

Abstract
Repeated stimulation of mu-opioid receptors (MOR), by a MOR selective agonist DAMGO induces Type II priming, a form of nociceptor neuroplasticity, which has two components: opioid-induced hyperalgesia (OIH) and prolongation of prostaglandin-E2 (PGE2)-induced hyperalgesia. We report that intrathecal antisense knockdown of the MOR in nociceptors, prevented the induction of both components of Type II priming. Type II priming was also eliminated by SSP-saporin, which destroys the peptidergic class of nociceptors. Since the epidermal growth factor receptor (EGFR) participates in MOR signaling, we tested its role in Type II priming. The EGFR inhibitor, tyrphostin AG 1478, prevented the induction of prolonged PGE2-induced hyperalgesia, but not OIH, when tested out to 30 days after DAMGO. However, even when repeatedly injected, an EGFR agonist did not induce hyperalgesia or priming. A phosphopeptide, which blocks the interaction of Src, focal adhesion kinase (FAK) and EGFR, also prevented DAMGO-induced prolongation of PGE2 hyperalgesia, but only partially attenuated the induction of OIH. Inhibitors of Src and mitogen-activated protein kinase (MAPK) also only attenuated OIH. Inhibitors of matrix metalloproteinase, which cleaves EGF from membrane protein, markedly attenuated the expression, but did not prevent the induction, of prolongation of PGE2 hyperalgesia. Thus, while the induction of prolongation of PGE2-induced hyperalgesia at the peripheral terminal of peptidergic nociceptor is dependent on Src, FAK, EGFR, and MAPK signaling, Src, FAK, and MAPK signaling is only partially involved in the induction of OIH.
Keywords: Hyperalgesic priming, Hyperalgesia, Epidermal growth factor receptor (EGFR), Mu-opioid receptor (MOR), Chronic pain
Introduction
Hyperalgesic priming refers to a neuroplastic change in nociceptors that has been shown to contribute to the transition from acute to chronic pain [3; 41; 47]. We have recently described a second form of hyperalgesic priming (Type II), induced by repeated exposure to DAMGO, a mu-opioid receptor (MOR) agonist [5; 8]. Type II priming is characterized not only by prolongation of prostaglandin E2 (PGE2)-induced hyperalgesia, as observed in Type I priming, but also by the onset of opioid-induced hyperalgesia (OIH) [5; 31]. A second important difference between these two types of priming is that Type I is induced by the activation of protein kinase epsilon (PKCε) [3; 23; 41] and maintained by protein translation [23] in the terminals of nonpeptidergic, IB4-positive nociceptors [30], while the maintenance of Type II priming is dependent on the simultaneous activation of Src tyrosine kinase (Src) and mitogen-activated protein kinase (MAPK) [8].
Mechanisms mediating hyperalgesic priming can be divided into those mediating induction, expression [5] and maintenance [8]. In this study, we investigated the mechanisms mediating the induction of OIH and the prolongation of PGE2-induced hyperalgesia in Type II hyperalgesic priming. In particular, we examined the role of the interaction of MOR, a Gαi-protein coupled receptor (GPCR) with epidermal growth factor receptor (EGFR), a receptor tyrosine kinase (RTK) that has been demonstrated to have crosstalk with opioid receptors [10; 16; 42]. We also investigated second messenger, downstream of MOR, involved in the induction of OIH and prolongation of PGE2 hyperalgesia in Type II hyperalgesic priming.
Materials and Methods
Animals
Male Sprague–Dawley rats (240–400 g, Charles River Laboratories, Hollister, CA, USA), housed three per cage, under a 12-hour light/dark cycle, in a humidity and temperature controlled animal care facility at the University of California, San Francisco, were used in the presented experiments. Water and food were available ad libitum. Nociceptive evaluations were performed between 10:00 A.M. and 5:00 P.M. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California at San Francisco and adhered to the National Institutes of Health Guide for the care and use of laboratory animals.
Testing mechanical nociceptive threshold
An Ugo Basile Analgesymeter® (Randall-Selitto paw-withdrawal test, Stoelting, Chicago, IL, USA) was used to quantify mechanical nociceptive threshold, by the application of a linearly increasing mechanical force to the dorsum of the rat's hind paw, as previously described [5; 8; 27; 54; 55]. Rats were placed in acrylic restrainers for 30 minutes prior to experiments. The restrainers provided proper ventilation and allowed the extension of the hind legs from lateral ports in the cylinder during assessment of nociceptive threshold. The nociceptive threshold was defined as the force (expressed in grams) at which the rat withdrew its paw. Baseline paw-pressure nociceptive threshold was defined as the mean of the three readings taken before the test agents were injected. Only one paw was used in an experiment, and each experiment was performed on a separate group of rats. To minimize experimenter bias, individuals conducting the behavioral experiments (D.A. and L.F.F) were blinded to experimental interventions.
Drug administration
The following drugs were used in this study: Prostaglandin-E2 (PGE2, a direct-acting hyperalgesic agent that sensitizes nociceptors), DAMGO ([D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt, a MOR agonist), salirasib (a RAS inhibitor), GW5074 (a cRaf1 kinase inhibitor), SU 6656 (a Src family kinase inhibitor), and Ilomastat (an inhibitor of a wide variety of matrix metalloproteases/MMPs), all from Sigma-Aldrich (St. Louis, MO, USA); recombinant rat EGF protein (an EGF ligand [EGFR agonist]) from Abcam (Cambridge, MA, USA); MMP-9 inhibitor from Calbiochem (Billerica, MA, USA); Tyrphostin AG 1478 hydrochloride (an EGFR inhibitor), U0126 (MAPK/ERK inhibitor), and N-Acetyl-O-phosphono-Tyr-Glu-Glu-Ile-Glu (peptide SH2; a phosphopeptide ligand for the Src SH2 domain that blocks Src interactions with EGFR and FAK) from Tocris (Avonmouth, Bristol, UK).
The PGE2 stock solution (1 μg/μL), initially prepared in ethanol 100%, was diluted in saline (0.9% NaCl), yielding a final ethanol concentration <1%. DAMGO, recombinant rat EGF protein and peptide SH2 were dissolved in saline. All other drugs were dissolved in DMSO 100% (Sigma-Aldrich) and further diluted in saline containing 2% of Tween 80 (Sigma-Aldrich). The final concentration of Tween 80 and DMSO was ∼2%. Intradermal administration of drugs was performed on the dorsum of the hind paw, using a 30-gauge hypodermic needle adapted to a 50 μL Hamilton syringe by a segment of PE/10 polyethylene tubing (Becton Dickinson, Franklin Lakes, NJ, USA). The administration of all drugs, except PGE2, recombinant rat EGF and DAMGO, was preceded by a hypotonic shock (injection of 1 μL of distilled water, separated from the drug in the same syringe by a bubble, avoiding their mixing) in order to facilitate their entry into the nerve terminal through the cell membrane [12; 13].
Mu-opioid receptor antisense
To investigate the role of MOR in induction of DAMGO-induced Type II priming, oligodeoxynucleotides (ODN) antisense (AS) for MOR mRNA was used [8; 33; 49]. The AS-ODN sequence for MOR, 5′-CGC-CCC-AGC-CTC-TTC-CTC-T-3′, (Invitrogen Life Technologies, Carlsbad, CA, USA) was directed against a unique region of rat MOR (UniProtKB database entry P33535 [OPRM_RAT] antisense sequence to block translation and downregulate the gene expression of all 8 known isoforms [MOR]). The ODN mismatch (MM) sequence, 5′-CGC-CCC-GAC-CTC-TTC-CCT-T-3′ for MOR, was a scrambled version of the antisense sequence that has the same base pairs and GC ratio, but scrambled nucleotide order, with little or no homology to any mRNA sequences posted at GeneBank.
