

Abstract
Next-generation sequencing (NGS), also known as high-throughput sequencing, is changing the field of microbial genomics research. NGS allows for a more comprehensive analysis of the diversity, structure and composition of microbial genes and genomes compared to the traditional automated Sanger capillary sequencing at a lower cost. NGS strategies have expanded the versatility of standard and widely used typing approaches based on nucleotide variation in several hundred DNA sequences and a few gene fragments (MLST, MLVA, rMLST and cgMLST). NGS can now accommodate variation in thousands or millions of sequences from selected amplicons to full genomes (WGS, NGMLST and HiMLST). To extract signals from high-dimensional NGS data and make valid statistical inferences, novel analytic and statistical techniques are needed. In this review, we describe standard and new approaches for microbial sequence typing at gene and genome levels and guidelines for subsequent analysis, including methods and computational frameworks. We also present several applications of these approaches to some disciplines, namely genotyping, phylogenetics and molecular epidemiology.
Keywords: Bacteria, epidemiology, MLST, pathogen, typing, WGS
1. Introduction
Microbial typing techniques are greatly enhancing our insights into microbial population epidemiology and microbial diversity and are widely used in diagnostics, genomics and pathogenesis related with microbiology research (Boers et al., 2012; Van Belkum, 2002). In fact, our ability to accurately distinguish among strains of infectious pathogens is crucial for efficient epidemiological and surveillance analysis, studying microbial population structure and dynamics and, ultimately, developing improved public health control strategies (Cooper and Feil, 2004). To achieve these goals, several molecular typing methods have been proposed that can identify isolates worldwide (global epidemiology) and/or in localized disease outbreaks (local epidemiology); see (Foley et al., 2009) for a review.
Since 1998, the established standard for molecular typing is Multilocus Sequence Typing (MLST) (Maiden et al., 1998), which has proven to be an effective method for characterizing bacterial Isolates. However, next-generation sequencing (NGS) technologies are drastically changing the field of microbial genomics research (Forde and O’Toole, 2013). These new sequencing methods supply a range of applications (Glenn, 2011), allowing for a more comprehensive and in depth analysis of the diversity, structure and content of microbial genomes compared to traditional automated Sanger capillary sequencers at a substantially lower cost (Mardis, 2008; Metzker, 2010). The reader is referred to the following reviews for further information about NGS technologies (Goodwin et al., 2016; Mardis, 2013, 2017; Metzker, 2010). As a consequence, in March 2017, a total of 246,189 (complete and draft) bacterial and 6,615 viral genomes have been deposited in the genomes online database GOLD (Mukherjee et al., 2017), of which 1,041 correspond to metagenomic studies (i.e., the study of microbial communities directly in their natural environments). NGS platforms have proven to be effective tools for the re-sequencing and de novo sequencing reference microbial species and strains (pathogens and underrepresented taxa), but also assembling genomes of entire microbial communities of unculturable microbes (microbiotas) (Kyrpides et al., 2014; Sangwan et al., 2016; Sharon and Banfield, 2013).
Non-cultured organisms represent the vast majority of the total microbial diversity which exists in the world (Pace, 2009). Microbial genomic studies usually focus on microbial diversity and structure at the species (or strain) and community levels through targeted sequencing of gene amplicons (e.g., housekeeping genes, 16S/18S rRNA, ITS) or shotgun sequencing of (nearly) full genomes (Caporaso et al., 2012; Chun and Rainey, 2014; Kwong et al., 2015; MacCannell, 2013; Petrosino et al., 2009; Vincent et al., 2016).
NGS strategies have expanded the versatility of widely used typing approaches, such as MLST, to accommodate high-throughput data (e.g., NGMLST and HiMLST). The drawback is that this massive amount of data comes in the form of short reads with relatively high sequencing errors; hence one needs to invest heavily in computational analysis. Additionally, novel analytic and statistical techniques that can handle these data had to be developed. Over the last five years new typing approaches that take advantage of parallel amplicon and whole genome sequencing have been proposed.
In this review, we describe and compare classical (e.g., MLST) and recent (e.g., HiMLST) DNA typing approaches (Section 2) of microbial sequence typing. Then we present statistical methods and computational tools for the analysis of nucleotide, locus, and genome data generated via Sanger and NGS platforms (Section 3). In the last section (Section 4), we present several applications of these DNA-based approaches to the fields of genotyping, phylogenetics and molecular epidemiology. We also refer the reader to other reviews on MLST for complementary information (Boers et al., 2012; Cooper and Feil, 2004; Jolley et al., 2012; Jolley and Maiden, 2014; Larsen et al., 2012; Maiden, 2006; Pérez-Losada et al., 2017; Pérez-Losada et al., 2006; Pérez-Losada et al., 2013; Sullivan et al., 2005; Urwin and Maiden, 2003).
2. DNA-based typing approaches
2.1 Standard typing approaches: MLST and MLVA
Multilocus Sequence Typing (MLST) examines nucleotide variation in sequences of internal fragments of usually seven housekeeping genes, i.e., those encoding fundamental metabolic functions; although the number of genes may vary in dependence of the strains, species, and other particularities of the studied sample. For each gene, then the different sequences present within a species are assigned as distinct alleles and, for each isolate, the alleles at each of the seven loci define the allelic profile or sequence type (ST). Each isolate is therefore unambiguously characterized by a series of seven integers, which correspond to the alleles at the seven housekeeping loci.
MLST is widely used for molecular typing (Jolley and Maiden, 2014; Maiden, 2006; Maiden et al., 2013; Pérez-Losada et al., 2013). This was made possible by three advances in molecular microbiology (Maiden, 2006) involving: 1) bacterial evolution and population biology knowledge; 2) high-throughput nucleotide sequencing; and 3) genetic sequence databases. The bacterial population studies undertaken from the 1980s onwards were central to the development of MLST. Those studies showed that genetic exchange among bacteria was more common than previously thought, leading to a reassessment of the role of sexual processes in the structuring of bacterial populations. Using sequence data, it has been shown that recombination (mosaic genes) was not only frequent in genes under diversifying selection (e.g., antigen-encoding and antibiotic resistant genes), but also in genes under purifying selection (housekeeping genes) (see Maiden, 2006). This suggested that the clonal model (variation can only arise by mutation) was not universal and led to the proposal of new non-clonal or panmictic (variation is mainly generated by recombination) and partially clonal models of bacterial population structure (Feil and Enright, 2004; Spratt and Maiden, 1999). Consequently, typing methods needed to accommodate and distinguish among a broader spectrum of population structures, hence providing not only discriminatory power but also information about the clonal structure of the organism under study. Therefore, only molecular techniques that can contrast results across independent markers (such as MLST) would be adequate for bacterial typing and population genetic analyses.
The length of the nucleotide sequence amplified for each locus is generally in the range of 400–600 bp and is determined largely by the parameters of automated sequencing instruments available at the time. Most MLST nucleotide sequence data are generated by Sanger sequencing, however this technology is being replaced by high-throughput technologies such as pyrosequencing (Boers et al., 2012; Margulies et al., 2005), sequencing-by-synthesis (Illumina/Ion Torrent) and single-molecule sequencing (PacBio/Nanopore) (Chen et al., 2015; Pérez-Losada et al., 2013) for targeted-amplicon and whole-genome sequencing. These technologies can generate accurate read lengths of ~150 bp to 10 kb (Illumina and PacBio, respectively) and up to 25–50 million paired-end reads (Illumina MiniSeq/MiSeq platforms) per run. Moreover, the design of barcoded primers allows simultaneous and efficient sequencing of homologous products from hundreds of samples in the same run (Kozich et al., 2013; Rao et al., 2016; Taylor et al., 2016).
One of the goals of the MLST approach was the development of online platforms containing MLST databases to which public health officials and researchers could both have access and contribute and from which clinical, epidemiological and population studies could benefit (Maiden, 2006; Maiden et al., 1998; Urwin and Maiden, 2003). The first MLST online platforms were based on single databases implemented in the MLSTdB software (Chan et al., 2001), but as MLST schemes began to expand several limitations became apparent: redundant information (each record contained the ST designation and the allelic profile), isolate bias (single databases were dominated by specific studies), and access (all databases were stored at a single location). To overcome these limitations, a new network-based database software, MLSTdBNet (Jolley et al., 2004) was developed and implemented on the PubMLST site (http://pubmlst.org/). This site includes two databases: i) a profiles database with the sequences of each MLST allele for each locus linked to an allele number, and ii) an allelic profiles database with the corresponding ST designations. The profile database can then interact with other isolate databases. For each scheme on the PubMLST site there is a PubMLST isolate database that aims to include at least one isolate for each ST. MLST databases are hence different to other repository databases such as GenBank, not only in organization but also in active curation for accuracy. It is important to highlight that MLST databases do not embody the global diversity of an organism but the extent of its diversity at the time of access. Moreover, stored data are unstructured and do not necessarily represent natural populations either. As high-throughput sequencing becomes more affordable, PubMLST is increasingly including whole genome sequences, e.g., BIGSdb (Jolley and Maiden, 2010; Larsen et al., 2012).
As the number of schemes available has increased, MLST has become the most commonly used method of pathogen typing. In comparison to older methods (serotyping; multilocus enzyme electrophoresis analysis), the use of genetic variation gives MLST the advantage of producing variable data (more resolution) that are universally comparable (within schemes), easily validated, and readily shared across laboratories. The use of sequencing makes MLST a broadly applicable methodology that can be fully automated and scalable from single isolates to thousands of samples. Importantly, the material needed for MLST analysis – DNA or dead cells – is easily transported among labs, without the problems associated with infective materials. Furthermore, the use of online databases to store and curate MLST schemes makes them a globally and highly accessible resource.
The number of loci that should be evaluated to confidently assign a ST has been minimized to reduce the expense and time required for characterization, with most studies using 6 – 10 loci. If performed manually, evaluating even these many loci can be time consuming. However, fully automated systems, e.g., robotics (Jefferies et al., 2003; Sullivan et al., 2006) provide a high-throughput pipeline for data collection that can run large volumes of samples with increased reliability. Likewise, commercial solutions such as Ion Torrent AmpliSeq panels targeting MLST schemes (www.ampliseq.com) can reduce costs down to cents per marker. As sequencing technology progresses, we expect the cost of automation to decrease, thus data interpretation rather than data generation will be the likely limiting factor in our understanding of pathogen population dynamics.
By focusing on sequence variation, MLST provides a highly replicable and reproducible typing method. Additionally, the focus on housekeeping-genes provides significant amounts of genetic data that can be used to calculate pathogen population genetic parameters at both local and global scales. Those parameters can then be used to construct more sophisticated models of pathogen evolution and epidemiology that will improve our understanding about the spread of disease. However, there is no single set of universal housekeeping genes that can be used for all pathogens as the recombination rates, substitution rates, and levels of selection vary across loci and species (Pérez-Losada et al., 2006). Therefore, a unique set of loci must be identified for each novel, un-typed pathogen under study. The rapid increase of available microbial genomes will make data mining for housekeeping genes more feasible, reducing the time and cost required for constructing new MLST schemes.
Currently, the main drawback of the MLST method is that the selection of housekeeping loci requires reference genomes (Parkhill et al., 2003). Moreover, not all pathogens are suitable for MLST methods. Some pathogens (e.g., M. tuberculosis, Y. pestis) exhibit very little variation throughout their entire genome, most likely representing “evolutionarily young” pathogens that have not yet accumulated sufficient genetic variation to differentiate strains. For typing these pathogens, more rapidly evolving loci (e.g., insertion sequences or antibiotic-resistance determinants) or more markers (e.g., genome-wide single nucleotide polymorphisms or SNPs) are needed. Conversely, some bacterial genomes have accumulated so much variation that MLST housekeeping genes do not provide adequate information for typing. As we advance MLST schemes in the genomic era, we should be able to combine information-rich and widely adopted MLST schemes with cost-effective whole-genome sequencing.
Given the above limitations of MLST, over the last few years other typing approaches have been developed based on similar principles. Multilocus Variable number of tandem repeats Analysis (MLVA) uses polymorphic repeated sequences (VNTR) instead of housekeeping genes. Comparative studies between MLVA and MLST have yielded similar results (e.g., van Cuyck et al., 2012) and in recently evolved species, the MLVA approach can provide higher discriminatory power (Marsh et al., 2010). Finally, to achieve even greater resolution, other approaches have been developed based on core/accessory genes or distributed genes among bacterial species with the same MLST profile (Hall et al., 2010; Leekitcharoenphon et al., 2012). This new approach could skip the laborious and time-consuming steps needed to develop bacteria-specific MLST schemes; this is, however, replaced by in-silico work.
