

Abstract
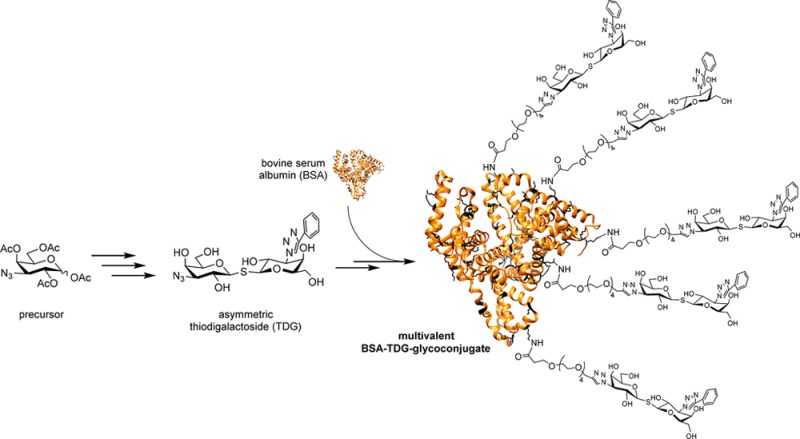
Galectin inhibitors are urgently needed to understand the mode of action and druggability of different galectins, but potent and selective agents still evade researchers. Small-sized inhibitors based on thiodigalactoside (TDG) have shown their potential while modifications at their C3 position indicated a strategy to improve selectivity and potency. Considering the role of galectins as glycoprotein traffic police, involved in multivalent bridging interactions, we aimed to create multivalent versions of the potent TDG inhibitors. We herein present for the first time the multivalent attachment of a TDG derivative using bovine serum albumin (BSA) as the scaffold. An efficient synthetic method is presented to obtain a novel type of neoglycosylated proteins loaded with different numbers of TDG moieties. A polyethylene glycol (PEG)-spacer is introduced between the TDG and the protein scaffold maintaining appropriate accessibility for an adequate galectin interaction. The novel conjugates were evaluated in galectin binding and inhibition studies in vitro. The conjugate with a moderate density of 19 conjugated TDGs was identified as one of the most potent multivalent Gal-3 inhibitors so far, with a clear demonstration of the benefit of a multivalent ligand presentation. The described method may facilitate the development of specific galectin inhibitors and their application in biomedical research.
A dense layer of carbohydrates is found on mammalian cells, and the variety of the attached glycans results in specific profiles for molecular recognition. This recognition involving the so-called “sugar code” is operational by reversible interaction of carbohydrate-binding proteins. Members of this protein class are described as lectins and fulfill a variety of effector functions in terms of cellular communication.1 Galectins as one subtype of lectins that can specifically recognize β-galactosides are found in fungi, invertebrates, and vertebrates.2 Fifteen different galectins have been identified in humans until now, and they play crucial roles in the organization of receptor-lectin complexes (lattices)3,4 and regulation of immune responses.5 Their concave-shaped groove for oligosaccharide binding, namely, the carbohydrate recognition domain (CRD), is highly preserved. Galectin-1 (Gal-1) and galectin-3 (Gal-3) are the most thoroughly studied galectins due to their involvement in angiogenesis, tumor progression, and metastasis.6−8 The participation in malignant processes makes them promising targets for anticancer therapy. In this regard, immense efforts have been spent on the synthesis of potent and specific ligands. The prevailing majority of drug discovery efforts have been focused on the synthetic modification of lactose (methyl β-lactoside, Kd = 220 μM for Gal-3, Kd = 190 μM for Gal-1) and N-acetyllactosamine (methyl β-LacNAc, Kd = 67 μM for Gal-3), which are the natural disaccharide ligands for both Gal-1 and Gal-3.9−11 Thiodigalactosides (TDG, Kd = 49 μM for Gal-3, Kd = 24 μM for Gal-1) were identified as more potent Gal-3 inhibitors with additional advantages such as their enhanced glycolytic stability while maintaining a similar binding mode compared to lactose and LacNAc.12,13 Especially, the introduction of triazole moieties at the C3′-position of the galactose molecules resulted in monovalent galectin inhibitors with outstanding high affinities showing Kd values in the low nanomolar range.14−17 Apart from designing small molecule inhibitors, a multivalency-based strategy was adopted to promote the interactions between ligands and protein, as this more closely mimics the natural way in which galectins interact with glycoproteins. So far, different scaffolds were reported to carry multiple galectin ligands.18−22 In our previous work, bovine serum albumin (BSA) was used as a plain protein carrier for neo-glycosylation.23,24N-hydroxysuccinimidyl (NHS)-esters, the most commonly used amine-reactive reagents, offer a larger variety of amine-reactive ligands for labeling proteins.25−27 Benefiting from this prior information, we successfully approached a type of neo-glycoproteins through conjugation of NHS functionalized-TDG to lysine residues of BSA. The most obvious finding to emerge from the present study is that the TDG-conjugates exhibit outstanding high inhibitory potencies despite a low or moderate number of attached ligands. Combining a highly potent monovalent ligand with a beneficial multivalent presentation resulted in some of the most effective Gal-3 inhibitors. Besides, the multivalent TDG-conjugates represent the first example of decorating a nonglycosylated carrier with TDG derivatives.
Results and Discussion
Synthesis of a Carboxy-Functionalized Thiodigalactoside Precursor
The synthesis of a carboxy-functionalized TDG precursor started as shown in Scheme 1. The key building block was the unsymmetrical thiodigalactoside (TDG) precursor 6 carrying phenyltriazole and azide at the 3- and 3′-positions. To construct TDG precursor 6, two building blocks, namely, tri-isopropylsilyl thio-glycoside (compound 4) and glycosyl halide (compound 2), were prepared using a published method of a one-pot desilylation and glycosyl thiol alkylation with glycosyl halide.28 Hence, compound 1, prepared from commercially available 1,2,5,6-diacetone-α-d-glucofuranoside through a known four-step reaction,29 was converted to 4. Meanwhile, copper-catalyzed azide–alkyne cycloaddition of compound 1 with phenylacetylene provided the corresponding triazole analog crude, which reacted with HBr to obtain the glycosyl halide compound 2 (79% yield in two steps). Desilylating and activating with TBAF turned compound 4 into a thiol nucleophile and thus replaced the anomeric bromide of compound 2 through an SN2 reaction. Purification by silica column chromatography gave the resulting compound 5 in 54% yield. After removing the acetyl protecting group, the resulting crude 6 was used for the next step without further purification (Scheme 1).
Scheme 1. Synthesis of Thiodigalactoside 6.

Reagents and conditions: (a) (i) Phenylacetylene, CuSO4, sodium ascorbate, DMF/H2O, 80 °C, microwave, (ii) HBr, CH2Cl2, r.t., 79% yield in two steps; (b) TiBr4, CH2Cl2/EtOAc, 25 °C, 67%; (c) TIPSSH, K2CO3, CH3CN, 25 °C, 30%; (d) TBAF, CH3CN, 25 °C, 62%; (e) NaOMe, CH3OH, 25 °C.
The synthesis of polyethylene glycol (PEG)-spacer (compound 7) was started from tetraethylene glycol. As previously reported, the reaction of tetraethylene glycol with an equal amount of propargyl bromide in the presence of NaH in THF at room temperature gave the monoalkyne terminated PEG4.30 Then the Michael addition of the resulting compound to tert-butyl acrylate in the presence of catalytic sodium metal gave compound 7 in 70% yield.31 Having assembled the important intermediates (compounds 6 and 7), the next objective was their CuAAC conjugation assisted by copper iodide. After purification with size-exclusion, compound 8 was obtained. Removal of the tert-butyl group from compound 8 gave carboxyl compound 9 (90% yield), which was transferred to the corresponding NHS-ester 10 through coupling with TSTU (Scheme 2). Compound 10 is prone to hydrolysis (e.g., during purification); thus, it was directly used for further reaction.