MM- or AS-ODNs were reconstituted in nuclease-free 0.9% NaCl, before use, and then injected intrathecally at a dose of 6 μg/μL in a volume of 20 μL (120 μg/20 μL). ODNs were injected for 3 consecutive days, once a day and, at the 4th day, repeated (hourly × 4) intradermal injections of DAMGO (1 μg) was performed and the mechanical nociceptive threshold evaluated 30 min after the 4th injection. Injections of MOR MM- or AS-ODN were performed for 2 more days (until day 5) and, on the 6th day (∼ 17 hours after the last injection of ODN), DAMGO (1 μg) or PGE2 (100 ng) was injected intradermally. DAMGO or PGE2 was injected again 5, 15 and 30 days after the last injection of ODN. Rats were anesthetized with isoflurane (2.5% in O2) and, the ODNs, administered using a microsyringe adapted to a 30-gauge needle, placed into the subarachnoid space, between the L4 and L5 vertebrae, as previously described [1]. The dose and volume of ODN injected were, respectively, 120 μg in 20 μL. During the injections, the sudden flick of the rat's tail confirmed the access to the subarachnoid space [39]. Rats recovered consciousness approximately 2 minutes after the injection. Of note, the attenuation of the expression of proteins involved in nociceptor sensitization by using intrathecal AS-ODNs has been previously demonstrated by several studies [5; 6; 8; 11; 25; 40; 46; 51-53].
Intrathecal administration of SSP-saporin
[Sar9, Met(O2)11]-substance P-saporin, a SP-positive nociceptor neurotoxin (SSP-Saporin, Advanced Targeting Systems, San Diego, CA) was prepared in saline (5 ng/μL), and 20 μL was injected intrathecally, 14 days before the nociceptive tests [4; 7]. The addition of [Sar9, Met(O2)11] to substance P conjugated to saporin makes the agent more stable and a more potent toxin than when substance P alone is bound to saporin. The pre-treatment interval and dose were established by Wiley and colleagues [59] and Choi and colleagues [18], who observed prominent loss of neurons expressing the neurokinin 1 (NK1) receptor in the laminae 1 of the lumbar dorsal horn after using this protocol [32; 34; 56; 58].
Induction of opioid-induced hyperalgesia and prolongation of PGE2-induced hyperalgesia
The repeated (hourly × 4) intradermal injections of the MOR agonist DAMGO (1 μg) produces opioid-induced hyperalgesia (OIH) and a latent state of hyper-responsiveness to subsequent injection of pro-algesic mediators [5; 8; 31], referred to as Type II hyperalgesic priming [5; 8]. This neuroplasticity is expressed as prolongation of PGE2-induced mechanical hyperalgesia, lasting more than 4 hours, as opposed to the injection of PGE2 in naive paws, in which hyperalgesia is no longer present by 2 hours [2]. To investigate the signaling pathways involved in the induction of Type II hyperalgesic priming (OIH and prolongation of PGE2-induced hyperalgesia) by repeated exposure to a MOR agonist, all inhibitors were injected 10 min before the first injection of DAMGO. The OIH produced by repeated injections of DAMGO, was evaluated 30 min after the 4th injection of DAMGO and 5, 15 and 30 days after the repeated injections of DAMGO, by a single injection of DAMGO at each time point. The presence of hyperalgesia, 30 min after the injection of DAMGO, is a characteristic of OIH. To evaluate the signaling pathways involved in the prolongation of PGE2-induced hyperalgesia, all inhibitors were injected 10 min before the first injection of DAMGO. PGE2 was injected intradermally 5, 15 and 30 days after repeated injections of DAMGO. Mechanical hyperalgesia was evaluated for after 30 min and again at 4 hours after the injection of PGE2; the presence of hyperalgesia at the 4th hour is characteristic of hyperalgesic priming [3; 5-8; 24; 41]. If inhibitors were able to attenuate the expression (see Fig. 1) of Type II hyperalgesic priming (OIH and prolongation of PGE2-induced hyperalgesia), we also evaluated their role 30 days after injection, when DAMGO or PGE2 was again injected. If at this time, the DAMGO-induced hyperalgesia or prolongation of PGE2-induced hyperalgesia was still not present at 30 min (for DAMGO) or the 4th hour (for PGE2), then the inhibitor was considered able to prevent the induction (see Fig. 1) of DAMGO-induced Type II hyperalgesic priming (OIH and/or prolongation of PGE2-induced hyperalgesia).
Figure 1.
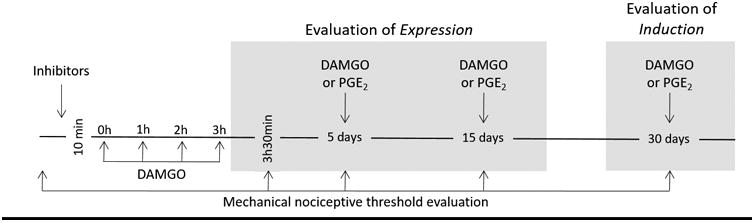
Schematic depicting the protocol used to evaluate the expression and induction of Type II hyperalgesic priming (OIH and prolongation of PGE2-induced hyperalgesia) induced by repeated exposure to DAMGO.
Data analysis
Data from all experiments are presented as mean ± SEM of n independent observations; the dependent variable was change in mechanical paw-withdrawal threshold, expressed as percentage change from baseline. Statistical evaluations were made using GraphPad Prism 5.0 statistical software (GraphPad Software). A p value < 0.05 was considered statistically significant. To evaluate the role of second messengers in the induction of Type II priming, inhibitors were injected in only one paw. In the experiments in which the MM- or AS-ODN was injected intrathecally, only the left paws were used (6 rats per group). No significant difference in mechanical nociceptive threshold was observed before the repeated injections of DAMGO and 5, 15 or 30 days later, when PGE2 or DAMGO was administered (average mechanical nociceptive threshold before repeated (hourly × 4) injections of DAMGO: 132.7 ± 1.11 g; average mechanical nociceptive threshold before PGE2 or DAMGO injection: 129.3 ± 1.23 g; n = 132 paws (= 132 rats); paired Student's t test, t(131) = 1.017, p = 0.4219). As indicated in the figure legends, Student's t test or two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, was performed to compare the magnitude of the hyperalgesia induced by the 4th injection of DAMGO or by the subsequent injection of PGE2 or DAMGO, in the different groups, or to compare the effect produced by different treatments on the DAMGO-induced OIH (evaluated 30 min after the 4th injection or 5, 15 and 30 days after injection) or the prolongation of PGE2-induced hyperalgesia (evaluated 4 hours after its injection at days 5, 15 and 30 after repeated injections of DAMGO) with the control (vehicle) groups.
Results
Mu-opioid receptor (MOR) dependence
Oligodeoxynucleotides (ODNs) mismatch (MM) or antisense (AS) to MOR mRNA were used to study the role of MOR, in the peripheral terminal of the nociceptor, in the induction of Type II hyperalgesic priming (OIH and prolongation of PGE2 hyperalgesia). Rats were treated daily with MM- or AS-ODN, by intrathecal administration, for 3 consecutive days and on the 4th day, ∼17 hours after an injection of ODN, rats received repeated (hourly × 4) intradermal injections of DAMGO. In the group treated with antisense, DAMGO-induced hyperalgesia was prevented when the mechanical nociceptive threshold was evaluated 30 min after the injection of 4th dose of DAMGO (Fig. 2A). MM- or AS-ODN was injected for 2 more days (days 4 and 5) and on the 6th day, ∼17 hours after the last injection of ODN, a single injection of DAMGO was administered. DAMGO did not induce hyperalgesia in the AS-ODN-treated group (Fig. 2A). When DAMGO was injected again 5 (Fig. 2B), 15 (Fig. 2C) and 30 (Fig. 2D) days after the last injection of MOR ODN, the mechanical hyperalgesia evaluated 30 min after its injection was not present in AS-ODN-treated rats. Another group of rats was pretreated with either MM- or AS-ODN for 5 consecutive days and on the 4th day, received repeated (hourly × 4) intradermal injections of DAMGO. On the 6th day, ∼17 hours after the injection of ODN, an intradermal injection of PGE2 was performed. In the AS-ODN-treated group, prolongation of PGE2 hyperalgesia was not present (Fig. 2E). The prolongation of PGE2 hyperalgesia was also not present, 5 (Fig. 2F), 15 (Fig. 2G) and 30 (Fig. 2H) days after the last injection of MOR AS-ODN. Taken together these findings indicate that DAMGO acts at MOR to induce both components of Type II priming (OIH and prolongation of PGE2 hyperalgesia).