2.2 NGS-based approaches: WGS, SNP, rMLST, cgMLST, HiMLST, NGMLST and metaMLST
Whole-genome sequencing (WGS) has emerged as a powerful technology for the comparison of isolates in outbreak analysis. The applications of WGS in clinical and public health microbiology have already been demonstrated in proof-of-concept studies conducted retrospectively or in response to an emerging outbreak (Didelot et al., 2012; Walker et al., 2013). Few studies so far have used WGS in prospective surveillance and in typing of bacteria (Dallman et al., 2015; den Bakker et al., 2014). Salipante et al. (2015) explored the utility of WGS as a strain typing approach for the clinical laboratory using a single, universal protocol (encompassing library preparation, sequencing, and data analysis) for 3 distinct bacterial species: methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus, and multidrug-resistant Acinetobacter baumannii. They showed that WGS was highly reproducible, which enabled a functional, quantitative definition for determining clonality. Then, Kwong et al. (2016b) compared routine prospective WGS to other conventional typing methods, including MLST and MLVA, for the national epidemiologic surveillance of Listeria monocytogenes. MLST inferred in silico from the WGS data was highly concordant (>99%) with laboratory typing performed in parallel. However, WGS could identify distinct nested clusters within groups of isolates that were otherwise indistinguishable using traditional typing methods. As in previous studies, WGS provided a greater level of discrimination, than that from conventional typing, for surveillance or inferring linkage to point source outbreaks.
Single nucleotide polymorphism (SNP) is a variation in a single nucleotide that occurs at a specific position in the genome, where each variation is present to some appreciable degree within a population. SNP-typing is widely used for bacteria and has also been improved through using genome sequences. When coupled with NGS of DNA, genome-wide screening of SNPs is a powerful discriminatory technique that enables the identification of strain-specific genetic markers. This approach is Genome-wide SNP typing has been, for example, applied to the genotyping of strains and species of Bacillus anthracis and B. cereus (Kuroda et al., 2010), Streptococcus suis (Chen et al., 2013) or Coxiella burnetii (Huijsmans et al., 2011) among others, outbreak attribution (e.g., Hendriksen et al., 2011), phylogeography of recently emerged diseases (e.g., Keim and Wagner, 2009), or genome-wide association studies of SNPs associated with acquisition of bacteremia in healthcare settings (e.g., Nelson et al., 2014). Pipelines for phylogenetic typing of SNP genotypes have also been developed to identify bacterial strains including metagenomic samples (Sahl et al., 2015).
Although MLST genotyping is a superb approach to characterize microbial species and strains, their methodological implementation can be costly, time-consuming, and laborious. To accelerate automation and expand the versatility of the current MLST method, other approaches that take advantage of NGS technology have been developed to produce millions of high-quality bases at low cost within a single sequence run. The Ribosomal Multilocus Sequence Typing method (rMLST) has been proposed to index the molecular variation of 53 genes encoding bacterial ribosome protein subunits (Jolley et al., 2012). This method pursues the integration of a taxonomic and typing method in a similar curated MLST scheme. Although more expensive, the rMLST is likely to provide better resolution than standard MLST methodologies. Likewise, core-genome (cg) MLST has been developed to overcome a lack of resolution of MLST schemes for certain taxa. By collecting a sample of genome sequences representing extant diversity, the cgMLST scheme applies more than 1,000 genes to create sequence types with increased resolution for clonal populations of bacteria (de Been et al., 2015).
Boers et al. (2012) developed High-throughput MLST (HiMLST), which uses the Genome Sequencer Junior (Roche) and generates up to 70,000 amplicon reads with average read lengths of around 400–500 bases. This long-read sequencing performance, in combination with a sample pooling strategy that uses “bar-coded” amplicons for parallel analysis of pooled samples, allow the generation of MLST profiles from multiple bacterial isolates in a single NGS-run. They then demonstrate the successful parallel sequencing of MLST alleles that were amplified by PCR from 96 bacterial isolates from four species in single NGS-runs. Roche 454 sequencing (pyrosequencing) technology has been discontinued and replaced by the Illumina MiSeq platform (sequencing-by-synthesis). The latest Illumina MiSeq chemistry enables up to 15 Gb of output with 25 million sequencing reads and 2×300 bp read lengths (i.e., partially overlapping contigs of 400–500 bp). Such a vast improvement in sequencing depth promises a substantial reduction of labor and costs compared to traditional Sanger and Roche 454 approaches for amplicon sequencing.
More recently, Chen et al. (2015) developed a high-throughput Next-Generation sequencing MLST (NGMLST) approach and an automated software program for data analysis, MLSTEZ. Essentially, they coupled an efficient multiplex PCR approach with PacBio circular consensus sequencing technology, which can generate relatively inexpensive single-molecule consensus reads of 1–2 kbp with lower error rates (after multiple sequencing cycles). The software MLSTEZ can then automatically identify the barcodes and primers used in the PCR, correct sequencing errors, generate the MLST profile for each isolate, and predict potentially heterozygous loci. Next, they compared NGMLST to conventional MLST. The major advantages of the NGMLST approach at the time of analyses were: (i) the employment of multiplex PCR greatly reduces the amount of labor; (ii) PacBio greatly extends the maximum read length of target loci or genes from 500-bp to 2-kb without requiring fragmentation into shorter sequences; (iii) the NGMLST workflow is optimized to reduce unnecessary steps; (iv) MLSTEZ can be easily implemented and does not require technical expertise or a background in bioinformatics; and (v) for analysis of hybrid isolates, unlike most programs, MLSTEZ can detect heterozygous loci and sequence their alleles.
Finally, Zolfo et al. (2017) developed a software tool for microbial typing of haploid organisms called MetaMLST that combines the effectiveness of the MLST approach with the high throughput of metagenomics. MetaMLST overcomes the computational limitations and lowers the limit of detection (i.e., strain level) compared to metagenomic assembly. The authors then tested the pipeline on synthetic and spiked-in real metagenome datasets and showed that MetaMLST reconstructed the MLST sequences with high accuracy at low coverage (as low as 1×). When applied to real biological samples, MetaMLST also showed higher sensitivity than assembly-based approaches allowing the identification of pathogenic strains in epidemic outbreaks.
3. DNA-based typing analysis
The analysis of MLST data is usually based on two main strategies (Fig. 1). The first one relies on the consideration of allele and ST designations to estimate relatedness among isolates (allele-based methods), hence nucleotide differences among alleles are considered as an ST unit. The other one relies on the direct application of nucleotide sequences to estimate relatedness and population parameters (nucleotide-based methods). The allele-based approach has been adopted from the analysis of Multilocus Enzyme Electrophoresis data and therefore, these were the first methods applied to the analysis of MLST data (Enright and Spratt, 1999; Maiden et al., 1998). The allele-based approach is thought to work well in non-clonal organisms (e.g., Helicobacter pylori), while nucleotide-based approaches are preferable for clonal organisms (e.g., Escherichia coli) (Maiden, 2006). In practice, most microbes show some degree of clonality (clonal complex) in their populations and, therefore, we suggest that both types of analysis should be considered in population and epidemiological studies (e.g., Loubna et al., 2010). In this section, we present a brief description of the most commonly used approaches for analyzing MLST data, including allele, nucleotide, SNP and WGS data.
Figure 1.

Proposed workflow for typing analysis of microbial DNA sequences using allele- and nucleotide-base approaches. The workflow shows data and tasks in boxes, and databases and computer programs in circles. We have highlighted in light red our preferred approach and computer programs.
3.1 Allele-based methods
Allele-based methods consider the allele as the unit of analysis and, consequently, these methods first require assigning an allele number to each DNA sequence from each locus (Fig. 1). This is done by matching the study sequences against those stored in public MLST databases. If no match is found a number is assigned following the order of discovery. Several computational programs have been developed to perform this task, although Sequence Typing Analysis and Retrieval System (STARS) seems to be the most functional and popular (Sullivan et al., 2005). The STARS interface was specifically designed for typing and allows the assembly of many sequences at once.
Once alleles have been assigned, data are entered in MLST sites of curated databases (Aanensen and Spratt, 2005; Jolley et al., 2004; Jolley and Maiden, 2006) to acquire an ST profile. At this point, exploratory analysis (e.g., allele and profile frequencies, polymorphism estimates, codon usage) of the data can be performed. The software package Sequence Type Analysis and Recombinational Tests (START2) can perform all of these tasks (Jolley et al., 2001). Next, relatedness among STs can be displayed with heuristic approaches of cluster reconstruction such as Based Upon Related Sequences Types (eBURST) (Feil et al., 2004), the simple Unweighted Pair Group Method with Arithmetic Mean (UPGMA) or network-based methods such as NeighborNet (Bryant and Moulton, 2004) or split decomposition (Bandelt and Dress, 1992).
The method eBURST is based on a simple model of clonal expansion and diversification (Feil et al., 2004). It first identifies mutually exclusive groups of related STs and next, it tries to identify the founding ST of each group. Bootstrap estimates can also be calculated to assess confidence in the grouping. The algorithm then predicts the descent from the predicted founding ST to the other STs in the group and finally displays a radial diagram centered on the predicted founding ST. A globally optimized version (goeBURST) identifies alternative patterns of descent using a graphic matroid approach (Francisco et al., 2009). Recently, a new approach (PHYLOViZ) allows for the integration of allelic profiles from MLST or MLVA methods (although SNP data can also be included) and the associated epidemiological data (Francisco et al., 2012). PHYLOViZ uses goeBURST for representing the estimated evolutionary relationships among strains.
The traditional UPGMA method relies on a matrix of distances to estimate isolate relatedness. Distances are calculated for each pair of STs considering the number of allelic differences and next, groups can be sequentially clustered in order of similarity (i.e., allelic matches) to generate a phylogenetic tree. Other distance-based methods, such as NeighborNet and split decomposition, provide clustering through phylogenetic networks, considering conflicting signatures or alternative evolutionary histories (Fitch, 1997). Today these networks are widely used to analyze MLST data (e.g., Jolley and Maiden, 2014; Maiden et al., 2013). Additional distance and parsimony methods have been proposed to estimate relatedness based on allele frequencies, but note that distance methods generally outperform parsimony methods (Wiens, 2000).
Allele-based methods have the advantage of simplicity and speed (especially relevant when dealing with large datasets), which are crucial for efficient epidemiological surveillance and public health management, but disregard much of the evolutionary information contained at the nucleotide level. A larger and more sophisticated plethora of nucleotide-based methods exist to estimate isolate relationships and key parameters of population genetics.
3.2 Nucleotide-based methods
The first step for the analysis of nucleotide data is usually the generation of a multiple sequence alignment (MSA) (identification and phasing of the homologous nucleotide sites). Since the loci used for MLST usually evolve slowly and code for proteins, this step becomes straightforward, especially at the amino acid level (Fig. 1). If needed, several fast, accurate and user-friendly aligning methods are implemented in MAFFT (Katoh and Standley, 2013), MUSCLE (Edgar, 2004), and TranslatorX for translated-alignment of coding sequences (Abascal et al., 2010).
Once an MSA has been generated, the substitution model of evolution that best fits the data can be determined. Over the past two decades, substitution models have increased in complexity by incorporating more informative parameters to better fit with real observations (Arenas, 2015b). The consideration of the best fitting substitution model is critical because it can affect diverse subsequent phylogenetic and population analyses (Lemmon and Moriarty, 2004). Therefore, substitution model choice is required and is usually assessed with evolutionary frameworks such as jModelTest2 (Darriba et al., 2012). This framework implements confidence sets of models (model averaging) (Posada and Buckley, 2004) and several criteria for model selection such as Akaike Information Criterion (AIC), Bayesian Information Criterion (BIC), Decision Theory (DT) and hierarchical Likelihood Ratio Test (hLRT). Although AIC is the most broadly used criterion for evaluating model fit, BIC and DT should be preferred (Luo et al., 2010). Additionally, methods for co-inferring nucleotide partitions and substitution models (Lanfear et al., 2016), as well as for inferring substitution models under a Bayesian framework (Bouckaert and Drummond, 2017) have been developed.
3.2.1 Phylogenetic relatedness
Phylogenetic reconstruction methods can be classified into two categories, those that proceed algorithmically through distances (e.g., UPGMA and Neighbor-joining -NJ) and those based on optimality criteria. Here we focus on methods that implement maximum likelihood and Bayesian optimality criteria and allow for the consideration of multiple data partitions each under a best-fit model, a feature particularly important for analyzing MLST data and that can improve the accuracy of phylogenetic inferences (Zoller et al., 2015).
