

Abstract
Background:
Idiopathic inflammatory myopathies (IIMs) are a group of chronic, autoimmune disorders which include a new entity, necrotizing autoimmune myopathy (NAM). NAM lacks inflammation and presents with markedly elevated creatinine phosphokinase (CPK) levels. It is associated with connective tissue diseases (CTDs), statin use, malignancies, and most cases are idiopathic.
Objectives:
The objectives of this study are to describe the clinicopathologic features in muscle biopsy-proven cases of NAM. To emphasize the role of laboratory parameters such as CPK levels and myositis profile in the diagnosis of NAM.
Materials and Methods:
This is a retrospective study including 15 patients of NAM diagnosed on muscle biopsy over a period of 2 years. The slides of the biopsies were reviewed, and clinical data, electromyography findings, and CPK levels were obtained. Myositis profile was done.
Results:
Necrotizing myopathy accounted for 13.63% (15 cases) of total inflammatory myopathies (110 cases) in the study. These were grouped into CTD-associated NAM, statin-associated NAM, paraneoplastic NAM and idiopathic NAM which was the common type. All cases presented with progressive proximal muscle weakness and had markedly elevated CPK levels. Anti-3-hydroxy-3-methyl-glutaryl-coenzyme A reductase and antisignal recognition particle antibodies were seen to be positive in six patients. Muscle biopsies showed predominant fiber necrosis with significant fiber degeneration and regeneration in the absence of inflammation. All patients received immunotherapy with significant improvement was seen in six patients with two mortalities.
Conclusion:
Necrotizing myopathy is a new addition to the spectrum of IIM. Clinicopathologic correlation is important for appropriate diagnosis. It is found to be refractory to corticosteroids monotherapy. The course of illness is not uniform, and in some patients, there can be rapid worsening with mortality.
Keywords: 3-Hydroxy-3-methyl-glutaryl-coenzyme A reductase, muscle biopsy, necrotizing myopathy, signal recognition particle antibodies, statins
INTRODUCTION
Idiopathic inflammatory myopathies (IIMs) are a group of chronic, autoimmune disorders which include dermatomyositis (DM), polymyositis (PM), and inclusion body myositis (IBM).[1] Muscle biopsy features have become an essential part of diagnosis of IIMs. Tony Amato proposed a new classification criterion for the IIMs, which were designed on behalf of the muscle study group that includes a new entity named necrotizing autoimmune myopathy (NAM).[1] Though grouped as a part of IIM, NAM peculiarly lacks any inflammation on muscle biopsy, and usually, patients have creatinine phosphokinase (CPK) elevation of more than 10 times normal. More than being a well-defined entity, NAM is like a clinicopathologic syndrome owing to its various associations. It has been shown to be associated with connective tissue diseases (CTDs), statin use, malignancies, and in rest, it can be idiopathic. The literature about NAM is still evolving; however, it has been shown to be distinct from other forms of IIM with respect to acute/subacute and progressive course of illness, marked elevation in CPK, and specific pathologic features. There are no planned treatment strategies, and therapeutic aspects are mainly based on the published data. It is important to be aware of the entity to differentiate it from other forms of IIM. These were initially confused with PM lacking inflammation or some forms of muscular dystrophies. The largest data of 63 patients of NAM have been reported from Mayo clinic eliciting the elaborate clinicopathologic and prognostic details.[2] They have concluded that NAM remains refractory to corticosteroid monotherapy and more immunotherapeutic agents are required. Literature about NAM in Indian patients is mostly as case reports.[3,4] In this article, we have attempted to describe the clinicopathologic features of 15 patients of NAM where the diagnosis was essentially based on muscle biopsy analysis.
