

Abstract
Newborn screening (NBS) for cystic fibrosis (CF) has been gradually established in several countries, but scant data are available on its long-term effects on survival. Our objective was to evaluate the long-term effects of CF NBS on survival.
586 patients, diagnosed and followed between 1971 and 2014 at the Verona CF Centre were analysed. Eligibility was confirmed in 342 cases diagnosed by NBS, 101 with meconium ileus and 143 through symptoms (44 out of 143 were NBS false negatives). The primary end-point was the 30-year overall survival in patients diagnosed by NBS. Patients were grouped according to the number of hospitalisations for respiratory or nutritional symptoms in the first 3 years of life: 0 (mild), 1–2 (moderate) and ≥3 (severe). Survival in NBS and symptoms groups was compared.
The 30-year survival probability of the NBS group was 80.1% (95% CI 71.4–86.4%); in the symptoms group it was 71.0% (95% CI 62.2–78.2%). The 20-year survival was significantly higher in the NBS versus symptoms group in the severe (85% versus 64%, p=0.007) and moderate (94% versus 86%, p=0.016) groups. An adjusted Cox-model estimation confirmed differences in both the groups.
Poor outcome associated with early severe presentation of CF is tempered by NBS.
Short abstract
Cystic fibrosis newborn screening maximises survival in patients with early severe presentation of the disease http://ow.ly/lCOJ30iGHI6
Introduction
Newborn screening (NBS) programmes for cystic fibrosis (CF) have been implemented since the early 1970s, using measurement initially of meconium proteins and later of immunoreactive trypsinogen, usually in combination with mutation analysis [1–2]. CF NBS has been established gradually across North America, Oceania and most of Europe [3–6], based on the assumption that presymptomatic detection permits early access to specialised medical care, and thus results in less morbidity and longer life expectancy. In fact, several benefits have been associated with early identification of CF, including better growth and lung function, less intensive therapeutic burden and reduced cost of care [7–11]. However, few data have been published about long-term effects of CF NBS on the most relevant outcome measure, survival [12]. This is partly due to methodological limitations intrinsic to the study of screened populations and to the relatively few areas were CF NBS has been running for a period long enough to assess its effects on survival. One of these regions is in northeastern Italy, where since 1973 the Verona Cystic Fibrosis Centre (VR-CFC) has implemented a neonatal screening programme, later progressively expanded to all neonates born in the area served by the centre.
The availability of data on a cohort of CF patients extending for >40 years allowed us to estimate the long-term survival of those diagnosed at birth by NBS and compare long-term survival probability for different degrees of disease severity, both in neonates diagnosed at birth and in infants diagnosed later because of symptoms.
The VR-CFC was established in the 1960s and serves two northeastern regions of Italy (Veneto and Trentino Alto Adige) with a population of ∼7 million [13]. The first CF NBS programme in this area began in 1973 measuring albumin levels in dried meconium by semiquantitative radial immunodiffusion assay. In 1981 immunoreactive trypsinogen (IRT) measurement was adopted; the protocol involved measuring IRT levels at birth and then retesting hypertrypsinaemic newborns at the age of 1 month. Since the early 1990s, the screening strategy has involved a three-tier system, whose progressive steps are IRT, mutation analysis with complementary meconium lactase determination and sweat testing. If the IRT measurement is >99.5th percentile, lactase determination and mutation analysis are performed; if lactase exceeds its threshold level and/or the mutation analysis detects one or two mutations, the infant is sweat tested. If the IRT measurement is >99.9th percentile and lactase determination and mutation analysis are negative, IRT is resampled and retested by 2 months of age and when this second sample is above an adjusted cut-off the infant is called for a sweat test. The number of mutations tested has gradually increased over the years; presently 66 mutations are included in the NBS panel, covering 98% of the affected alleles in the local population [14–17]. Sweat testing in NBS positives is centralised in the VR-CFC laboratory. The coverage of the whole neonatal population was gradual and completed by the mid-1980s. All CF patients born in the area are referred to and followed at the VR-CFC.
Methods
Study population
This retrospective, observational study includes the VR-CFC population with available information born between December 1971 and April 2014.
Patients were considered eligible for analysis if they had a positive NBS test and/or characteristic symptoms plus sweat chloride ≥60 mmol·L−1. 25 patients diagnosed through symptoms were excluded due to missing information.
