

Abstract
Investigation of natural products is an attractive strategy to identify novel compounds for cancer prevention and treatment. Numerous studies have shown the efficacy and safety of natural products, and they have been widely used as alternative treatments for a wide range of illnesses, including cancers. However, it remains unknown whether natural products affect homologous recombination (HR)-mediated DNA repair and whether these compounds can be used as sensitizers with minimal toxicity to improve patients’ responses to radiation therapy, a mainstay of treatment for many human cancers. In this study, in order to systematically identify natural products with an inhibitory effect on HR repair, we developed a high-throughput image-based HR repair screening assay and screened a chemical library containing natural products. Among the most interesting of the candidate compounds identified from the screen was β-thujaplicin, a bioactive compound isolated from the heart wood of plants in the Cupressaceae family, can significantly inhibit HR repair. We further demonstrated that β-thujaplicin inhibits HR repair by reducing the recruitment of a key HR repair protein, Rad51, to DNA double-strand breaks. More importantly, our results showed that β-thujaplicin can radiosensitize cancer cells. Additionally, β-thujaplicin sensitizes cancer cells to PARP inhibitor in different cancer cell lines. Collectively, our findings for the first time identify natural compound β-thujaplicin, which has a good biosafety profile, as a novel HR repair inhibitor with great potential to be translated into clinical applications as a sensitizer to DNA-damage-inducing treatment such as radiation and PARP inhibitor. In addition, our study provides proof of the principle that our robust high-throughput functional HR repair assay can be used for a large-scale screening system to identify novel natural products that regulate DNA repair and cellular responses to DNA damage-inducing treatments such as radiation therapy.
Keywords: β-thujaplicin, Radiosensitizer, Homologous recombination, DNA repair, PARP inhibitor
1. Introduction
Radiotherapy is the primary treatment for many kinds of solid tumors, however, some cancer types are considered radioresistant, which limit the usage of radiotherapy in those cancers. Osteosarcoma is the most common histological type of primary malignant bone tumor and is most prevalent in younger populations [1]. Although the combination of neoadjuvant chemotherapy and radical surgery is the standard treatment, the treatment of osteosarcoma is still challenging for those patients suffering from local and distant relapse [2,3]. Radiotherapy is an alternative therapy for osteosarcoma with locally relapsed or recurrent disease [4,5]. The role of radiotherapy is still controversial because it is generally considered radioresistant with poorly understood mechanism [4]. Thus, identifying novel radiosensitizing combination therapies for the treatment of radioresistant cancers such as osteosarcoma, is still of great importance for improving the effectiveness of the radiotherapy.
Response to radiation therapy is highly heterogeneous and can differ according to the type of radiation used and intrinsic tumor features, including genetic background, protein expression, the hypoxia status of tumor cells, and the efficiency of repair of DNA double-strand breaks (DSBs), a major form of DNA damage caused by radiation therapy [6]. The initial efforts to improve the clinical efficacy of radiation therapy resulted in combined-modality approaches, including radio-chemotherapy [7]. In the past few decades, to improve the effectiveness of radiation therapy, extensive research has been conducted to search for radiosensitizers that can selectively increase the lethal effects of radiation in tumors. Among the wide ranges of potential radiosensitizers investigated, well-recognized reagents includes gold nanoparticles, hypoxia-specific cytotoxins, drugs involved in DNA repair, drugs involved in cell signaling, and growth factors [8]. Experimental and preclinical studies have demonstrated the efficacy of radiosensitizers in improving response to radiation therapy, and several inhibitors have been or are currently being tested as potential radiosensitizers in clinical trials, including poly(ADP-ribose) polymerase inhibitors(PARPi) [9–11], proteasome inhibitors [12–14], and epidermal growth factor receptor inhibitors [15]. Recently some strategies have been investigated as the radiosensitizers for osteosarcoma, histone deacetylase inhibitors (HDACi),DNA-PKcs inhibitors are considered as attractive options for the radiosensitization of this cancer type [16,17]. Given the high cost of targeted therapeutic drugs as radisensitizers, further exploration of novel agents with promising radiosensitization effect, lower cost and fewer side effects is still clinically important. Therefore, identification of novel, affordable and safe radiosensitizers remains a critical clinical need in radiation therapy for solid tumors, especially for those typical type of radioresistant tumors like osteosarcoma.
Ionizing radiation (IR) induces cell death through DNA damage. The most toxic form of DNA damage caused by IR is DNA DSBs. Unrepaired DSBs directly contribute to the tumoricidal effect of radiation therapy, which mainly targets cells in the S/G2/M phases. To repair damaged DNA and maintain genomic integrity, cells have evolved two major mechanisms to repair DSBs, non-homologous end joining (NHEJ) and homologous repair(HR) [18]. In contrast to NHEJ, HR-mediated DNA repair (HR repair) utilizes genetic information contained in the homologous sequences to repair DSBs. Thus, HR is considered an error-free, high-fidelity repair mechanism, which are predominates in the S/G2/M phases when DSBs occur at DNA replication forks, NHEJ is otherwise predominant in all interphase cells [19,20]. Given the fundamental role of HR repair in repairing DSBs in cells in the S/G2/M phases, HR repair deficiency not only predisposes to cancer development (e.g., breast and ovarian cancer risk is increased in people with mutations in BRCA1 and BRCA2, which encode DNA repair proteins) but also sensitizes cancer cells to DNA damage-inducing therapy such as radiation therapy [18,21,22]. Moreover, in both experimental and clinical studies, chemical inhibitors that target kinases involved in HR repair, such as ATM, ATR, CHK1, and CHK2, have been shown the capacity to significantly enhance the sensitivity of cancer cells to DNA damage-inducing treatments, including radiation therapy and chemotherapy [23–26]. Although these findings suggest that novel agents targeting HR repair would be effective radiosensitizers, there is no such HR repair inhibitor clinically available to radiosensitize various types of tumors. In order to identify chemical modulators of HR repair, we developed a high-throughput image-based assay as a functional assay of measuring HR repair capacity.