Figure 2. Induction of Type II hyperalgesic priming (OIH and prolongation of PGE2-induced hyperalgesia) is mu-opioid receptor (MOR) dependent.

Rats were treated daily with an intrathecal injection of MM-ODN (120 μg/20 μL/day; black bars) or AS-ODN (120 μg/20 μL/day; dotted bars) for mu-opioid receptor (MOR) mRNA for 3 consecutive days. The average baseline mechanical nociceptive threshold, before ODNs, was 131.3 ± 1.9 g for the MM-ODN group and 132.1 ± 1.8 g for the AS-ODN group.
Upper panel (A – D). On the 4th day, ∼17 hours after the last injection of MM- or AS-ODN, repeated (hourly × 4) intradermal injections of DAMGO (1 μg) were administered and, the mechanical nociceptive threshold evaluated 30 min after the 4th injection of DAMGO. Average baseline mechanical nociceptive threshold, before repeated injections of DAMGO, was 133.3 ± 1.1 g for the MM-ODN group and 131.2 ± 1.5 g for the AS-ODN group (A). In the AS-ODN-treated group, hyperalgesia induced by the 4th injection of DAMGO (OIH) was prevented (t(10) = 17.51, *** p < 0.0001, when MOR MM-ODN- and MOR AS-ODN-treated groups was compared at 30 min after the 4th injection of DAMGO; unpaired Student's t test). Treatment with intrathecal injections of MOR MM- or AS-ODN was then continued for 2 additional days and, on the 6th day (∼17 hours after the last injection of MM- or AS-ODN), DAMGO (1 μg) was injected intradermally on the dorsum of the hind paw and the mechanical nociceptive threshold evaluated 30 min after injection. The average baseline mechanical nociceptive threshold, before a single injection of DAMGO, was 130.7 ± 1.9 g for the MM-ODN group and 129.8 ± 2.2 for the AS-ODN group. When compared to MOR MM-ODN-treated group, DAMGO-induced hyperalgesia was completely blocked in the MOR AS-ODN-treated group (F(1,20) = 53.57, *** p < 0.0001, when MOR MM-ODN-treated and AS-ODN-treated group was compared 30 min after the injection of DAMGO; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Five (B), 15 (C) and 30 (D) days after the last injection of MOR MM- or AS-ODN, when DAMGO (1 μg) was intradermally again injected, DAMGO-induced hyperalgesia at 30 min was still blocked in the MOR AS-ODN-treated group (F(1,10) = 34.16, *** p = 0.0002, for 5 days (B), F(1,10) = 46.10, *** p < 0.0001 for 15 days (C), and F(1,10) = 56.50, *** p < 0.0001 for 30 days (D) when the MOR MM-ODN-treated and MOR AS-ODN-treated groups were compared, 30 min after injection of DAMGO; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Of note, the average baseline mechanical nociceptive threshold, before DAMGO, was 131.9 ± 1.6 g (15 days) and 130.7 ± 2.2 g (30 days) for the MM-ODN group and 130.8 ± 1.5 g (15 days) and 129.9 ± 1.9 g (30 days) for the AS-ODN group.
Lower panel (E – H). On the 6th day (∼17 hours after the last injection of MOR MM- or AS-ODN, and 2 days after having received repeated injections of DAMGO), groups of rats were treated with an intradermal injection of PGE2 (100 ng). The average baseline mechanical nociceptive threshold, before repeated injections of DAMGO, was 129.8 ± 1.9 g for the MM-ODN group and 131.8 ± 1.3 g for the AS-ODN group; before the injection of PGE2, was 128.5 ± 2.4 g for the MM-ODN group and 129.9 ± 1.9 g for the AS-ODN group. (E). Treatment with MOR AS-ODN was able to prevent the prolongation of PGE2 hyperalgesia at the 4th hour, compared to the MOR MM-ODN-treated group (F(2,20) = 159.45, *** p < 0.0001, when MOR MM-ODN-treated group is compared to MOR AS-ODN-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Five (F) and 15 (G) days after the last injection of MOR MM- or AS-ODN, PGE2 (100 ng) was again injected. The prolongation of PGE2 hyperalgesia at the 4th hour was still blocked in the group that had been treated with MOR AS-ODN (F(2,20) = 259.52, *** p < 0.0001, for 5 days (F) and, F(2,20) = 202.30, *** p < 0.0001 for 15 days (G), when MOR MM-ODN-treated group is compared to MOR AS-ODN-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Of note, the average baseline mechanical nociceptive threshold, before PGE2, was 130.7 ± 1.2 g (15 days) and 129.7 ± 1.9 g (30 days) for the MM-ODN group and 129.9 ± 2.1 g (15 days) and 131.2 ± 1.6 g (30 days) for the AS-ODN group. H. Thirty days after the last injection of ODN, when the average baseline mechanical nociceptive threshold was 130.1 ± 1.3 g for the MM-ODN group and 132.1 ± 1.1 g for the AS-ODN group, PGE2 was again injected intradermally. The prolongation of PGE2-induced hyperalgesia was again markedly attenuated at the 4th hour in the MOR AS-ODN-treated group (F(2,20) = 108.36, *** p < 0.0001, when MOR MM-ODN-treated group is compared to MOR AS-ODN-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These findings indicate that MOR is necessary for the induction of Type II priming (OIH and prolongation of PGE2 hyperalgesia). n = 6 rats/6 paws per treated-group.
Lesion of IB4-negative nociceptors prevents priming
We previously demonstrated that IB4-saporin, which destroys nonpeptidergic IB4-positive nociceptors and mediates Type I priming [7; 30], did not attenuate DAMGO-induced Type II priming (OIH or prolongation of PGE2 hyperalgesia) [5]. In rats pretreated with SSP-saporin, which destroys IB4-negative peptidergic nociceptors, repeated (hourly × 4) injections of DAMGO did not induce hyperalgesia, evaluated after its 4th injection, or produce prolongation of PGE2 hyperalgesia, when tested 5 days later (Fig. 3). These data support the suggestion that Type II hyperalgesic priming occurs in peptidergic, IB4-negative nociceptors.
Figure 3. Lesion of IB4-negative nociceptors prevents Type II priming.

Rats were treated with vehicle (20 μL; black bars) or SSP-saporin (100 ng/20 μL; gray bars) by intrathecal injection. The average baseline mechanical nociceptive threshold, before treatments, was 128.3 ± 1.8 g for the vehicle group and 130.3 ± 1.5 g for the SSP-saporin group. Two weeks later, when the average baseline mechanical nociceptive threshold was 130.5 ± 1.5 g for the vehicle group and 131.1 ± 1.8 g for the SSP-saporin group, DAMGO (1 μg) was repeatedly (hourly × 4) injected on the dorsum of the hind paw. In the SSP-saporin-treated group, the 4th injection of DAMGO did not induce hyperalgesia (t(10) = 7.481, *** p < 0.0001, when vehicle- and SSP-saporin-treated groups are compared 30 min after the 4th injection of DAMGO; unpaired Student's t test). Five days later, when mechanical nociceptive threshold was not different from the pre-DAMGO baseline (129.1 ± 2.2 g, t(5) = 1.581; p = 0.1747, for the vehicle-treated group; 130.8 ± 1.0 g, t(5) = 1.536; p = 0.1852, for the SSP-saporin-treated group, when the mechanical nociceptive threshold is compared before and 5 days after DAMGO; paired Student's t test), PGE2 (100 ng) was injected and the mechanical nociceptive threshold evaluated 30 min and 4 hours later. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test showed PGE2 hyperalgesia at 30 min in both groups, with no significant (ns) difference between the groups. However, the prolongation of PGE2 hyperalgesia was markedly attenuated in the SSP-saporin-treated group at the 4th hour (F(2,14) = 45.53, *** p < 0.0001, when vehicle-treated group is compared to SSP-saporin-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that IB4-negative nociceptors are necessary for OIH and the prolongation of PGE2 hyperalgesia induced by repeated exposure to DAMGO. n = 6 paws per group.