Variables and statistical analysis
Demographic and diagnostic data were obtained from the VR-CFC database, where lifelong clinical information is recorded for all patients.
The following variables were analysed: sex, birth cohort (1971–1980, 1981–1990, 1991–2000 and 2001–2014), pancreatic status and diagnosis (NBS, symptoms and meconium ileus).
The primary end-point was overall survival probability in the NBS group at 30 years of age. Secondary end-points were 1) effect of mode of diagnosis (NBS versus symptoms) on survival; 2) effect of birth cohort, sex and pancreatic status at birth on survival probability at 20 and 30 years of age in the NBS group; and 3) 20-year survival probability of patients with meconium ileus.
In consideration of the high variability of CF phenotypes, the comparison between NBS and symptoms groups was performed in clusters, taking into account severity in the first years of life: patients were considered “severely affected” if they had been admitted to the VR-CFC or to another hospital because of acute respiratory or severe nutritional symptoms three or more times in the first 3 years of life (severe group); patients who had needed one/two hospitalisations were considered moderately affected (moderate group). Patients who had never been hospitalised because of CF-related clinical symptoms in their first 3 years of life were classified as mild (mild group). The patients with meconium ileus were analysed separately.
To evaluate the robustness of our results, we calculated other survival end-points: the cumulative incidence of CF-related death considering post-transplant deaths as competing events and the probability of death or of being lung transplanted.
Overall survival considering death due to any cause as event was computed using a Kaplan–Meier estimator. In addition, event-free survival was estimated, considering death due to any cause or transplant as events. Difference between groups was tested by log-rank testing. Cumulative incidence was used to estimate the probability of CF-related death, considering death due to CF as event of interest and post-transplant death as a competing event. Difference between groups was tested using the Gray test. The impact on survival of method of diagnosis (NBS versus symptoms), sex (male versus female), pancreatic status (pancreatic insufficiency versus pancreatic sufficiency) and birth cohort (1971–1990 versus 1991–2014) was assessed in the univariate analysis using the Kaplan–Meier estimator; variables with a p-value <0.2 were included in the multivariate Cox regression model. All analyses were performed using SAS (version 9.4; SAS Institute, Cary, NC, USA). A p-value <0.05 was considered statistically significant.
Results
586 patients (287 males) were included. Of these, 443 were diagnosed at birth (342 through NBS and 101 because of meconium ileus) and 143 through symptoms. In the symptom group, 44 (19%) patients had been screened at birth, but not detected by NBS: 13 patients were born between 1971 and 1980; 19 between 1981 and 1990; nine between 1991 and 2000; and three after 2001.
Follow-up data were as of October 31, 2016; at this time, 33 (7%) patients and 26 (5%) patients had been lost to follow-up from 5 and 10 years, respectively.
Median age at diagnosis was 1.8 years (0–37.7 years) for symptoms and 1 month (0–3 months) for NBS.
In the NBS group, 220 (64%) patients were born after 1991; 281 (82%) were pancreatic insufficient; the information was missing for one patient. The pancreatic insufficiency/sufficiency ratio varied slightly over time: ≤1980 93%/7%, 1981–1990 87%/13%, 1991–2000 85%/15% and 2001–2013 72%/28% (meconium ileus group not included). The main characteristics of the patients are reported in table 1.
TABLE 1.