Different from previous HR repair screenings [27], by using this powerful and robust assay, we analyzed the NCI chemical libraries which containing both mechanical compounds and natural products. More specifically, compounds in natural product library were isolated or produced from plants or chemically synthesized to be identical to products produced from plants. As well known,chemotherapeutic approaches have adapted noticeable amount of natural products in cancer treatment due to their highly selective anti-cancer effect, examples are bleomycin, mitomycin, paclitaxel, camptothecin, irrinotecan and daunorubicin, etc., and some of them(like paclitaxel) have been reported the efficacy of radiosensitizing efficiency [28]. However, the potential clinical application of natural products as radiosensitizers has not been well studied. How these compounds may have an impact on response to radiation therapy still remains largely unknown.
In this study, we analyzed the NCI natural products and mechanical compounds libraries using a high-throughput image-based functional HR repair assay. We identified one of the natural product β-thujaplicin, also known as hinokitiol, has significant inhibition on HR repair pathway. We further validated the hypothesis that natural product β-thujaplicin can be used as radiosensitizer and has synthetically lethality with PARP inhibitor.
2. Materials and methods
2.1. Cell culture and irradiation
U2OS human osteosarcoma cells were purchased from the American Type Culture Collection. These cells were maintained in McCoy’s 5A modified medium (Cellgro, 10-050-CV) supplemented with 10% FBS. Breast cancer MDA-AB-231 and HS578T cells were maintained in RPMI 1640 (Corning,10-040-CV) and DMEM (Corning, 10-017-CV) supplemented with 10% FBS. Cells were incubated at 37 °C in a humidified incubator with 5% CO2. Cells were irradiated with a RAD SOURCE RS-2000 X-ray irradiator (Suwanee, GA) at the indicated doses.
2.2. Compound library and antibodies
The compound library was obtained from the National Cancer Institute and was composed of 933 compounds including natural product compounds and bioactive compounds (Natural Compounds Set III and 1 mM Mechanistic Diversity Set II). Monoclonal antibodies for western blotting against phosphorylated RPA (p-RPA),RPA,and CTIP were purchased from Bethyl (Montgomery,TX); anti-p-CHK1 and anti-CHK1 antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA); anti-BRCA1 antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas,TX);and anti-tubulin antibody was purchased from Sigma (St. Louis, MO). Goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP were purchased from Santa Cruz Biotechnology, Inc. Primary antibody for immunofluorescent staining against Rad51 (SC-8349) and 53BP1 (SC-22760) were purchased from Santa Cruz Biotechnology, Inc., anti-p-RPA32 (A300-245A)was purchased from Bethyl, anti-γ-H2AX (16-202A) was purchased from EMD Millipore. Secondary antibody Alex Fluor-conjugated goat anti-rabbit IgG (A21206) and secondary antibody Alex Fluor-conjugated goat anti-mouse IgG (A-11001) were purchased from Life Technology (Waltham, MA).
2.3. Plasmids and transfection
The DR-GFP recombination substrate construct was a kind gift of Dr. Maria Jasin (Memorial Sloan-Kettering Cancer Center,New York, NY). U2OS cells containing a single copy of the HR repair reporter substrate DR-GFP in a random locus were generated as previously described [29]. Positive clones have a single integrated copy of the reporter and exhibit two specific bands after digestion with HindIII and probing with the indicated Southern probe. FuGENE 6 (Promega, E2691) was used for all plasmid transfections following the manufacturer’s protocols. Six hours after transfection, cells were dissociated in trypsin and re-suspended in fresh McCoy’s 5A cell culture medium and ready to be used for compound screening.
2.4. High-throughput compounds screening
For high-throughput compounds screening, the plate and liquid handling were performed using a high-throughput screening platform composed of an EL406 washer dispenser (Biotek, Winooski, VT) and a JANUS automated liquid handling workstation (PerkinElmer, Billerica, MA). Cell seeding was performed in black 96-well imaging plates (Greiner Bio-One, Monroe, NC) at a density of 5 × 102 cells/well. On the same day, screening compounds were dispensed into assay plates at a final concentration of 10 μM using the JANUS automated liquid handling workstation. These assay plates were incubated at 37 °C for 3 days. On day 4, chemical treatment was withdrawn via discarding medium. Cells were fixed with 0.4% PFA at 4°. Then cells in assay plates were exposed to DAPI for 20 min. Images were acquired and staining intensity was quantified using an ImageXpress Micro XLS widefield high-content analysis system (Molecular Devices, Sunnyvale, CA). Eight fields of images per well were acquired at a 10× objective in two wavelengths: wavelengths1 (W1), wavelengths2 (W2), respectively. Images were analyzed using a modified multi-wavelength cell scoring analysis module of MetaXpress software (Molecular Devices, Sunnyvale, CA) as previously described [30]. The DAPI signal was measured at W1 detecting all cell nuclei. The GFP signal was measured at W2. The quantitative data image analysis algorithm included all nuclei mean integrated intensities, all nuclei mean areas, positive W2, % positive W2, all W2 mean stain integrated intensities, all W2 mean stain areas.
2.5. Secondary validation assay
The positive hits identified from the primary screening were analyzed using a secondary validation assay. Briefly, cells were seeded in 96-well plates at a density of 5 × 102 cells/well in a total volume of 100 μL in triplicate in each experiment, and nontransfected cells were used for control. On the same day, cells were treated with DMSO or compounds identified as positive hits in the primary screening at indicated concentrations (0.5, 1, 2, 5, and 10 μM). Seventy-two hours later, on day 4, chemical treatment was withdrawn via application of discard medium. Cells were fixed with 0.4% PFA at 4°. Then cells in assay plates were exposed to DAPI for 20 min. Images were acquired and staining intensity was quantified and analyzed as previously described [30].