MATERIALS AND METHODS
The study included 15 patients of NAM diagnosed on muscle biopsy over a period of 2 years from January 2015 to February 2017. The biopsies were done from clinically weak muscle, and slides were reviewed with the help of Hemotoxylin and Eosin and enzyme histochemistry including adenosine triphosphatase, nicotinamide adenine dinucleotide-reduced form, and succinate dehydrogenase. Special stains such as modified Gomori's trichrome and acid phosphatase were also performed. Immunohistochemistry was performed for major histocompatibility complex (MHC) Class I antigens in all the cases. Clinical data, electromyography findings, and CPK levels were obtained from the medical records. Myositis profile was done, which includes myositis-specific antibodies such as anti-tRNA synthetases such as histidyl tRNA synthetase (Jo-1), anti-3-Hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), signal recognition particle (SRP), threonyl tRNA synthetase (PL-7), alanyl tRNA synthetase (PL-12), glycyl tRNA synthetase (EJ), isoleucyl tRNA synthetase (OJ), and anti Sjögren's syndrome A antigen-antibody (Ro 52).
RESULTS
The total number of inflammatory myopathies diagnosed on muscle biopsy in this period was 110. These included DM (46), PM (6), IBM (19) and necrotizing myopathy (15), nonspecific myositis (22), and granulomatous myositis (2). Necrotizing myopathy accounted for 13.63% of total inflammatory myopathies in the study period. These were grouped into CTD-associated NAM (n = 2), statin-associated NAM (n = 3), paraneoplastic NAM (n = 1), and idiopathic NAM (n = 9).
Demographic data
The 15 patients included six males and nine females in the age range from 21 to 67 years. Median age at presentation was 44.5 years. There was slight female preponderance with M:F of 1:2.5. The clinical, demographic features with treatment and follow-up details of all 16 patients are provided in Table 1.
Table 1.
The clinical features, laboratory findings, treatment and follow up details of all patients
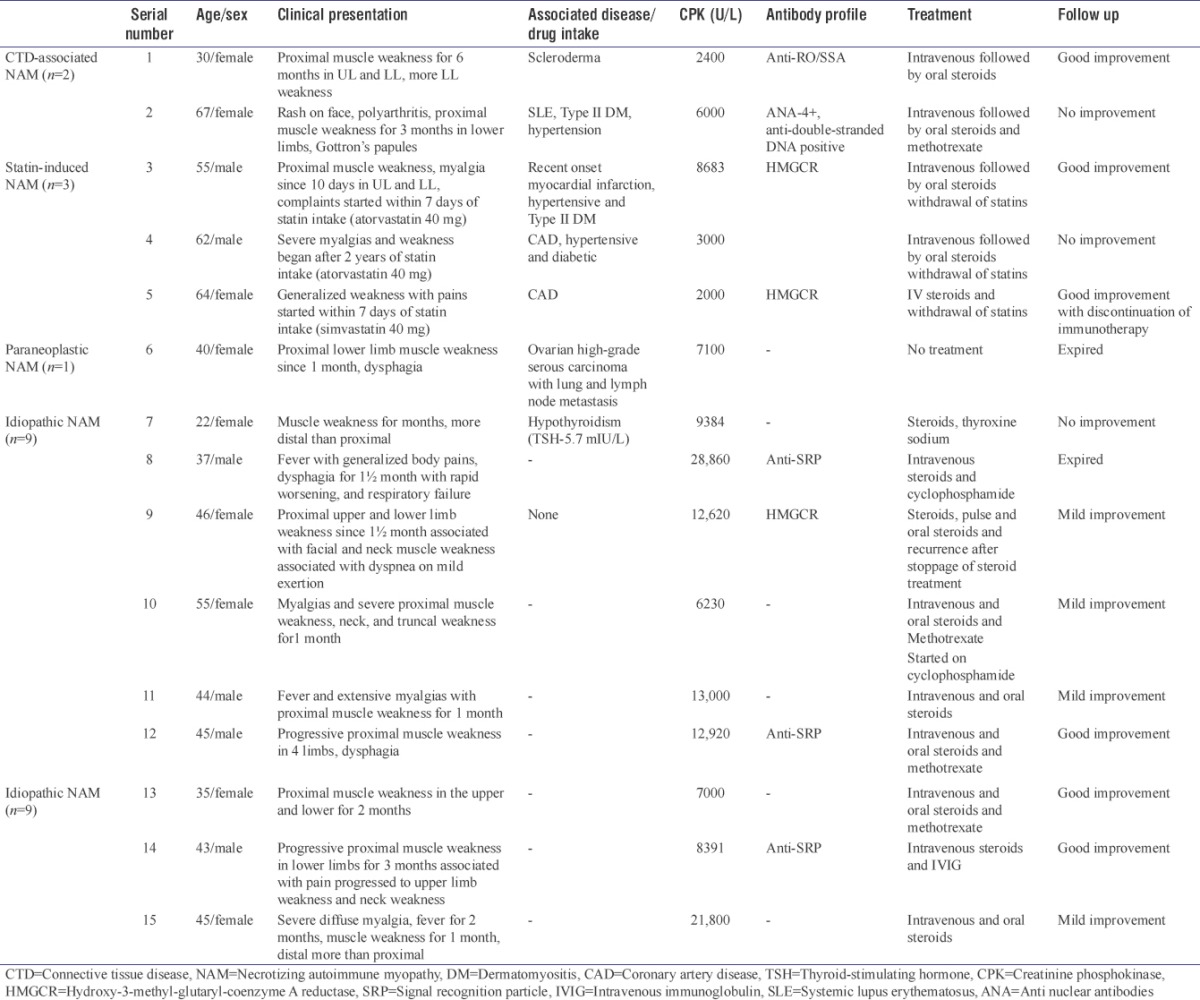
Clinical data
All cases presented with progressive muscle weakness, proximal more than distal associated with diffuse myalgia. There was the inability to get up from squatting and do overhead activities. Associated dysphagia was present in only two patients with one having facial and neck muscle weakness. The disease onset was acute to subacute in all and ranged from 10 days to 4 months.
Connective tissue disease-associated necrotizing autoimmune myopathy
This comprised of one patient of scleroderma and systemic lupus erythematosus (SLE) each. The SLE patient had mostly articular and cutaneous symptoms and did not show evidence of lupus nephritis.
Statin-induced necrotizing autoimmune myopathy