Overall survival of children with cystic fibrosis diagnosed by neonatal screening or symptoms or with meconium ileus
| Patients | Events# | 20-year survival % (95% CI) | 30-year survival % (95% CI) | p-value¶ | |
| NBS | 342 (100) | 35 | 92.35 (88.03–95.16) | 80.11 (71.42–86.40) | |
| Sex | |||||
| Male | 167 (49) | 15 | 91.86 (85.26–95.58) | 84.95 (74.51–91.35) | 0.3 |
| Female | 175 (51) | 20 | 92.90 (86.14–96.43) | 73.70 (57.27–84.61) | |
| Pancreatic status at birth+ | |||||
| Sufficient | 60 (18) | 1 | 96.15 (75.69–99.45) | 96.15 (75.69–99.45) | 0.10 |
| Insufficient | 281 (82) | 34 | 91.75 (86.96–94.84) | 78.34 (68.99–85.18) | |
| Birth cohort | |||||
| 1971–1980 | 30 (9) | 10 | 74.90 (54.33–87.20) | 67.20 (46.26–81.47) | 0.2 |
| 1981–1990 | 92 (27) | 18 | 93.34 (85.78–96.95) | 79.89 (67.80–87.83) | |
| 1991–2000 | 119 (35) | 6 | 96.40 (90.68–98.63) | ||
| 2001–2014 | 101 (29) | 1 | |||
| Symptoms | 143 (100) | 50 | 82.98 (75.49–88.35) | 71.03 (62.21–78.15) | |
| Sex | |||||
| Male | 74 (52) | 27 | 76.93 (65.10–85.19) | 69.58 (56.69–79.32) | 0.43 |
| Female | 69 (48) | 23 | 89.29 (78.83–94.75) | 72.82 (59.89–82.19) | |
| Pancreatic status at birth+ | |||||
| Sufficient | 48 (34) | 0 | 0.00 | 0.00 | <0.001 |
| Insufficient | 94 (66) | 49 | 75.40 (65.07–83.06) | 58.50 (47.32–68.10) | |
| Birth cohort | |||||
| 1971–1980 | 96 (67) | 46 | 77.08 (67.32–84.27) | 65.22 (54.70–73.87) | 0.06 |
| 1981–1990 | 31 (22) | 4 | 96.55 (77.95–99.51) | 84.69 (63.79–94.05) | |
| 1991–2000 | 10 (7) | 0 | 100.00 | ||
| 2001–2011 | 6 (4) | 0 | |||
| Meconium ileus | 101 (100) | 18 | 90.64 (82.04–95.24) | 71.77 (56.47–82.49) |
Data are presented as n (%) or n, unless otherwise stated. NBS: newborn screening. #: death due to any cause is considered an event; ¶: log-rank test; +: data were missing in one case.
Primary end-point
The 30-year survival probability of the NBS group was 80.1% (95% CI 71.4–86.4%); the median follow-up was 20.7 years (95% CI 18.7–21.9 years). The long-term survival probability did not reach 50%, thus the median survival time could not be estimated.
Secondary end-points
Classification of patients according to early severity
The NBS cohort included 95 severely affected, 80 moderately affected and 149 mildly affected patients. 66 patients were not included because they were aged <3 years and two were excluded because they died aged <3 years.
The symptoms group contained 34 severe patients diagnosed at a median age of 8.2 months (range 0.5 months–7.9 years), 40 moderate patients diagnosed at a median age of 16.1 months (range 24 days to 7.5 years) and 58 mild patients (median age at diagnosis of 6.6 years, range 0–37.7 years). Data from 11 patients in the symptoms group were not considered for analysis, due to missing data on hospitalisations in early childhood (n=9); age <3 years (n=1); and death aged <3 years (n=1).
Survival of NBS versus symptoms groups
In the severe group the 20-year survival was 85% in the NBS group versus 64% in the symptoms group (p=0.007); in the moderate group it was 94% in the NBS group versus 86% in the symptoms group (p=0.016); in the mild group it was 98% in the NBS group versus 93% in the symptoms group (p=0.6) (table 2). The difference between the NBS and symptoms groups remained statistically significant in the multivariate analysis, both in the severe (hazard ratio (HR) 2.2, 95% CI 1.2–4.0; p=0.01) and moderate (HR 3.9, 95% CI 1.2–12.4; p=0.02) subgroups.
TABLE 2.
