher A, Gertzen CG, Senger J, Zwinderman MR, Marek M, Romier C, Dekker FJ, Kurz T, Jung M. Design, multicomponent synthesis and anticancer activity of a focused histone deacetylase (HDAC) inhibitor library with peptoid-based cap groups. J Med Chem. 2017;60:5493–5506. doi: 10.1021/acs.jmedchem.7b00197. [DOI] [PubMed] [Google Scholar]
- 17.Duvic M, Vu J. Update on the treatment of cutaneous T-cell lymphoma (CTCL): focus on vorinostat. Biologics: targets therapy. 2007;1:377. [PMC free article] [PubMed] [Google Scholar]
- 18.Bertino EM, Otterson GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Invest Drugs. 2011;20:1151–1158. doi: 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- 19.Rashidi A, Cashen AF. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Future Oncol. 2015;11:1659–1664. doi: 10.2217/fon.15.62. [DOI] [PubMed] [Google Scholar]
- 20.Greig SL. Panobinostat: a review in relapsed or refractory multiple myeloma. Targeted oncology. 2016;11:107–114. doi: 10.1007/s11523-015-0413-6. [DOI] [PubMed] [Google Scholar]
- 21.Traoré MD, Zwick V, Simões-Pires CA, Nurisso A, Issa M, Cuendet M, Maynadier M, Wein S, Vial H, Jamet H. Hydroxyl ketone-based histone deacetylase inhibitors to gain insight into class I HDAC selectivity versus that of HDAC6. ACS Omega. 2017;2:1550–1562. doi: 10.1021/acsomega.6b00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Micelli C, Rastelli G. Histone deacetylases: structural determinants of inhibitor selectivity. Drug Discovery Today. 2015;20:718–735. doi: 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Zhan P, Itoh Y, Suzuki T, Liu X. Strategies for the discovery of target-specific or isoform-selective modulators. J Med Chem. 2015;58:7611–7633. doi: 10.1021/acs.jmedchem.5b00229. [DOI] [PubMed] [Google Scholar]
- 24.Baud MG, Haus P, Leiser T, Meyer-Almes FJ, Fuchter MJ. Highly ligand efficient and selective N-2-(Thioethyl) picolinamide histone deacetylase inhibitors inspired by the natural product Psammaplin A. Chem Med Chem. 2013;8:149–156. doi: 10.1002/cmdc.201200450. [DOI] [PubMed] [Google Scholar]
- 25.Kinzel O, Llauger-Bufi L, Pescatore G, Rowley M, Schultz-Fademrecht C, Monteagudo E, Fonsi M, Gonzalez Paz O, Fiore F, Steinkühler C. Discovery of a potent class I selective ketone histone deacetylase inhibitor with antitumor activity in vivo and optimized pharmacokinetic properties. J Med Chem. 2009;52:3453–3456. doi: 10.1021/jm9004303. [DOI] [PubMed] [Google Scholar]
- 26.McClure JJ, Zhang C, Inks ES, Peterson YK, Li J, Chou CJ. Development of allosteric hydrazide-containing class I histone deacetylase inhibitors for use in acute myeloid leukemia. J Med Chem. 2016;59:9942–9959. doi: 10.1021/acs.jmedchem.6b01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steele N, Plumb J, Vidal L, Tjørnelund J, Knoblauch P, Buhl-Jensen P, Molife R, Brown R, De Bono J, Evans T. Pharmacokinetic and pharmacodynamic properties of an oral formulation of the histone deacetylase inhibitor Belinostat (PXD101) Cancer Chemother Pharmacol. 2011;67:1273–1279. doi: 10.1007/s00280-010-1419-5. [DOI] [PubMed] [Google Scholar]
- 28.Rubin EH, Agrawal NG, Friedman EJ, Scott P, Mazina KE, Sun L, Du L, Ricker JL, Frankel SR, Gottesdiener KM. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res. 2006;12:7039–7045. doi: 10.1158/1078-0432.CCR-06-1802. [DOI] [PubMed] [Google Scholar]