All three cases had a history of coronary artery disease/myocardial infarction and were on statins for the same. Although the dose of statins was same in all three, the duration onset of muscle necrosis was variable. Generalized myalgia was the common symptom apart from proximal weakness.
Paraneoplastic necrotizing autoimmune myopathy
This case also presented with dysphagia and rash and was diagnosed with high-grade serous carcinoma ovary with lung and lymph node metastasis 4 months before the diagnosis of paraneoplastic NAM.
Idiopathic necrotizing autoimmune myopathy
Nine of these patients did not have any associated features and presented with isolated symptoms of muscle weakness and myalgias. The muscle weakness was mostly proximal associated with facial weakness in only one patient. One of the patients was hypothyroid on thyroxine sodium. One patient had a rapid progression of the muscle weakness with respiratory failure, was connected to ventilator and he expired within 12 days of admission. Postmortem examination revealed extensive fascicular necrosis of muscles with diffuse alveolar damage in bilateral lungs.
Laboratory data
All cases had elevated CPK levels ranging from 2000 U/L to 28,860 U/L with a median of 8683 U/L. Electromyoneurography recorded fibrillary potentials and was suggestive of primary muscle disease in 10 cases and was normal in one case. Of these, two patients of statin-associated NAM showed myotonic discharges.
The details of the myositis profile are provided in Table 1. Among the antibodies specific for NAM, HMGCR was identified in three patients, of which two were associated with statin intake.
Muscle biopsy
Muscle biopsies in all the patients showed predominant fiber necrosis with significant fiber degeneration and regeneration. Myophagocytosis was seen. Perivascular or endomysial inflammation, perifascicular atrophy, or rimmed vacuoles were not seen in any of the biopsies. Immunohistochemistry with MHC Class I antigens were performed in all and showed sarcolemmal expression in few non necrotic fibers. Acid phosphatase highlighted the presence of macrophages around necrotic fibers. The muscle biopsy findings are highlighted in Figure 1.
Figure 1.

(a and b) Presence of necrotic muscle fibers on muscle biopsy (H and E, ×40). (c) Many regenerating fibers around the necrotic fibers (H and E, ×100). (d and e) Macrophages around the necrotic muscle fibers take up red color (acid phosphatase ×100). (f) Major histocompatibility complex Class I antigen expression in the muscle fibers
Treatment and follow-up
All patients received intravenous methyl prednisolone 1 g/day for 3 days followed by oral steroids (tablet Wysolone 1 mg/kg). Steroid-sparing agents (methotrexate) were added while tapering the dose of steroids. The patients were followed after 4 weeks. Two patients expired irrespective of immunosuppressive medications. Good improvement was seen in 6 whereas in others the weakness persisted.