Scheme 2. Preparation of Carboxy- (9) and NHS-Functionalized (10) TDG Derivatives.

Reagents and conditions: (a) Na, THF, 0–25 °C, 39%; (b) CuI, CH3OH, 25 °C, 64%; (c) TFA/DCM, 25 °C, 98%; (d) TSTU, DiPEA, DMF, 25 °C.
Neo-Glycoprotein Synthesis and Analysis
Compared to click chemistry32 and squarate linker chemistry,23,24,33−35 NHS-mediated coupling25,27,36 is another straightforward and convenient coupling strategy for the modification of protein carriers. The lysine residues of bovine serum albumin (BSA) react with the NHS ester moiety of the TDG derivative, but a crucial factor that needs to be taken into consideration is the distance between the TDGs and BSA. Benefiting from our previous work on chito-oligomer spacers in neo-glycoproteins,34 we concluded that a suitable spacer of a certain length would be recommended to maintain an appropriate ligand accessibility and proper galectin interaction of final products. As a logical consequence thereof, a PEG as placeholder with a similar spacing distance was incorporated into conjugation agent 9. Furthermore, the modification of carbohydrates or derivatives thereof with PEG as a biocompatible molecule is a commonly used technique.37,38 The NHS functionalized TDG 10 readily reacted with amino groups of BSA using a reaction buffer that contained 35 mM HEPES (pH 7.0). The total amount of compound 10 was divided into three and added batchwise after every 24 h. As a result, we obtained compound 11 that was verified by the TNBSA-assay23 to carry 7.0 ± 1.0 TDG moieties per BSA molecule (Scheme 3). The coupling efficiency was 7.8%. Shifting the pH to slightly higher values (pH 8.0–9.0) by the addition of triethylamine (TEA) gave compound 12. The TNBSA-assay confirmed that the number of attached TDGs was now 18.7 ± 1.6 corresponding to a coupling efficiency of 20.1%. The elevation of the pH may deprotonate the amino groups of lysine residues to a degree sufficiently high for fast and efficient coupling. In accordance with our previous findings, reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) confirmed the attachment of TDGs (Figure S15, Supporting Information).
Scheme 3. Loading of BSA Protein Carriers with NHS-Functionalized Compound 10 Resulting in Compounds 11 and 12.

Reagents and conditions: (a) compound 11: BSA (0.06 mM, 150 μL) in HEPES buffer (35 mM, pH 7.0), compound 10 in DMF (54 mM, 3 × 5 μL), 72 h, 4 °C; (b) compound 12: BSA (0.06 mM, 50 μL) in HEPES buffer (pH 8.0–9.0, adjusted with TEA); compound 10 in DMF (54 mM, 3 × 1.67 μL), 72 h, 4 °C.
Evaluation of Multivalent BSA-Conjugates as Galectin Ligands
Multivalent TDG-conjugates 11 and 12 were utilized as immobilized ligands in solid-phase binding assays for recombinant human His6-tagged galectin-1 (Gal-1) and galectin-3 (Gal-3) (Figure 1). Our binding studies revealed that Gal-1 bound ligand 11 in a 3-fold more efficient manner than Gal-3 did. Gal-1 showed higher affinity for conjugate 11 with a 6-fold reduced apparent dissociation constant Kd in comparison with Gal-3 with p < 0.001 (Student’s t test). However, Gal-3 showed an elevated capacity when binding to conjugate 11, indicated by the higher Bmax value (Table 1). This is putatively caused by cluster glycoside effects,39 which may lead to galectin oligomerization and thus to an increased binding signal (Figure 1). This effect might be more pronounced for Gal-3, which is generally considered to form higher oligomers when binding to multivalent ligands. Gal-1 aggregation is likely limited to the formation of dimers.
Figure 1.
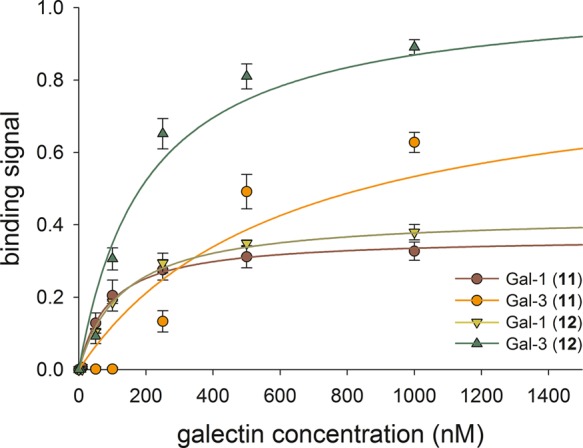
Behavior of recombinant human Gal-1 and Gal-3 for binding to immobilized neo-glycoconjugates 11 and 12. The subtracted blank value (no Gal-1 and/or Gal-3) was 0.047 ± 0.003.
Table 1. Binding Behavior of Gal-1 and Gal-3 Using Multivalent TDG Conjugates as Immobilized Ligands.
| galectin | ligand | apparent Kd (μM)a | Bmax (−)a | galectin binding efficiency [μM–1]b |
|---|---|---|---|---|
| Gal-1 | 11 | 0.090 ± 0.012 | 0.37 ± 0.01 | 4.1 ± 0.8 |
| Gal-1 | 12 | 0.131 ± 0.017 | 0.43 ± 0.01 | 3.3 ± 0.6 |
| Gal-3 | 11 | 0.616 ± 0.246 | 0.86 ± 0.12 | 1.4 ± 0.5 |
| Gal-3 | 12 | 0.199 ± 0.045 | 1.04 ± 0.06 | 5.2 ± 0.3 |
Determined in ELISA.
Ratio of Bmax and apparent Kd.
Conjugate 12 carries an increasing number of TDG derivatives and was therefore bound with a higher affinity and capacity by Gal-3. The lower apparent Kd value and raised Bmax value led to the highest binding efficiency observed for this binding assay (Table 1). The enhancement of Gal-3 binding efficiency for compound 12 was significant with p < 0.001 (Student’s t test). In contrast, the higher TDG loading of compound 12 did not affect Gal-1 binding much and the capacity (Bmax) was only slightly increased while the affinity was even slightly reduced. The corresponding binding efficiencies for Gal-1 toward conjugates 11 and 12 are similar within experimental error (Table 1).
To summarize these assay results, prototype Gal-1 and chimera-type Gal-3 bind compounds 11 and 12 with high affinity through recognizing the conjugated TDGs. Our findings corroborate previous studies, which reported on the interaction of both tested galectins with the corresponding monovalent and symmetrical TDG-based compounds by a fluorescence polarization assay.17 However, we did not detect any specificity differences as seen for previously synthesized neo-glycoproteins carrying poly-N-acetyllactosamine (poly-LacNAc) derivatives.24 In the present case, the TDG ligands may primarily interact with the conserved region in the galectin’s CRD as reported before and thus show only little variation.40,41 The conjugation of TDGs with larger aromatic substituents (e.g., 4-phenoxyphenyl)17 may help to design specialized glycoconjugates for Gal-3 selective targeting. In this context, multivalency could greatly maintain an outstanding strong Gal-3 interaction.