Survival analysis of cystic fibrosis (CF) patients according to clinical severity in the first 3 years of life and diagnosis
| Patients | Events |
10-year survival % (95% CI) |
15-year survival % (95% CI) |
20-year survival % (95% CI) |
p-value | |
| Overall survival# | ||||||
| Severe | ||||||
| NBS | 95 | 23 | 98.84 (92.03–99.84) | 92.56 (84.17–96.59) | 84.93 (74.29–91.41) | 0.007§ |
| Symptoms | 34 | 21 | 87.88 (70.86–95.27) | 72.73 (54.13–84.77) | 63.64 (44.95–77.46) | |
| Moderate | ||||||
| NBS | 80 | 4 | 100.00 | 98.46 (89.58–99.78) | 94.48 (83.72–98.20) | 0.016§ |
| Symptoms | 40 | 12 | 100.00 | 88.89 (73.05–95.68) | 85.93 (69.41–93.89) | |
| Mild | ||||||
| NBS | 149 | 6 | 99.29 (95.07–99.90) | 99.29 (95.07–99.90) | 98.21 (92.83–99.56) | 0.6§ |
| Symptoms | 58 | 12 | 96.55 (86.91–99.13) | 96.55 (86.91–99.13) | 92.80 (81.92–97.24) | |
| Cumulative incidence of CF-related death¶ | ||||||
| Severe | ||||||
| NBS | 95 | 13 | 1.16 (0.17–8.16) | 6.22 (2.66–14.55) | 12.36 (6.66–22.92) | 0.0001ƒ |
| Symptoms | 34 | 19 | 12.12 (4.84–30.38) | 27.27 (15.62–47.61) | 36.36 (23.16–57.11) | |
| Moderate | ||||||
| NBS | 80 | 3 | 0.00 | 0.00 | 3.98 (1.02–15.48) | 0.01ƒ |
| Symptoms | 40 | 10 | 0.00 | 11.11 (4.41–27.99) | 14.07 (6.24–31.75) | |
| Mild | ||||||
| NBS | 149 | 4 | 0.71 (0.10–5.00) | 0.71 (0.10–5.00) | 1.79 (0.44–7.28) | 0.2ƒ |
| Symptoms | 58 | 9 | 3.45 (0.88–13.46) | 3.45 (0.88–13.46) | 7.20 (2.80–18.53) | |
| Event-free survival+ | ||||||
| Severe | ||||||
| NBS | 95 | 33 | 98.84 (92.03–99.84) | 88.68 (79.34–93.95) | 79.76 (68.53–87.33) | 0.07§ |
| Symptoms | 34 | 25 | 87.88 (70.86–95.27) | 72.73 (54.13–84.77) | 63.64 (44.95–77.46) | |
| Moderate | ||||||
| NBS | 80 | 6 | 100.00 | 98.46 (89.58–99.78) | 92.68 (81.55–97.21) | 0.013§ |
| Symptoms | 40 | 15 | 100.00 | 88.89 (73.05–95.68) | 85.93 (69.41–93.89) | |
| Mild | ||||||
| NBS | 149 | 8 | 99.29 (95.07–99.90) | 99.29 (95.07–99.90) | 95.78 (88.94–98.43) | 0.8§ |
| Symptoms | 58 | 14 | 96.55 (86.91–99.13) | 96.55 (86.91–99.13) | 92.80 (81.92–97.24) |
Data are presented as n, unless otherwise stated. NBS: newborn screening. #: death due to any cause is considered an event; ¶: CF-related death is considered an event; +: death due to any cause and lung transplant were considered events; §: log-rank test; ƒ: Gray test.
In order to avoid a cohort effect, a further comparison between NBS and symptoms groups was performed only in patients born before 1990 and confirmed that NBS patients have a higher 20-year survival (significant difference for severe patients 86% versus 64%, p=0.01; and for moderate patients 95% versus 83%, p=0.02).
A non-statistically significant period effect was seen: 20-year survival was 74.9% in the 1971–1980 birth cohort, 93.3% in the 1981–1990 birth cohort and 96.4% in the 1991–2000 birth cohort (table 1).
In the NBS group, survival at 30 years was lower in females and in patients with pancreatic insufficiency (survival 73.7%, 95% CI 57.3–84.6% in females versus 86.0%, 95% CI 74.5–91.4% in males (p=0.3); and 78.3%, 95% CI 69.0–85.2% in patients with pancreatic insufficiency versus 96.2%, 95% CI 75.7–99.5% in patients with pancreatic sufficiency (p=0.1)).
The 20-year survival in the meconium ileus group was 90.6% (95% CI 82.0–95.2%).
Cumulative incidence of CF-related death was significantly lower in the severe (12% versus 36%, p=0.0001) and moderate (4% versus 14%, p=0.01) NBS groups versus symptoms groups. Similar differences between the two groups were found in survival when considering death or transplant as an event. These results are reported in figure 1a and b and in table 2.
FIGURE 1.