DISCUSSION
The classification of inflammatory myopathies has undergone several revisions since the earliest descriptions by Bohan and Peter.[5] However, unfortunately, there are still debates about the clinical utility of the classification criteria with respect to their diagnostic and prognostic specificity. The latest update of the classification has been made by the European neuromuscular muscle working group where the category of NAM was introduced for the first time.[1] The clinical criteria for the diagnosis of NAM include subacute or insidious onset of proximal muscle weakness, with neck flexor rather than extensor weakness, associated with an elevated serum creatine kinase (CK) level, and no ocular weakness. After its introduction, several case reports and series have been published which has increased awareness and importance of diagnosing NAM. In this study, we provide detailed data on clinical presentation, diagnosis with treatment, and outcome of various forms of NAM.
NAM is rare in children and is usually seen in adults around 45 years.[6,7] The median age at presentation in our study was 44 years with the youngest being 22 years old. The Mayo clinic study had a median age of 62 years at presentation which was slightly higher compared to our study.[2] However, idiopathic group in their study had a median age of 36 years which is comparable to our patients. The peculiar female preponderance seen in our patients is comparable to other studies.[6,7]
The clinical presentation with progressive muscle weakness has also been reported uniformly in all the published data. Slowly progressing forms of NAM can be mistaken for limb-girdle muscular dystrophies.[8] Dysphagia is reported as a significantly more common (69%) symptom in NAM by Hengstman et al., which was seen in only one case of NAM in our study.[6]
The connective tissue association of NAM is less common but comparable to our patients. Kassardjian et al. had only 3 CTDs among 63 patients of NAM.[2] Similarly, Christopher-Stine et al. found Antisynthetase antibodies in only four of the 38 patients with necrotizing myopathy.[9] O'Grady et al. report a very interesting patient of lupus nephritis with NAM who had positive anti-SRP antibodies and remained refractory to multiple immunosuppressant.[10] They have concluded that the myositis was related to SRP antibodies rather than SLE.[10] Nevertheless, it is important to consider a diagnosis of NAM when there is rapid progression in muscle weakness.[8] Paik et al. in their description of 44 patients of scleroderma with muscle biopsy have shown nonspecific myositis (35.7%) and necrotizing myopathy (21.4%) to be the most common pathologies.[11]
Statin myopathy is a form of toxic myopathy with a very low risk and an incidence of one in 10,000 persons/year. The onset may be delayed by up to 10 years following the initiation of statin and may occur several months after statin discontinuation.[12,13] Among the statins, atorvastatin is associated with greatest risk of adverse events and increased myotoxicity.[14] Two of the three patients of statin-associated NAM in our study were on atorvastatin. Basharat et al. have described 58 patients of HMGCR-associated NAM. They found that HMGCR-related NM had more prevalence of Type 2 diabetes and female preponderance, of which most patients used atorvastatin.[15] We found that two of the three of our patients were diabetic. HMGCR positivity is not seen in patients on statins without myopathy or self-limited myopathic process and suggests necrotizing myopathy. Although rare, possibility of myotoxicity secondary to statins has to be considered as these are the most commonly prescribed drugs used on a long-term basis.[16] Kassardjian et al.[2] have mentioned that statin-associated NAM with HMGCR positivity shows milder course which is comparable to our patients who showed significant improvement with immunotherapy which was subsequently discontinued in one patient.
Majority of the patients did not have any associated factors and were labeled as idiopathic NAM. The same has been observed in the study by Kassardjian et al. where 32 of the 63 patients fell into idiopathic category.[2] Apart from associations with statins and CTD, it is also important to rule out thyroid disorders and alcoholism since these can cause toxic myopathies which have similar picture on muscle biopsy. However, these are mostly present as rhabdomyolysis rather than inflammatory myopathies which show marked elevation in CPK and renal failure.[13,14,17] One of our patients was found to be hypothyroid. Cross-reactivity of antithyroid autoantibodies or auto-reactive T-cells with muscle tissue may play a significant role in these cases of idiopathic NAM.[15,18,19]
Paraneoplastic NAM is a rare new entity seen in less than one percent of patients with cancer, and prognosis depends on underlying malignancy.[20,21] The most common underlying malignancies are of gastrointestinal origin; however, other primaries such as ovary, lung, and breast are also found to be associated with necrotizing myopathies.[2,21] Min et al. have reported a similar patient like ours with Grade 3 serous carcinoma of ovary with necrotizing myopathy.[22] Myopathy can precede or succeed the diagnosis of malignancy. It is important to remember this rare paraneoplastic association of NAM.