Neo-Glycoproteins Acting as Gal-3 Inhibitors
The univalent TDG derivative 9 and multivalent glycoconjugates 11 and 12 were evaluated in terms of their capability to block the binding of Gal-3 to immobilized asialofetuin (ASF). The ASF glycoprotein is used as a standard galectin ligand as reported before.23,24,34,42,43 Gal-3 was incubated together with increasing amounts of inhibitors 9, 11, or 12 aiming at a complete prevention of ASF–galectin interaction. Nonmodified BSA was utilized as negative control and confirmed to be not interfering with Gal-3 binding to ASF. Hence, we conclude that the observed inhibition phenomena were only due to the TDG cargo of compounds 11 and 12. As depicted by Figure 2, a complete inhibition of Gal-3 binding to ASF was reached when using inhibitor 9 (>25 μM) and conjugates 11 and 12 (>200 nM). The resulting sigmoidal inhibition curves were the basis for the calculation of the IC50 value, defined as the inhibitor concentration at which half-maximal inhibition was reached (Table 2). The inhibition strength of compound 9 was in the low micromolar range and fits the range of N′,N″-diacetyllactosamine (LacdiNAc)-LacNAc tetrasaccharide, which was identified as a specific ligand of Gal-3.23 However, we assume that the NHS-/PEG-modification of one C3 atom (asymmetric character) of 9 may reduce its affinity when applied as nonconjugated Gal-3 inhibitor. When comparing previous relevant studies, it becomes clear that Gal-3 has a much higher affinity for the symmetrically modified TDG. When both of TDG’s galactose moieties carry selected substituents such as C(3)-benzamides16,41 or C(3)-triazoles,14,17 the Gal-3 affinity is optimal showing apparent Kd values between 22 and 360 nM in a fluorescence polarization assay.
Figure 2.

Competitive inhibition of Gal-3 (5.56 μM) binding to ASF using monovalent carboxy-functionalized compound 9 (A) and multivalent TDG-conjugates 11 and 12 at indicated concentration (B). Refer to Scheme 2 and Scheme 3 for compound structures. The subtracted blank value (no Gal-3) was 0.091 ± 0.005.
Table 2. Inhibition Constants and Inhibitory Potencies of TDG Derivative 9 and Multivalent TDG-Conjugates 11 and 12.
| inhibitor | IC50 (nM)a | number of loaded TDG moieties (−)b | relative inhibitory potency | relative inhibitory potency per glycan |
|---|---|---|---|---|
| 9 | 9030 ± 27 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 |
| 11 | 19.40 ± 1.09 | 7.0 ± 1.0 | 465.5 ± 27.5 | 66.5 ± 13.4 |
| 12 | 1.88 ± 0.38 | 18.7 ± 1.6 | 4803.2 ± 985.2 | 256.9 ± 74.7 |
ELISA.
TNBSA-assay.
Nevertheless, the conjugation of TDG derivative 9 to nonglycosylated serum protein scaffold gave conjugates 11 and 12. Their use as inhibitors of Gal-3 binding resulted in extraordinarily high inhibitory potencies and low IC50 values (Table 2). Even though the cargo of compound 11 was only a quantity of seven TDG derivatives, the inhibition strength was increased 465-fold compared to univalent compound 9. This corresponds to an improvement factor of 66 per loaded TDG. The impact of the multivalent ligand presentation was even more pronounced for glycoconjugate 12 presenting a higher number (n = 18.7) of TDG derivatives. Here, the determined IC50 value is reduced by more than 4800-fold compared with that of compound 9, representing an improvement factor per TDG of 256.
Our findings suggest that multivalent conjugates 11 and 12, but not monovalent compound 9, inactivate more Gal-3 molecules than the amount of presented TDG derivatives, as seen before.22 On the one hand, both multivalent inhibitors may induce the formation of Gal-3 complexes, cross-linked by their N-termini.44 On the other hand, type-C Gal-3 self-association is most likely. Here, the nonoccupied CRD of Gal-3 molecules interact with already TDG-bound Gal-3 leading to an oligomerization and stacking as reported before.45 To the best of our knowledge, the tremendously diminished IC50 value makes multivalent glycoconjugate 12 one of the most effective Gal-3 inhibitors. The multivalent design promotes the cluster glycoside effect resulting in a highly efficient entrapment of Gal-3.1,39,46
Neo-glycoproteins with a cargo of different poly-LacNAc derivatives were recently synthesized and applied as Gal-3 inhibitors.23 Thus, we may use them as a reference to evaluate the presented results. In particular, those BSA neo-glycoconjugates bearing the LacNAc-LacNAc (n = 7.5) or LacdiNAc-LacNAc (n = 7.4) glycans are ideal benchmarks because of an equal modification density with regard to conjugate 11. In that case only moderate inhibition strengths were observed, with IC50 values of 850 nM ([LacNAc-LacNAc]n=7.4-BSA) and 1100 nM ([LacdiNAc-LacNAc]n=7.4-BSA), respectively.23 Clearly the TDG ligand has a potency advantage but the implementation of the PEG-spacer may also be a favorable feature in terms of ligand accessibility and flexibility.38,47 To evaluate conjugate 12, previously synthesized neo-glycoproteins were used as ideal references again. Conjugates with LacNAc-LacNAc (n = 17.8), LacdiNAc-LacNAc (n = 18.0),23 or derivatized poly-LacNAc hexasaccharides of equal modification density (n = 16–19)24 were prepared and thoroughly studied in terms of galectin interaction. The respective inhibition constants ranged between 60 and 90 nM23 and 37 and 76 nM.24 Based on the outstanding low IC50 (1.88 nM), the potency of conjugate 12 is at least more than 20-fold elevated in comparison with the most potent reference neo-glycoproteins.
TDG derivatives have been validated to be valuable inhibitors for galectin research. The aromatic groups on the C3 and C3′ positions of TDG tune galectin selectivity and affinity. We herein report on the synthesis of an asymmetrical TDG structure that can be used to yield multivalent compounds through conjugating to a protein scaffold. To obtain the key precursor, a straightforward approach was used to lead to the NHS functionalized-TDG derivative. Subsequent reaction with BSA gave multivalent TDG-glycoconjugates. Weak alkaline pH, adjusted by TEA, was crucial for an effective conjugation. To the best of our knowledge, this is the first example of conjugating a TDG derivative to a nonglycosylated carrier. The multivalent presentation on conjugates 11 and 12 unlocks TDG’s full potential. Extraordinarily high multivalency factors were obvious that resulted in one of the most effective inhibition of Gal-3 in vitro until now. The result is clearly a combination of the binding properties of the monovalent ligand and the multivalent display by the BSA. As previously noted, potent galectin inhibition cannot be achieved with very weak or nonbinding ligands, conjugated to BSA.34 Furthermore, we note that, while a multivalent scaffold can enhance existing binding potency, the specificity at the multivalent level remains the same.48 In other systems, very strong multivalency effects have been reported leading to picomolar inhibition, usually involving the simultaneous binding of ligands to nearby binding sites.49 This chelation type mechanism is less likely to contribute to the present system, due to the monovalent nature of the nonaggregated protein. Considering this, other modes of action such as statistical rebinding or aggregation usually lead to smaller effects,46 which makes the present results more notable. Furthermore, this work shows that the multivalent inhibitor is able to inhibit far more Gal-3 molecules than its number of attached ligands. This feature is a likely consequence of aggregation phenomena, blocking Gal-3 binding sites, previously observed for Gal-3 and named type-C-self-association.45 Systems such as the present, capable of nucleating the process, may lead us to a full understanding of this phenomenon. In the present system, the PEG-spacer likely helps to make the TDGs accessible for the interacting galectins. The multivalent TDG-modified conjugates (11, 12) have the ideal properties for a putative biomedical application because of (i) the serum protein scaffold has the approved quality to be applied to the bloodstream; (ii) the PEG-spacer is biocompatible, sustained, and safe; and (iii) the TDG derivative is considered chemically stable. Hence, cell culture in vitro experiments (inhibition of Gal-3 induced angiogenesis) are planned in the due course in order to elucidate the power of the synthesized conjugate. In vivo applications may follow. The conjugation of TDGs with different functional groups on C3 and C3′ position (e.g., 4-phenoxyphenyl) are planned to modulate the inhibition potency and tune the galectin specificity on a multivalent level.
