

Abstract
We report a strategy to introduce a reactive electrophile into proteins through the conversion of a chemically inert group into a bioreactive one in response to an inducer molecule. This strategy was demonstrated by an oxidation-induced and proximity-enhanced protein-protein crosslinking in the presence of a large access of free nucleophiles.
Graphical abstract
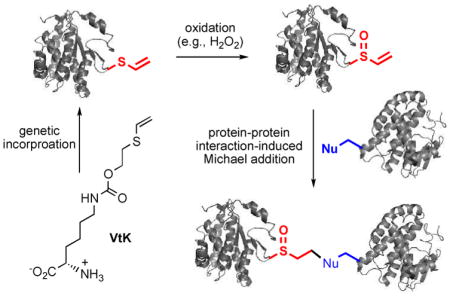
Oxidation-induced and proximity-enhanced protein-protein crosslinking through a genetically encoded vinylthioether probe.
The introduction of bioorthogonal functional groups into proteins has become an indispensable chemical biology tool.1–5 Recently, mild bioreactive electrophiles, such as haloalkanes,6–8 isothiocyanate,9 and vinylsulfonamide,10 have also been incorporated into proteins to react with nucleophilic amino acid residues, such as cysteine and lysine. This has enabled a number of new chemistry in live cells,11 which could complement the currently available bioorthogonal chemistry. As a new approach, the genetic incorporation of bioreactive chemical probes could have a very broad application in both biochemical research and biomedical applications.
While most of the bioreactive chemical probes were directly introduced into proteins and underwent spontaneous reactions with nucleophiles,6–10 an elegant combination of photoswitchable and electrophilic groups enabled a photoresponsive and proximity-enhanced reaction.12–13 Besides photo-crosslinking groups,14–19 this is the only example of artificially introduced bioreactive chemical probes whose reaction can be induced with an exogenously added signal. Such a strategy can potentially provide spatiotemporal resolution for biological studies. Here we report a new approach to introduce a non-natural bioreactive probe into biomolecules. It consists of two steps (Fig. 1): (1) the introduction of a bio-inert functional group; and (2) the in situ generation of a bioreactive functional group in response to an inducer molecule. More specifically, this masked reactive probe was based on the redox chemistry of sulfur. A non-bioreactive vinylthioether group was introduced into proteins through unnatural amino acid mutagenesis. In the presence of reactive oxygen species (e.g., H2O2), vinyl sulfoxide, a mild Michael acceptor, was generated to enable proximity-dependent reactions, such as protein-protein crosslinking. The features of this new approach include: (1) no undesirable side reactions during the genetic incorporation of the probe; (2) proximity-enhanced reactivity in the presence of large amount of free nucleophiles; and (3) cellular environment-induced reactivity that could be useful for the study of oxidative stress-associated biological processes. Given the importance of reactive oxygen species in diseases, our strategy may be useful for generating novel protein-based therapeutics that covalently bind to a specific target protein.
Fig. 1.

The overall design of the oxidation-induced and proximity-enhanced protein-protein crosslinking. Protein 1 and protein 2 interact to each other. The generation of vinyl sulfoxide from vinylthioether in the presence of H2O2 was confirmed by mass spectrometry (Fig. S10 of ESI).
To demonstrate our inducible strategy in introducing a bioreactive group into proteins, we synthesized an unnatural amino acid, N6-((2-(vinylthio)ethoxy)carbonyl)-L-lysine (VtK; Fig. 2A), that contains the desirable vinylthioether group. In order to identify an aminoacyl-tRNA synthetase (aaRS) that can charge an amber suppressor tRNA with VtK, we constructed an aaRS library that was derived from Methanosarcina barrkeri pyrrolysyl-tRNA synthetase. In this library, four amino acid residues, Leu270, Tyr271, Leu274, and Cys313, were randomized. After consecutive rounds of positive and negative selections, three hits were obtained including VtKRS-1, VtKRS-2, and VtKRS-3 (Table S1 of ESI).
Fig. 2.

Genetic incorporation of N6-((2-(vinylthio)ethoxy)carbonyl)-L-lysine (VtK). (A) The structure of VtK; (B) SDS-PAGE analysis of the expression of a myoglobin mutant (Mb-Lys99TAG) with and without VtK; (C) Fluorescence readings of cells expressing VtKRS-1 and sfGFP-Asn149TAG mutant. Fluorescence intensity was normalized to cell growth. All protein expressions were conducted either in the presence or in the absence of 1.0 mM VtK.