Maximum likelihood (ML) inference attempts to identify the topology that explains the evolution of the aligned sequences, under a given substitution model of evolution, with the highest likelihood (Felsenstein, 1981). Several evolutionary frameworks implement phylogenetic tree inference (Yang and Rannala, 2012), but only a few consider partitions evolving under different substitution models. RAxML (Stamatakis, 2006) implements the ML criterion efficiently and can handle large datasets (more than 1,000 taxa with more than 20,000,000 bp) (Stamatakis et al., 2012). Confidence in the estimated relationships (i.e., clade support) is usually assessed with a non-parametric bootstrap procedure (Felsenstein, 1985), which should be repeated more than 1,000 times to achieve reasonable precision. Thus, RAxML is widely used to analyze MLST data (e.g., Zolfo et al., 2017). Another ML framework oriented to analyzing large datasets and heterogeneous evolution among partitions is FastTree2 (Price et al., 2010). This program can internally optimize phylogenetic tree inferences through the joint application of several approaches (NJ, minimum-evolution and ML) and it is also frequently considered to analyze MLST data (Kwong et al., 2016b; Skarp-de Haan et al., 2014). Interestingly, FastTree can infer phylogenetic trees with similar accuracy to RAxML but several orders of magnitude faster (Liu et al., 2011).
Bayesian inference (BI) combines the prior probability of a phylogeny with the likelihood of it producing a posterior probability distribution of trees, which can be interpreted as the probability of those trees (or tree) being correct (Huelsenbeck et al., 2001). Clade support is estimated by summarizing this distribution of trees through consensus analysis. Bayesian phylogenies are inferred using Metropolis-coupled Markov chain Monte Carlo (MCMC) methods and are implemented in programs such as MrBayes (Ronquist et al., 2012), RevBayes (Höhna et al., 2016), and BEAST (Bouckaert et al., 2014; Drummond et al., 2012). The output of the BI analysis must be evaluated to assure that the MCMC chains are well mixed and converged; such tasks can be performed with Tracer (Rambaut and Drummond, 2009). Importantly, the best fitting substitution model can vary across partitions. For this concern, an interesting program is PhyloBayes (Lartillot et al., 2009), which implements priors for the internal assignment of across-partition heterogeneity (Lartillot and Philippe, 2004). Importantly for many datasets, Bayesian approaches can perform phylogenetic inferences accounting for longitudinal sampling (Navascués et al., 2010; Rieux and Balloux, 2016) and relaxed molecular clocks, considering much more realistic evolutionary scenarios (Drummond et al., 2006).
Often gene trees differ even when sampled from the same population. This can be the result of molecular processes (e.g., recombination) or stochastic variation (e.g., lineage sorting). Whatever the case, it may be necessary to check if individual gene topologies are significantly different as ignoring these processes may lead to biased evolutionary inferences (Arenas and Posada, 2010b; Mallo et al., 2015; Schierup and Hein, 2000b). Multiple ML topological tests have been developed for such purposes and several are implemented in CONSEL (Shimodaira and Hasegawa, 2001). In the subsection WGS-based analyses, we describe the inference of phylogenetic trees from genome-scale data (i.e., accounting for genomic evolutionary events).
3.2.2 Population dynamics
The evolution of DNA sequences in natural populations can be described with parameters such as recombination, mutation, population growth and selection rates. Consequently, the accurate estimation of these parameters is key for understanding the dynamics and evolutionary process of populations, epidemiology, the potential and mode for obtaining antibiotic and immune system resistance, and ultimately the design of efficient public health control strategies (Omenn, 2010). Population parameters are efficiently estimated with explicit statistical models of evolution such as the coalescence (Kingman, 1982) and, therefore, most well-established evolutionary frameworks are based on such models, although programs vary in how they handle these parameters.
The rate of evolution quantifies the extent of genetic change over time. Traditionally, this parameter was assumed to be constant over time (strict molecular clock); however, recently, various data have been collected that violate this assumption (e.g., Bello et al., 2007). The rate of evolution can be accurately estimated with Bayesian approaches (i.e., implemented in BEAST) that account for variation through time with models of a relaxed molecular clock (Drummond et al., 2006).
Genetic recombination influences biological evolution at many levels (i.e., by increasing genetic diversity Spencer et al., 2006) and affects the estimation of other evolutionary parameters and processes (i.e., selection (Anisimova et al., 2003; Arenas and Posada, 2010a) or ancestral sequence reconstruction (Arenas and Posada, 2010b). Comprehensive assessments of statistical methods for detecting and estimating recombination rates were presented in Martin et al. (2011) and Posada et al. (2002). These studies concluded that one should not rely on a single method to detect or estimate recombination. With this in mind, software packages such as RDP (Martin et al., 2015) have been developed to implement a variety of methods for the same dataset. RDP includes 12 methods to estimate recombination and allows the user to draw conclusions based on the outcome of all those analyses. Another ML method to detect recombination is GARD (Kosakovsky Pond et al., 2006), which outperformed previously developed methods. GARD is implemented in the HYPHY package (Pond and Muse, 2005) and in the Datamonkey webserver (Delport et al., 2010). In HYPHY, GARD requires a multiprocessor machine and in Datamonkey, it can only analyze small datasets. Other programs such as LAMARC (Kuhner, 2006), LDhat (McVean et al., 2004), CodABC (Arenas et al., 2015), and OmegaMap (Wilson and McVean, 2006) can be used to estimate recombination rates and, therefore, quantify the amount of observed recombination (Pérez-Losada et al., 2007). Conveniently, these methods can also estimate the substitution rate because of the relationships between the population recombination rate (ρ = 2nNrl, where n = 1 or 2 for haploid or diploid populations, N is the effective population size, r is the recombination rate per site per generation and l is the alignment length) and the population substitution rate (θ = 2nNμl, where μ is substitution rate per site per generation) through the effective population size. Note also that the observed recombination can be influenced by the substitution rate –under low substitution levels, recombination can be underestimated due to lack of genetic information (Posada and Crandall, 2001).
Another key parameter for characterizing microbial population dynamics is the population growth rate, which influences the variation of genetic diversity over time. Population growth rate can be estimated under a certain demographic model (e.g., exponential) or without dependence on a model (e.g., Minin et al., 2008). The latter approach is implemented in BEAST. Exponential growth rate can also be estimated with LAMARC (under both ML and Bayesian approaches) and approximate Bayesian computation (ABC) approaches (e.g., Alves et al., 2016). ABC is a statistical approach to perform model selection and parameter estimation. Essentially, it performs computer simulations according to user-specified prior distributions, calculation of descriptive summary statistics from real and simulated data (to summarize the data) and a rejection or a multiple regression analysis to obtain a posterior distribution for each studied model or parameter. ABC is widely used in population genetics (Beaumont et al., 2002) but it can also be applied to other areas such as ecology (Beaumont, 2010). Advantages of using ABC are: 1) user-specified evolutionary models that can be more realistic than those considered in ML or Bayesian methods; 2) not need to compute a likelihood function, which for many scenarios cannot be designed or be computationally intractable; and 3) co-estimation of several parameters (Arenas, 2015a; Beaumont, 2010; Bertorelle et al., 2010). A disadvantage is that ABC assumes the internally performed computer simulations are realistic. Interestingly, ABC can outperform ML methods (Lopes et al., 2014) if it is correctly designed (i.e., incorporating informative summary statistics, realistic computer simulations or narrow prior distributions that include the “true” value). In pathogen populations, selection is usually estimated from protein-coding sequences with the nonsynonymous (dN) to synonymous (dS) substitution ratio dN/dS (ω). Here, ω > 1 indicates positive or diversifying selection, ω < 1 indicates negative or purifying selection and, ω ≈ 1 indicates lack of selection (neutral evolution). Accurate estimation of ω can be obtained with ML (e.g., Pond and Frost, 2005b), Bayesian (e.g., Wilson and McVean, 2006) and ABC (e.g., Lopes et al., 2014) approaches under the assumption of an explicit model of codon substitution. Such codon models can be very complex, allowing, for example, ω to vary across codon sites and/or tree branches under diverse probability distributions (Pond and Frost, 2005a; Yang and Nielsen, 2002). In this concern, ω can be estimated per site (Pond and Frost, 2005b) or branch (Pond and Frost, 2005a), although these estimates require many taxa to provide enough information (Pond and Frost, 2005b). The estimation of ω is implemented in a variety of computational frameworks such as PAML (Yang, 2007), HYPHY or CodABC. Importantly, if recombination is suspected in the data, it should be incorporated when estimating ω to avoid identifying false positively selected sites (Anisimova et al., 2003; Arenas and Posada, 2010a, 2014a). If recombination is detected, it is possible to co-estimate recombination and selection rates simultaneously with frameworks such as OmegaMap or CodABC, or account for the former while estimating the latter (e.g., HYPHY).
Other key aspects in microbial dynamics include time of emergence (e.g., pathogen outbreaks) and geographical distribution of pathogens. New probabilistic models based on the Bayesian approaches have recently been developed for inference and hypothesis testing of divergence times, ancestral locations and historical patterns of migration (i.e., phylogeographic history) (Lemey et al., 2010). Such models are implemented in BEAST and SPREAD (Bielejec et al., 2011) and the outputs can be visualized with virtual globe software such as Google Earth (www.google.com/earth/).
3.3 SNPs-based analyses
Typing data can also be presented as strings of SNPs (Maiden et al., 2013). These strings can be used to obtain evolutionary relatedness and to analyze the evolution and dynamics of populations by the estimation of population genetics parameters (Fig. 2). The main disadvantage from using SNPs instead of nucleotide sequences is the amount of information. With nucleotide sequences we have four character states (A, C, G and T), while for SNPs we only have two states (0 and 1). Although any genomic region can be presented as a sequence of nucleotides or a string of SNPs, one should consider that more information usually leads to more accurate results. The methods applied to the analysis of SNPs are not better or worse than the methods applied to DNA, the key point is the amount of information available from the data. For example, four states data allow for the implementation of more realistic substitution models of evolution than those based on two states data, thus leading to more accurate inferences. Advantages of working with two-state SNP data include fast computational time of analysis and potentially less homoplasy (Pearson et al., 2009). However, SNPs can present low mutation rates and therefore accurate analyses may require large sequences (Pearson et al., 2009).
Figure 2.
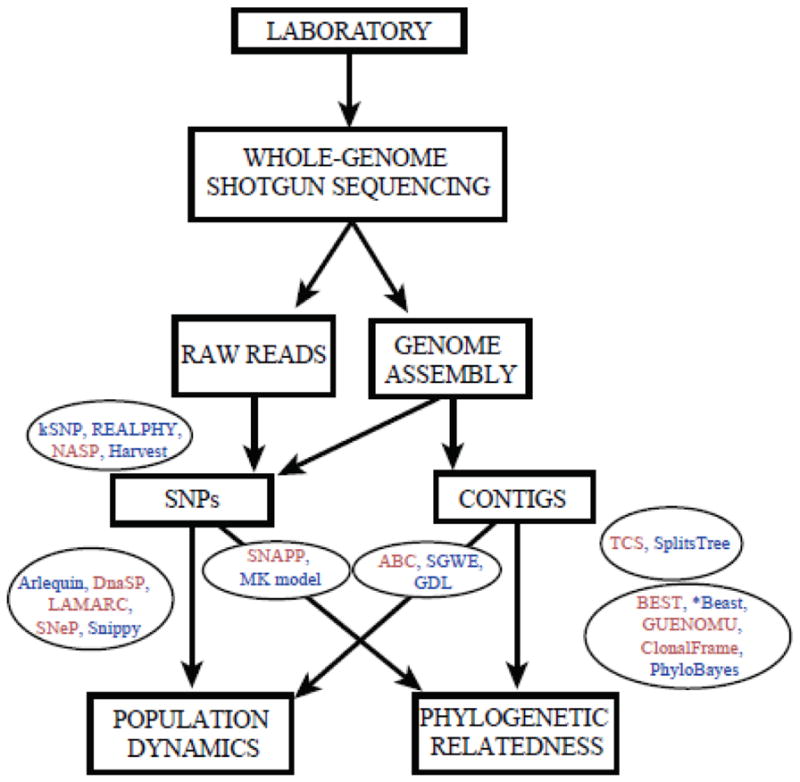
Proposed workflow for typing analysis of microbial DNA sequences using Whole-Genome Shotgun sequences (WGS). Current methods can take either SNPs or gene regions (contigs) for assessing phylogenetic relatedness and/or population dynamic patterns. The workflow shows data and tasks in boxes, and databases and computer programs in circles. We have highlighted in light red our preferred computer programs.