2.6. HR repair assay
In the HR repair assay system, the DR-GFP reporter substrate includes the SceGFP region, which contains an I-SceI endonuclease site within the coding region, and the iGFP region, which contains homologous sequence or the SceGFP region. Expression of I-SceI induces a single DSB in the genome. When this DSB is repaired by HR, the expression of green fluorescence protein (GFP) is restored and indicates the efficiency of HR repair. Six hours after transfection with an I-SceI transfection vector, cells were treated with β-thujaplicin at different concentrations (5, 10, 20 μM) or DMSO for 48 h and then were analyzed to detect GFP-positive cells using a flow cytometer with CellQuest software (BD Biosciences, Franklin Lakes, NJ) at the Flow Cytometry and Cellular Imaging Facility at The University of Texas MD Anderson Cancer Center. Three independent experiments were performed, mean values and standard deviations (SDs) were calculated. For the PARP inhibitor combination assay,cells transfected with an I-SceI transfection vector were treated with DMSO, 5 μM β-thujaplicin, 0.1 μM BMN673 and the combination of 5 μM β-thujaplicin and 0.1 μM BMN673 for 48 h and were analyzed to detect GFP-positive cells as prescribed above.
2.7. Colony formation assay
For colony formation assay, 500 U2OS cells were seeded into 6-well plates in triplicate and then treated the next day with β-thujaplicin at different concentrations (0.2, 0.5, 1, 2, and 5 μM) or DMSO. On the third day, cells were exposed to IR at a dose of 0.5, 1, or 1.5 Gy or not exposed to IR. Cells were incubated for 2 weeks to allow colonies to form. For the PARP inhibitor combination assay, 500 U2OS cells were seeded into 6-well plates, DMSO, 0.1 μM BMN-673, 5 μM β-Thujaplicin and the combination of the 0.1 μM BMN-673 and 5 μM β-Thujaplicin were added into the plates for 10 days.500 MDA-AB-231 and HS578T cells were seeded into 6 well plates, DMSO, 5 nM BMN-673, 10 μMβ-Thujaplicin and the combination of the 5 nM BMN-673 and 10 μM β-Thujaplicin were added into the plates for 7 days. Colonies were stained with 0.001% crystal violet and counted by NIH Image J software (National Institutes of Health, Bethesda, MD), all experiments were repeated for three times.
2.8. Western blotting
U2OS cells were treated with β-thujaplicin at different concentrations (0.5, 1, 2, 5 μM) or DMSO for 72 h. Cells were harvested and washed in phosphate-buffered saline (PBS), and whole cellular extracts were incubated with urea buffer (8 M urea, 50 mM Tris-HCl [pH 7.4], and 150 mM 2-mercaptoethanol) for 30 min on ice. Lysates were cleared by centrifugation, and proteins were separated by gel electrophoresis. Membranes were blocked in PBS–0.1% Tween-20 (PBST) with 5% (w/v) nonfat, dry milk for 1 h at room temperature. Membranes were then incubated with 1:1000 dilutions of primary antibodies (anti-p-RPA, anti-RPA, anti-Rad51, anti-CTIP, anti-BRCA1 and anti-tubulin) overnight at 4°. Subsequently, membranes were washed with PBST and incubated with secondary antibodies (1:1000) diluted in PBST with 5% nonfat, dry milk. Membranes were washed in PBST, and bound antibody was detected by enhanced chemiluminescence (GE Healthcare, Pittsburgh, PA).
2.9. Immunofluorescent staining
For detection of DNA damage–induced foci of Rad51, 53BP1, γ-H2AX and p-RPA32, immunofluorescent staining was carried out essentially as described previously [29,31]. Specifically, to detect Rad51 foci, U2OS cells grown on coverslips were treated with DMSO or 10 μM β-thujaplicin for 16 h and then exposed to IR (10 Gy). Eight h after IR exposure, cells were exposed to cytoskeleton buffer, stripping buffer, and fixing buffer. Then cells were spread onto slides for staining. Rabbit anti-Rad51 antibody (1:500) was used to detect Rad51 foci. Cells were incubated with primary antibodies overnight at 4 °C, and cells were incubated with secondary antibody Alex Fluor-conjugated goat anti-rabbit IgG for 2 h at room temperature. Slides were mounted in medium containing DAPI and analyzed under a fluorescence microscope. The number of foci per cell was scored for at least 50 cells per sample. To detect 53BP1 and γ-H2AX foci, U2OS cells were treated with DMSO or 5 μM β-Thujaplicin 16 h before exposed to IR (10 Gy) and the staining was performed 2 h after IR. Rabbit anti-53BP1 antibody (1:500) was used to detect 53BP1 foci. Mouse anti-γ-H2AX antibody (1:500) was used to detect γ-H2AX foci. Primary antibodies were incubated for overnight at 4°, secondary antibody was incubated for 2 h at room temperature. To detectp-RPA32, β-thujaplicin at a concentration of 1, 2, 5, or 10 μM or DMSO was added into cells, and the staining was performed 48 h later. Rabbit anti-p-RPA32 antibody (1:500) was used to detect p-RPA32 foci,the same procedures as described above were performed.
2.10. Contact inhibition cell cycle and HR repair analysis
U2OS cells were cultured at 37 °C incubator until they reach 90% confluency, then DMSO, 1, 2, 5, 10 μM β-Thujaplicin were added into U2OS cells 48 h before fixing into 70% ethanol at −20 ° overnight, then cells were washed with PBS twice, suspended and stained with PI Staining solution (50 ug/ml PI and RNase in PBS), cell cycle analysis were measured using FACS (Beckman Coulter, USA). Additionally, cells were transfected with an I-SceI transfection vector and treated with β-thujaplicin at different concentrations (5,10, 20 μM) or DMSO for 48 h, after cell reached the 90% confluency, cells were harvested and analyzed to detect GFP-positive cells using a flow cytometer with Cell Quest software.
2.11. Apoptosis assay
Annexin V/PI apoptosis kit (Invitrogen, USA) was applied to detect the apoptosis. Cells were treated with β-Thujaplicin at the concentration of 10 μM and 20 μM for 48 h, gently trypsinized, then washed with cold PBS twice, cells were resuspended in 500 μL Annexin V binding buffer, 5 μL Annexin V and 5 μL propidium iodide (PI) were added into each sample, incubated in the dark room for 5 min at room temperature, the Annexin-V and PI binding were analyzed using fluorescence-activated cell sorting (FACS) (Beckman Coulter, USA). Cells without β-Thujaplicin and cells treated with IR for 48 h were served as negative and positive control respectively.