EGFR in DAMGO-induced prolongation of PGE2 hyperalgesia
We next determined if inhibition of EGFR is able to prevent DAMGO-induced prolongation of PGE2 hyperalgesia. Pre-administration of the EGFR inhibitor (tyrphostin AG 1478), prevented the prolongation of PGE2 hyperalgesia at the 4th hour when tested 5 (Fig. 4A), 15 (Fig. 4B) or 30 (Fig. 4C) days after repeated injections of DAMGO. These findings indicate that induction of DAMGO-induced prolongation of PGE2 hyperalgesia is EGFR dependent. An EGFR agonist (EGF ligand) alone was, however, unable to induce either hyperalgesia or prolongation of PGE2 hyperalgesia (Fig. 4D).
Figure 4. MOR-EGFR crosstalk in the induction of prolongation of PGE2 hyperalgesia.

Rats received an intradermal injection of vehicle (5 μL; black bars) or EGFR inhibitor (Tyrphostin AG 1478, 1 μg; dotted bars). The average baseline mechanical nociceptive threshold, before treatments, was 130.1 ± 1.4 g for the vehicle group and 132.0 ± 1.3 g for the EGFR inhibitor group. Ten minutes later, repeated (hourly × 4) intradermal injections of DAMGO (1 μg) were performed on the dorsum of the hind paw. Five days later, when mechanical threshold was not different from the pre-DAMGO baseline (128.8 ± 2.1 g, t(5) = 0.3162; p = 0.7646, for vehicle-treated group; 130.7 ± 2.0 g, t(5) = 2.169; p = 0.0822, for EGFR inhibitor-treated group, when the mechanical nociceptive threshold is compared before and 5 days after DAMGO; paired Student's t test), PGE2 (100 ng) was injected at the same site and mechanical nociceptive threshold evaluated 30 min and 4 hours later. In the group previously treated with EGFR inhibitor, the prolongation of PGE2 hyperalgesia was almost completely eliminated at the 4th hour (F(2,16) = 175.68, *** p < 0.0001, when vehicle-treated group is compared to EGFR inhibitor treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). B – C. When PGE2 (100 ng) was again injected on the dorsum of the hind paw, 15 (B) or 30 (C) days after repeated exposure to DAMGO, the prolongation of PGE2-induced hyperalgesia was not present at the 4th hour in the EGFR inhibitor-treated group (F(2,16) = 98.89, *** p < 0.0001 for 15 days (B) and, F(2,16) = 120.35, *** p < 0.0001 for 30 days (C), when the vehicle-treated group is compared to EGFR inhibitor-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These data support a role of crosstalk between mu-opioid and EGF receptors in the induction of the prolongation of PGE2 hyperalgesia by repeated exposure to DAMGO. Of note, the average baseline mechanical nociceptive threshold, before PGE2, was 129.7 ± 1.7 g (15 days) and 131.4 ± 1.8 g (30 days) for the vehicle group and 131.1 ± 1.1 g (15 days) and 130.6 ± 1.2 g (30 days) for the EGFR inhibitor group. D. A different group of rats, with average baseline mechanical nociceptive threshold of 130.9 ± 2.0 g, was treated with repeated (hourly × 4) intradermal injections of the EGFR agonist (EGF ligand; 1 μg/5 μL; gray bars) and the mechanical nociceptive threshold evaluated 30 min after each injection. No change in nociceptive threshold was observed after the 1st (not significant, ns, 130.3 ± 2.2 g, t(5) = 1.267, p = 0.2610), 2nd (ns, 130.0 ± 1.6 g, t(5) = 1.066, p = 0.3352), 3rd (ns, 128.0 ± 2.1 g, t(5) = 2.236, p = 0.0756) or 4th (ns, 128.3 ± 1.9 g, t(5) = 2.214, p = 0.0778) injection of EGFR agonist (when the mechanical nociceptive threshold before the 1st injection and 30 min after each injection of EGFR agonist, is compared; paired Student's t test). Five days (5 d) later, when the mechanical threshold was not different from the pre-EGFR agonist baseline (132.3 ± 1.7 g, t(5) = 2.485; p = 0.0555, when the mechanical nociceptive threshold is compared before and 5 days after repeated injections of EGFR agonist; paired Student's t test), PGE2 (100 ng) was injected at the same site, on the dorsum of the hind paw, and the mechanical nociceptive threshold evaluated 30 min and 4 hours after injection. PGE2 induced hyperalgesia 30 min after injection (t(5) = 10.03, *** p = 0.0002, when mechanical thresholds before and 30 min after PGE2, were compared; paired Student's t test), but not at the 4th hour (t(5) = 0.7895, p = 0.4656, when mechanical thresholds before and 4 hours after PGE2, were compared; paired Student's t test), indicating that repeated activation of EGF receptor does not induce hyperalgesic priming. n = 6 paws per group.
Src, FAK, and EGFR signaling in the induction of prolongation of PGE2 hyperalgesia
We next examined signaling pathways downstream of EGFR that mediate the EGFR-dependent induction of prolonged PGE2 hyperalgesia by repeated exposure to DAMGO. SH2, a peptide that blocks the interaction of Src, FAK and EGFR, was able to prevent the development of the prolongation of PGE2-induced hyperalgesia when tested 5 (Fig. 5A), 15 (Fig. 5B) and 30 (Fig. 5C) days after repeated exposure to DAMGO. These findings indicate that signaling via Src, FAK, and EGFR is required for the induction of the prolongation of PGE2 hyperalgesia by a MOR agonist.
Figure 5. EGFR signaling in the induction of prolongation of PGE2 hyperalgesia.

Rats were treated intradermally with vehicle (5 μL; black bars) or phosphopeptide SH2 (SH2, which blocks the Src interaction's with FAK and EGFR; 1 μg, withe bars). The average baseline mechanical nociceptive threshold, before treatments, was 131.3 ± 1.8 g for the vehicle group and 129.7 ± 1.7 g for the SH2 group. Starting ten minutes later, repeated (hourly × 4) intradermal injections of DAMGO (1 μg) were performed on the dorsum of the hind paw. Five days later, when the mechanical nociceptive threshold was not different from the pre-DAMGO baseline (129.3 ± 1.2 g, t(5) = 0.2758; p = 0.738, for the vehicle-treated group; 128.7 ± 2.1 g, t(5) = 0.1644; p = 0.8759, for the SH2-treated group; when the mechanical nociceptive threshold is compared before and 5 days after DAMGO; paired Student's t test), PGE2 (100 ng) was injected at the same site on the dorsum of the hind paw and the mechanical nociceptive threshold evaluated 30 min and 4 hours later. In the SH2-treated group, the prolongation of PGE2 hyperalgesia was inhibited at the 4th hour (F(2,30) = 144.07, *** p < 0.0001, when vehicle-treated group was compared to SH2-treated group at the 4th hour after the injection of PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). B. Fifteen days later, when the average baseline mechanical nociceptive threshold was 131.5 ± 1.0 g for the vehicle group and 128.9 ± 1.9 g for the SH2 group, PGE2 (100 ng) was again injected intradermally. We observed that the 4th hour of PGE2-induced hyperalgesia was still attenuated in the SH2-treated group (F(2,30) = 309.92, *** p < 0.0001, when SH2-treated group was compared to vehicle-treated group at the 4th hour after PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). C. Thirty days after repeated (hourly × 4) injections of DAMGO, when the average baseline mechanical nociceptive threshold was 131.7 ± 1.7 g for the vehicle group and 129.3 ± 1.4 g for the SH2 group, PGE2 (100 ng) was again injected intradermally. In the SH2-treated group the prolongation of PGE2 hyperalgesia was completely inhibited (F(2,30) = 168.26, *** p < 0.0001, when SH2-treated group is compared to vehicle-treated group at the 4th hour after PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), demonstrating that the interaction of Src with FAK and EGFR plays a role in the induction of prolongation of PGE2 hyperalgesia by repeated exposure to DAMGO. n = 6 paws per group.