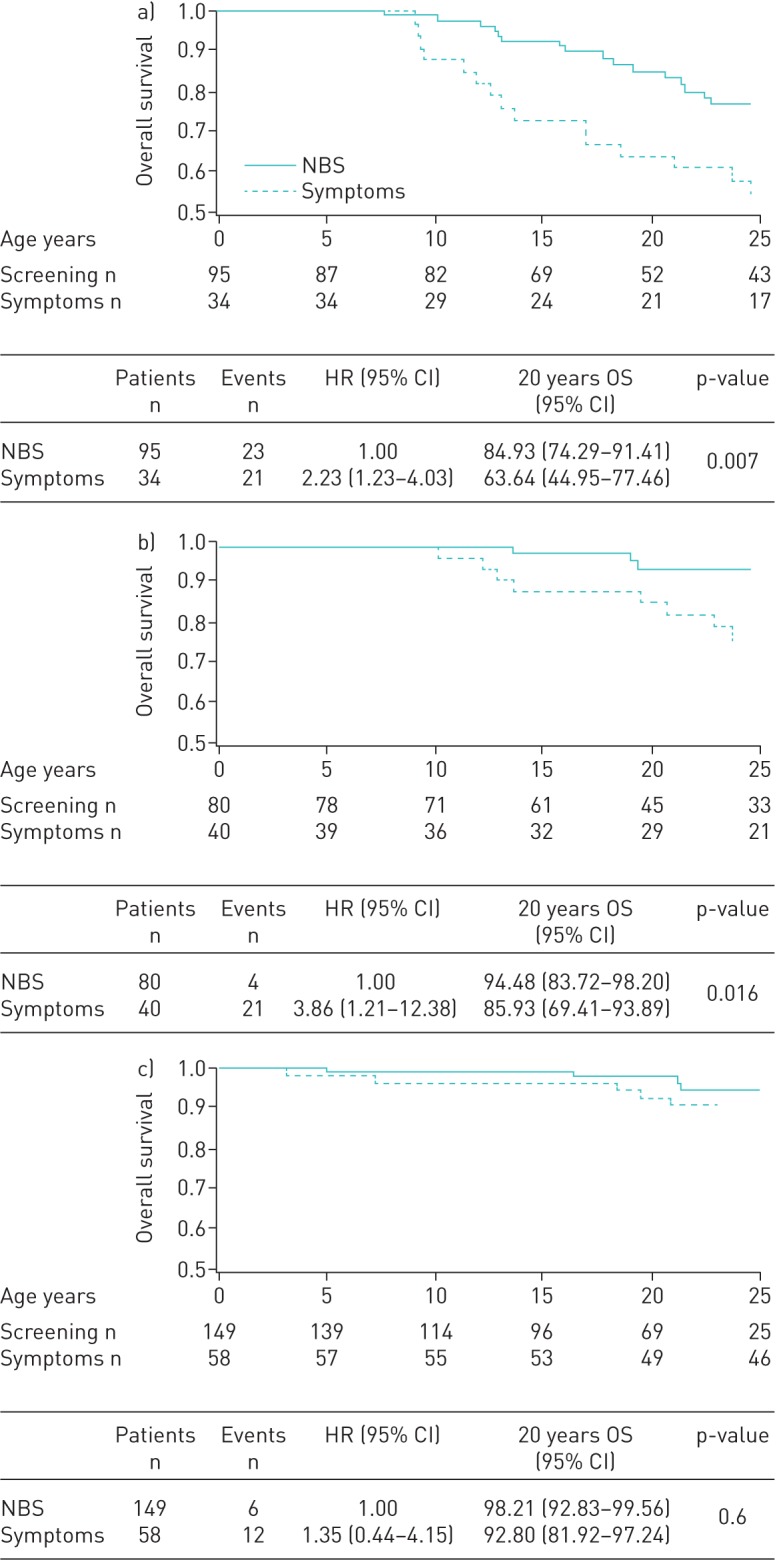
The 20-year overall survival was significantly higher in the newborn screening (NBS) group than in the symptoms group for each of the subgroups defined according to severity. In a) the severe group it was 85% in the NBS versus 64% in the symptoms group (p=0.007); in b) the moderate group it was 94% in the NBS versus 86% in the symptoms group (p=0.016); and in c) the mild group it was 98% in the NBS versus 93% in the symptoms group (p=0.6). Survival probability is presented on a scale from 0.5 to 1.0 for readability. HR: hazard ratio; OS: overall survival.