2.12. Data analysis
All experiements were repeated for three times and data were summarized as mean ± SD. Differences among groups were analyzed by using one-tailed Student’s t-tests and two-way analysis of variance. Statistical significant was defined as P < 0.05. Calculations were performed with GraphPad Prism software (GraphPad Software Inc., LaJolla, CA). Protein levels and colony numbers were quantified using NIH Image J software.
3. Results
3.1. Development of a high-throughput image-based screening assay to identify chemical regulators of HR repair
We utilized DR-GFP cells to develop a high-throughput assay for functional identification of HR repair inhibitors. We previously established a DR-GFP cell line, which contains a DR-GFP HR reporter plasmid [29,31,32]. This plasmid was stably integrated into the cellular genome. It contains two mutant copies of GFP, which abolish GFP expression. The first mutant GFP contains a recognition site for I-SceI. The second mutant GFP is truncated but contains a homologous sequence for the first GFP. When cells are transfected with I-SceI plasmid, expression of I-SceI enzyme results in a DSB in the first GFP. If this DSB is successfully repaired by HR repair utilizing the homologous sequence in the second GFP, GFP expression is restored. Thus, in this assay, the HR repair capacity is proportional to the percentage of GFP-positive cells (Fig. 1A). We adopted this DR-GFP cell-based functional HR repair assay with a high-throughput imaging platform (Fig. 1B).
Fig. 1.
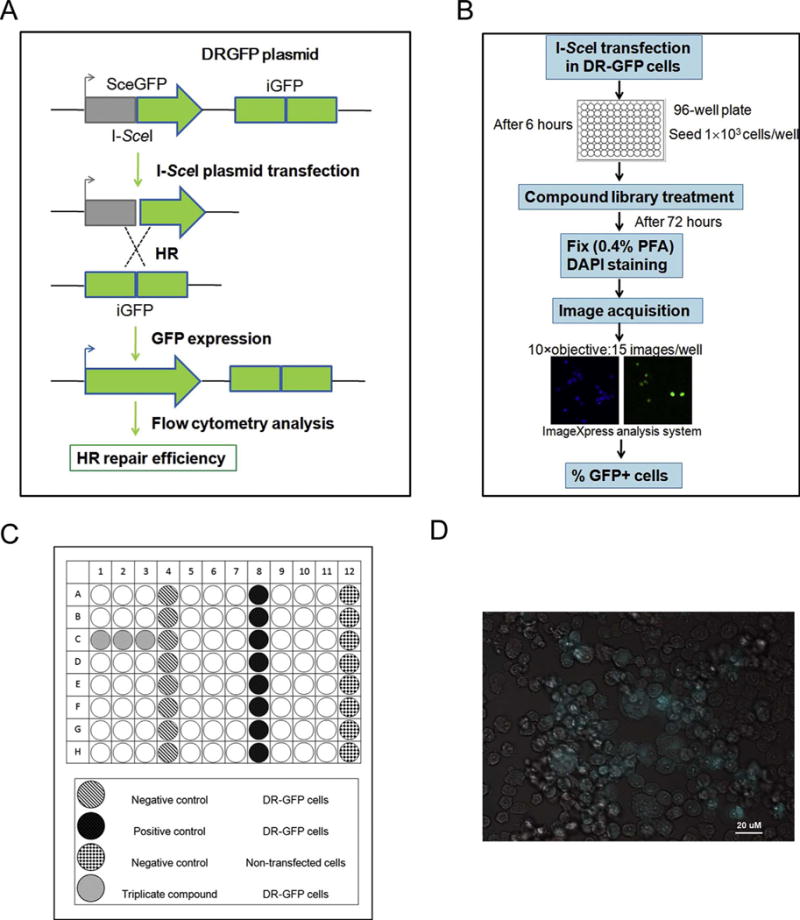
Designation of high-throughput image-based screening assay.
A. Schematic diagram of DR-GFP cell-based functional HR repair assay.
B. Schematic representation of the high-throughput screening.
C. Plate layout designation.
D. Automated imaging of U2OS cells engineered to permit assessment of the efficiency of HR repair. Cells expressing green fluorescent protein (GFP) indicating successful HR repair.
As shown in Fig. 1C, we designed a plate layout to test each compound in triplicate. The negative control (DMSO) and positive control (ATM inhibitor) were arranged in separate columns, as were the nontransfected DR-GFP cells. As described previously [29], in this system, the efficiency of HR repair can be assessed by the expression of GFP. The nuclear (DAPI) and GFP fluorescence signals were acquired by high-content analysis system at two different wavelengths,commensurate with DAPI and GFP dyes. The GFP positive cells are shown in green color, overlaid a phase contrast image (Fig. 1D).
3.2. Quality control and hit selection of high throughput image-based screening assay
In our screening assay, we normalized the data using a vehicle treatment approach and calculate the fold change in the values of wavelength 2 (GFP, W2) as a percentage of negative controls in each plate. We used ATM inhibitor as our positive control and we used vehicle DMSO as our negative control. All controls were included in each plate in 8 replicates, and served as reference points to assess both inner-plate and inter-plate variability. For the quality control of the screen extensive downstream analysis measures were taken. These included the assessment of the statistical variability due to non-specific background via eight replicates of vehicle-treated non-transfected cells, in comparison to DR-GFP containing, vehicle-treated cells. Secondly, signal intensities from DR-GFP transfected, vehicle-treated cells were compared to those from DR-GFP containing, ATM inhibitor-treated cells in eight replicates of the positive control. Thus, the signal variability and Z-scores within natural product plates, as well as among all plates were calculated. (Fig. 2A–B).
Fig. 2.