Induction of OIH
Since our previous [5; 8] and current findings demonstrate the participation of Src, MAPK, FAK and EGFR in the induction of prolongation of PGE2-induced hyperalgesia by repeated exposure to DAMGO, we tested inhibitors for these second messengers in OIH. MAPK, Src and EGFR inhibitors, and SH2 were able to block the hyperalgesia induced by the 4th injection of DAMGO (Fig. 6A). Five (Fig. 6A) and 15 (Fig. 6B) days after the treatment with these inhibitors, we observed that in the Src inhibitor- and SH2-treated groups DAMGO was unable to induce hyperalgesia, while in MAPK and EGFR inhibitors-treated groups, DAMGO-induced hyperalgesia was only partially developed. When DAMGO was again injected 30 days (Fig. 6C) after treatment with these inhibitors, a partial attenuation of DAMGO-induced hyperalgesia was still observed in the MAPK and Src inhibitors and SH2-treated groups (Fig. 6C), while no significant attenuation was observed between vehicle- and EGFR inhibitor-treated groups. These findings suggest a partial contribution of Src, FAK, and MAPK to the induction of OIH.
Figure 6. Second messengers in induction of OIH.

Rats were treated intradermally with vehicle (5 μL), MAPK inhibitor (U0126, 1 μg; dark gray bars), Src inhibitor (SU 6656, 1 μg; light gray bars), EGFR inhibitor (tyrphostin AG 1478; 1 μg; white bars) or phosphopeptide SH2 (SH2, which blocks the interaction of Src with FAK and EGFR; 1 μg; dotted bars). The average baseline mechanical nociceptive threshold, before treatments, was 132.3 ± 1.8 g for the vehicle group, 129.9 ± 1.8 g for the MAPK inhibitor, 133.3 ± 1.3 g for the Src inhibitor group, 130.7 ± 1.1 g for the EGFR inhibitor group, and 130.3 ± 2.0 g for the SH2 group. Starting ten minutes later, repeated (hourly × 4) intradermal injections of DAMGO (1 μg) were performed on the dorsum of the hind paw and the mechanical nociceptive threshold evaluated 30 min after the 4th injection of DAMGO. A. The hyperalgesia induced by the 4th injection of DAMGO was prevented by all inhibitors (F(4,40) = 65.72, *** p < 0.001, when vehicle-treated group is compared to all inhibitor-treated groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). Five days later, when the mechanical thresholds were not different from the pre-DAMGO baseline (130.3 ± 1.4 g, t(5) = 2.215; p = 0.0776, for the vehicle-; 130.7 ± 1.2 g, t(5) = 0.5407; p = 0.6119, for MAPK inhibitor; 129.9 ± 1.0 g, t(5) = 1.064; p = 0.3358, for Src inhibitor-; 128.5 ± 2.2 g, t(5) = 1.766; p = 0.1377, for EGFR inhibitor-; and 129.9 ± 1.8 g, t(5) = 0.8596; p = 0.4293, for the SH2-treated group, when the mechanical nociceptive threshold is compared before and 5 days after DAMGO; paired Student's t test), DAMGO (1 μg) was injected at the same site on the dorsum of the hind paw and the mechanical nociceptive threshold evaluated 30 min later. DAMGO-induced hyperalgesia was partially attenuated in the groups previously treated with inhibitors for MAPK (* p < 0.05) and EGFR (** p < 0.01) and completely inhibited in the Src inhibitor- and SH2-treated groups (F(2,40) = 38.15, *** p < 0.001, when vehicle-treated group is compared to inhibitors-treated groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). B. Fifteen days later, when the average baseline mechanical nociceptive threshold was 127.3 ± 2.6 g for the vehicle group, 132.3 ± 1.7 g for the MAPK inhibitor group, 134.3 ± 2.4 g for the Src inhibitor group, 134.3 ± 2.5 g for the EGFR inhibitor group, and 133.3 ± 1.9 g for the SH2 group, DAMGO (1 μg) was injected again on the dorsum of the hind paw. An attenuation on DAMGO-induced hyperalgesia was observed in the groups previously treated with MAPK and EGFR inhibitors (** p < 0.01, when vehicle-treated group is compared to MAPK and EGFR inhibitor-treated groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). However, a marked inhibition of DAMGO-induced hyperalgesia was observed in the groups pretreated with the Src inhibitor and SH2 (F(4,20) = 27.13, *** p < 0.001, when vehicle-treated group is compared to Src inhibitor- and SH2-treated groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). C. To verify that pretreatment with these inhibitors is able to prevent the induction of OIH, DAMGO (1 μg) was injected again, 30 days after the repeated (hourly × 4) injections of DAMGO. Of note, the average baseline mechanical nociceptive threshold, 30 days after treatments, was 132.0 ± 1.7 g for the vehicle group, 134.0 ± 2.0 g for the MAPK inhibitor, 132.8 ± 1.1 g for the Src inhibitor group, 129.7 ± 2.1 g for the EGFR inhibitor group, and 132.7 ± 1.7 g for the SH2 group. At this time, only a partial attenuation of DAMGO-induced hyperalgesia was observed in the groups previously treated with the MAPK and Src inhibitors, and SH2, when compared to the vehicle-treated group (F(4,40) = 3.47, ** p < 0.01, when vehicle-treated group is compared to MAPK or Src inhibitors- or SH2-treated groups; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). However, no significant (ns) difference was observed between vehicle- and EGFR inhibitor-treated groups (ns, two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These findings indicate a partial contribution of a Src, FAK and MAPK signaling in induction of OIH by repeated exposure to DAMGO. n = 6 paws per group.
Matrix metalloprotease in the expression of DAMGO-induced prolongation of PGE2 hyperalgesia
Since it has previously been shown that MOR signals to EGFR by activating MMP, which cleaves EGF from a membrane surface precursor molecule [9; 42], we next tested if matrix metalloproteases (MMPs) play a role in MOR-EGFR crosstalk associated with the induction of prolongation of PGE2-induced hyperalgesia, induced by repeated exposure to DAMGO. Ilomastat, which inhibits a wide variety of MMPs, and a MMP-9 selective inhibitor, were both able to block the prolongation of PGE2-induced hyperalgesia when PGE2 was administered 5 days after the repeated exposure to DAMGO (Fig. 7A). However, when PGE2 was again injected, 15 days after the repeated exposure to DAMGO, the prolongation of PGE2-induced hyperalgesia at the 4th hour was present (Fig. 7B). These findings indicate that MMPs play a role in the expression, but not the induction, of DAMGO-induced prolongation of PGE2 hyperalgesia.
Figure 7. Role of matrix metalloproteinases in DAMGO-induced prolongation of PGE2 hyperalgesia.