Anti-HMGCR and anti-SRP antibodies have emerged as two most important serologic indicators of NAM of which HMGCR is more specific whereas SRP has also been reported to be associated with other autoimmune conditions. We detected HMGCR in association with statins and SRP in the idiopathic group which is similar to that reported in literature.[2] Both HMGCR and SRP antibodies correlate with CK levels and degree of muscle weakness, thus establishing their role in the pathogenesis of NAM. Anti-SRP antibody-positive patients are more susceptible to the development of interstitial lung disease whereas anti-HMGCR antibody-positive patients have a higher incidence of malignant disease, especially within 3 years of diagnosis of NAM.[23,24,25] Therefore, screening for cancers is recommended in patients positive for these antibodies.
Multiple immunosuppressive agents needed to be used to treat NAM. Similar to the study by Kassardjian et al.,[2] we observed that most of the patients required two immunotherapeutic agents. The follow-up was not uniform with most patients showing only mild improvement in muscle weakness. Only one patient of statin NAM was able to discontinue the immunotherapy. Probably, it is essential to use aggressive immunotherapy in the early phase for better outcome and preventing relapse. Corticosteroids and immunosuppressive therapy such as azathioprine, mycophenolate, methotrexate, or cyclosporine, and more recently, monoclonal antibodies such as rituximab have also been used for the treatment of myopathies including necrotizing myopathy.[26,27]
CONCLUSION
In this study, we have attempted to put forth the clinicopathologic features of necrotizing autoimmune myopathies awareness of which is important as an aggressive form of IIM. Muscle biopsy helps in making a correct diagnosis as well as to rule out other clinical mimics including dystrophies. To the best of our knowledge, this is the first case series from India concentrating on NAM.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, the Netherlands. Neuromuscul Disord. 2004;14:337–45. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol. 2015;72:996–1003. doi: 10.1001/jamaneurol.2015.1207. [DOI] [PubMed] [Google Scholar]
- 3.Babu SS, Khanna L, Saran RK, Mittal G, Peter S, Sebastian I, et al. Necrotizing autoimmune myopathy. Ann Indian Acad Neurol. 2016;19:288–90. doi: 10.4103/0972-2327.176864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel S, Rohatgi A, Gupta P. Statin-triggered immune-mediated necrotizing myopathy. Neurol India. 2016;64:562–4. doi: 10.4103/0028-3886.181571. [DOI] [PubMed] [Google Scholar]
- 5.Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:403–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 6.Hengstman GJ, ter Laak HJ, Vree Egberts WT, Lundberg IE, Moutsopoulos HM, Vencovsky J, et al. Anti-signal recognition particle autoantibodies: Marker of a necrotising myopathy. Ann Rheum Dis. 2006;65:1635–8. doi: 10.1136/ard.2006.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: Clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002;73:420–8. doi: 10.1136/jnnp.73.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki S, Satoh T, Sato S, Otomo M, Hirayama Y, Sato H, et al. Clinical utility of anti-signal recognition particle antibody in the differential diagnosis of myopathies. Rheumatology (Oxford) 2008;47:1539–42. doi: 10.1093/rheumatology/ken325. [DOI] [PubMed] [Google Scholar]