Methods
Preparation of Compound 1 (2,4,6-Tri-O-acetyl-3-deoxy-3-(4-phenyl-1H-1,2,3-triazol)-α-d-galactopyranosyl bromide)
Compound 1 (300 mg, 0.80 mmol), sodium l-ascorbate (237.6 mg, 1.2 mmol), CuSO4·5H2O (100 mg, 0.40 mmol), Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 4.2 mg, 0.0096 mmol), and phenylacetylene (176.4 μL, 1.6 mmol) were dissolved in DMF (13.5 mL) and H2O (1.5 mL). The reaction was performed under microwave irradiation at 80 °C for 40 min. Subsequently, the solvent was evaporated and the residue was dissolved in CH2Cl2 (100 mL), washed with H2O (1 × 100 mL) and brine (1 × 100 mL), dried over Na2SO4, filtered, and concentrated. The crude product was obtained as a white solid (310 mg) and used for next reaction directly. To a solution of crude (310 mg) in dry CH2Cl2 (50 mL) the HBr (33% HBr in acetic acid, 2.0 mL) was dropwise added under N2 atmosphere. The solution was sealed and stirred overnight at room temperature. A saturated NaHCO3 solution (50 mL) was added to quench the reaction and then the organic layer was washed with H2O (1 × 50 mL) and brine (1 × 50 mL), dried with Na2SO4, and filtered. The residue was purified by silica chromatography (hexanes:EtOAc = 1:1) and gave the product 2 as a light yellow solid (312 mg, two step yield 79%). 1H NMR (400 MHz, CDCl3) δ 7.85–7.70 (m, 3H, ar, triazole), δ 7.41 (dd, J = 8.3, 6.8 Hz, 2H, ar), 7.37–7.28 (m, 1H, ar), 6.86 (d, J = 3.8 Hz, 1H, H1), 5.79 (dd, J = 11.4, 3.8 Hz, 1H, H2), 5.63 (dd, J = 3.1, 1.3 Hz, 1H, H4), 5.32 (d, J = 3.0 Hz, 1H, H3), 4.67–4.58 (m, 1H, H5), 4.22 (dd, J = 11.6, 6.3 Hz, 1H, H6a), 4.11 (dd, J = 11.6, 6.3 Hz, 1H, H6b), 2.05, 2.04, and 1.93 (3s, each 3H, OCH3).
13C NMR (101 MHz, CDCl3) δ 170.25, 169.55, 168.94, 128.89, 128.49, 125.70, 119.33, 88.52, 77.17, 71.34, 67.76, 66.85, 60.86, 58.66, 31.40, 29.67, 20.58, 20.45, 20.34.
HRMS (EI, m/z) calculated for C20H22BrN3O7H+ ([M + H]+): 496.0714, found 496.0707.
Preparation of Compound 3 (2,4,6-tri-O-acetyl-3-azido-α-d-galactopyranosyl bromide)
Compound 1 (1.2 g, 3.2 mmol) was dissolved in CH2Cl2 (50 mL) and EtOAc (5.0 mL) and then titanium tetrabromide (TiBr4, 2.4 g, 6.4 mmol) was added slowly. The reaction mixture was stirred under sealed conditions overnight at room temperature. NaOAc (2.0 g, 24 mmol) was added to quench the reaction and washed with H2O (3 × 50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Silica chromatography (Hexanes:EtOAc = 3:1) yielded 7 as a clear oil (855 mg, 67.4%). 1H NMR (400 MHz, CDCl3) δ 6.68 (d, J = 3.9 Hz, 1H, H1), 5.48 (dd, J = 3.4, 1.4 Hz, 1H, H4), 4.98–4.89 (m, 1H, H2), 4.39 (td, J = 6.8, 6.1, 1.5, 0.7 Hz, 1H, H5), 4.17 (dd, J = 11.5, 6.1 Hz, 1H, H6a), 4.11 (dd, J = 10.6, 3.3 Hz, 1H, H3), 4.03 (dd, J = 11.5, 6.8 Hz, 1H, H6b), 2.16 (s, 3H, C(O)CH3), 2.14 (s, 3H, C(O)CH3), 2.05 (s, 3H, C(O)CH3).
13C NMR (101 MHz, CDCl3) δ 170.27, 169.75, and 169.58 (C(O)CH3), 88.19 (C-1), 71.29 (C-5), 69.47 (C-2), 67.05 (C-4), 60.95 (C-6), 58.32 (C-3), 20.69, 20.60, 20.50 (C(O)CH3).
Preparation of Compound 4 (Tri-isopropylsilyl 3-azido-2,4,6-tri-O-acetyl-1-thio-β-d-galactopyranoside)
To a solution of 3 (770 mg, 1.95 mmol) in dry CH3CN (10 mL) N2 gas was purged for 10 min, then K2CO3 (809 mg, 5.86 mmol) was added followed by tri-isopropylsilylthiol (TIPSSH, 628 μL, 2.93 mmol), and the reaction was stirred for 3 h at room temperature. After complete conversion of the starting material according to TLC monitoring, the solvent was evaporated and the residue was dissolved in CH2Cl2 (20 mL), washed with H2O (2 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Silica chromatography (hexanes:EtOAc = 4:1) yielded 4 as a white solid (300 mg, 30%).
1H NMR (400 MHz, CDCl3) δ 5.42 (dd, J = 3.5, 1.0 Hz, 1H, H4), 5.20 (t, J = 9.8 Hz, 1H, H2), 4.60 (d, J = 9.5 Hz, 1H, H1), 4.11 (dd, J = 11.5, 5.6 Hz, 1H, H6a), 4.00 (dd, J = 11.6, 7.1 Hz, 1H, H6b), 3.79 (ddd, J = 6.9, 5.5, 1.1 Hz, 1H, H5), 3.53 (dd, J = 10.1, 3.4 Hz, 1H, H3), 2.15, 2.12, 2.02 (3S, 9H, 3 COCH3), 1.25 (m, 3H, -SiC3H3), 1.14–1.07 (m, 18H, -SiC3H3C6H18).
13C NMR (101 MHz, CDCl3) δ 170.57, 170.25, 169.39 (3 COCH3), 80.37 (C-1), 75.47 (C-5), 62.87 (C-3), 72.17 (C-2), 68.12 (C-4), 62.13 (C-6), 20.84, 20.76, 20.51 (3COCH3), 18.52, 18.22 (6 −SiCHCH3), 12.76 (3 −SiCH).
HRMS (EI, m/z): calculated for C21H37N3O7SSiNa+ ([M + Na]+): 526.2014, found 526.2011.
Preparation of Compound 5 (3-azido-3′-phenyl-2,2′,4,4′,6,6′-hexa-O-acetyl β-d-thiodigalactoside)
The solution of 2 (190 mg, 0.38 mmol) in dry CH3CN (10 mL) was added by 4 (193 mg, 0.38 mmol), N2 gas was purged for 10 min through the solution, and tetra-n-butylammoniumfluoride (TBAF, 1 M in THF, 460 μL) was added. Following complete conversion of the starting material after 5 min according to TLC analysis, the solvent was evaporated and silica chromatography (hexanes:EtOAc = 1:1 → 1:2) gave compound 5 (160 mg, 54%) as a white solid.