Rats received an intradermal injection of vehicle (5 μL; black bars), ilomastat (a general MMP inhibitor, 1 μg; gray bars) or MMP-9 inhibitor (1 μg; dotted bars) followed, 10 min later, by repeated (hourly × 4) injections of DAMGO (1 μg) at the same site. The average baseline mechanical nociceptive threshold, before treatments, was 129.4 ± 2.2 g for the vehicle group, 131.3 ± 1.9 g for the MMP inhibitor group, and 130.7 ± 1.1 g for the MMP-9 inhibitor group. Five days later, when the mechanical nociceptive threshold was not different from the pre-DAMGO baseline (127.0 ± 2.5 g, t(5) = 1.395; p = 0.2215, for the vehicle-treated group; 132.3 ± 1.7 g, t(5) = 0.4416; p = 0.6772, for the general MMP inhibitor-treated group, 127.7 ± 2.2 g, t(5) = 0.2548; p = 0.8090, for the MMP-9 inhibitor-treated group, when the mechanical nociceptive threshold is compared before and 5 days after DAMGO; paired Student's t test), PGE2 (100 ng) was injected intradermally on the dorsum of the hind paw and, a significant inhibition of PGE2 hyperalgesia at the 4th hour was observed in both MMP and MMP-9 inhibitor-treated groups (F(2,30) = 223.71, *** p < 0.0001, when inhibitor-treated groups are compared to vehicle-treated group at the 4th hour after PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). B. However, at day 15, when the average baseline mechanical nociceptive threshold was 128.9 ± 1.5 g for the vehicle group, 130.3 ± 2.3 g for the MMP inhibitor, and 129.9 ± 2.1 g for the MMP-9 inhibitor group, PGE2 (100 ng) was again injected intradermally. We observed that prolongation of PGE2 hyperalgesia at the 4th hour was only weakly attenuated in both the general MMP and the MMP-9 inhibitor-treated groups (F(2,30) = 7.89, * p = 0.0088, when inhibitors-treated groups are compared to vehicle-treated group at the 4th hour after PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that MMPs play a role in the expression, but not induction, of DAMGO-induced prolongation of PGE2 hyperalgesia. n = 6 paws per group.
MOR-EGFR crosstalk
We also evaluated the role of two other signaling molecules downstream of MOR, RAS and cRaf1 [60] in the induction of the prolongation of PGE2 hyperalgesia. Inhibitors of RAS (salirasib) and cRaf1 (GW5074) did not prevent the prolongation of PGE2 hyperalgesia induced by repeated exposure to DAMGO (Fig. 8).
Figure 8. Second messengers involved in MOR-EGFR crosstalk.

Rats were treated intradermally with vehicle (5 μL; black bars), salirasib (RAS inhibitor, 1 μg; gray bars) or GW5074 (cRaf1 inhibitor, 1 μg; white bars) on the dorsum of the hind paw. The average baseline mechanical nociceptive threshold, before treatments, was 127.7 ± 2.4 g for the vehicle group, 132.7 ± 2.2 g for the RAS inhibitor group, and 129.3 ± 2.8 g for the cRaf1 inhibitor group. Ten minutes later, repeated (hourly × 4) injections of DAMGO (1 μg) was performed at the same site on the dorsum of the hind paw. Five days later, PGE2 (100 ng) was injected at the same site and the mechanical nociceptive threshold evaluated 30 min and 4 hours later. Compared to vehicle-treated group, in all inhibitors treated-groups PGE2 was able to induce hyperalgesia 30 min after its injection (data not shown). A small attenuation in the prolongation of PGE2 hyperalgesia at the 4th hour was observed only in the group previously treated with cRaf1 inhibitor (F(5,30) = 3.10, * p = 0.0226, when inhibitor-treated groups were compared to vehicle-treated group at the 4th hour after PGE2; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). The RAS inhibitor and vehicle did not inhibit the prolongation of PGE2 hyperalgesia at the 4th hour. These findings indicate that RAS and cRaf1 do not play a role in the induction of prolongation of PGE2 hyperalgesia.
Discussion
DAMGO-induced Type II hyperalgesic priming consists of two phenomena: opioid-induced hyperalgesia (OIH) and prolongation of PGE2-induced hyperalgesia. Induction of both phenomena are dependent on the action of DAMGO at the mu-opioid receptor (MOR), since AS-ODN against MOR, prevented the induction of both components of Type II priming. These findings agree with a recent report, which found that activation of MOR expressed in primary afferent nociceptors initiates OIH [19]. We also found that intrathecal administration of SSP-saporin, which destroys peptidergic nociceptors (which contain MOR [50]), prevented DAMGO-induced OIH and the prolongation of PGE2-induced hyperalgesia. Thus, DAMGO induces Type II priming by action at the MOR in peptidergic IB4-negative nociceptors.
Chronic use of opioids modifies multiple MOR signaling mechanisms, including receptor phosphorylation, change in downstream signaling pathways, and receptor multimerization and trafficking, which may contribute to OIH [48]. Previous studies have shown crosstalk between receptor tyrosine kinase (RTK) and opioid receptors, which mediate changes in opioid receptor signaling [10; 16; 42]. We found that an EGFR inhibitor (tyrphostin AG 1478) was able to prevent the prolongation of PGE2 hyperalgesia, when tested out to 30 days after repeated exposure to DAMGO, indicating that the induction of the prolongation of PGE2-induced hyperalgesia is EGFR dependent. The EGFR inhibitor was, however, not able to prevent the induction of OIH. On the other hand, while EGFR was necessary for the induction of prolongation of PGE2 hyperalgesia, an EGFR agonist (EGF ligand) alone was not sufficient to induce hyperalgesia and/or hyperalgesic priming. Our result is similar to those reported in a recent study [38], in which the late-phase of the formalin test was not enhanced by intrathecal treatment with an EGF ligand, indicating that EGF ligand is not able to induce hypersensitivity.
It has been demonstrated that stimulation of several GPCRs, including opioid receptors, can transactivate EGFR signaling [10; 16; 29; 42]. RTK transactivation by GPCRs occurs via activation of membrane-bound matrix metalloproteinases (MMPs), which play a role in the processing of EGF-like precursor molecules expressed in the plasma membrane [9; 14; 38; 45]. Src may also activate metalloproteinases [43], which in turn cleaves EGF from a precursor molecule in the cell membrane. However, we found that MMPs are only involved in the expression, not the induction, of prolongation of PGE2 hyperalgesia. Therefore, we sought out a role of ligand-independent intracellular signaling pathways (i.e., Src family proteins) [15; 21; 28; 36; 44]. Importantly, a phosphopeptide ligand for the Src SH2 domain, which blocks the interaction of Src with FAK and EGFR, was able to prevent the induction of prolonged PGE2 hyperalgesia by repeated exposure to DAMGO. Thus, chronic exposure to opioids is able to increase Src activity, which phosphorylates EGFR in its cytosolic domain [57]. Still, how MOR, EGFR, Src, and FAK interact in the induction of prolongation of PGE2-induced hyperalgesia by DAMGO, remains to be established.