Discussion
This study shows that in a region where CF NBS has been running for >40 years, survival of patients is substantially improved and NBS had a long-term beneficial effect more evident in patients exhibiting severe disease since their first years of life. Survival is the most significant outcome measure in CF and has improved in several countries over time [18–21]. A longer life expectancy could be explained by a variety of factors including better and centralised treatment in multidisciplinary CF-focused centres, a higher number of milder cases due to expanded genetic analysis or to improved diagnostic capacity, increased awareness of the disease and the implementation of NBS. NBS allows early diagnosis of CF and referral to a specialised CF centre where the affected infants receive more aggressive and specific treatments compared to children similarly affected by CF but undiagnosed, who are treated according to more common, milder therapeutic approaches. The CF standards of care include chest physiotherapy; assessment of nutritional status; and pancreatic enzyme replacement therapy evaluated by a specialised team which maximises the possibilities of preventing chronic pulmonary colonisation and of reducing lung damage [22]; all effects expected to lead ultimately to improvements in survival. Nevertheless, the role of NBS in improving 30-year survival has proved difficult to appraise. To our knowledge the only evidence has been provided by a survival estimate that used 1986–2000 United States registry data and concluded that survival following NBS diagnosis was ≥40 years [23].
In more recent years, the study registered a trend towards a relative increase of pancreatic-sufficient patients. This may be explained by the evolution of the screening protocol. The measurement of meconium albumin in the 1970s was strongly biased towards detection of pancreatic-insufficient patients, whereas since the 1990s the introduction of genetic analysis and the gradual increase in the number of tested mutations allowed the detection of more pancreatic-sufficient patients.
The Wisconsin randomised trial on newborn screening for CF showed that screened neonates exhibit better nutritional status in the first years of life [24–26], but no output is available from this cohort on long-term survival. Data from Australia comparing a younger group of patients detected through NBS and an older group diagnosed because of symptoms showed improved survival in the former [21], but individuals diagnosed more recently may have benefited from treatments unavailable in previous years. The present study avoided a period effect by comparing NBS and symptoms groups born in the same period.
The survival probability of CF patients screened at birth in our region (30 years survival 80.1%), is one of the highest worldwide and presumably is at least partially connected with early intervention. Such a long lifespan prevented a proper calculation of the median survival time for the NBS group, which may be expected to be >50 years. The survival difference between NBS and symptoms groups reached statistical significance in moderate and severe groups; these results have been confirmed in the multivariate model.
In addition, the study tested the hypothesis that NBS could have its major effects in individuals exhibiting a more severe disease since early life. Stratification of CF patients by mutation does not necessarily identify patients with comparable severity, since the high variability in disease expression of CF is only partially accounted for by the cystic fibrosis transmembrane conductance regulator (CFTR) genotype [27]. We classified patients using early clinical manifestations as expressed by the number of hospitalisations in the first 3 years of life. Our results confirm that early pulmonary manifestations or severe nutritional symptoms and greater treatment burden are associated with earlier mortality in CF [8, 24]. Severe disease in both NBS and symptoms groups was associated with earlier mortality, but NBS patients had better survival probability, which strongly supports the rationale for screening at birth for CF. The median age at diagnosis of severe symptoms group was quite low (8.2 months), suggesting that in CF critical events are taking place in the first months of life. This is in line with bronchoscopy data that show that severe CF patients exhibit intense inflammatory activity and structural lung damage since the first trimesters of life [28], and with those from the Wisconsin study that show the relevance of early malnutrition [24].
Other studies have found a correlation between early and later severity of symptoms in CF [25, 26]. Dijk et al. [12] showed that patients diagnosed after the introduction of NBS have an improved survival at 25 years. Our data confirmed and extended these results, comparing groups born in the same period, avoiding the influence of the introduction of new therapeutic treatment and showing that the advantage in the NBS group is greater for patients with severe disease. Clinical manifestations connected with the impairment of CFTR function extend along a spectrum ranging from CF with pancreatic insufficiency to CFTR-related disorders, the latter being considerably milder than the former. Early pulmonary events taking place in CF have been reviewed recently by Ranganathan et al. [29].
Our results, highlighting a lower efficacy of CF NBS in the less severe forms of disease, suggest that children diagnosed with presumably mild conditions, such as CF-related metabolic syndromes [30], will derive little long-term benefit from early detection.
The study has several strengths. We did not introduce any selection bias, as the NBS programme of our region gradually extended, since the early 1970s, to cover the entire area and virtually all CF patients born in the region [16]. The progressive introduction of NBS explains the contraction of the number of patients diagnosed by symptoms in more recent years. All patients have been followed in a single centre since birth or diagnosis and the number of patients lost to follow-up is limited. Only patients with a clear diagnosis of CF were considered (sweat chloride >60 mEq·L−1), thus excluding borderline or atypical cases.