NGS has transformed microbiology, making genomic analyses possible for a huge number of species and pathogenic strains. However, converting millions of sequencing reads per sample into meaningful data is not trivial, and analytical choices made for genome assembly, sequence alignment, and SNP calling will impact final outcomes. SNP calling from microbial genomes includes some major challenges, such as reference genome selection, presence of rare polymorphisms within a culture, and use of de novo genome assemblies (Olson et al., 2015). Given the plethora of methods available to call SNPs from microbial genomes (for a review see Nielsen et al., 2011), optimization of bioinformatics pipelines for specific organisms and/or experiments is frequently required (Olson et al., 2015). A first step for any evolutionary analysis based on SNP data consists of sequence alignment with a reference sequence (i.e., by mapping read sequences to a reference genome with tools such as Snippy (Kwong et al., 2016b) and a posterior refinement of the alignment (Chang et al., 2009; Wegrzyn et al., 2009). However, some pipelines can infer SNPs directly from K-mers without the use of genomic references (Gardner et al., 2015). These tasks are crucial and errors can lead to biased estimates (Castro-Nallar et al., 2015; Pettengill et al., 2014).
3.3.1 Phylogenetic relatedness
To perform phylogenetic inferences, SNP sequences can be used to construct a distance matrix that generates (i.e., with hierarchical clustering) a phylogenetic tree (e.g., Gardy et al., 2011; Hendriksen et al., 2011). Commonly used frameworks to perform this inference are GARLI (Bazinet et al., 2014), RAxML, FastTree, SplitsTree (Huson and Bryant, 2006) and MrBayes. Another interesting program is SNAPP (Bryant et al., 2012), which performs phylogenetic inferences from SNP data under a Bayesian approach similar to that implemented in BEAST for DNA sequences. Alternatively, researchers use substitution models originally designed for analysis of morphological data such as the MK model (Lewis, 2001). Interestingly, the program BioNumerics (http://www.applied-maths.com) implements the different steps to properly analyze SNP data including align with a reference sequence, filter out SNPs and create a phylogenetic inference. Additional packages (Fig. 2) for performing phylogenetic inference from SNP data are NASP, which is useful for phylogenomic data and supports a variety of input formats (Sahl et al., 2016), REALPHY, which is useful for raw sequencing reads (Bertels et al., 2014), kSNP, which can analyze genomes without requiring a genome alignment or a reference genome (Gardner et al., 2015) and Harvest, which uses genomic data while accounting for core-genome alignment, variant calls and recombination (Treangen et al., 2014).
3.3.2 Population dynamics
Genetic diversity can easily be estimated from SNP data with traditional programs such as Arlequin (Excoffier and Lischer, 2010) and DNAsp (Rozas, 2009). Note that these programs implement different summary statistics to measure genetic diversity (i.e., number of alleles, heterozygosity and pairwise differences) and genetic differentiation (i.e., FST and FCT).
Population genetics parameters such as effective population size, population growth rate and migration rate can also be estimated from SNP data. The traditional procedure to estimate the effective population size was based on estimates of linkage disequilibrium (LD; the non-random association between alleles at different loci in a population) (Do et al., 2014; Hill, 1981). However, some evolutionary processes, such as admixture and genetic drift (Wang, 2005), hitchhiking during selective sweeps and background selection (Charlesworth et al., 1997), can affect the estimation of LD. Consequently, computer programs that consider these processes are recommended, e.g., SNeP (Barbato et al., 2015). Additionally, ML and Bayesian approaches can be used to estimate population parameters such as population growth rate, recombination rate and migration rate (Kuhner, 2006), e.g., the program LAMARC estimates all three parameters from SNP data.
ABC can also be applied to estimate population parameters from SNP data (Cornuet et al., 2014; Theunert et al., 2012). A variety of computer simulators (i.e., SIMCOAL2 (Laval and Excoffier, 2004) and SPLATCHE2 (Ray et al., 2010)) implement the simulation of SNPs under diverse evolutionary scenarios (i.e., complex demographics or migration) that the user has to specify. Indeed, as noted above, summary statistics required for ABC can be obtained from SNP data with programs such as Arlequin.
3.4 GWS-based analyses
Genome-wide sequences can help resolve complex evolutionary problems. However, large datasets can enhance systematic errors leading to statistically well-supported incorrect estimates (Kumar et al., 2011; Phillips et al., 2004; Rodríguez-Ezpeleta et al., 2007). Importantly, the impact of various evolutionary processes specific to GWS evolution (i.e., horizontal gene transfer (HGT), gene duplication and loss (GDL), incomplete lineage sorting (ILS) or heterogeneous evolution among genomic regions) should be examined as otherwise they may bias analyses (Galtier and Daubin, 2008; Jeffroy et al., 2006; Mallo et al., 2015). We next describe potential ways to explore the effect of these processes on analyses (see Fig. 2).
3.4.1 Phylogenetic relatedness
A variety of coalescent approaches have been developed to deal with stochastic variation in gene trees from multilocus molecular data and to infer species trees (Fig. 2). Among these, BEST (Liu, 2008) and *BEAST (Heled and Drummond, 2010) incorporate the effect of ILS by implementing multispecies coalescent into a Bayesian hierarchical model. Unfortunately, these programs require extensive computational time (Ogilvie et al., 2016). Concatenation methods, that assume all recovered gene trees share a common evolutionary history (DeGiorgio and Degnan, 2010; Larget et al., 2010), can be an alternative; however, subsampled datasets analyzed with *BEAST yielded more reliable results than full datasets analyzed with concatenation methods (Ogilvie et al., 2016). A few algorithms have also explored additional processes occurring in phylogenomic datasets (ILS, GDL or HGT) (Rasmussen and Kellis, 2012; Szöllősi et al., 2012; Yu et al., 2013) although they have not been implemented in analytical programs. To this end, a hierarchical Bayesian model that jointly considers ILS, GDL and HGT was recently developed (Martins et al., 2016) and implemented in the program GUENOMU (de Oliveira Martins and Posada, 2017). This framework has the benefits of not requiring identification of orthologs and allows incorporation of multiple individuals from the same species.
When estimating evolutionary relationships among microbes using long DNA sequences, the impact of recombination becomes a significant issue. If recombination is substantial, the evolutionary history of those sequences is no longer captured by a bifurcating model, and therefore a tree representation may fail to accurately portray the genealogy (Schierup and Hein, 2000a). Under such circumstances, two strategies can be considered:
Inference of a phylogenetic network (Huson and Bryant, 2006). Woolley et al. (2008) have revised the most common algorithms for building phylogenetic networks and concluded that the union of maximum parsimonious (UMP) trees (Cassens et al., 2005) performed the best. The computer programs TCS (Templeton et al., 1992) and SplitsTree also performed well for inferring network genealogies. Finally, Didelot and Falush (2007) have developed a Bayesian coalescent approach (ClonalFrame) that takes homologous recombination into account while inferring clonal relationships between the members of a sample. Phylogenetic networks are also useful for identifying the presence of clusters and their genetic relationships (Huson and Bryant, 2006).
Inference of a phylogenetic tree for each recombinant fragment (Arenas and Posada, 2010b). This methodology is based on two steps. First, recombination breakpoints are detected using programs such as RDP or Hyphy (with GARD, see above). Second, a specific phylogenetic tree is inferred for each recombinant fragment. This strategy is especially useful for performing posterior evolutionary analyses, such as ancestral sequence reconstruction (e.g., Arenas and Posada, 2010b) or molecular adaptation (e.g., Pérez-Losada et al., 2011; Pérez-Losada et al., 2009) accounting for recombination.
The substitution process along the genome can be highly heterogeneous where different genomic regions fit with different substitution models of evolution (Arbiza et al., 2011). Since the selection of the best fitting substitution model is crucial for accurate phylogenetic inferences (Lemmon and Moriarty, 2004), phylogenetic programs (e.g., RAxML, MrBayes and PhyloBayes) that consider heterogeneous substitution across sites or regions are preferable.
3.4.2 Population dynamics
The methods above described for analyzing population dynamics data based on short nucleotide sequences could be extended to analyze GWS data. However, genomes can experience unique evolutionary events such as duplications, insertions, deletions, inversions, translocations or gene-gene interactions. These events are difficult to model, leading to inefficient or intractable ML functions (Marjoram et al., 2003; Wegmann et al., 2009). To deal with complex evolutionary scenarios, ABC can serve as an alternative (Arenas, 2015a). Fortunately, some frameworks (see Fig. 2) have recently been developed to simulate GWS data accounting for complex evolutionary genomic events: 1) the program ALF, for example, can simulate genome evolution accounting for GDL, gene fusion and fission, lateral gene transfer (LGT), genome rearrangement and speciation under a birth-death process (Dalquen et al., 2012); 2) the program SGWE can simulate genome evolution accounting for heterogeneous recombination and substitution rates and combine different gene trees into a species tree (Arenas and Posada, 2014b); 3) prior distributions and summary statistics for the ABC analysis could be used for the whole genome but also for specific genomic regions (Arenas, 2015a). Unfortunately, there is not yet an ABC framework available to analyze GWS data, but it is possible to design an ABC method by combining simulations of GWS data, estimation of summary statistics and rejection or multiple regression approaches (e.g., Csilléry et al., 2012; Wegmann et al., 2010).
4. Microbial sequence typing applications
Most contemporary studies use more than one approach to typing, epidemiology, and phylogenetic inference, with the aim of maximizing compatibility with current and past data and genetic resolution down to the strain level (e.g., MLST, SNPs, and WGS Castro-Nallar et al., 2015). Moreover, considering the current antibiotic crises highlighted by recent reports by the World Health Organization, researchers are also turning to in silico MLST schemes from whole-genome sequences to assign new sequence types to clinically important isolates while appreciating the value of genome sequences to typing. Additionally, with constantly decreasing sequencing costs, genome-scale microbial typing studies are becoming more affordable. The analysis of WGS data tends to lead to high statistical confidence (P value). However, as indicated above, increasing reports are showing highly significant P values for contrasting phylogenetic hypotheses depending on the evolutionary model and inference method used. Additionally, genomes can experience unique evolutionary events (e.g., duplications, translocations, etc) that can also bias epidemiological and phylogenetic inferences. Therefore, when applying WGS-base typing approaches, emphasizing effect size and biological relevance, rather than the P value, may help to alleviate systematic error (Kumar et al., 2011). Similarly, using ABC methods instead of ML functions to estimate population parameters may also help to accommodate complex evolutionary scenarios. Since the use of genome wide sequence data is a trend that will likely continue, we want to highlight here again that the application of standard methods for phylogenetic and population dynamics analysis to WGS data is potentially problematic given the intrinsic limitations of these gene-based approaches.
In the following sections, we show current examples of modern use of molecular typing for both epidemiology and phylogenetic inference. Other examples can be found in previous studies cited in the Introduction of this review.
4.1 Molecular epidemiology
There has been increasing attention paid to tracking and identifying sources of (opportunistic) pathogens in hospital-based settings in the last few years. One such opportunistic pathogen is Klebsiella pneumoniae, a human commensal whose hyper-virulent and multidrug resistance members have emerged worldwide. Recently, Yang et al. (2017) studied the short-term evolution and transmission of K. pneumoniae over a 40-week period in a hospital from the US, finding that isolates showed low intra-host diversity (up to 15 single nucleotide variants). This level of resolution proved sufficient for following the transmission chain back to its source (endoscopic device); this was only possible due to the use of WGS, with the added benefit of detecting the blaCTX-M-15 gene (involved in Carbapenem resistance) in 27 out of 32 isolates. Other studies compared the power of electrophoresis-based methods to WGS making evident that previously clonal isolates are distinguishable through WGS (Salipante et al., 2015). Similarly, Mathers et al. (2015) presented a five-year single-institution outbreak investigation revealing the molecular epidemiology of K. pneumoniae and where they could follow isolates both by sequence type (in silico determined) and SNP dataset derived from the core genome. The authors highlighted the practicality of linking MLST types with antimicrobial resistance determinants and the power of a whole-genome SNP dataset for increased phylogenetic resolution. The combined use of WGS and MLST can provide valuable information regarding origin, clinical phenotype, and potential treatment of nosocomial infectious disease.