We further characterized VtKRS-1 (Leu274Ser and Cys313Val) by conducting protein expression with a myoglobin variant containing an amber mutation at position Lys99 (Mb-Lys99TAG). As shown in Fig. 2B, full-length myoglobin was only observed in the presence of VtK. The site-specific incorporation of VtK was also confirmed by mass spectrometry analysis (Fig. S1 of ESI). The mutant myoglobin (Mb-Lys99VtK) gave an observed average mass of 18485.1 Da (with the N-terminal methionine) and 18354.7 Da (without the N-terminal methionine), which were in close agreement with the calculated masses of 18485.1 Da (with the N-terminal methionine) and 18353.9 Da (without the N-terminal methionine). Besides myoglobin, we also examined protein expression of a super-folder GFP mutant (sfGFP-Asn149TAG). The protein yield was 25.3 mg/L. As shown in Fig. 2C, full-length sfGFP was detected only in cells supplemented with VtK, while no sfGFP fluorescence was observed otherwise. All above data confirmed that the incorporation efficiency and fidelity of VtK were excellent for the evolved VtKRS-1 mutant, which was used for all subsequent studies.
In order to generate vinyl sulfoxide from vinylthioether, we focused on catalytic oxidations using H2O2 as the final oxidant. After an initial catalyst screening, we chose sodium tungstate (Na2WO4) as the catalyst, which has been employed in Noyori Oxidation20 and considered as a physiologically harmless catalyst.21–22 It was reported that tungstate could be used to oxidize sulfide into sulfone and sulfoxide in the presence of a high22 and a low23 concentration of H2O2, respectively. In our experiments, the formation of sulfoxide was confirmed in the presence of 2.5 or 5 mM H2O2 (Fig. S10 of ESI). While it could be potentially toxic to cells at high concentrations, tungstate has been used as an insulin mimetic and displays a small toxicity profile.24 Although cautious are recommended for certain live cell experiments, we expect that Na2WO4 is suitable for studies where crosslinked proteins are formed in live cells but analyzed under cell-free conditions (e.g., mass spectrometry).
To examine the generation and the reactivity of vinyl sulfoxide in the context of protein, sfGFP-Asn149VtK was incubated with 5 mM H2O2, and varied concentrations of Na2WO4. In addition, N-dansylethylene-diamine (DNSEDA), a free amine-containing fluorescence probe that is reactive towards vinyl sulfoxide, was included in the reaction mixture as the labeling reagent. As shown in Fig. 3, fluorescence labeling of sfGFP-Asn149VtK by DNSEDA was clearly detected in the presence of both H2O2 and Na2WO4. The intensity of fluorescent signal decreased when the concentration of Na2WO4 was lowered from 0.5 to 0.05 mM. As controls, no detectable fluorescence labeling was observed when H2O2 or Na2WO4 or both were omitted from the reaction mixture. As another control, no apparent fluorescence signal was detected when wild-type sfGFP was used instead of sfGFP-Asn149VtK.
Fig. 3.

Small molecule labeling of protein through the oxidation-induced Michael addition reaction. Following labelling reactions, protein samples (wild-type sfGFP or sfGFP-Asn149VtK mutant) were denatured by heating, then analyzed by SDS-PAGE. The top panel shows Coomassie blue stained gel and the bottom panel shows the fluorescent image of the same gel before Coomassie blue treatment. DNSEDA, N-dansylethylene-diamine.