Admittedly, this study has limitations, one of them being that hospitalisations were physician-determined outcomes. However, the Verona CF centre medical team was very stable and homogeneous, all hospitalisations were supervised by the centre director and all doctors were paediatricians dedicated to CF care. Infants or children who had been hospitalised in other institutions were referred to our centre as soon as a suspicion of CF was raised, according to Italian rules which entitle only accredited CF centres to ascertain a diagnosis of CF.
The study is retrospective and based on a clinical and empirical definition of severity. To our knowledge, the categorisation of disease severity we used had never been used before. It was mainly based on the authors' clinical experience.
Since the small numbers presenting with symptoms in the most recent cohort could represent a bias, we performed a further comparison including only patients born before 1990; the results confirmed that NBS groups have a higher 20-year survival in patients with severe and moderate disease.
More sensitive and quantitative methods to identify severity of CF in infants, such as lung high-resolution computed tomography and infant pulmonary function tests were not available in the years considered by this study [28]. Unscreened patients with severe disease who died without a diagnosis of CF may have been lost, thus underestimating the beneficial effect of NBS. The validity of the results might be limited to the area under study and not necessarily be general. Patients born in the past 40 years only partially benefited from new treatments and the availability of new disease-modifying compounds might increase life expectancy of CF patients dramatically, thus limiting the advantages of neonatal screening.
Survival data originated by this study strongly support the role of NBS and could be used for decision-making in health policy assessments.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
FIGURE S1 Cumulative incidence of death due to cystic fibrosis 00109-2017_figureS1
TABLE S1 Univariate and multivariate analysis for severe and moderate groups 00109-2017_tableS1
Acknowledgements
The authors wish to thank Philip M. Farrell (University of Wisconsin School of Medicine and Public Health, Madison, WI, USA) for his suggestions and revision of the manuscript.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Author contributions: all authors conceived the study, interpreted the data, drafted and critically revised the manuscript, approved the final version for publication and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. G. Tridello analysed the data.
Conflict of interest: None declared.
References
- 1.Prosser R, Owen H, Bull F, et al. Screening for cystic fibrosis by examination of meconium. Arch Dis Child 1974; 49: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crossley JR, Elliott RB, Smith PA. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet 1979; 1: 472–474. [DOI] [PubMed] [Google Scholar]
- 3.Southern KW, Munck A, Pollitt R, et al. A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros 2007; 6: 57–65. [DOI] [PubMed] [Google Scholar]
- 4.Castellani C, Southern KW, Brownlee K, et al. European best practice guidelines for cystic fibrosis neonatal screening. J Cyst Fibros 2009; 8: 153–173. [DOI] [PubMed] [Google Scholar]
- 5.Massie RJ, Curnow L, Glazner J, et al. Lessons learned from 20 years of newborn screening for cystic fibrosis. Med J Aust 2012; 196: 67–70. [DOI] [PubMed] [Google Scholar]
- 6.Hale JE, Parad RB, Dorkin HL, et al. Cystic fibrosis newborn screening: using experience to optimize the screening algorithm. J Inherit Metab Dis 2010; 33: Suppl. 2, S255–S261. [DOI] [PubMed] [Google Scholar]
- 7.Sims EJ, Mugford M, Clark A, et al. Economic implications of newborn screening for cystic fibrosis: a cost of illness retrospective cohort study. Lancet 2007; 369: 1187–1195. [DOI] [PubMed] [Google Scholar]
- 8.Sims EJ, Clark A, McCormick J, et al. Cystic fibrosis diagnosed after 2 months of age leads to worse outcomes and requires more therapy. Pediatrics 2007; 119: 19–28. [DOI] [PubMed] [Google Scholar]