K. pneumoniae is one of six pathogens which are leading cause of nosocomial infections throughout the world (Pendleton et al., 2013). Although K. pneumoniae is traditionally acquired through nosocomial infection, it has elicited the interest of researchers to unravel potential zoonotic transmission pathways. Davis et al. (2015) compared K. pneumoniae isolated from retail meat from grocery stores and from human urine and blood specimens (both sources from Flagstaff, AZ). By combining traditional MLST and WGS, they observed that meat source isolates were more likely multidrug resistant than clinical isolates, even though isolates from both sources shared MLST profiles and were phylogenetically intermingled. Their results suggested potential food-borne transmission routes that carry the risk of spreading multidrug resistance into the general population. For a review on K. pneumoniae population genomics see Wyres and Holt (2016) and for a review on food animal production and antibiotic resistance, see Silbergeld et al. (2008).
The spread of multidrug resistance (MDR) has also been studied for old foes including the causative agent of Typhoid Fever. Wong et al. (2015) explored the intra- and inter-continental spread of a H58 MDR Salmonella Typhi clade using WGS. Based on a dataset with more than 20 thousand SNPs, the authors showed that multiple transfers from Asia to Africa have occurred and are still occurring and that MDR isolates are replacing drug sensitive isolates. Interestingly, another study by the same group did not find this clade in Nigeria, where multiple introductions could better explain the Salmonella genotypes present (Wong et al., 2016b). Overall, these and other similar studies highlight the need for unbiased sampling in molecular epidemiology studies, as often studies select isolates for sequencing and typing based on pathogenicity and convenience, which tend to overlook much of the variation needed for informed public health policy decisions (Holt et al., 2008).
Another Enterobacteriaceae that has been studied by modern typing methods is Escherichia coli, like in the German outbreak of May–July 2011 (Rohde et al., 2011). Within 24 hours, the DNA sequences of E. coli infecting patient zero (TY2482) were assembled and MLST genes identified, which allowed researchers to rapidly genotype the causative strain and find the close phylogenetic relationship between this new strain and a previous one reported in 2001 in Germany. Later whole-genome comparisons showed that TY2482 was nearly identical to an African strain that may (or may not) harbor the Shiga toxin gene (Mossoro et al., 2002). While traditional typing approaches pointed to the outbreak strain as being enterohemorrhagic E. coli, it was only after more detailed whole-genome inspection that researchers discovered it corresponded to an enteroaggregative E. coli harboring two conjugative plasmids, a small plasmid, and a stx2 prophage (Rohde et al., 2011). Along these same lines, linking outbreaks from different localities has been possible due to the increased resolution that WGS allows. For example, a small E. coli outbreak was reported in southwest France in June 2011, which was indistinguishable from the German one by traditional methods. Researchers could only separate the two variants by WGS, revealing that the German outbreak isolates were limited in genetic diversity (2 SNPs from four individuals) compared to the French isolates (19 SNPs from seven individuals) (Grad et al., 2012). Therefore, slow-evolving pathogens or pathogen outbreaks over short periods of time are difficult to type and present challenges for traditional MLST approaches.
Similarly, Neisseria gonorrhea represents an extremely slow-evolving pathogen. De Silva et al. (2016) studied patients infected with N. gonorrhea from diverse UK locations and found that in 26% of the infections, N. gonorrhea isolates differed by zero nucleotide substitutions at the genome level; however, in 76% of the infections, contact-tracing demonstrated local, national, and international transmissions. Epidemiological and population dynamic patterns have also been inferred from MLST data, where a combination of several housekeeping and hypervariable genes were used for increased resolution at the local level (Pérez-Losada et al., 2005). Other studies have contrasted traditional typing with WGS for populations from different localities but also different epidemiological properties. For instance, Didelot et al. (2016) analyzed 237 isolates from predominantly heterosexual men from populations in Sheffield and London, respectively. Interestingly, all isolates resolved into a single sequence type per population (ST12 and ST225, respectively) by the most widely used tool for N. gonorrhea detection, multi-antigen sequence typing (for in silico version see Kwong et al., 2016a). In contrast, WGS could resolve relationships among isolates at the intra- and inter-specific levels (less than 200 substitutions genome-wide) (Didelot et al., 2016).
4.2 Genotyping and phylogenetic inference
Molecular markers can be used for both genotyping and inferring evolutionary relationships. More comprehensive genotyping frameworks link genetic variation with phylogenetic placement to obtain more information regarding origin and pathogenicity. For pathogens with limited genetic diversity such as Salmonella Typhi, new genotyping frameworks have been developed where researchers have identified genome-wide SNPs that link isolates to geographic source populations (Roumagnac et al., 2006; Wong et al., 2016a). Wong et al. (2016a) used nearly 2,000 isolates from over 60 countries to identify 68 phylogenetically informative SNPs. Using this framework, the authors predicted geographic origin at the country level for a subset of novel isolates, paving the way for future developments aimed at increasing accuracy and empowering clinicians and public health officials.
Another group of enteric pathogens, Shigella spp., have been studied regarding their evolutionary history and adaptation to human hosts. Four species exist that cause dysentery: S. sonnei, S. flexneri, S. boydii, and S. dysenteriae, all of which are phylogenetically nested within the E. coli clade. Yang et al. (2007) tested the monophyly and phylogenetic relationships of Shigella spp. by using up to 23 housekeeping chromosomal genes from Shigella and E. coli. Their results supported the hypothesis of multiple independent origins (probably four) of Shigella members from diverse E. coli strains. This would explain why Shigella spp. harbor diverse genomes but a similar phenotype.
In particular, S. sonnei is a human pathogen that diverged less than 500 years ago (Holt et al., 2012). The researchers collected samples from four continents and sequenced 132 genomes from S. sonnei isolates from 1943 to 2008, which allowed them to detect more than 10,000 SNPs for increased phylogenetic resolution. Interestingly, the authors defined four lineages from the SNP phylogeny that correlated with more traditional typing methods such as Biotypes and CRISPR types (Nastasi et al., 1993; Touchon et al., 2011), suggesting that for S. sonnei, traditional typing methods provide sufficient resolution to distinguish lineages even over short periods of time.
Similar approaches have been applied to S. dysenteriae type 1 (dysenteriae bacillus), a pathogen responsible for major epidemics during the 20th century. Njamkepo et al. (2016) reconstructed the historical spread and geographic distribution of S. dysenteriae by sequencing 331 isolates collected from 1915 to 2011 (66 countries) and found that the global spread of the bacterium predates the First World War and the global expansion of S. sonnei (Holt et al., 2012; Njamkepo et al., 2016). While these studies cannot establish causal relationships, the major expansions from Europe to the rest of the world coincide with periods of intense European migration due to colonialism. It is important to note that while traditional typing techniques and WGS were congruent in S. sonnei, the opposite was true for S. dysenteriae, where single lineages were separated by tens or hundreds of SNPs. See (The et al., 2016) for a review of Shigella spp. evolution, adaptation, and historical geographic spread.
Phylogenetic relationships of other globally important pathogens have also been elucidated using molecular typing methods. Vibrio cholerae epidemics have been characterized by several waves of global transmission with the latest Haiti outbreak belonging to the seventh cholera pandemic (caused by V. cholerae El Tor biotype of serogroup O1). In the Fall of 2010, the Haiti outbreak was reported and initial Pulse-Field Gel Electrophoresis (PFGE; a technique used for the separation of large DNA molecules by applying to a gel matrix an electric field that periodically changes direction) typing indicated that different Haiti isolates were indistinguishable. Using WGS, the authors concluded that Haiti isolates were related and were not identical to isolates from India and Cameroon (Reimer et al., 2011). Then, Chin et al. (2011) established an association between Haiti and Bangladesh isolates by means of WGS. More importantly, Chin et al. refuted the hypothesis that the Haiti outbreak was related to indigenous V. cholerae, as it was presented by Hasan et al. (2012), who reported both V. cholerae O1 and non-O1/O139 early in the Haiti cholera epidemic with samples collected from victims of 18 towns of Haiti. Katz et al. (2013) sequenced longitudinal samples from the Haiti outbreak and concluded that it corresponded to a single source introduction. Hendriksen et al. (2011) demonstrated that the Nepalese isolates formed a monophyletic group with other isolates from Haiti and Bangladesh, findings that were consistent with PFGE patterns and antibiotic susceptibility tests. We refer the reader to other reviews on the Haiti outbreak (Frerichs et al., 2012; Orata et al., 2014).
As V. cholerae remains a highly relevant human pathogen, the development of higher resolution tools and more comprehensive sampling will aid researchers in establishing global routes of transmission and large-scale patterns of gene flow. For instance, Mutreja et al. (2011) collected global genomic data on V. cholerae and developed high-resolution markers (SNPs) to genotype V. cholerae lineages. Using phylogenetic inference and SNP markers, they showed that the seventh V. cholerae pandemic has spread from the Bay of Bengal on multiple overlapping waves, some of which reached such faraway places as Haiti.
5. Conclusions and future prospects
MLST is a flexible approach for characterizing bacteria and some eukaryotes. It has become a standard mainly due to the existence of comprehensive databases and its broad implementation in clinical practice and molecular diagnostics. MLST is widely used in basic research labs (PCR + Sanger) and core sequencing facilities performing genome sequencing. MLST has broadened its basic scheme from using housekeeping genes to incorporate more and new molecular markers, such as ribosomal proteins (rMLST) and polymorphic repeated sequences (MLVA); most recently, it has begun integrating draft and full genomes (cgMLST).
Over the last five years, whole-genome sequencing (WGS) has emerged as a powerful technology for microbial sequence typing and is increasingly applied in clinical and public health microbiology. New MLST-genome strategies that take advantage of NGS technology to accelerate and automate WGS will expand the power and versatility of DNA typing. Such strategies will also allow for calculations of more accurate and robust estimates of phylogenies and population genetic parameters under more complex (realistic) statistical models using, for example, Bayesian phylogenomics and approximate Bayesian computation. Those statistical frameworks will also integrate epidemiologic and geographic information, allowing the estimation of the spatial and temporal dynamics of pathogens. Several examples of WGS typing have already been published assessing the time of emergence, origin, and dissemination of pathogen outbreaks and the spread of antibiotic resistance.
While the exploitation of WGS for typing microbial diversity presents great opportunities, it also brings major challenges; some of these challenges are computational, such as genome data analysis, sharing and storage, but others are conceptual, such as relating WGS data to typing and microbial taxonomy. The genomic era opens the door to new types of holistic microbiology research, i.e., a systems biology (aka ecology) framework, in which taxonomic, epidemiological and evolutionary information are integrated with other Omic information from both the microbial communities and the host. In coming years, one can only assume that classical or expanded forms of MLST will remain a key component of the microbial genomicist’s toolkit used for understanding the diversity and dynamics of infectious diseases.
Highlights.
Next-generation sequencing (NGS) is changing the field of microbial genomics research.
NGS strategies have expanded the versatility of MLST typing approaches.
We describe standard and new approaches of DNA sequence typing in microbiology.
We provide guidelines for DNA sequence typing analysis, including methods and computational frameworks.
We present several applications of standard and new typing approaches to microbiology.