1H NMR (400 MHz, CDCl3) δ = 7.80 (s, 1H, triazole), 7.77–7.71 (m, 2H, ar), 7.43–7.28 (m, 3H, ar), 5.75–5.68 (m, 1H, H-2′), 5.61 (d, 1H, J = 3.2 Hz, H-4′), 5.47 (d,1H, J4,3 = 3.4 Hz, J4,5 = 1.1 Hz, H-4), 5.21 (dd, 1H, J3′,2′ = 8.6 Hz, J3′,4′ = 2.5 Hz, H-3′), 5.17 (m, 1H, H-2), 4.98 (1H, d, J1′,2′ = 9.8 Hz, H-1′), 4.84 (1H, J1,2 = 10.0 Hz, H-1), 4.11 (m, 5H, H-5′, H-6ab, H-6a′b′), 3.89 (1H, td, J5,4 = 1.2 Hz, J5,6ab = 6.4 Hz, H-5), 3.67 (1H, J3,2 = 10.1 Hz, J3,4 = 3.4 Hz, H-3), 2.15, 2.13, 2.08, 2.05, 2.04, and 2.02 (6s, total 18H, C(O)CH3).
13C NMR (101 MHz, CDCl3) δ 170.34, 170.25, 169.86, 169.54, 169.37, 168.70, 147.92, 129.96, 128.90, 128.47, 125.68, 118.26, 82.11, 81.51, 77.34, 77.02, 76.70, 75.53, 68.71, 68.43, 67.66, 66.33, 62.94, 62.78, 61.56, 61.38, 20.76, 20.68, 20.62, 20.61, 20.47, 20.38.
HRMS (EI, m/z) calculated for C32H38N6O14SH+ ([M + H]+): 763.2239, found 763.2277.
Preparation of Compound 6 (3-(4-phenyl-1H-1,2,3-triazol)-3′-azido-thiodigalactoside)
NaOMe (40 mg, 2.5 mmol) was added in the solution of compound 5 (120 mg, 0.16 mmol) in CH3OH (5.0 mL) and the mixture was stirred for 6 h at room temperature. The solution was neutralized with DOWEX-H+ resin, filtered, and evaporated. Crude 6 was obtained as a white solid and used in the next step without further purification.
Preparation of Compound 7
To a solution 3,6,9,12-tetraoxapentadec-14-yn-1-ol of (200 mg, 0.86 mmol) in 5 mL of THF was added sodium (0.6 mg, 0.025 mmol). When the sodium was dissolved, tert-butyl acrylate (0.125 mL, 0.86 mmol) was added. The solution was stirred for 20 h at room temperature and H2O (1 mL) was added to quench the reaction. After removal of the solvent, the residue was suspended in brine and extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4 before the solvent was removed. The resulting oil was purified by silica chromatography (hexanes:EtOAc = 1:1 → 1:2) to give compound 7 (120 mg, 39%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 4.17 (d, J = 2.4 Hz, 2H), 3.73–3.53 (m, 18H), 2.46 (t, J = 6.6 Hz, 2H), 2.40 (t, J = 2.4 Hz, 1H), 1.41 (s, 9H).
13C NMR (101 MHz, CDCl3) δ 170.82, 80.42, 74.43, 70.57, 70.56, 70.53, 70.46, 70.36, 70.33, 69.07, 69.07, 66.85, 58.34, 36.24, 28.08, 28.05.
Preparation of Compound 8
Compound 7 (33 mg, 0.092 mmol) and compound 6 crude (48 mg) were dissolved into CH3CN (2.0 mL) and then CuI (18 mg, 0.093 mmol) was added into the solution. The resulting mixture was heated under microwave irradiation to 80 °C for 90 min. After complete conversion of the starting material according to TLC monitoring, the mixture was concentrated in vacuo, and then H2O (1.0 mL) was added. A clear solution was obtained after centrifuge, which was purified by size-exclusion chromatography (Bio-Gel P2 fine; column 2.5 cm × 120 cm; flow rate 0.3 mL/min; elution with H2O/n-Butanol = 95/5). The fractions containing the product were pooled and freeze-dried to give compound 8 (52 mg, 0.060 mmol, 64%) as a white fluffy solid.
1H NMR (500 MHz, D2O) δ 8.55 (s, 1H), 8.26 (s, 1H), 7.87 (d, J = 7.7 Hz, 2H), 7.55 (t, J = 7.6 Hz, 2H), 7.48 (t, J = 7.5 Hz, 1H), 6.81–6.69 (m, 2H), 5.14 (dd, J = 9.8, 7.4 Hz, 2H), 5.02 (td, J = 13.7, 10.5, 2.9 Hz, 2H), 4.80–4.73 (m, 2H), 4.74 (s, 2H), 4.46 (dt, J = 27.5, 10.3 Hz, 3H), 4.26 (dd, J = 22.4, 3.0 Hz, 2H), 4.06 (td, J = 8.5, 4.4 Hz, 3H), 3.89–3.76 (m, 4H), 3.75 (t, J = 2.4 Hz, 2H), 3.66 (dq, J = 7.8, 4.3, 3.5 Hz, 16H), 3.40–3.26 (m, 2H), 3.00–2.88 (m, 4H), 2.56 (t, J = 6.0 Hz, 2H), 1.45 (s, 9H).
13C NMR (126 MHz, D2O, extracted from HSQC) δ 121.43, 124.35, 125.65, 126.36, 125.65, 129.20, 129.23, 128.75, 84.13, 84.12, 66.86, 66.97, 63.11, 66.85, 67.89, 79.54, 79.55, 61.03, 66.44, 61.05, 61.06, 69.53, 69.52, 69.50, 70.21, 42.99, 42.99, 35.06, 27.18, 27.17, 27.18.
HRMS (EI, m/z) calculated for C38H58N6O15SNa+ ([M + Na]+): 893.3579, found 893.3595.
Preparation of Compound 9
Compound 8 (52 mg, 0.060 mmol) was added into TFA/CH2Cl2 (10 mL, 1:1) and the solution was stirred for 2 h at room temperature. After being fully evaporated, the residue was purified by size-exclusion chromatography (Bio-Gel P2 fine; column 2.5 cm × 120 cm; flow rate 0.3 mL/min; elution with H2O/n-Butanol = 95/5). The fractions containing the product were pooled and freeze-dried to give compound 9 (48 mg, 0.059 mmol, 98%) as a white solid.
1H NMR (400 MHz, D2O) δ 8.40 (s, 1H), 8.11 (s, 1H), 7.75–7.68 (m, 2H), 7.44–7.37 (m, 2H), 7.36–7.28 (m, 1H), 4.99 (dd, J = 9.8, 6.1 Hz, 2H), 4.87 (ddd, J = 11.9, 10.7, 3.0 Hz, 2H), 4.59 (s, 2H), 4.31 (dt, J = 20.7, 10.2 Hz, 2H), 4.16–4.06 (m, 2H), 3.91 (td, J = 7.6, 4.4 Hz, 2H), 3.74–3.60 (m, 5H), 3.60–3.48 (m, 17H), 2.31 (t, J = 6.7 Hz, 2H).
13C NMR (101 MHz, D2O) δ 180.11, 163.09, 162.74, 147.43, 143.78, 129.19, 128.82, 125.65, 124.36, 121.38, 117.70, 114.80, 84.10, 79.48, 69.48, 69.45, 69.43, 69.17, 68.85, 67.98, 67.93, 67.84, 66.94, 66.83, 66.77, 66.74, 63.04, 61.00, 37.52.
HRMS (EI, m/z): calculated for C34H50N6O15SH+ ([M + H]+): 815.3128, found 815.3135.
Preparation of Compound 10
Compound 9 (22 mg, 0.027 mmol) was dissolved in anhydrous DMF (0.5 mL) and DiPEA (4.69 μL, 0.027 mmol) was added, followed by addition of TSTU (8.1 mg, 0.027 mmol). The resulting mixture was stirred for 30 min, and then TLC and HPLC showed that the starting material was fully converted. To avoid hydrolysis, the crude 10 was used for labeling the protein directly.