Next, we examined a proximity-enhanced protein-protein crosslinking using this oxidation-induced Michael addition reaction. We employed glutathione S-transferase (GST), an obligate dimer, as a model system in this study. Two mutants, GST-Asp61VtK and GST-Val62VtK, were constructed by genetic incorporation of VtK at positions 61 and 62 of GST, respectively. Since the two sites are at the edge of the GST dimer interface, the incorporation of VtK is unlike to significantly affect GST dimerization. According to the crystal structure of the GST dimer (PDB: 1Y6E),25 there are three nucleophiles in the close proximity of Asp61 and Val62, including Lys63, Cys84 and Lys86 (Fig. S2 of ESI). Once being incorporated at position 61 or 62 in one GST monomer, VtK should be in a close proximity of Cys84 and Lys86 from the other monomer. We envisage that the crosslinking reaction can happen as long as the protein-protein interaction does not prevent a direct contact between the two reactants. Given that the crystal data may not accurately reflect the actual GST dimer interface in the solution phase, we cannot directly predict whether Cys84 or Lys86 or both can readily react with VtK to afford a crosslinked GST dimer. While Lys63 is right next to Val62 and Asp61 on the same GST monomer, its side chain points toward an opposite direction relative to that of Asp61 and Val62. We hypothesize that Lys63 is unlikely to affect the crosslinking of the GST dimer. To test our hypothesis, we generated two mutants, GST-Asp61VtK-Lys63Gln and GST-Val62VtK-Lys63Gln, where Lys63 was mutated into non-nucleophilic Gln. In comparison to GST-Asp61VtK and GST-Val62VtK, the two Lys63Gln mutants did not lead to any increase in the crosslinking efficiency (Fig. S3 of ESI). In fact, slightly lower yield of the crosslinked GST dimer were observed (Fig. S3 of ESI).
The crosslinking experiments were conducted in phosphate buffer (pH 7.4) in the presence of 5 mM H2O2 and 0.5 mM Na2WO4 (as catalyst). After 60 min of incubation, the reaction mixture was treated with 10 mM tris(2-carboxyethyl)phosphine (TCEP) for 90 min. This step was used to eliminate any potential complications (such as oligomerization) by removing excess H2O2 and to reduce any potential disulfide bonds. As shown in Fig. 4, we observed 58% and 49% dimerization for the GST-Asp61VtK and GST-Val62VtK mutant, respectively. As a control, no significant crosslinking was detected for the wild-type GST under the same reaction conditions. The efficiency of oxidation-induced dimerization of GST was comparable to that of photo-crosslinking with genetically encoded benzophenone-containing probe (~50%).14
Fig. 4.
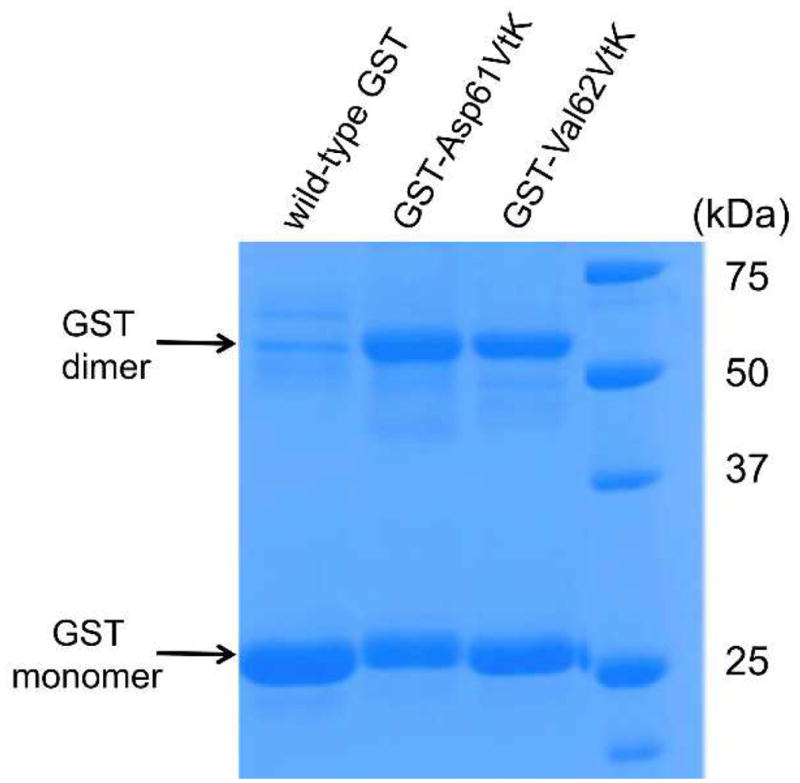
Crosslinking of GST dimer through the oxidation-induced Michael addition reaction. All GST samples (~50 μM) were incubated in the presence of 5 mM H2O2 and 0.5 mM Na2WO4 at 37 °C for 60 min. TCEP (10 mM) was added after the oxidation reaction before SDS-PAGE analysis.