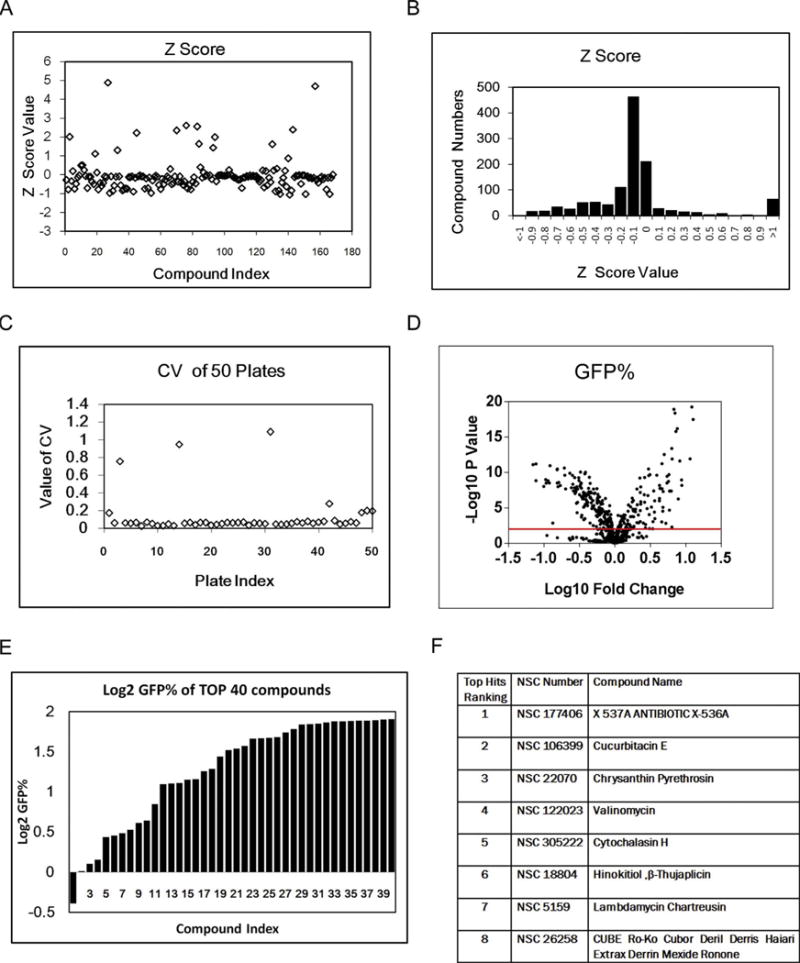
Performace of high-throughput image-based screening assay.
A. Z score scatter plot of screening in NCI Natural Compounds Set III plates.
B. Z score distribution of screening (NCI Natural Compounds Set III and Mechanistic Diversity Set II plates).
C. CV scatter plot of 50 plates in the screening (NCI Natural Compounds Set III and Mechanistic Diversity Set II plates).
D. Volcano plot of screening results (NCI Natural Compounds Set III and Mechanistic Diversity Set II plates).
E. Top 40 hits waterfall plot in the primary screening (NCI Natural Compounds Set III and Mechanistic Diversity Set II plates).
F. The compound list of top 8 positive hits in NCI Natural Compounds Set III plates.
To assess assay performance, the coefficient of variation (CV) were monitored across all plates of the screen. From the first fifty screen plates,all CVs, except those from only four plates, were smaller than 20%, suggesting that variations among different plates were at an acceptable level (Fig. 2C). We also checked the plates with the very high CVs individually and eliminated them from the screen analysis. The screening results were also presented in a volcano plot (Fig. 2D). Positive hits were defined as compounds that reduced the percentage of GFP-positive cells compared to the percentage in nontreated control cells and had p values less than 0.01 in the statistical analysis (Fig. 2D). We used mean+/−3sd as our cut-off for selection of top hits and selected positive hits by Z score. The screening results of top 40 positive hits were presented in a bar histogram (Fig. 2E) and a table presenting the compound names of first eight hits of natural product collection was shown in Fig. 2F.
3.3. Validation of β-thujaplicin as an HR repair inhibitor by secondary screening
To validate the positive hits in the primary screening, we performed a secondary validation assay to determine the dose-dependent effects of positive hits on HR repair. We treated DR-GFP cells with increasing doses of hit compounds ranging from 0 μM to 10 μM, cells were incubated with hit compounds for 72 h, the same amount of time as in the primary screening. The representative three validation assay results, including β-thujaplicin, deoxycytidine and alzolastone,are shown in Fig. 3A–C. We selected β-thujaplicin as our top candidate based on the marked suppression of HR of GFP, the existing literature reporting the biological activities and clinical usage safety of this potential candidate,and the most promising translational research potential [33–35].
Fig. 3.
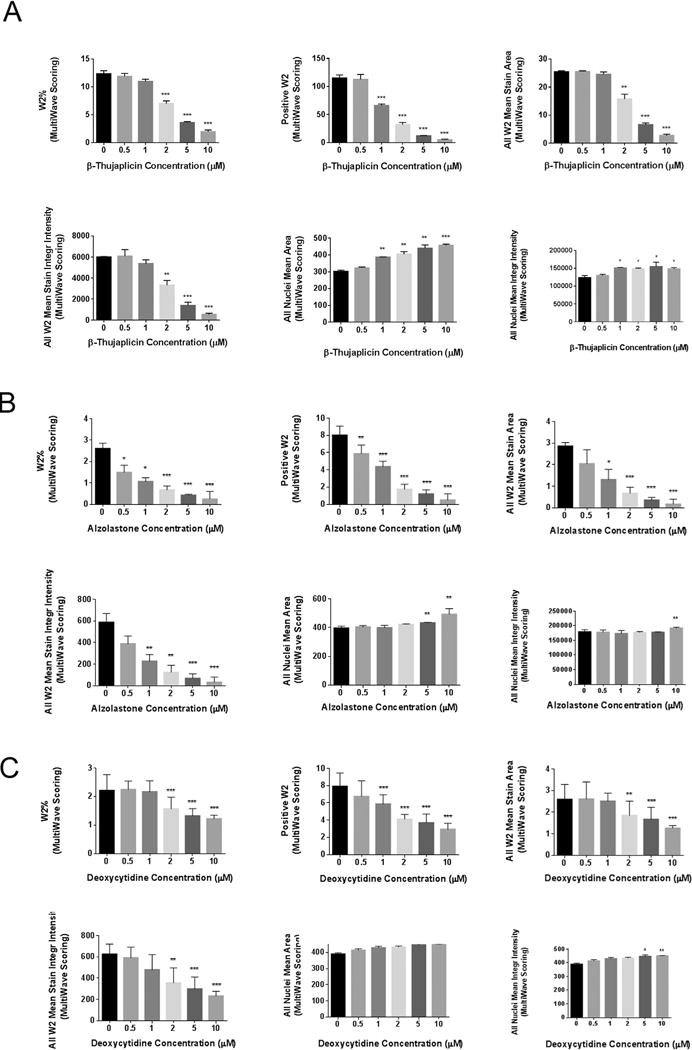
HR repair inhibitor hits validation in a secondary screening assay.