Preparation of Multivalent TDG-Conjugates 11 and 12
A volume of 150 μL BSA (60 μM in 35 mM HEPES buffer, pH 7.0) was mixed with 5 μL of the coupling agent (compound 10 crude, 54 mM in DMF) was added into the solution and the reaction mixture was incubated at 4 °C. After 24 and 48 h, additional volumes of 5 μL compound 10 crude were added. As to synthesis of conjugate 12, the pH of the BSA solution was elevated to pH 9.0 using TEA before adding the conjugation agent. Conjugates 11 and 12 were isolated and rinsed with H2O using VivaSpin 500 centrifugal concentrators (Sartorius Stedim Biotech, Goettingen, Germany) with an MWCO of 10 kDa. The protein concentration was determined by Bradford reagent (Carl Roth, Karlsruhe, Germany) according to manufacturer’s instruction.
Galectin Preparation
Constructs for human His6-tagged galectin-1 (Gal-1) and His6-tagged galectin-3 (Gal-3) were used from previous investigations. With regard to galectin-1, the full-length sequence was cloned into His6-tag providing pETDuet-1 (Novagen, Darmstadt, Germany) using restriction sites BamHI and SgsI, followed by the introduction of the C2S mutation to increase construct stability.50 The full-length sequence of galectin-3 was cloned into pETDuet-1 (Novagen, Darmstadt, Germany) using restriction sites SgsI and EcoRI.42 The expression of both galectins was performed in recombinant E. coli Rosetta (DE3) pLysS cells, which were cultivated in 1 L TB medium (5 L baffled flask) containing appropriate antibiotics (80 rpm, 100 μg/mL ampicillin, 34 μg/mL chloramphenicol). After an optical density (OD600 nm) of 0.5–0.8 was reached the temperature was decreased from 37 to 25 °C and isopropyl-1-thio-β-d-galactopyranoside (IPTG, 0.5 mM) was added for inducing protein expression. After 24 h post-induction, the cells were harvested by centrifugation (7000 rpm, 30 min, 4 °C) and stored at −20 °C. With regard to galectin purification, bacteria were suspended in lysis buffer (50 mM HEPES, 500 mM NaCl, 20 mM imidazole, pH 7.5) and sonicated on ice (two cycles, 30 s each). After removal of cell debris by centrifugation (15 000 rpm, 30 min, 4 °C), supernatant was filtered through 0.8 μm syringe filter. HisTrap HP 5 mL columns were used (GE Healthcare) according to the manufacturer’s instructions for galectin enrichment. Elution of His6-tagged galectins was achieved by augmenting the imidazole concentration in one step to 500 mM. Isolated galectins were dialyzed against phosphate buffered saline (PBS, 50 mM NaH2PO4, 150 mM NaCl, pH 7.5) supplemented with 2 mM ethylenediaminetetraacetic acid (EPBS) using SnakeSkin Dialysis Tubing (10 kDa MWCO, ThermoFisher Scientific). Durability of Gal-1 was increased by supplementing storage buffer with 20% (v/v) glycerol.
Galectin Binding In Vitro
F16 Maxisorp NUNC-Immuno Modules (Thermo Scientific, Roskilde, Denmark) were immobilized with appropriate amounts of 11, 12, or nonmodified BSA (0.1 μM in PBS, 50 μL, 5 pmol per well, 18 h). Immobilization and all further steps were performed at room temperature. After three washing steps using PBS supplemented with 0.05% (v/v) Tween 20 (PBST), residual unoccupied binding sites were blocked with PBS containing 2% (w/v) BSA. An additional 3-fold PBST washing step followed. Gal-1 and Gal-3 were added at different concentrations (1 5000 nM, 50 μL, 1 h). An additional 3-fold PBST washing step followed. The addition of peroxidase conjugated anti-His6-IgG2a from mouse (Roche Diagnostics, 1:4000 in PBS, 50 μL, 12.5 mU/mL, 1 h) enabled the detection of His6-tagged galectins. An additional 3-fold PBST washing step followed. Reaction of IgG-conjugated peroxidase was initiated by addition of 3,3′5,5′-tetramethylbenzidine (TMB) One (Kem-En-Tec, Taastrup, Denmark) substrate solution (50 μL). The reaction was stopped by addition of 3 M HCl (50 μL). Spectra Max Plus (Molecular Devices, Biberach, Germany) plate reader was utilized to measure the optical density at 450 nm, which is a measure for galectin binding. Blank values (no Gal-1 and/or Gal-3) were subtracted from all measurement values. SigmaPlot 10 was used to match the data points by nonlinear regression and to extract the respective kinetic parameters (eq 1).
| 1 |
where Y is binding signal; Bmax is maximal binding signal; X is galectin concentration; Kd is half-maximal apparent affinity constant [nM].
Galectin Inhibition In Vitro
F16 Maxisorp NUNC-Immuno Modules (Thermo Scientific, Roskilde, Denmark) were immobilized with appropriate amounts of asialofetuin as standard glycoprotein ligand (0.1 μM in PBS, 50 μL, 5 pmol per well, 18 h). Immobilization and all further steps were performed at room temperature. After three washing steps using PBS supplemented with 0.05% (v/v) Tween 20 (PBST), residual unoccupied binding sites were blocked with PBS containing 2% (w/v) BSA. An additional 3-fold PBST washing step followed. As a next step, monovalent inhibitor 9 and multivalent inhibitors 11 and 12 (5 μL each) of different concentration were given in the wells and Gal-3 (45 μL, 5.56 μM) was added for 1 h. Signals for Gal-3 binding to ASF decreased as more inhibitor was present. Blank values (no Gal-3) were subtracted from all measurement values showing that almost complete inhibition of Gal-3 molecules was reached. SigmaPlot 10 was used to match the data points by nonlinear regression and extract the respective kinetic parameters, illustrated by the Hill Equation (three parameters, eq 2).
| 2 |
where Y is binding signal; top is binding signal without inhibitor; X is inhibitor concentration; IC50 is inhibitor concentration for half-maximal (50%) inhibition [nM].
Acknowledgments
Lothar Elling and Dominic Laaf gratefully acknowledge the financial support by German Research Foundation (DFG, project EL 135/12-1). Hao Zhang gratefully acknowledge the financial support by a scholarship (file No. 201306210049) from the China Scholarship Council (http://www.csc.edu.cn/).
Glossary
Abbreviations
- TDG
thiodigalactoside
- NHS
N-hydroxysuccinimide
- Gal-3
galectin-3
- LacNAc
N-acetyllactosamine
- LacdiNAc
N′,N″-diacetyllactosamine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00047.