- 9.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL, et al. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–66. doi: 10.1002/art.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Grady J, Harty L, Mayer N, Critcher V, Ryan J. Immune-mediated necrotizing myopathy, associated with antibodies to signal recognition particle, together with lupus nephritis: Case presentation and management. J Clin Med Res. 2015;7:490–4. doi: 10.14740/jocmr2133w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paik JJ, Wigley FM, Lloyd TE, Corse AM, Casciola-Rosen L, Shah AA, et al. Spectrum of muscle histopathologic findings in forty-two scleroderma patients with weakness. Arthritis Care Res (Hoboken) 2015;67:1416–25. doi: 10.1002/acr.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khattri S. Statin-associated necrotizing myopathy in an older woman. J Musculoskelet Med. 2012;29:112–3. [Google Scholar]
- 13.Albayda J, Christopher-Stine L. Identifying statin-associated autoimmune necrotizing myopathy. Cleve Clin J Med. 2014;81:736–41. doi: 10.3949/ccjm.81a.13158. [DOI] [PubMed] [Google Scholar]
- 14.Tiniakou E, Mammen AL. Necrotizing myopathy caused by central hypothyroidism. Muscle Nerve. 2015;52:152–3. doi: 10.1002/mus.24637. [DOI] [PubMed] [Google Scholar]
- 15.Basharat P, Lahouti AH, Paik JJ, Albayda J, Pinal-Fernandez I, Bichile T, et al. Statin-induced anti-HMGCR-associated myopathy. J Am Coll Cardiol. 2016;68:234–5. doi: 10.1016/j.jacc.2016.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA, et al. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41:185–90. doi: 10.1002/mus.21486. [DOI] [PubMed] [Google Scholar]
- 17.Pasnoor M, Barohn RJ, Dimachkie MM. Toxic myopathies. Neurol Clin. 2014;32:647–70. doi: 10.1016/j.ncl.2014.04.009. viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaheen D, Kim CS. Myositis associated with the decline of thyroid hormone levels in thyrotoxicosis: A syndrome? Thyroid. 2009;19:1413–7. doi: 10.1089/thy.2009.0014. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Li H, Kai C, Deng J. Polymyositis associated with hypothyroidism or hyperthyroidism: Two cases and review of the literature. Clin Rheumatol. 2011;30:449–58. doi: 10.1007/s10067-010-1570-8. [DOI] [PubMed] [Google Scholar]
- 20.Acciavatti A, Avolio T, Rappuoli S, Foderi L, Soldati V, Franchi M, et al. Paraneoplastic necrotizing myopathy associated with adenocarcinoma of the lung – A rare entity with atypical onset: A case report. J Med Case Rep. 2013;7:112. doi: 10.1186/1752-1947-7-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu J. Malignancy-associated myositis. Brain Nerve. 2010;62:427–32. [PubMed] [Google Scholar]
- 22.Min KJ, Ouh YT, Hong HR, So KA, Hong JH, Lee JK, et al. Muscle weakness and myalgia as the initial presentation of serous ovarian carcinoma: A case report. J Ovarian Res. 2014;7:43. doi: 10.1186/1757-2215-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinal-Fernandez I, Parks C, Werner JL, Albayda J, Paik J, Danoff SK, et al. Longitudinal course of disease in a large cohort of myositis patients with autoantibodies recognizing the signal recognition particle. Arthritis Care Res (Hoboken) 2017;69:263–70. doi: 10.1002/acr.22920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe Y, Uruha A, Suzuki S, Nakahara J, Hamanaka K, Takayama K, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry. 2016;87:1038–44. doi: 10.1136/jnnp-2016-313166. [DOI] [PubMed] [Google Scholar]
- 25.Allenbach Y, Keraen J, Bouvier AM, Jooste V, Champtiaux N, Hervier B, et al. High risk of cancer in autoimmune necrotizing myopathies: Usefulness of myositis specific antibody. Brain. 2016;139:2131–5. doi: 10.1093/brain/aww054. [DOI] [PubMed] [Google Scholar]
- 26.Sampson JB, Smith SM, Smith AG, Singleton JR, Chin S, Pestronk A, et al. Paraneoplastic myopathy: Response to intravenous immunoglobulin. Neuromuscul Disord. 2007;17:404–8. doi: 10.1016/j.nmd.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Samuels N, Applbaum YH, Esayag Y. Paraneoplastic necrotizing myopathy and dermatomyositis in a patient with rectosigmoid carcinoma. Rheumatol Int. 2013;33:1619–21. doi: 10.1007/s00296-011-2304-1. [DOI] [PubMed] [Google Scholar]