A. Secondary screening validation of positive hit β-thujaplicin.
B. Secondary screening validation of positive hit Alzolastone.
C. Secondary screening validation of positive hit Deoxycytidine.
The multiwave scoring of W2 percentage, the number of W2 positve cells, the W2 mean stain area, and the W2 mean stain integrity intensity decreased significantly with increasing hits’ concentration(0, 0.5, 1, 2, 5, 10 μM). (W2 indicating wavelength 2 using by the analysis system to detect GFP signal by microscopy). All nuclei mean area and all nuclei mean integrity intensity increased significantly with increasing hits’ concentration. Error bars represent SD.*P < 0.05, **P < 0.01, ***P < 0.001.
We adapted four GFP signal-associated parameters to confirm β-thujaplicin’s HR inhibitory function, including “the percentage of W2 cells”, “the number of W2 cells”, “all W2 mean stain area” (indicating the mean of GFP stained area of all fields in each well acquired by wavelength 2 in analysis system) and “all W2 mean stain integrity intensity” (indicating the mean of GFP stained integrity intensity of all fields in each well acquired by wavelength 2 in analysis system). Following treatment with β-thujaplicin for 72 h, the percentage of GFP-positive cells and the number of GFP-positive cells decreased as β-thujaplicin concentration increased. At the doses of 2 μM, 5 μM and 10 μM, the percentage of GFP-positive cells upon compound treatment were significantly lower than values in the untreated control cells. At the doses of 1 μM, 2 μM, 5 μM and 10 μM, the number of GFP-positive cells in treated cells were significantly lower than values in the untreated control cells. The other two parameters we chose to measure GFP signal were “all W2 mean stain area” and “all W2 mean stain integrity intensity,” which also decreased as β-thujaplicin concentration increased, with significant differences noted at the concentrations of 2 μM, 5 μM and 10 μM.
Notably, parameters chosen to represent all cell nuclei signal, “all nuclei mean area” (indicating the mean of DAPI stained area of all fields in each well in analysis system) and “all nuclei mean integrity intensity” (indicating the mean of DAPI stained integrity intensity of all fields in each well in analysis system), increased significantly with increasing doses of β-thujaplicin.
3.4. Confirmation of β-thujaplicin’s inhibitory effect on HR repair
In our screening system, we used image-based screening to detect GFP expression restored by HR repair in DR-GFP cells. To confirm β-thujaplicin’s role in HR repair inhibition, we analyzed the percentage of cells with I-SceI-induced GFP expression using flow cytometry. Representative flow cytometry profiles are shown in Fig. 4A. We observed a significant decrease in GFP-positive cells after cells were treated with different dosages of β-thujaplicin. The percentage of GFP-positive cells was 16.29% in control cells and 4.38% in cells treated with 20 μM β-thujaplicin, indicating defective HR repair after β-thujaplicin treatment. The quantitative summary of three independent experiments (Fig. 4B) showed that the fold change of GFP-positive cells decreased in a dose-dependent manner after β-thujaplicin treatment, with significant differences noted between control cells and treated cells at concentrations of at least 10 μM. In this assay, we observed that β-thujaplicin at 10 μM caused a 50% reduction of HR repair efficiency (Fig. 4B), while the HR efficiency of compounds imatinib identified as negative hits in our screening shows no change at all in this HR repair assay(Fig. 4C).
Fig. 4.
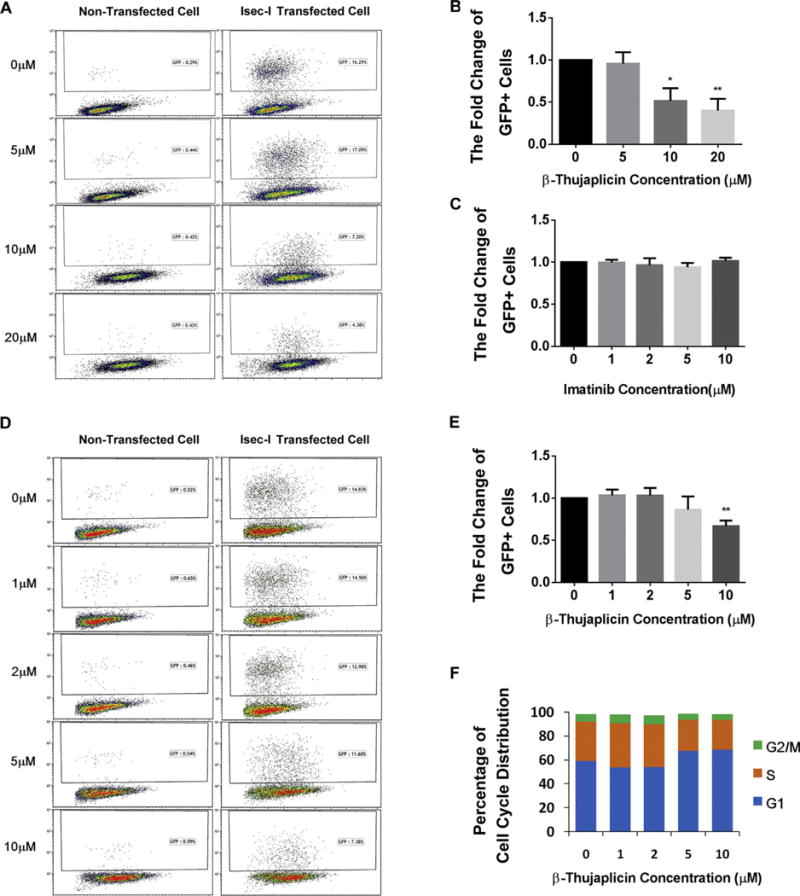
Final confirmation of β-thujaplicin’s HR repair inhibition effect by flow cytometry.