Acknowledgments
MP-L was funded by a DC D-CFAR Research Award from the District of Columbia Developmental Center for AIDS Research (P30AI087714), by a University Facilitating Fund award from George Washington University, the Margaret Q. Landenberger Research Foundation and a K12 Career Development Program 5 K12 HL119994 award. MA was supported by the “Ramón y Cajal” grant RYC-2015-18241 from the Spanish Government. ECN was funded by “CONICYT + PAI/CONCURSO NACIONAL APOYO AL RETORNO DE INVESTIGADORES/AS DESDE EL EXTRANJERO, CONVOCATORIA 2014 + FOLIO 82140008” and “FONDECYT de iniciación 11160905”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aanensen DM, Spratt BG. The multilocus sequence typing network: mlst. net. Nucleic Acids Res. 2005;33:W728–W733. doi: 10.1093/nar/gki415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010:gkq291. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves I, Arenas M, Currat M, Hanulova AS, Sousa VC, Ray N, Excoffier L. Long-distance dispersal shaped patterns of human genetic diversity in Eurasia. Mol Biol Evol. 2016;33:946–958. doi: 10.1093/molbev/msv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164:1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiza L, Patricio M, Dopazo H, Posada D. Genome-wide heterogeneity of nucleotide substitution model fit. Genome Biol Evol. 2011;3:896–908. doi: 10.1093/gbe/evr080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M. Advances in computer simulation of genome evolution: toward more realistic evolutionary genomics analysis by approximate Bayesian computation. J Mol Evol. 2015a;80:189–192. doi: 10.1007/s00239-015-9673-0. [DOI] [PubMed] [Google Scholar]
- Arenas M. Trends in substitution models of molecular evolution. Front Genet. 2015b;6:319. doi: 10.3389/fgene.2015.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M, Lopes JS, Beaumont MA, Posada D. CodABC: a computational framework to coestimate recombination, substitution, and molecular adaptation rates by approximate bayesian computation. Mol Biol Evol. 2015;32:1109–1112. doi: 10.1093/molbev/msu411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M, Posada D. Coalescent simulation of intracodon recombination. Genetics. 2010a;184:429–437. doi: 10.1534/genetics.109.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M, Posada D. The effect of recombination on the reconstruction of ancestral sequences. Genetics. 2010b;184:1133–1139. doi: 10.1534/genetics.109.113423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M, Posada D. The influence of recombination on the estimation of selection from coding sequence alignments. Natural Selection: Methods and Applications. 2014a:112–125. [Google Scholar]
- Arenas M, Posada D. Simulation of Genome-Wide Evolution under Heterogeneous Substitution Models and Complex Multispecies Coalescent Histories. Mol Biol Evol. 2014b;31:1295–1301. doi: 10.1093/molbev/msu078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Dress AW. Split decomposition: a new and useful approach to phylogenetic analysis of distance data. Mol Phylogenet Evol. 1992;1:242–252. doi: 10.1016/1055-7903(92)90021-8. [DOI] [PubMed] [Google Scholar]
- Barbato M, Orozco-terWengel P, Tapio M, Bruford MW. SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet. 2015:6. doi: 10.3389/fgene.2015.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazinet AL, Zwickl DJ, Cummings MP. A gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst Biol. 2014;63:812–818. doi: 10.1093/sysbio/syu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA. Approximate Bayesian computation in evolution and ecology. Annu Rev Ecol Evol Syst. 2010;41:379–406. [Google Scholar]
- Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–2035. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello G, Casado C, García S, Rodríguez C, del Romero J, Carvajal-Rodriguez A, Posada D, López-Galíndez C. Lack of temporal structure in the short term HIV-1 evolution within asymptomatic naive patients. Virology. 2007;362:294–303. doi: 10.1016/j.virol.2006.11.039. [DOI] [PubMed] [Google Scholar]
- Bertels F, Silander OK, Pachkov M, Rainey PB, van Nimwegen E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol Biol Evol. 2014;31:1077–1088. doi: 10.1093/molbev/msu088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorelle G, Benazzo A, Mona S. ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Mol Ecol. 2010;19:2609–2625. doi: 10.1111/j.1365-294X.2010.04690.x. [DOI] [PubMed] [Google Scholar]
- Bielejec F, Rambaut A, Suchard MA, Lemey P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2912. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boers SA, van der Reijden WA, Jansen R. High-throughput multilocus sequence typing: bringing molecular typing to the next level. PloS one. 2012;7:e39630. doi: 10.1371/journal.pone.0039630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR, Drummond AJ. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol Biol. 2017;17:42. doi: 10.1186/s12862-017-0890-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol. 2012;29:1917–1932. doi: 10.1093/molbev/mss086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol. 2004;21:255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassens I, Mardulyn P, Milinkovitch MC. Evaluating intraspecific “network” construction methods using simulated sequence data: do existing algorithms outperform the global maximum parsimony approach? Syst Biol. 2005;54:363–372. doi: 10.1080/10635150590945377. [DOI] [PubMed] [Google Scholar]
- Castro-Nallar E, Hasan NA, Cebula TA, Colwell RR, Robison RA, Johnson WE, Crandall KA. Concordance and discordance of sequence survey methods for molecular epidemiology. PeerJ. 2015;3:e761. doi: 10.7717/peerj.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MS, Maiden MC, Spratt BG. Database-driven multi locus sequence typing (MLST) of bacterial pathogens. Bioinformatics. 2001;17:1077–1083. doi: 10.1093/bioinformatics/17.11.1077. [DOI] [PubMed] [Google Scholar]
- Chang HW, Chuang LY, Cheng YH, Ho CH, Wen CH, Yang CH. Seq-SNPing: multiple-alignment tool for SNP discovery, SNP ID identification, and RFLP genotyping. OMICS. 2009;13:253–260. doi: 10.1089/omi.2008.0058. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Nordborg M, Charlesworth D. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet Res. 1997;70:155–174. doi: 10.1017/s0016672397002954. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhang W, Zheng H, Lan R, Wang H, Du P, Bai X, Ji S, Meng Q, Jin D. Minimum core genome sequence typing of bacterial pathogens: a unified approach for clinical and public health microbiology. J Clin Microbiol. 2013;51:2582–2591. doi: 10.1128/JCM.00535-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Frazzitta AE, Litvintseva AP, Fang C, Mitchell TG, Springer DJ, Ding Y, Yuan G, Perfect JR. Next generation multilocus sequence typing (NGMLST) and the analytical software program MLSTEZ enable efficient, cost-effective, high-throughput, multilocus sequencing typing. Fungal Genet Biol. 2015;75:64–71. doi: 10.1016/j.fgb.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P. The origin of the Haitian cholera outbreak strain. N Engl J Med. 2011;364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Rainey FA. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int J Syst Evol Microbiol. 2014;64(Pt 2):316–324. doi: 10.1099/ijs.0.054171-0. [DOI] [PubMed] [Google Scholar]
- Cooper JE, Feil EJ. Multilocus sequence typing--what is resolved? Trends Microbiol. 2004;12:373–377. doi: 10.1016/j.tim.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A. DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics. 2014;30:1187–1189. doi: 10.1093/bioinformatics/btt763. [DOI] [PubMed] [Google Scholar]
- Csilléry K, François O, Blum MG. abc: an R package for approximate Bayesian computation (ABC) Methods Ecol Evol. 2012;3:475–479. doi: 10.1016/j.tree.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Dallman TJ, Byrne L, Ashton PM, Cowley LA, Perry NT, Adak G, Petrovska L, Ellis RJ, Elson R, Underwood A, Green J, Hanage WP, Jenkins C, Grant K, Wain J. Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin Infect Dis. 2015;61:305–312. doi: 10.1093/cid/civ318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalquen DA, Anisimova M, Gonnet GH, Dessimoz C. ALF—a simulation framework for genome evolution. Mol Biol Evol. 2012;29:1115–1123. doi: 10.1093/molbev/msr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GS, Waits K, Nordstrom L, Weaver B, Aziz M, Gauld L, Grande H, Bigler R, Horwinski J, Porter S. Intermingled Klebsiella pneumoniae populations between retail meats and human urinary tract infections. Clin Infect Dis. 2015;61:892–899. doi: 10.1093/cid/civ428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Been M, Pinholt M, Top J, Bletz S, Mellmann A, van Schaik W, Brouwer E, Rogers M, Kraat Y, Bonten M, Corander J, Westh H, Harmsen D, Willems RJL. A core genome MLST scheme for high-resolution typing of Enterococcus faecium. J Clin Microbiol. 2015 doi: 10.1128/JCM.01946-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Martins L, Posada D. Species Tree Estimation from Genome-wide Data with Guenomu. Bioinformatics: Volume I: Data, Sequence Analysis, and Evolution. 2017:461–478. doi: 10.1007/978-1-4939-6622-6_18. [DOI] [PubMed] [Google Scholar]
- De Silva D, Peters J, Cole K, Cole MJ, Cresswell F, Dean G, Dave J, Thomas DR, Foster K, Waldram A. Whole-genome sequencing to determine transmission of Neisseria gonorrhoeae: an observational study. Lancet Infect Dis. 2016;16:1295–1303. doi: 10.1016/S1473-3099(16)30157-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio M, Degnan JH. Fast and consistent estimation of species trees using supermatrix rooted triples. Mol Biol Evol. 2010;27:552–569. doi: 10.1093/molbev/msp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Poon AF, Frost SD, Pond SLK. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Bakker HC, Allard MW, Bopp D, Brown EW, Fontana J, Iqbal Z, Kinney A, Limberger R, Musser KA, Shudt M, Strain E, Wiedmann M, Wolfgang WJ. Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg Infect Dis. 2014;20:1306–1314. doi: 10.3201/eid2008.131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Dordel J, Whittles LK, Collins C, Bilek N, Bishop CJ, White PJ, Aanensen DM, Parkhill J, Bentley SD. Genomic Analysis and Comparison of Two Gonorrhea Outbreaks. mBio. 2016;7:e00525–00516. doi: 10.1128/mBio.00525-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, Vaughan A, O’Connor L, Golubchik T, Batty EM, Piazza P, Wilson DJ, Bowden R, Donnelly PJ, Dingle KE, Wilcox M, Walker AS, Crook DW, Peto TE, Harding RM. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012;13:R118. doi: 10.1186/gb-2012-13-12-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KT, Lee JH, Lee HK, Kim J, Park KD. Estimation of effective population size using single-nucleotide polymorphism (SNP) data in Jeju horse. J Anim Sci Technol. 2014;56:28. doi: 10.1186/2055-0391-56-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright MC, Spratt BG. Multilocus sequence typing. Trends Microbiol. 1999;7:482–487. doi: 10.1016/s0966-842x(99)01609-1. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Feil EJ, Enright MC. Analyses of clonality and the evolution of bacterial pathogens. Curr Opin Microbiol. 2004;7:308–313. doi: 10.1016/j.mib.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Fitch WM. Networks and viral evolution. J Mol Evol. 1997;44:S65–S75. doi: 10.1007/pl00000059. [DOI] [PubMed] [Google Scholar]
- Foley SL, Lynne AM, Nayak R. Molecular typing methodologies for microbial source tracking and epidemiological investigations of Gram-negative bacterial foodborne pathogens. Infect Genet Evol. 2009;9:430–440. doi: 10.1016/j.meegid.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Forde BM, O’Toole PW. Next-generation sequencing technologies and their impact on microbial genomics. Brief Funct Genomics. 2013;12:440–453. doi: 10.1093/bfgp/els062. [DOI] [PubMed] [Google Scholar]
- Francisco AP, Bugalho M, Ramirez M, Carrico JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics. 2009;10:152. doi: 10.1186/1471-2105-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carrio JA. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics. 2012;13:87. doi: 10.1186/1471-2105-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerichs RR, Keim PS, Barrais R, Piarroux R. Nepalese origin of cholera epidemic in Haiti. Clin Microbiol Infect. 2012;18:E158–E163. doi: 10.1111/j.1469-0691.2012.03841.x. [DOI] [PubMed] [Google Scholar]
- Galtier N, Daubin V. Dealing with incongruence in phylogenomic analyses. Philos Trans R Soc Lond B Biol Sci. 2008;363:4023–4029. doi: 10.1098/rstb.2008.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SN, Slezak T, Hall BG. kSNP3. 0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics. 2015:btv271. doi: 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Johnston JC, Sui SJH, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–739. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad YH, Lipsitch M, Feldgarden M, Arachchi HM, Cerqueira GC, FitzGerald M, Godfrey P, Haas BJ, Murphy CI, Russ C. Genomic epidemiology of the Escherichia coli O104: H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A. 2012;109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG, Ehrlich GD, Hu FZ. Pan-genome analysis provides much higher strain typing resolution than multi-locus sequence typing. Microbiology. 2010;156:1060–1068. doi: 10.1099/mic.0.035188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan NA, Choi SY, Eppinger M, Clark PW, Chen A, Alam M, Haley BJ, Taviani E, Hine E, Su Q. Genomic diversity of 2010 Haitian cholera outbreak strains. Proc Natl Acad Sci U S A. 2012;109:E2010–E2017. doi: 10.1073/pnas.1207359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27:570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen RS, Price LB, Schupp JM, Gillece JD, Kaas RS, Engelthaler DM, Bortolaia V, Pearson T, Waters AE, Upadhyay BP. Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. MBio. 2011;2:e00157–00111. doi: 10.1128/mBio.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG. Estimation of effective population size from data on linkage disequilibrium. Genet Res. 1981;38:209–216. [Google Scholar]
- Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F. RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model-specification language. Syst Biol. 2016:syw021. doi: 10.1093/sysbio/syw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KE, Baker S, Weill FX, Holmes EC, Kitchen A, Yu J, Sangal V, Brown DJ, Coia JE, Kim DW. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat Genet. 2012;44:1056–1059. doi: 10.1038/ng.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, Goodhead I, Rance R, Baker S, Maskell DJ, Wain J. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat Genet. 2008;40:987–993. doi: 10.1038/ng.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Huijsmans CJ, Schellekens JJ, Wever PC, Toman R, Savelkoul PH, Janse I, Hermans MH. Single-nucleotide-polymorphism genotyping of Coxiella burnetii during a Q fever outbreak in The Netherlands. Appl Environ Microbiol. 2011;77:2051–2057. doi: 10.1128/AEM.02293-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Jefferies J, Clarke SC, Diggle MA, Smith A, Dowson C, Mitchell T. Automated pneumococcal MLST using liquid-handling robotics and a capillary DNA sequencer. Mol Biotechnol. 2003;24:303–307. doi: 10.1385/MB:24:3:303. [DOI] [PubMed] [Google Scholar]
- Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: the beginning of incongruence? Trends Genet. 2006;22:225–231. doi: 10.1016/j.tig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MC. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Chan MS, Maiden MC. mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics. 2004;5:86. doi: 10.1186/1471-2105-5-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Feil EJ, Chan MS, Maiden MC. Sequence type analysis and recombinational tests (START) Bioinformatics. 2001;17:1230–1231. doi: 10.1093/bioinformatics/17.12.1230. [DOI] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. AgdbNet–antigen sequence database software for bacterial typing. BMC bioinformatics. 2006;7:314. doi: 10.1186/1471-2105-7-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. Using MLST to study bacterial variation: prospects in the genomic era. Future Microbiol. 2014;9:623–630. doi: 10.2217/fmb.14.24. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LS, Petkau A, Beaulaurier J, Tyler S, Antonova ES, Turnsek MA, Guo Y, Wang S, Paxinos EE, Orata F. Evolutionary dynamics of Vibrio cholerae O1 following a single-source introduction to Haiti. MBio. 2013;4:e00398–00313. doi: 10.1128/mBio.00398-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim PS, Wagner DM. Humans, evolutionary and ecologic forces shaped the phylogeography of recently emerged diseases. Nat Rev Microbiol. 2009;7:813. doi: 10.1038/nrmicro2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman JFC. The coalescent. Stoch Process Their Appl. 1982;13:235–248. [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22:3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner MK. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22:768–770. doi: 10.1093/bioinformatics/btk051. [DOI] [PubMed] [Google Scholar]
- Kumar S, Filipski AJ, Battistuzzi FU, Kosakovsky Pond SL, Tamura K. Statistics and truth in phylogenomics. Mol Biol Evol. 2011;29:457–472. doi: 10.1093/molbev/msr202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Serizawa M, Okutani A, Sekizuka T, Banno S, Inoue S. Genome-wide single nucleotide polymorphism typing method for identification of Bacillus anthracis species and strains among B. cereus group species. J Clin Microbiol. 2010;48:2821–2829. doi: 10.1128/JCM.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JC, da Silva AG, Dyet K, Williamson DA, Stinear TP, Howden BP, Seemann T. NGMASTER: in silico multi-antigen sequence typing for Neisseria gonorrhoeae. Microb Genom. 2016a:2. doi: 10.1099/mgen.0.000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JC, McCallum N, Sintchenko V, Howden BP. Whole genome sequencing in clinical and public health microbiology. Pathology. 2015;47:199–210. doi: 10.1097/PAT.0000000000000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JC, Mercoulia K, Tomita T, Easton M, Li HY, Bulach DM, Stinear TP, Seemann T, Howden BP. Prospective Whole-Genome Sequencing Enhances National Surveillance of Listeria monocytogenes. J Clin Microbiol. 2016b;54:333–342. doi: 10.1128/JCM.02344-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, Amann R, Beck BJ, Chain PS, Chun J. Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:e1001920. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2016:msw260. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- Larget BR, Kotha SK, Dewey CN, Ané C. BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics. 2010;26:2910–2911. doi: 10.1093/bioinformatics/btq539. [DOI] [PubMed] [Google Scholar]
- Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Ponten T, Ussery DW, Aarestrup FM, Lund O. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Laval G, Excoffier L. SIMCOAL 2.0: a program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics. 2004;20:2485–2487. doi: 10.1093/bioinformatics/bth264. [DOI] [PubMed] [Google Scholar]
- Leekitcharoenphon P, Lukjancenko O, Friis C, Aarestrup FM, Ussery DW. Genomic variation in Salmonella enterica core genes for epidemiological typing. BMC genomics. 2012;13:88. doi: 10.1186/1471-2164-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Moriarty EC. The importance of proper model assumption in bayesian phylogenetics. Syst Biol. 2004;53:265–277. doi: 10.1080/10635150490423520. [DOI] [PubMed] [Google Scholar]
- Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol. 2001;50:913–925. doi: 10.1080/106351501753462876. [DOI] [PubMed] [Google Scholar]
- Liu K, Linder CR, Warnow T. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PloS one. 2011;6:e27731. doi: 10.1371/journal.pone.0027731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics. 2008;24:2542–2543. doi: 10.1093/bioinformatics/btn484. [DOI] [PubMed] [Google Scholar]
- Lopes JS, Arenas M, Posada D, Beaumont MA. Coestimation of recombination, substitution and molecular adaptation rates by approximate Bayesian computation. Heredity. 2014;112:255–264. doi: 10.1038/hdy.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loubna T, Pérez-Losada M, Gu W, Yang Y, Xue L, Crandall KA, Viscidi RP. Population dynamics of Neisseria gonorrhoeae in Shanghai, China: A comparative study. BMC Infect Dis. 2010;10:13. doi: 10.1186/1471-2334-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A, Qiao H, Zhang Y, Shi W, Ho SY, Xu W, Zhang A, Zhu C. Performance of criteria for selecting evolutionary models in phylogenetics: a comprehensive study based on simulated datasets. BMC Evol Biol. 2010;10:242. doi: 10.1186/1471-2148-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCannell D. Bacterial strain typing. Clinics in laboratory medicine. 2013;33:629–650. doi: 10.1016/j.cll.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC, Van Rensburg MJJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Micro. 2013;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo D, Sánchez-Cobos A, Arenas M. Diverse Considerations for Successful Phylogenetic Tree Reconstruction: Impacts from Model Misspecification, Recombination, Homoplasy, and Pattern Recognition. Pattern Recognition in Computational Molecular Biology: Techniques and Approaches. 2015:439–456. [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem. 2013;6:287–303. doi: 10.1146/annurev-anchem-062012-092628. [DOI] [PubMed] [Google Scholar]
- Mardis ER. DNA sequencing technologies: 2006–2016. Nat Protoc. 2017;12:213–218. doi: 10.1038/nprot.2016.182. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjoram P, Molitor J, Plagnol V, Tavaré S. Markov chain Monte Carlo without likelihoods. Proc Natl Acad Sci U S A. 2003;100:15324–15328. doi: 10.1073/pnas.0306899100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JW, O’Leary MM, Shutt KA, Sambol SP, Johnson S, Gerding DN, Harrison LH. Multilocus Variable-Number Tandem-Repeat Analysis and Multilocus Sequence Typing Reveal Genetic Relationships among Clostridium difficile Isolates Genotyped by Restriction Endonuclease Analysis. J Clin Microbiol. 2010;48:412–418. doi: 10.1128/JCM.01315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Posada D. Analysing recombination in nucleotide sequences. Mol Ecol Resour. 2011;11:943–955. doi: 10.1111/j.1755-0998.2011.03026.x. [DOI] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LDO, Mallo D, Posada D. A Bayesian supertree model for genome-wide species tree reconstruction. Syst Biol. 2016;65:397–416. doi: 10.1093/sysbio/syu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers AJ, Stoesser N, Sheppard AE, Pankhurst L, Giess A, Yeh AJ, Didelot X, Turner SD, Sebra R, Kasarskis A. Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: insights into endemicity from whole-genome sequencing. Antimicrob Agents Chemother. 2015;59:1656–1663. doi: 10.1128/AAC.04292-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Minin VN, Bloomquist EW, Suchard MA. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol. 2008;25:1459–1471. doi: 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossoro C, Glaziou P, Yassibanda S, Lan NTP, Bekondi C, Minssart P, Bernier C, Le Bouguénec C, Germani Y. Chronic diarrhea, hemorrhagic colitis, and hemolytic-uremic syndrome associated with HEp-2 adherent Escherichia coli in adults infected with human immunodeficiency virus in Bangui, Central African Republic. J Clin Microbiol. 2002;40:3086–3088. doi: 10.1128/JCM.40.8.3086-3088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Verezemska O, Isbandi M, Thomas AD, Ali R, Sharma K, Kyrpides NC. Genomes OnLine Database (GOLD) v. 6: data updates and feature enhancements. Nucleic Acids Res. 2017;45:D446–D456. doi: 10.1093/nar/gkw992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastasi A, Pignato S, Mammina C, Giammanco G. rRNA gene restriction patterns and biotypes of Shigella sonnei. Epidemiol Infect. 1993;110:23–30. doi: 10.1017/s0950268800050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navascués M, Depaulis F, Emerson BC. Combining contemporary and ancient DNA in population genetic and phylogeographical studies. Mol Ecol Resour. 2010;10:760–772. doi: 10.1111/j.1755-0998.2010.02895.x. [DOI] [PubMed] [Google Scholar]
- Nelson CL, Pelak K, Podgoreanu MV, Ahn SH, Scott WK, Allen AS, Cowell LG, Rude TH, Zhang Y, Tong A. A genome-wide association study of variants associated with acquisition of Staphylococcus aureus bacteremia in a healthcare setting. BMC Infect Dis. 2014;14:83. doi: 10.1186/1471-2334-14-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njamkepo E, Fawal N, Tran-Dien A, Hawkey J, Strockbine N, Jenkins C, Talukder KA, Bercion R, Kuleshov K, Kolínská R. Global phylogeography and evolutionary history of Shigella dysenteriae type 1. Nat Microbiol. 2016;1:16027. doi: 10.1038/nmicrobiol.2016.27. [DOI] [PubMed] [Google Scholar]
- Ogilvie HA, Heled J, Xie D, Drummond AJ. Computational performance and statistical accuracy of* BEAST and comparisons with other methods. Syst Biol. 2016:syv118. doi: 10.1093/sysbio/syv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ND, Lund SP, Colman RE, Foster JT, Sahl JW, Schupp JM, Keim P, Morrow JB, Salit ML, Zook JM. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front Genet. 2015:6. doi: 10.3389/fgene.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn GS. Evolution and public health. Proc Natl Acad Sci U S A. 2010;107:1702–1709. doi: 10.1073/pnas.0906198106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orata FD, Keim PS, Boucher Y. The 2010 cholera outbreak in Haiti: how science solved a controversy. PLoS Pathog. 2014;10:e1003967. doi: 10.1371/journal.ppat.1003967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace NR. Mapping the tree of life: progress and prospects. Microbiology and molecular biology reviews: MMBR. 2009;73:565–576. doi: 10.1128/MMBR.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- Pearson T, Okinaka RT, Foster JT, Keim P. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect Genet Evol. 2009;9:1010–1019. doi: 10.1016/j.meegid.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- Pérez-Losada M, Arenas M, Castro-Nallar E. Multilocus Sequence Typing of Pathogens: Methods, Analyses, and Applications. In: Tibayrenc M, editor. Genetics and Evolution of Infectious Diseases. 2. Elsevier; Amsterdam, Netherlands: 2017. [Google Scholar]