Analytical methods, synthetic details, MS spectra, and NMR data (PDF)
Author Contributions
§ Hao Zhang and Dominic Laaf contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Cecioni S.; Imberty A.; Vidal S. (2015) Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 115, 525–561. 10.1021/cr500303t. [DOI] [PubMed] [Google Scholar]
- Barondes S. H.; Cooper D. N.; Gitt M. A.; Leffler H. (1994) Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 269, 20807–20810. [PubMed] [Google Scholar]
- Elola M. T.; Blidner A. G.; Ferragut F.; Bracalente C.; Rabinovich G. A. (2015) Assembly, organization and regulation of cell-surface receptors by lectin–glycan complexes. Biochem. J. 469, 1–16. 10.1042/BJ20150461. [DOI] [PubMed] [Google Scholar]
- Nabi I. R.; Shankar J.; Dennis J. W. (2015) The galectin lattice at a glance. J. Cell Sci. 128, 2213–2219. 10.1242/jcs.151159. [DOI] [PubMed] [Google Scholar]
- Chaudhari A. D.; Gude R. P.; Kalraiya R. D.; Chiplunkar S. V. (2015) Endogenous galectin-3 expression levels modulate immune responses in galectin-3 transgenic mice. Mol. Immunol. 68, 300–311. 10.1016/j.molimm.2015.09.015. [DOI] [PubMed] [Google Scholar]
- D’Haene N.; Sauvage S.; Maris C.; Adanja I.; Le Mercier M.; Decaestecker C.; Baum L.; Salmon I. (2013) VEGFR1 and VEGFR2 Involvement in Extracellular Galectin-1- and Galectin-3-Induced Angiogenesis. PLoS One 8, e67029. 10.1371/journal.pone.0067029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuna-Costa A.; Gomes A. M.; Kozlowski E. O.; Stelling M. P.; Pavão M. S. G. (2014) Extracellular Galectin-3 in Tumor Progression and Metastasis. Front. Oncol. 4, 138. 10.3389/fonc.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funasaka T.; Raz A.; Nangia-Makker P. (2014) Galectin-3 in angiogenesis and metastasis. Glycobiology 24, 886–891. 10.1093/glycob/cwu086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharaman J.; Kanigsberg A.; Slaaby R.; Leffler H.; Barondes S. H.; Rini J. M. (1998) X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J. Biol. Chem. 273, 13047–13052. 10.1074/jbc.273.21.13047. [DOI] [PubMed] [Google Scholar]
- Tejler J.; Salameh B.; Leffler H.; Nilsson U. J. (2009) Fragment-based development of triazole-substituted O-galactosyl aldoximes with fragment-induced affinity and selectivity for galectin-3. Org. Biomol. Chem. 7, 3982–3990. 10.1039/b909091f. [DOI] [PubMed] [Google Scholar]
- Sörme P.; Arnoux P.; Kahl-Knutsson B.; Leffler H.; Rini J. M.; Nilsson U. J. (2005) Structural and thermodynamic studies on cation-Pi interactions in lectin-ligand complexes: high-affinity galectin-3 inhibitors through fine-tuning of an arginine-arene interaction. J. Am. Chem. Soc. 127, 1737–1743. 10.1021/ja043475p. [DOI] [PubMed] [Google Scholar]
- Cumpstey I.; Carlsson S.; Leffler H.; Nilsson U. J. (2005) Synthesis of a phenyl thio-[small beta]-d-galactopyranoside library from 1,5-difluoro-2,4-dinitrobenzene: discovery of efficient and selective monosaccharide inhibitors of galectin-7. Org. Biomol. Chem. 3, 1922–1932. 10.1039/b502354h. [DOI] [PubMed] [Google Scholar]
- Leffler H.; Barondes S. H. (1986) Specificity of binding of three soluble rat lung lectins to substituted and unsubstituted mammalian beta-galactosides. J. Biol. Chem. 261, 10119–10126. [PubMed] [Google Scholar]
- Delaine T.; Collins P.; MacKinnon A.; Sharma G.; Stegmayr J.; Rajput V. K.; Mandal S.; Cumpstey I.; Larumbe A.; Salameh B. A.; Kahl-Knutsson B.; van-Hattum H.; van Scherpenzeel M.; Pieters R. J.; Sethi T.; Schambye H.; Oredsson S.; Leffler H.; Blanchard H.; Nilsson U. J. (2016) Galectin-3-Binding Glycomimetics that Strongly Reduce Bleomycin-Induced Lung Fibrosis and Modulate Intracellular Glycan Recognition. ChemBioChem 17, 1759–1770. 10.1002/cbic.201600285. [DOI] [PubMed] [Google Scholar]
- Salameh B. A.; Cumpstey I.; Sundin A.; Leffler H.; Nilsson U. J. (2010) 1H-1,2,3-Triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 18, 5367–5378. 10.1016/j.bmc.2010.05.040. [DOI] [PubMed] [Google Scholar]
- Salameh B. A.; Leffler H.; Nilsson U. J. (2005) 3-(1,2,3-Triazol-1-yl)-1-thio-galactosides as small, efficient, and hydrolytically stable inhibitors of galectin-3. Bioorg. Med. Chem. Lett. 15, 3344–3346. 10.1016/j.bmcl.2005.05.084. [DOI] [PubMed] [Google Scholar]
- van Hattum H.; Branderhorst H. M.; Moret E. E.; Nilsson U. J.; Leffler H.; Pieters R. J. (2013) Tuning the Preference of Thiodigalactoside- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1. J. Med. Chem. 56, 1350–1354. 10.1021/jm301677r. [DOI] [PubMed] [Google Scholar]
- André S.; Kaltner H.; Furuike T.; Nishimura S.-I.; Gabius H.-J. (2004) Persubstituted Cyclodextrin-Based Glycoclusters as Inhibitors of Protein–Carbohydrate Recognition Using Purified Plant and Mammalian Lectins and Wild-Type and Lectin-Gene-Transfected Tumor Cells as Targets. Bioconjugate Chem. 15, 87–98. 10.1021/bc0340666. [DOI] [PubMed] [Google Scholar]
- André S.; Pieters R. J.; Vrasidas I.; Kaltner H.; Kuwabara I.; Liu F.-T.; Liskamp R. M. J.; Gabius H.-J. (2001) Wedgelike Glycodendrimers as Inhibitors of Binding of Mammalian Galectins to Glycoproteins, Lactose Maxiclusters, and Cell Surface Glycoconjugates. ChemBioChem 2, 822–830. . [DOI] [PubMed] [Google Scholar]
- Marchiori M. F.; Pires Souto D. E.; Oliveira Bortot L.; Francisco Pereira J.; Kubota L. T.; Cummings R. D.; Dias-Baruffi M.; Carvalho I.; Campo V. L. (2015) Synthetic 1,2,3-triazole-linked glycoconjugates bind with high affinity to human galectin-3. Bioorg. Med. Chem. 23, 3414–3425. 10.1016/j.bmc.2015.04.044. [DOI] [PubMed] [Google Scholar]
- Tejler J.; Tullberg E.; Frejd T.; Leffler H.; Nilsson U. J. (2006) Synthesis of multivalent lactose derivatives by 1,3-dipolar cycloadditions: selective galectin-1 inhibition. Carbohydr. Res. 341, 1353–1362. 10.1016/j.carres.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Vrasidas I.; André S.; Valentini P.; Böck C.; Lensch M.; Kaltner H.; Liskamp R. M.; Gabius H. J.; Pieters R. J. (2003) Rigidified multivalent lactose molecules and their interactions with mammalian galectins: a route to selective inhibitors. Org. Biomol. Chem. 1, 803–810. 10.1039/b210923a. [DOI] [PubMed] [Google Scholar]
- Böcker S.; Laaf D.; Elling L. (2015) Galectin Binding to Neo-Glycoproteins: LacDiNAc Conjugated BSA as Ligand for Human Galectin-3. Biomolecules 5, 1671–1696. 10.3390/biom5031671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaf D.; Bojarova P.; Pelantova H.; Kren V.; Elling L. (2017) Tailored Multivalent Neo-Glycoproteins: Synthesis, Evaluation, and Application of a Library of Galectin-3-Binding Glycan Ligands. Bioconjugate Chem. 28, 2832–2840. 10.1021/acs.bioconjchem.7b00520. [DOI] [PubMed] [Google Scholar]