A. β-thujaplicin decreases HR repair efficiency. Representative flow cytometry profiles show a significant decrease of GFP-positive cells after treatment with different doses of β-thujaplicinas (0, 5, 10, 20 μM) indicated by I-SceI HR reporter assay.
B. Quantitation of β-thujaplicin’s inhibitory effect on HR. β-thujaplicin decreases the fold change of GFP positive cells significantly at the concentration of 10 and 20 μM. Error bars represent SD. *P < 0.05, **P < 0.01.
C. Quantitation of negative control imatinib’s effect on HR. Imatinib doesn’t change the fold change of GFP positive cells at different doses. Error bars represent SD. *P < 0.05, **P < 0.01.
D. In contact inhibition assay (add β-thujaplicin or DMSO when cells reach 90% confluency), β-thujaplicin decreases the fold change of GFP positive cells. Representative flow cytometry profiles show a significant decrease of the fold change of GFP positive cells after treatment with different doses of β-thujaplicinas (0, 1, 2, 5, 10 μM) indicated by I-SceI HR reporter assay.
E. Quantitation of β-thujaplicin’s inhibitory effect on HR. β-thujaplicin decreases the fold change of GFP positive cells significantly at the concentration of 10 μM. Error bars represent SD. *P < 0.05, **P < 0.01.
F. In contact inhibition assay(add β-thujaplicinor DMSO when cells reach 90% confluency),cell cycle analysis shows that β-thujaplicin slightly increase the G1 cell cycle distribution at the concentration of 5 μM and 10 μM, but doesn’t cause further increased G1 cell cycle arrest.
To improve that β-thujaplicin’s HR repair reduction effect is not caused by fluctuations in cell cycle progression,we also conducted the contact inhibition assay to test β-thujaplicin’s effect on cell cycle progression as well as HR repair. In contact inhibition assay,the results show that β-thujaplicin inhibits the fold change of GFP-positive cells at the concentration of 10 μM (Fig. 4D–E),at the same time,it may slightly increase the G1 cell cycle distribution at the concentration of 5 μM and 10 μM,but doesn’t cause G1 cell cycle arrest (Fig. 4F).
3.5. β-thujaplicin induces phosphorylation of RPA
When DSBs are repaired by the HR repair pathway, multiple nucleases, including CTIP, EXO1, BLM, and DNA2, are required to initiate the resectionof the DSB ends and generate a stretch of single-strand DNA (ssDNA). This ssDNA at the broken ends of DSBs serves as a platform to recruit ssDNA binding protein RPA. RPA is activated at DSB ends by phosphorylation, and activation of RPA then facilitates the recruitment of additional key repair proteins, such as Rad51. The recruitment of Rad51 replaces RPA and forms Rad51-coated ssDNA filaments, which initiate the homology search and recombination process of HR repair. Thus, defective Rad51 recruitment can result in persistent binding of RPA to ssDNA, which leads to RPA phosphorylation [36,37]. As expected, when cells were treated with different concentrations of β-thujaplicin, we observed an increasing foci formation of p-RPA (Fig. 5A–C). It is worthy of note that β-thujaplicin at a concentration of 10 μM induced p-RPA in the absence of exogenous DNA damage, as shown in Fig. 5D, E. At a concentration of 10 μM, β-thujaplicin activated p-RPA protein expression and induced a significant seven fold increase in p-RPA protein level (Fig. 5D, E). The protein levels of other proteins, including RPA, Rad51, CTIP, and BRCA1, exhibited no significant changes after β-thujaplicin treatment. We reasoned that β-thujaplicin may have perturbed HR repair, most likely due to the presence of endogenous DNA damage. Collectively, these data showed that the effect of β-thujaplicin on RPA activation was consistent with the reduced Rad51 foci formation observed in cells treated with β-thujaplicin as shown in Fig. 6A–C.
Fig. 5.
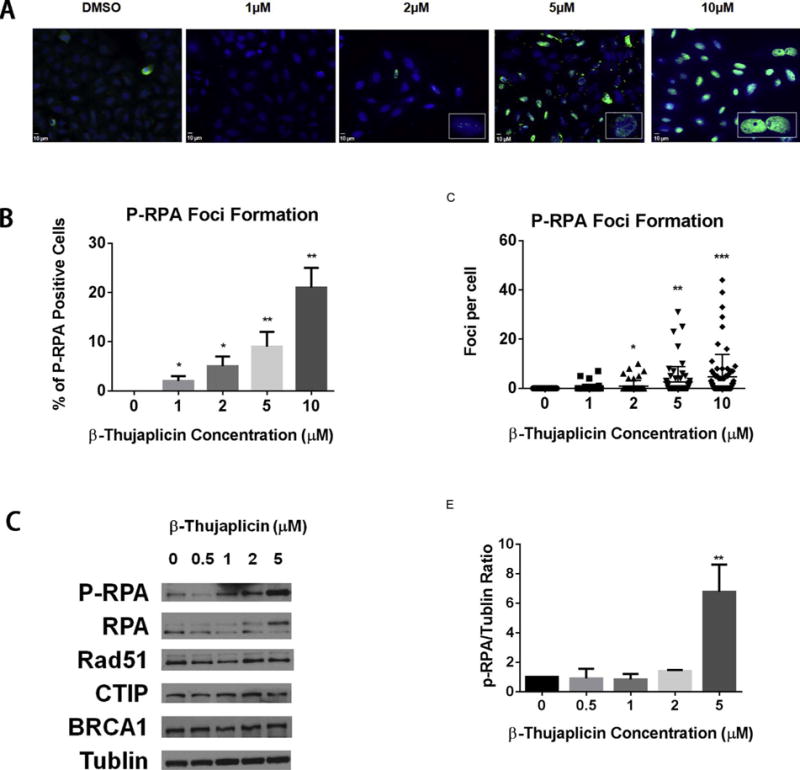
β-thujaplicin induces phosphorylation of RPA.