- Pérez-Losada M, Browne EB, Madsen A, Wirth T, Viscidi RP, Crandall KA. Population genetics of microbial pathogens estimated from Multilocus Sequence Typing (MLST) data. Infect Genet Evol. 2006;6:97–112. doi: 10.1016/j.meegid.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M, Cabezas P, Castro-Nallar E, Crandall KA. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect Genet Evol. 2013;16:38–53. doi: 10.1016/j.meegid.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Pérez-Losada M, Jobes DV, Sinangil F, Crandall KA, Arenas M, Posada D, Berman PW. Phylodynamics of HIV-1 from a phase III AIDS vaccine trial in Bangkok, Thailand. PloS one. 2011;6:e16902. doi: 10.1371/journal.pone.0016902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M, Porter ML, Tazi L, Crandall KA. New methods for inferring population dynamics from microbial sequences. Infect Genet Evol. 2007;7:24–43. doi: 10.1016/j.meegid.2006.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M, Posada D, Arenas M, Jobes DV, Sinangil F, Berman PW, Crandall KA. Ethnic differences in the adaptation rate of HIV gp120 from a vaccine trial. Retrovirology. 2009;6:67. doi: 10.1186/1742-4690-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M, Viscidi RP, Demma JC, Zenilman J, Crandall KA. Population genetics of Neisseria gonorrhoeae in a high-prevalence community using a hypervariable outer membrane porB and 13 slowly evolving housekeeping genes. Mol Biol Evol. 2005;22:1887–1902. doi: 10.1093/molbev/msi184. [DOI] [PubMed] [Google Scholar]
- Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettengill JB, Luo Y, Davis S, Chen Y, Gonzalez-Escalona N, Ottesen A, Rand H, Allard MW, Strain E. An evaluation of alternative methods for constructing phylogenies from whole genome sequence data: a case study with Salmonella. PeerJ. 2014;2:e620. doi: 10.7717/peerj.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Delsuc F, Penny D. Genome-scale phylogeny and the detection of systematic biases. Mol Biol Evol. 2004;21:1455–1458. doi: 10.1093/molbev/msh137. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SD. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 2005a;22:478–485. doi: 10.1093/molbev/msi031. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005b;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Muse SV. HyPhy: hypothesis testing using phylogenies, Statistical methods in molecular evolution. Springer; 2005. pp. 125–181. [Google Scholar]
- Posada D, Buckley TR. Model selection and model averaging in phylogenetics: Advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53:793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci U S A. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA, Holmes EC. Recombination in evolutionary genomics. Annu Rev Genet. 2002;36:75–97. doi: 10.1146/annurev.genet.36.040202.111115. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ. Tracer: MCMC trace analysis tool, 1.5 ed. Institute of Evolutionary Biology; Edinburgh: 2009. p. http://tree.bio.ed.ac.uk/software/tracer/ [Google Scholar]
- Rao PN, Uplekar S, Kayal S, Mallick PK, Bandyopadhyay N, Kale S, Singh OP, Mohanty A, Mohanty S, Wassmer SC. A method for amplicon deep sequencing of drug resistance genes in Plasmodium falciparum clinical isolates from India. J Clin Microbiol. 2016;54:1500–1511. doi: 10.1128/JCM.00235-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MD, Kellis M. Unified modeling of gene duplication, loss, and coalescence using a locus tree. Genome Res. 2012;22:755–765. doi: 10.1101/gr.123901.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray N, Currat M, Foll M, Excoffier L. SPLATCHE2: a spatially explicit simulation framework for complex demography, genetic admixture and recombination. Bioinformatics. 2010;26:2993–2994. doi: 10.1093/bioinformatics/btq579. [DOI] [PubMed] [Google Scholar]
- Reimer AR, Van Domselaar G, Stroika S, Walker M, Kent H, Tarr C, Talkington D, Rowe L, Olsen-Rasmussen M, Frace M. Comparative genomics of Vibrio cholerae from Haiti, Asia, and Africa. Emerg Infect Dis. 2011;17:2113. doi: 10.3201/eid1711.110794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieux A, Balloux F. Inferences from tip-calibrated phylogenies: a review and a practical guide. Mol Ecol. 2016;25:1911–1924. doi: 10.1111/mec.13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ezpeleta N, Brinkmann H, Roure B, Lartillot N, Lang BF, Philippe H. Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol. 2007;56:389–399. doi: 10.1080/10635150701397643. [DOI] [PubMed] [Google Scholar]
- Rohde H, Qin J, Cui Y, Li D, Loman NJ, Hentschke M, Chen W, Pu F, Peng Y, Li J. Open-source genomic analysis of Shiga-toxin–producing E. coli O104: H4. N Engl J Med. 2011;365:718–724. doi: 10.1056/NEJMoa1107643. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumagnac P, Weill FX, Dolecek C, Baker S, Brisse S, Chinh NT, Le TAH, Acosta CJ, Farrar J, Dougan G. Evolutionary history of Salmonella typhi. Science. 2006;314:1301–1304. doi: 10.1126/science.1134933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J. DNA sequence polymorphism analysis using DnaSP. Bioinformatics for DNA sequence analysis. 2009:337–350. doi: 10.1007/978-1-59745-251-9_17. [DOI] [PubMed] [Google Scholar]
- Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, Driebe EM, Drees KP, Hicks ND, Williamson CHD. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb Genom. 2016:2. doi: 10.1099/mgen.0.000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl JW, Schupp JM, Rasko DA, Colman RE, Foster JT, Keim P. Phylogenetically typing bacterial strains from partial SNP genotypes observed from direct sequencing of clinical specimen metagenomic data. Genome Med. 2015;7:52. doi: 10.1186/s13073-015-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol. 2015;53:1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangwan N, Xia F, Gilbert JA. Recovering complete and draft population genomes from metagenome datasets. Microbiome. 2016;4:8. doi: 10.1186/s40168-016-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000a;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000b;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, Banfield JF. Genomes from metagenomics. Science. 2013;342:1057–1058. doi: 10.1126/science.1247023. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Silbergeld EK, Graham J, Price LB. Industrial food animal production, antimicrobial resistance, and human health. Annu Rev Public Health. 2008;29:151–169. doi: 10.1146/annurev.publhealth.29.020907.090904. [DOI] [PubMed] [Google Scholar]
- Skarp-de Haan CP, Culebro A, Schott T, Revez J, Schweda EK, Hänninen ML, Rossi M. Comparative genomics of unintrogressed Campylobacter coli clades 2 and 3. BMC genomics. 2014;15:129. doi: 10.1186/1471-2164-15-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, Donnelly P, Bentley D, McVean G. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt BG, Maiden MC. Bacterial population genetics, evolution and epidemiology. Philos Trans R Soc Lond B Biol Sci. 1999;354:701–710. doi: 10.1098/rstb.1999.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Aberer AJ, Goll C, Smith SA, Berger SA, Izquierdo-Carrasco F. RAxML-Light: a tool for computing terabyte phylogenies. Bioinformatics. 2012;28:2064–2066. doi: 10.1093/bioinformatics/bts309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan C, Jefferies J, Diggle M, Clarke S. Automation of MLST using third-generation liquid-handling technology. Mol Biotechnol. 2006;32:219–225. doi: 10.1385/MB:32:3:219. [DOI] [PubMed] [Google Scholar]
- Sullivan CB, Diggle MA, Clarke SC. Multilocus sequence typing: Data analysis in clinical microbiology and public health. Mol Biotechnol. 2005;29:245–254. doi: 10.1385/MB:29:3:245. [DOI] [PubMed] [Google Scholar]
- Szöllősi GJ, Boussau B, Abby SS, Tannier E, Daubin V. Phylogenetic modeling of lateral gene transfer reconstructs the pattern and relative timing of speciations. Proc Natl Acad Sci U S A. 2012;109:17513–17518. doi: 10.1073/pnas.1202997109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Walters WA, Lennon NJ, Bochicchio J, Krohn A, Caporaso JG, Pennanen T. Accurate Estimation of Fungal Diversity and Abundance through Improved Lineage-Specific Primers Optimized for Illumina Amplicon Sequencing. Appl Environ Microbiol. 2016;82:7217–7226. doi: 10.1128/AEM.02576-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The HC, Thanh DP, Holt KE, Thomson NR, Baker S. The genomic signatures of Shigella evolution, adaptation and geographical spread. Nat Rev Genet. 2016;14:235–250. doi: 10.1038/nrmicro.2016.10. [DOI] [PubMed] [Google Scholar]
- Theunert C, Tang K, Lachmann M, Hu S, Stoneking M. Inferring the history of population size change from genome-wide SNP data. Mol Biol Evol. 2012;29:3653–3667. doi: 10.1093/molbev/mss175. [DOI] [PubMed] [Google Scholar]
- Touchon M, Charpentier S, Clermont O, Rocha EP, Denamur E, Branger C. CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection. J Bacteriol. 2011;193:2460–2467. doi: 10.1128/JB.01307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin R, Maiden MC. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11:479–487. doi: 10.1016/j.tim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Van Belkum A. Molecular typing of micro-organisms: at the centre of diagnostics, genomics and pathogenesis of infectious diseases? J Med Microbiol. 2002;51:7–10. doi: 10.1099/0022-1317-51-1-7. [DOI] [PubMed] [Google Scholar]
- van Cuyck H, Pichon B, Leroy P, Granger-Farbos A, Underwood A, Soullie B, Koeck JL. Multiple-locus variable-number tandem-repeat analysis of Streptococcus pneumoniae and comparison with multiple loci sequence typing. BMC Microbiol. 2012;12:241. doi: 10.1186/1471-2180-12-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ. Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. J Microbiol Methods. 2016 doi: 10.1016/j.mimet.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–146. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Estimation of effective population sizes from data on genetic markers. Philos Trans R Soc Lond B Biol Sci. 2005;360:1395–1409. doi: 10.1098/rstb.2005.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann D, Leuenberger C, Excoffier L. Efficient approximate Bayesian computation coupled with Markov chain Monte Carlo without likelihood. Genetics. 2009;182:1207–1218. doi: 10.1534/genetics.109.102509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L. ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC bioinformatics. 2010;11:116. doi: 10.1186/1471-2105-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn JL, Lee JM, Liechty J, Neale DB. PineSAP—sequence alignment and SNP identification pipeline. Bioinformatics. 2009;25:2609–2610. doi: 10.1093/bioinformatics/btp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens JJ. Reconstructing phylogenies from allozyme data: comparing method performance with congruence. Biol J Linnean Soc. 2000;70:613–632. [Google Scholar]
- Wilson DJ, McVean G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics. 2006;172:1411–1425. doi: 10.1534/genetics.105.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VK, Baker S, Connor TR, Pickard D, Page AJ, Dave J, Murphy N, Holliman R, Sefton A, Millar M. An extended genotyping framework for Salmonella enterica serovar Typhi, the cause of human typhoid. Nat Commun. 2016a:7. doi: 10.1038/ncomms12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VK, Baker S, Pickard DJ, Parkhill J, Page AJ, Feasey NA, Kingsley RA, Thomson NR, Keane JA, Weill FX. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter-and intracontinental transmission events. Nat Genet. 2015;47:632–639. doi: 10.1038/ng.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VK, Holt KE, Okoro C, Baker S, Pickard DJ, Marks F, Page AJ, Olanipekun G, Munir H, Alter R. Molecular Surveillance Identifies Multiple Transmissions of Typhoid in West Africa. PLoS Negl Trop Dis. 2016b;10:e0004781. doi: 10.1371/journal.pntd.0004781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley SW, Posada D, Crandall KA. A comparison of phylogenetic network methods using computer simulation. PLOS Comput Biol. 2008;3:e1913. doi: 10.1371/journal.pone.0001913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyres KL, Holt KE. Klebsiella pneumoniae Population Genomics and Antimicrobial-Resistant Clones. Trends Microbiol. 2016;24:944–956. doi: 10.1016/j.tim.2016.09.007. [DOI] [PubMed] [Google Scholar]
- Yang J, Nie H, Chen L, Zhang X, Yang F, Xu X, Zhu Y, Yu J, Jin Q. Revisiting the molecular evolutionary history of Shigella spp. J Mol Evol. 2007;64:71–79. doi: 10.1007/s00239-006-0052-8. [DOI] [PubMed] [Google Scholar]
- Yang S, Hemarajata P, Hindler J, Li F, Adisetiyo H, Aldrovandi G, Sebra R, Kasarskis A, MacCannell D, Didelot X. Evolution and Transmission of Carbapenem-resistant Klebsiella pneumoniae Expressing the blaoxa-232 Gene During an Institutional Outbreak Associated With Endoscopic Retrograde Cholangiopancreatography. Clin Infect Dis. 2017;64:894–901. doi: 10.1093/cid/ciw876. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yang Z, Rannala B. Molecular phylogenetics: principles and practice. Nat Rev Genet. 2012;13:303–314. doi: 10.1038/nrg3186. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ristic N, Nakhleh L. Fast algorithms and heuristics for phylogenomics under ILS and hybridization. BMC bioinformatics. 2013;14:S6. doi: 10.1186/1471-2105-14-S15-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolfo M, Tett A, Jousson O, Donati C, Segata N. MetaMLST: multi-locus strain-level bacterial typing from metagenomic samples. Nucleic Acids Res. 2017;45:e7–e7. doi: 10.1093/nar/gkw837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller S, Boskova V, Anisimova M. Maximum-likelihood tree estimation using codon substitution models with multiple partitions. Mol Biol Evol. 2015:msv097. doi: 10.1093/molbev/msv097. [DOI] [PMC free article] [PubMed] [Google Scholar]