- Cheng K.; El-Boubbou K.; Landry C. C. (2012) Binding of HIV-1 gp120 Glycoprotein to Silica Nanoparticles Modified with CD4 Glycoprotein and CD4 Peptide Fragments. ACS Appl. Mater. Interfaces 4, 235–243. 10.1021/am2013008. [DOI] [PubMed] [Google Scholar]
- Jin X.; Newton J. R.; Montgomery-Smith S.; Smith G. P. (2009) A generalized kinetic model for amine modification of proteins with application to phage display. BioTechniques 46, 175–182. 10.2144/000113074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil U. S.; Qu H.; Caruntu D.; O’Connor C. J.; Sharma A.; Cai Y.; Tarr M. A. (2013) Labeling Primary Amine Groups in Peptides and Proteins with N-Hydroxysuccinimidyl Ester Modified Fe(3)O(4)@SiO(2) Nanoparticles Containing Cleavable Disulfide-bond Linkers. Bioconjugate Chem. 24, 1562. 10.1021/bc400165r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S.; Nilsson U. J. (2014) Tri-isopropylsilyl thioglycosides as masked glycosyl thiol nucleophiles for the synthesis of S-linked glycosides and glyco-conjugates. Org. Biomol. Chem. 12, 4816–4819. 10.1039/C4OB00741G. [DOI] [PubMed] [Google Scholar]
- Lowary T. L.; Hindsgaul O. (1994) Recognition of synthetic O-methyl, epimeric, and amino analogues of the acceptor alpha-L-Fuc p-(1-->2)-beta-D-Gal p-OR by the blood-group A and B gene-specified glycosyltransferases. Carbohydr. Res. 251, 33–67. 10.1016/0008-6215(94)84275-2. [DOI] [PubMed] [Google Scholar]
- Sung S. R.; Han S. C.; Jin S.-H.; Lee J. W. (2011) Convergent Synthesis and Characterization of Dumbbell Type Dendritic Materials by Click Chemistry. Bull. Korean Chem. Soc. 32, 3933–3940. 10.5012/bkcs.2011.32.11.3933. [DOI] [Google Scholar]
- Seitz O.; Kunz H. (1997) HYCRON, an Allylic Anchor for High-Efficiency Solid Phase Synthesis of Protected Peptides and Glycopeptides. J. Org. Chem. 62, 813–826. 10.1021/jo960743w. [DOI] [Google Scholar]
- Wang H.; Huang W.; Orwenyo J.; Banerjee A.; Vasta G. R.; Wang L.-X. (2013) Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 21, 2037–2044. 10.1016/j.bmc.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischöder T.; Laaf D.; Dey C.; Elling L. (2017) Enzymatic Synthesis of N-Acetyllactosamine (LacNAc) Type 1 Oligomers and Characterization as Multivalent Galectin Ligands. Molecules 22, 1320. 10.3390/molecules22081320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaf D.; Bojarová P.; Mikulová B.; Pelantová H.; Křen V.; Elling L. (2017) Two-Step Enzymatic Synthesis of β-D-N-Acetylgalactosamine-(1 → 4)-D-N-Acetylglucosamine (LacdiNAc) Chitooligomers for Deciphering Galectin Binding Behavior. Adv. Synth. Catal. 359, 2101–2108. 10.1002/adsc.201700331. [DOI] [Google Scholar]
- Tietze L. F.; Schroter C.; Gabius S.; Brinck U.; Goerlach-Graw A.; Gabius H. J. (1991) Conjugation of p-aminophenyl glycosides with squaric acid diester to a carrier protein and the use of neoglycoprotein in the histochemical detection of lectins. Bioconjugate Chem. 2, 148–153. 10.1021/bc00009a003. [DOI] [PubMed] [Google Scholar]
- Jin R.; Greenwald A.; Peterson M. D.; Waddell T. K. (2006) Human Monocytes Recognize Porcine Endothelium via the Interaction of Galectin 3 and α-GAL. J. Immunol. 177, 1289–1295. 10.4049/jimmunol.177.2.1289. [DOI] [PubMed] [Google Scholar]
- Alcantar N. A.; Aydil E. S.; Israelachvili J. N. (2000) Polyethylene glycol-coated biocompatible surfaces. J. Biomed. Mater. Res. 51, 343–351. . [DOI] [PubMed] [Google Scholar]
- Giorgi M. E.; Agusti R.; de Lederkremer R. M. (2014) Carbohydrate PEGylation, an approach to improve pharmacological potency. Beilstein J. Org. Chem. 10, 1433–1444. 10.3762/bjoc.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist J. J.; Toone E. J. (2002) The cluster glycoside effect. Chem. Rev. 102, 555–578. 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- Blanchard H.; Bum-Erdene K.; Hugo M. W. (2014) Inhibitors of Galectins and Implications for Structure-Based Design of Galectin-Specific Therapeutics. Aust. J. Chem. 67, 1763–1779. 10.1071/CH14362. [DOI] [Google Scholar]
- Öberg C. T.; Leffler H.; Nilsson U. J. (2011) Inhibition of galectins with small molecules. Chimia 65, 18–23. 10.2533/chimia.2011.18. [DOI] [PubMed] [Google Scholar]
- Kupper C. E.; Böcker S.; Liu H.; Adamzyk C.; van de Kamp J.; Recker T.; Lethaus B.; Jahnen-Dechent W.; Neuss S.; Müller-Newen G.; Elling L. (2013) Fluorescent SNAP-tag galectin fusion proteins as novel tools in glycobiology. Curr. Pharm. Des. 19, 5457–5467. 10.2174/1381612811319300017. [DOI] [PubMed] [Google Scholar]
- Laaf D.; Steffens H.; Pelantová H.; Bojarová P.; Křen V.; Elling L. (2017) Chemo-Enzymatic Synthesis of Branched N-Acetyllactosamine Glycan Oligomers for Galectin-3 Inhibition. Adv. Synth. Catal. 359, 4015–4024. 10.1002/adsc.201700969. [DOI] [Google Scholar]
- Ahmad N.; Gabius H.-J.; André S.; Kaltner H.; Sabesan S.; Roy R.; Liu B.; Macaluso F.; Brewer C. F. (2004) Galectin-3 Precipitates as a Pentamer with Synthetic Multivalent Carbohydrates and Forms Heterogeneous Cross-linked Complexes. J. Biol. Chem. 279, 10841–10847. 10.1074/jbc.M312834200. [DOI] [PubMed] [Google Scholar]
- Lepur A.; Salomonsson E.; Nilsson U. J.; Leffler H. (2012) Ligand Induced Galectin-3 Protein Self-association. J. Biol. Chem. 287, 21751–21756. 10.1074/jbc.C112.358002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters R. J. (2009) Maximising multivalency effects in protein-carbohydrate interactions. Org. Biomol. Chem. 7, 2013–2025. 10.1039/b901828j. [DOI] [PubMed] [Google Scholar]
- Liese S.; Netz R. R. (2015) Influence of length and flexibility of spacers on the binding affinity of divalent ligands. Beilstein J. Org. Chem. 11, 804–816. 10.3762/bjoc.11.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parera Pera N.; Branderhorst H. M.; Kooij R.; Maierhofer C.; van der Kaaden M.; Liskamp R. M. J.; Wittmann V.; Ruijtenbeek R.; Pieters R. J. (2010) Rapid Screening of Lectins for Multivalency Effects with a Glycodendrimer Microarray. ChemBioChem 11, 1896–1904. 10.1002/cbic.201000340. [DOI] [PubMed] [Google Scholar]
- Haataja S.; Verma P.; Fu O.; Papageorgiou A. C.; Pöysti S.; Pieters R. J.; Nilsson U. J.; Finne J. (2018) Rationally Designed Chemically Modified Glycodendrimer Inhibits Streptococcus suis Adhesin SadP at Picomolar Concentrations. Chem. - Eur. J. 24, 1905–1912. 10.1002/chem.201704493. [DOI] [PubMed] [Google Scholar]
- Witten K. G.; Rech C.; Eckert T.; Charrak S.; Richtering W.; Elling L.; Simon U. (2011) Glyco-DNA–Gold Nanoparticles: Lectin-Mediated Assembly and Dual-Stimuli Response. Small 7, 1954–1960. 10.1002/smll.201100492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.