Since Src, MAPK and FAK participate in the prolongation of PGE2 hyperalgesia produced by repeated exposure to DAMGO [8], we tested the effect of inhibitors for these second messengers on DAMGO-induced OIH. Thirty days after DAMGO (hourly × 4) was injected, all inhibitors, injected prior to DAMGO (induction protocol), only partially attenuated DAMGO-induced hyperalgesia, indicating that the mechanism involved in the induction of OIH differs substantially from that involved in the prolongation of PGE2 hyperalgesia. Thus, while both are produced by action of DAMGO at the MOR, the mechanisms involved in the induction of OIH and prolongation of PGE2 hyperalgesia are, at least in part, different. Changes in MOR signaling produced by chronic use of opioids cannot be entirely explained by the typical GPCR signaling pathway and it is possibly a result from shifts in intracellular signaling. In this direction, a previous study suggested a crucial role of Src mediating MOR phosphorylation, producing long-term changes in the signals downstream the receptor [60]. Similarly, a switch in intracellular signaling produced by phosphorylated GPCR activation involving the participation of β-arrestins has also been observed. In a 2005 study, Lefkowitz and Shenoy suggested that the binding of β-arrestins to phosphorylated GPCR receptors was not just a mechanism that resulted in signaling termination, but could also produce changes in the receptor, allowing it to recruit messengers that were not previously activated directly by the GPCR, such as Src [35], and to interact with downstream effectors such as MAP kinases [37]. This could explain, for example, events such as biased agonism [35] or, in our case, Type II hyperalgesic priming [22]. In fact, a previous study has suggested a novel, non-canonical signaling pathway for MOR observed after chronic exposure to opioid agonists, in which phosphorylation of MOR led to the recruitment and activation of cRaf1 and RAS in a Src kinase-dependent manner [60]. Interaction between GPCRs and MAPK cascades has been shown to either depend on RAS (possibly involving Src) or to be RAS independent, in which case it would require the GPCR to transactivate RTKs such as EGFR [20]. However, although Src, MAPK and EGFR do play a role in our model, our results are not totally compatible with this mechanism, since we found that inhibitors for RAS and cRaf1 did not affect the induction of prolongation of PGE2 hyperalgesia.
The prolongation of PGE2 hyperalgesia in Type I priming is mediated by an autocrine mechanism involving the release of cAMP from the peripheral terminal of the primary afferent nociceptor, its metabolism to adenosine by two ectonucleotidases (i.e., ecto-5′-phosphodiesterase and ecto-5′-nucleotidase [17; 26], and the action of the end product of this metabolic pathway, adenosine, at A1-adenosine receptors on the peripheral terminal of the nociceptor, to produce PKCε-dependent prolonged mechanical hyperalgesia [26]. Thus, it would appear that the coupling of receptors to second messengers that mediate hyperalgesia differs between Type I and Type II priming, coupling to PKCε in Type I hyperalgesic priming [26] and to PKA in Type II [5]. Type I and Type II priming occur in different populations of nociceptors, Type I in IB4-positive (nonpeptidergic) [7; 30] and Type II in IB4-negative (peptidergic) nociceptors. Additionally, the induction of Type I priming can also be prevented by an injection of a protein translation inhibitor (i.e., cordycepin) in the peripheral terminal of the nociceptor [7; 23], which failed to prevent the induction of DAMGO-induced Type II priming [5].
In conclusion, our results demonstrate a crucial role of MOR, in IB4-negative peptidergic nociceptors, in the induction of Type II priming (OIH and prolongation of PGE2 hyperalgesia), which is partially dependent of Src, FAK and MAPK signaling to develop OIH and completely dependent on Src, FAK, EGFR and MAPK to induce prolongation of PGE2 hyperalgesia. The current findings are summarized in Figure 9. Understanding the mechanisms responsible for the induction of Type II hyperalgesic priming, a form of neuroplasticity in the peripheral terminal of the primary afferent nociceptor, may provide useful information for the design of drugs with improved therapeutic profiles to treat neuroplasticity induced by chronic use of opioids.
Figure 9. Schematic of mechanisms involved in induction of Type II hyperalgesic priming (OIH and prolongation of PGE2-induced hyperalgesia).
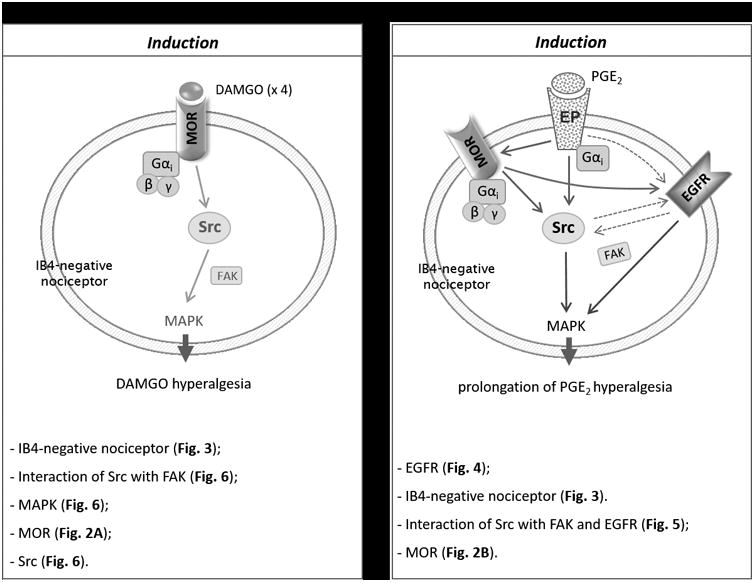
Chronic opioid use changes MOR signaling, that may reflect intracellular signal switching. Our findings show that repeated (hourly × 4) intradermal injections of a MOR selective agonist DAMGO induces Type II hyperalgesic priming (OIH [A] and prolongation of PGE2-induced hyperalgesia [B]), in peptidergic IB4-negative nociceptors. Induction of both components present in Type II priming are MOR dependent, which activates diverse downstream second messengers. A. Activation of MOR (a Gαi-protein-coupled receptor) by repeated (hourly × 4) injections of DAMGO, stimulates Src and FAK, leading to activation of MAPK, which ultimately produces sensitization in the peripheral terminal of IB4-negative nociceptor. B. After the peripheral terminal of IB4-negative nociceptors have received repeated injections of DAMGO, PGE2 was injected, which activates a signaling cascade, involving EGFR, Src and FAK, leading to stimulation of MAPK, which ultimately prolongs PGE2-induced hyperalgesia. Thus, while the induction of OIH is partially attenuated by the inhibition of Src, FAK and MAPK signaling (A), the prolongation of PGE2 hyperalgesia is completely dependent on Src, FAK, EGFR and MAPK signaling (B). Schematics summarize the signaling pathways involved in the induction of OIH (A) and prolongation of PGE2 hyperalgesia (B) induced by repeated exposure to DAMGO. Abbreviations: βγ, G-protein βγ subunit; DAMGO, [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin acetate salt (a mu-opioid receptor agonist); EGFR, epidermal growth factor receptor; EP, prostaglandin receptor; FAK, focal adhesion kinase; Gαi, G-protein αi subunit; IB4, isolectin B4; MAPK, mitogen-activated protein kinase; MOR, mu-opioid receptor; PGE2, prostaglandin-E2; Src, proto-oncogene tyrosine-protein kinase.