We also examined a range of different reaction conditions. Firstly, we conducted the reaction in the presence of different concentrations of H2O2 and Na2WO4. While higher concentrations of H2O2 (5 mM) and Na2WO4 (0.5 mM) yielded slightly larger amount of dimers, lower concentrations of H2O2 (2.5 mM) and Na2WO4 (0.25 mM) worked similarly well (Fig. S4 of ESI). Next we examined different reaction time and found that a 10 min oxidation was good enough for a 40% and 19% dimerization of GST-Asp61VtK and GST-Val62VtK mutant (with 2.5 mM H2O2 and 0.25 mM Na2WO4), respectively (Fig. S5 of ESI). Finally, we examined the reaction at two different pH values. Similar crosslinking efficiency was observed at pH 7 and pH 8 (Fig. S6 of ESI).
To test the oxidation-induced Michael addition reaction in a more complex biological environment, we conducted this reaction in cell lysate and were able to detect crosslinked GST dimers for both the GST-Asp61VtK and the GST-Val62VtK mutants (lanes 2 and 5 in Fig. 5). In addition, we examined GST-Asp61VtK-Lys63Gln and GST-Val62VtK-Lys63Gln mutants. Again, the mutation at Lys63 position did not increase the crosslinking efficiency, which indicates that Lys63 did not react intramolecularly with the in situ generated Michael acceptor. Interestingly, we only observed a slight decrease in the crosslinking efficiency when one potential nucleophile, Lys86, was mutated (lanes 4 and 7 of Fig. 5). This result indicates that Cys84 is likely the main nucleophile in the Michael addition reaction. Indeed, when Cys84 was mutated to alanine, no apparent crosslinking product was detected (lanes 5 and 11 in Fig. S7 of ESI). We expect that both the nucleophilicity and the spatial arrangement of the two amino acid residues (Cys84 and Lys86) affect the crosslinking reaction. In terms of nucleophilicity, cysteine is a stronger nucleophile than Lys according to the Pearson HSAB theory. It is expected that cysteine reacts with vinyl sulfoxide at a faster rate than that of weaker nucleophiles (e.g., Lys and His). However, it has been observed that these amino acid residues can display very different trends of reactivity in proteomes.26 Since a primary amine could readily react with vinyl sulfoxide (Fig. 3) and the Cys84Ala mutant showed no apparent crosslinking product (Fig. 5), we hypothesize that the spatial constrain, rather than the nucelophilicity, was more important for the observed crosslinking reaction. A more rigorous investigation will be needed in the future to fully understand the role of the two factors in this crosslinking reaction.
Fig. 5.

Crosslinking of GST dimer in cell lysate. His-tagged GST variants were detected by Western blot using anti-His antibody. All GST-containing cell lysates were incubated in the presence of 50 mM H2O2 and 0.25 mM Na2WO4 at 37 °C. TCEP (50 mM) was added after the oxidation reaction. Lane 1, wild-type GST; lane 2, GST-Asp61VtK; lane 3, GST-Asp61VtK- Lys63Gln; lane 4, GST-Asp61VtK-Lys86Gln; lane 5, GST-Val62VtK; lane 6, GST-Val62VtK- Lys63Gln, lane 7, GST-Val62VtK-Lys86Gln; lane M, molecular weight marker.
Finally, we sought to further demonstrate the proximity-enhanced nature of this chemistry by conducting the reactions with cell lysate in the presence of a large access of free cysteine. To this end, the reaction was conducted in the presence of 50 mM cysteine. As shown in Fig. S7 of ESI, no apparent difference in crosslinking efficiency was observed with or without free cysteine.
In conclusion, we have demonstrated an oxidation-induced protein crosslinking reaction through an in situ generation of a mild Michael acceptor from a biochemically inert vinylthioether in the presence of H2O2. This strategy provides a new approach to introduce bioreactive probes into proteins without potential side reactions during the incorporation process. Such oxidation-induced protein crosslinking has potential to be employed in the oxidative stress-associated biological studies or the generation of novel protein-based therapeutics in the future.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (grant 1R01AI111862 to J.G. and W.N.), National Science Foundation (grant 1553041 to J.G.). The authors thank Drs. Sophie Alvarez and Mike Naldrett (Proteomics and Metabolomics Facility) for mass spectrometry analysis. Dedicated to Professor Jin-Pei Cheng on the occasion of his 70th birthday.