- 9.Lai HJ, Cheng Y, Farrell PM. The survival advantage of patients with cystic fibrosis diagnosed through neonatal screening: evidence from the United States Cystic Fibrosis Foundation registry data. J Pediatr 2005; 147: S57–S63. [DOI] [PubMed] [Google Scholar]
- 10.Farrell PM, Lai HJ, Li Z, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! J Pediatr 2005; 147: S30–S36. [DOI] [PubMed] [Google Scholar]
- 11.Farrell PM, Kosorok MR, Laxova A, et al. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. N Engl J Med 1997; 337: 963–969. [DOI] [PubMed] [Google Scholar]
- 12.Dijk FN, McKay K, Barzi F, et al. Improved survival in cystic fibrosis patients diagnosed by newborn screening compared to a historical cohort from the same centre. Arch Dis Child 2011; 96: 1118–1123. [DOI] [PubMed] [Google Scholar]
- 13.Assael BM, Castellani C, Ocampo MB, et al. Epidemiology and survival analysis of cystic fibrosis in an area of intense neonatal screening over 30 years. Am J Epidemiol 2002; 156: 397–401. [DOI] [PubMed] [Google Scholar]
- 14.Scotet V, Assael BM, Duguépéroux I, et al. Time trends in birth incidence of cystic fibrosis in two European areas: data from newborn screening programs. J Pediatr 2008; 152: 25–32. [DOI] [PubMed] [Google Scholar]
- 15.Castellani C, Picci L, Tamanini A, et al. Association between carrier screening and incidence of cystic fibrosis. JAMA 2009; 302: 2573–2579. [DOI] [PubMed] [Google Scholar]
- 16.Castellani C, Bonizzato A, Cabrini G, et al. Newborn screening strategy for cystic fibrosis: a field study in an area with high allelic heterogeneity. Acta Paediatr 1997; 86: 497–502. [DOI] [PubMed] [Google Scholar]
- 17.Bonizzato A, Bisceglia L, Marigo C, et al. Analysis of the complete coding region of the CFTR gene in a cohort of CF patients from north-eastern Italy: identification of 90% of the mutations. Hum Genet 1995; 95: 397–402. [DOI] [PubMed] [Google Scholar]
- 18.Cystic Fibrosis Canada. Canadian Cystic Fibrosis Patient Data Registry 2010 Report www.cysticfibrosisdata.org/LiteratureRetrieve.aspx?ID=132669
- 19.Dodge JA, Lewis PA, Stanton M, et al. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007; 29: 522–526. [DOI] [PubMed] [Google Scholar]
- 20.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry, 2012 Annual Data Report Bethesda, Cystic Fibrosis Foundation, 2013. [Google Scholar]
- 21.Reid DW, Blizzard CL, Shugg DM, et al. Changes in cystic fibrosis mortality in Australia, 1979–2005. Med J Aust 2011; 195: 392–395. [DOI] [PubMed] [Google Scholar]
- 22.Coffey MJ, Whitaker V, Gentin N, et al. Differences in outcomes between early and late diagnosis of cystic fibrosis in the newborn screening era. J Pediatr 2017; 181: 137–145. [DOI] [PubMed] [Google Scholar]
- 23.Lai HJ, Cheng Y, Cho H, et al. Association between initial disease presentation, lung disease outcomes, and survival in patients with cystic fibrosis. Am J Epidemiol 2004; 159: 537–546. [DOI] [PubMed] [Google Scholar]
- 24.Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001; 107: 1–13. [DOI] [PubMed] [Google Scholar]
- 25.Shoff SM, Ahn HY, Davis L, et al. Temporal associations among energy intake, plasma linoleic acid, and growth improvement in response to treatment initiation after diagnosis of cystic fibrosis. Pediatrics 2006; 117: 391–400. [DOI] [PubMed] [Google Scholar]
- 26.Lai HJ, Shoff SM, Farrell PM, et al. Recovery of birth weight z score within 2 years of diagnosis is positively associated with pulmonary status at 6 years of age in children with cystic fibrosis. Pediatrics 2009; 123: 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knowles MR, Drumm M. The influence of genetics on cystic fibrosis phenotypes. Cold Spring Harb Perspect Med 2012; 2: a009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 2013; 368: 1963–1970. [DOI] [PubMed] [Google Scholar]
- 29.Ranganathan SC, Hall GL, Sly PD, et al. Early lung disease in infants and pre-school children with cystic fibrosis. What have we learned and what should we do about it? Am J Respir Crit Care Med 2017; 195: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren CL, Borowitz DS, Gonska T, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr 2017; 181S: S45–S51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
FIGURE S1 Cumulative incidence of death due to cystic fibrosis 00109-2017_figureS1
TABLE S1 Univariate and multivariate analysis for severe and moderate groups 00109-2017_tableS1