Acknowledgments
The authors would like to thank Marie Kern for technical assistance. This study was funded by a grant from the National Institutes of Health (NIH), NS084545.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39(3):497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- 2.Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19(6):2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20(12):4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araldi D, Ferrari LF, Green P, Levine JD. Marked sexual dimorphism in 5-HT1 receptors mediating pronociceptive effects of sumatriptan. Neuroscience. 2017;344:394–405. doi: 10.1016/j.neuroscience.2016.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araldi D, Ferrari LF, Levine JD. Repeated Mu-Opioid Exposure Induces a Novel Form of the Hyperalgesic Priming Model for Transition to Chronic Pain. J Neurosci. 2015;35(36):12502–12517. doi: 10.1523/JNEUROSCI.1673-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Araldi D, Ferrari LF, Levine JD. Adenosine-A1 receptor agonist induced hyperalgesic priming type II. Pain. 2016;157(3):698–709. doi: 10.1097/j.pain.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araldi D, Ferrari LF, Levine JD. Gi-protein-coupled 5-HT1B/D receptor agonist sumatriptan induces type I hyperalgesic priming. Pain. 2016;157(8):1773–1782. doi: 10.1097/j.pain.0000000000000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araldi D, Ferrari LF, Levine JD. Hyperalgesic Priming (Type II) Induced by Repeated Opioid Exposure: Maintenance Mechanisms. Pain. 2017 doi: 10.1097/j.pain.0000000000000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276(36):33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 10.Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. Mu opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: implications for mitogen-activated protein kinase signaling. Mol Pharmacol. 2003;64(6):1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- 11.Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci. 2012;32(6):2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217(4556):252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- 13.Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84(18):6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter G. EGF receptor transactivation mediated by the proteolytic production of EGF-like agonists. Sci STKE. 2000;2000(15):pe1. doi: 10.1126/stke.2000.15.pe1. [DOI] [PubMed] [Google Scholar]
- 15.Cattaneo F, Guerra G, Parisi M, De Marinis M, Tafuri D, Cinelli M, Ammendola R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int J Mol Sci. 2014;15(11):19700–19728. doi: 10.3390/ijms151119700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Long H, Wu Z, Jiang X, Ma L. EGF transregulates opioid receptors through EGFR-mediated GRK2 phosphorylation and activation. Mol Biol Cell. 2008;19(7):2973–2983. doi: 10.1091/mbc.E07-10-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiavegatti T, Costa VL, Jr, Araujo MS, Godinho RO. Skeletal muscle expresses the extracellular cyclic AMP-adenosine pathway. Br J Pharmacol. 2008;153(6):1331–1340. doi: 10.1038/sj.bjp.0707648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JI, Koehrn FJ, Sorkin LS. Carrageenan induced phosphorylation of Akt is dependent on neurokinin-1 expressing neurons in the superficial dorsal horn. Mol Pain. 2012;8:4. doi: 10.1186/1744-8069-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med. 2017;23(2):164–173. doi: 10.1038/nm.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379(6565):557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 21.Etkovitz N, Tirosh Y, Chazan R, Jaldety Y, Daniel L, Rubinstein S, Breitbart H. Bovine sperm acrosome reaction induced by G-protein-coupled receptor agonists is mediated by epidermal growth factor receptor transactivation. Dev Biol. 2009;334(2):447–457. doi: 10.1016/j.ydbio.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013;14(7):731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15(3):312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari LF, Khomula EV, Araldi D, Levine JD. Marked Sexual Dimorphism in the Role of the Ryanodine Receptor in a Model of Pain Chronification in the Rat. Sci Rep. 2016;6:31221. doi: 10.1038/srep31221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrari LF, Levine E, Levine JD. Role of a novel nociceptor autocrine mechanism in chronic pain. Eur J Neurosci. 2013;37(10):1705–1713. doi: 10.1111/ejn.12145. [DOI] [PubMed] [Google Scholar]
- 27.Ferrari LF, Levine JD. Plasma membrane mechanisms in a preclinical rat model of chronic pain. J Pain. 2015;16(1):60–66. doi: 10.1016/j.jpain.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013;280(21):5258–5268. doi: 10.1111/febs.12509. [DOI] [PubMed] [Google Scholar]
- 29.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20(13):1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 30.Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169(1):431–435. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30(13):4660–4666. doi: 10.1523/JNEUROSCI.5530-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22(20):9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khasar SG, Gold MS, Dastmalchi S, Levine JD. Selective attenuation of mu-opioid receptor-mediated effects in rat sensory neurons by intrathecal administration of antisense oligodeoxynucleotides. Neurosci Lett. 1996;218(1):17–20. doi: 10.1016/0304-3940(96)13111-6. [DOI] [PubMed] [Google Scholar]
- 34.Kras JV, Weisshaar CL, Pall PS, Winkelstein BA. Pain from intra-articular NGF or joint injury in the rat requires contributions from peptidergic joint afferents. Neurosci Lett. 2015;604:193–198. doi: 10.1016/j.neulet.2015.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308(5721):512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 36.Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331(2):222–231. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283(5402):655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 38.Martin LJ, Smith SB, Khoutorsky A, Magnussen CA, Samoshkin A, Sorge RE, Cho C, Yosefpour N, Sivaselvachandran S, Tohyama S, Cole T, Khuong TM, Mir E, Gibson DG, Wieskopf JS, Sotocinal SG, Austin JS, Meloto CB, Gitt JH, Gkogkas C, Sonenberg N, Greenspan JD, Fillingim RB, Ohrbach R, Slade GD, Knott C, Dubner R, Nackley AG, Ribeiro-da-Silva A, Neely GG, Maixner W, Zaykin DV, Mogil JS, Diatchenko L. Epiregulin and EGFR interactions are involved in pain processing. J Clin Invest. 2017 doi: 10.1172/JCI87406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mestre C, Pelissier T, Fialip J, Wilcox G, Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods. 1994;32(4):197–200. doi: 10.1016/1056-8719(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 40.Oliveira-Fusaro MC, Zanoni CI, Dos Santos GG, Manzo LP, Araldi D, Bonet IJ, Tambeli CH, Dias EV, Parada CA. Antihyperalgesic effect of CB1 receptor activation involves the modulation of P2X3 receptor in the primary afferent neuron. Eur J Pharmacol. 2017;798:113–121. doi: 10.1016/j.ejphar.2017.01.030. [DOI] [PubMed] [Google Scholar]
- 41.Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003;120(1):219–226. doi: 10.1016/s0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- 42.Phamduong E, Rathore MK, Crews NR, D'Angelo AS, Leinweber AL, Kappera P, Krenning TM, Rendell VR, Belcheva MM, Coscia CJ. Acute and chronic mu opioids differentially regulate thrombospondins 1 and 2 isoforms in astrocytes. ACS Chem Neurosci. 2014;5(2):106–114. doi: 10.1021/cn400172n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ. Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J Biol Chem. 2001;276(25):23155–23160. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- 44.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8(1):11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 45.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 46.Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J. Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res. 2012;1472:38–44. doi: 10.1016/j.brainres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 47.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32(12):611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: Cellular and molecular mechanisms. Neuroscience. 2016;338:160–182. doi: 10.1016/j.neuroscience.2016.06.029. [DOI] [PubMed] [Google Scholar]
- 49.Sanchez-Blazquez P, Garcia-Espana A, Garzon J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: role of delta-2 opioid receptors. J Pharmacol Exp Ther. 1997;280(3):1423–1431. [PubMed] [Google Scholar]
- 50.Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137(6):1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song MJ, Wang YQ, Wu GC. Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull. 2009;78(6):335–341. doi: 10.1016/j.brainresbull.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011;12:120. doi: 10.1186/1471-2202-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91(4):545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- 54.Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32(3):577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- 55.Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492(1-2):397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- 56.Vierck CJ, Jr, Kline RH, Wiley RG. Intrathecal substance p-saporin attenuates operant escape from nociceptive thermal stimuli. Neuroscience. 2003;119(1):223–232. doi: 10.1016/s0306-4522(03)00125-8. [DOI] [PubMed] [Google Scholar]
- 57.Wang Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress Challenges and Future Research. Int J Mol Sci. 2016;17(1) doi: 10.3390/ijms17010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weisshaar CL, Winkelstein BA. Ablating spinal NK1-bearing neurons eliminates the development of pain and reduces spinal neuronal hyperexcitability and inflammation from mechanical joint injury in the rat. J Pain. 2014;15(4):378–386. doi: 10.1016/j.jpain.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiley RG, Kline RHt, Vierck CJ., Jr Anti-nociceptive effects of selectively destroying substance P receptor-expressing dorsal horn neurons using [Sar9,Met(O2)11]-substance P-saporin: behavioral and anatomical analyses. Neuroscience. 2007;146(3):1333–1345. doi: 10.1016/j.neuroscience.2007.01.066. [DOI] [PubMed] [Google Scholar]
- 60.Zhang L, Loh HH, Law PY. A novel noncanonical signaling pathway for the mu-opioid receptor. Mol Pharmacol. 2013;84(6):844–853. doi: 10.1124/mol.113.088278. [DOI] [PMC free article] [PubMed] [Google Scholar]