Footnotes
Footnotes relating to the main text should appear here. These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data.
Electronic Supplementary Information (ESI) available: Full experimental details and additional data. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Ramil CP, Lin Q. Chem Commun. 2013;49:11007–11022. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang K, Chin JW. ACS Chem Biol. 2014;9:16–20. doi: 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]
- 3.Boutureira O, Bernardes GJL. Chem Rev. 2015;115:2174–2195. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 4.Gong YK, Pan LF. Tetrahedron Lett. 2015;56:2123–2132. [Google Scholar]
- 5.Oliveira BL, Guo Z, Bernardes GJL. Chem Soc Rev. 2017;46:4895–4950. doi: 10.1039/c7cs00184c. [DOI] [PubMed] [Google Scholar]
- 6.Xiang Z, Ren H, Hu YS, Coin I, Wei J, Cang H, Wang L. Nat Meth. 2013;10:885–888. doi: 10.1038/nmeth.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen XH, Xiang Z, Hu YS, Lacey VK, Cang H, Wang L. ACS Chem Biol. 2014;9:1956–1961. doi: 10.1021/cb500453a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, Wang L. Angew Chem Int Ed Engl. 2014;53:2190–2193. doi: 10.1002/anie.201308794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xuan W, Li J, Luo X, Schultz PG. Angew Chem. 2016;128:10219–10222. [Google Scholar]
- 10.Furman JL, Kang M, Choi S, Cao Y, Wold ED, Sun SB, Smider VV, Schultz PG, Kim CH. J Am Chem Soc. 2014;136:8411–8417. doi: 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L. New Biotechnology. 2017;38:16–25. doi: 10.1016/j.nbt.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, Wang L. Angew Chem, Int Ed. 2014;53:3932–3936. doi: 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoppmann C, Maslennikov I, Choe S, Wang L. J Am Chem Soc. 2015;137:11218–11221. doi: 10.1021/jacs.5b06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin JW, Martin AB, King DS, Wang L, Schultz PG. Proc Natl Acad Sci U S A. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG. J Am Chem Soc. 2002;124:9026–9027. doi: 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- 16.Tippmann EM, Liu W, Summerer D, Mack AV, Schultz PG. ChemBioChem. 2007;8:2210–2214. doi: 10.1002/cbic.200700460. [DOI] [PubMed] [Google Scholar]
- 17.Ai HW, Shen W, Sagi A, Chen PR, Schultz PG. ChemBioChem. 2011;12:1854–1857. doi: 10.1002/cbic.201100194. [DOI] [PubMed] [Google Scholar]
- 18.Chou C, Uprety R, Davis L, Chin JW, Deiters A. Chem Sci. 2011;2:480–483. [Google Scholar]
- 19.Zhang M, Lin SX, Song XW, Liu J, Fu Y, Ge X, Fu XM, Chang ZY, Chen PR. Nat Chem Biol. 2011;7:671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- 20.Sato K, Aoki M, Takagi J, Noyori R. J Am Chem Soc. 1997;119:12386–12387. [Google Scholar]
- 21.Spector WS, editor. Handbook of Toxicology. Vol. I. Acute Toxicities of Solids, Liquids, and Gases to Laboratory Animals. Vol. 1. W. B. Saunders Co; 1956. p. 280. [Google Scholar]
- 22.Noyori R, Aoki M, Sato K. Chem Commun. 2003:1977–1986. doi: 10.1039/b303160h. [DOI] [PubMed] [Google Scholar]
- 23.Karimi B, Ghoreishi-Nezhad M, Clark JH. Org Lett. 2005;7:625–628. doi: 10.1021/ol047635d. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Hernandez CJ, Llorens-Agost M, Calbo J, Murguia JR, Guinovart JJ. FEBS Lett. 2013;587:1579–1586. doi: 10.1016/j.febslet.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Rufer AC, Thiebach L, Baer K, Klein HW, Hennig M. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2005;61:263–265. doi: 10.1107/S1744309105004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weerapana E, Simon GM, Cravatt BF. Nat Chem Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.