- 29.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu J. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 32.Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S, Gale JP, Galbo T, Fisher SL, Meier BC. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in β-cell protection. ACS Chem Biol. 2016;11:363–374. doi: 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- 33.McClure JJ, Inks ES, Zhang C, Peterson YK, Li J, Chundru K, Lee B, Buchanan A, Miao S, Chou CJ. Comparison of the deacylase and deacetylase activity of zinc-dependent HDACs. ACS Chem Biol. 2017;12:1644–1655. doi: 10.1021/acschembio.7b00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Zhang Y, Jiang Y, Wu J, Inks ES, Chou CJ, Gao S, Hou J, Ding Q, Li J. Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: design, structure-activity relationship and anti-tumor activity study. Eur J Med Chem. 2017;134:185–206. doi: 10.1016/j.ejmech.2017.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Stowe RL, Pinello CE, Tian G, Madoux F, Li D, Zhao LY, Li JL, Wang Y, Wang Y. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem Biol. 2015;22:273–284. doi: 10.1016/j.chembiol.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson SD, Mitchell JR, Snodgrass W, Timbrell JA. Hepatotoxicity and metabolism of iproniazid and isopropylhydrazine. Journal of Pharmacology and Experimental Therapeutics. 1978;206:574–585. [PubMed] [Google Scholar]
- 37.Nolan CM, Goldberg SV, Buskin SE. Hepatotoxicity associated with isoniazid preventive therapy: a 7-year survey from a public health tuberculosis clinic. Jama. 1999;281:1014–1018. doi: 10.1001/jama.281.11.1014. [DOI] [PubMed] [Google Scholar]
- 38.Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, Reddy KR, Watkins PB, Navarro V, Barnhart H. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148:1340–1352e7. doi: 10.1053/j.gastro.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davidson B, Reich R, Risberg B, Nesland J. The biological role and regulation of matrix metalloproteinases (MMP) in cancer. Arkhiv patologii. 2002;64(3):47–53. [PubMed] [Google Scholar]
- 41.Wickström M, Larsson R, Nygren P, Gullbo J. Aminopeptidase N 9CD13) as a target for cancer chemotherapy. Cancer science. 2011;102:501–508. doi: 10.1111/j.1349-7006.2010.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68:7561–7569. doi: 10.1158/0008-5472.CAN-08-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, Tan R, Zhu X, Zhang Y, Tan Z, Su B, Li Y. Oncogenic K-ras confers SAHA resistance by up-regulating HDAC6 and c-myc expression. Oncotarget. 2016;7:10064. doi: 10.18632/oncotarget.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 45.Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc. 2011;54:10–14. [PubMed] [Google Scholar]
- 46.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 47.Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 48.Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild-type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011;18:2–11. doi: 10.1038/cgt.2010.63. [DOI] [PubMed] [Google Scholar]
- 49.Wade Harper J, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 50.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 51.Aurelio ON, Kong XT, Gupta S, Stanbridge EJ. p53 mutants have selective dominant-negative effects on apoptosis but not growth arrest in human cancer cell lines. Molecular and cellular biology. 2000;20:770–778. doi: 10.1128/mcb.20.3.770-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kajstura M, Halicka HD, Pryjma J, Darzynkiewicz Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytometry, Part A. 2007;71:125–131. doi: 10.1002/cyto.a.20357. [DOI] [PubMed] [Google Scholar]
- 53.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 54.Shirangi TR, Zaika A, Moll UM. Nuclear degradation of p53 occurs during down-regulation of the p53 response after DNA damage. FASEB J. 2002;16:420–422. doi: 10.1096/fj.01-0617fje. [DOI] [PubMed] [Google Scholar]
- 55.Zhang HG, Wang J, Yang X, Hsu HC, Mountz JD. Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene. 2004;23:2009–2015. doi: 10.1038/sj.onc.1207373. [DOI] [PubMed] [Google Scholar]
- 56.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’amelio M, Criollo A, Morselli E, Zhu C, Harper F. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 58.Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BV, Schwabe JW. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun. 2016;7:11262. doi: 10.1038/ncomms11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.