A. Immunofluorescence staining of p-RPA32 foci (green) in U2OS cells 48 hours following treatment with DMSO or β-thujaplicin (1, 2, 5, or 10 μM). The nucleus is counterstained with DAPI (blue).
B. Quantitation of percentage of p-RPA32 foci-positive cells shows β-thujaplicin significantly increases p-RPA32 foci formation at the concentration of 1, 2, 5, or 10 μM. Error bars represent SD from replicates. *P < 0.05, **P < 0.01.
C. Quantitation of p-RPA32 foci numbers per cell shows β-thujaplicin significantly increases p-RPA32 foci formation per cell at the concentration of 2, 5, or 10 μM. Error bars represent SD from replicates. *P < 0.05, **P < 0.01, ***P < 0.001.
D. Western blot analysis (p-RPA,RPA,Rad51,CTIP,BRCA1,Tublin) of U2OS cells pretreated with DMSO or β-thujaplicin at the indicated concentrations (0.5, 1, 2, 5 μM) for 72 h.
E. Quantitation of P-RPA protein level shows β-thujaplicin significantly increases the P-RPA protein level at the concentration of 5 μM. Error bars represent SD from replicates (*P < 0.05, **P < 0.01).
Fig. 6.
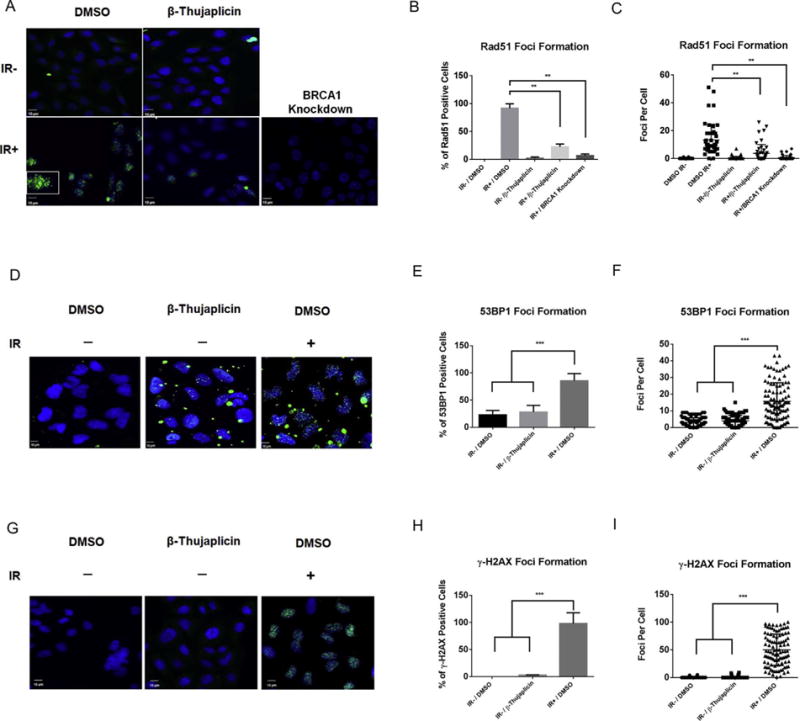
A–C β-thujaplicin inhibits radiation-induced Rad51 foci formation.
A. Immunofluorescence staining of Rad51 foci (green) in U2OS cells 8 h following treatment with β-thujaplicin (10 μM) followed 16 h later by exposure to IR (10 Gy). The nucleus is counterstained with DAPI (blue).
B. Quantitation of percentage of Rad51 foci-positive cells shows β-thujaplicin significantly decreases Rad51 foci formation in U2OS cells following treatment with β-thujaplicin (10 μM) compared to DMSO followed 16 h later by exposure to IR (10 Gy). Error bars represent SD from replicates. *P < 0.05, **P < 0.01.
C. Quantitation of Rad51 foci numbers per cell shows β-thujaplicin significantly decreases Rad51 foci formation in U2OS cells following treatment with β-thujaplicin (10 μM) compared to DMSO followed 16 h later by exposure to IR (10 Gy). Error bars represent SD from replicates. *P < 0.05, **P < 0.01. D–I. β-thujaplicin does not cause accumulation of DSBs.
D. Immunofluorescence staining of 53BP1 foci (green) in U2OS cells 2 h following treatment with β-thujaplicin (5 μM) or DMSO. The nucleus is counterstained with DAPI (blue). Positive control cells were exposed to IR (10 Gy).
E. Quantitation of percentage of 53BP1 foci-positive cells shows β-thujaplicin doesn’t increases 53BP1 foci formation in U2OS cells following treatment with β-thujaplicin (5 μM) compared to DMSO. Error bars represent SD from replicates.*P < 0.05, **P < 0.01, ***P < 0.001.
F. Quantitation of 53BP1 foci numbers per cell shows β-thujaplicin doesn’t increase 53BP1 foci formation in U2OS cells following treatment with β-thujaplicin (5 μM) compared to DMSO. Error bars represent SD from replicates.*P < 0.05, **P < 0.01, ***P < 0.001.
G. Immunofluorescence staining of γ-H2AX foci (green) in U2OS cells 2 h following treatment with β-thujaplicin (5 μM) or DMSO. The nucleus is counterstained with DAPI (blue). Positive control cells were exposed to IR (10 Gy). H. Quantitation of percentage of γ-H2AX foci-positive cells shows β-thujaplicin doesn’t increase γ-H2AX foci formation in U2OS cells following treatment with β-thujaplicin (5 μM) compared to DMSO. Error bars represent SD from replicates. *P < 0.05, **P < 0.01, ***P < 0.001.
I. Quantitation of γ-H2AX foci numbers per cell shows β-thujaplicin doesn’t increase γ-H2AX foci formation in U2OS cells following treatment with β-thujaplicin (5 μM) compared to DMSO. Error bars represent SD from replicates. *P < 0.05, **P < 0.01, ***P < 0.001.
3.6. β-thujaplicin inhibits recruitment of key protein in HR repair to DSBs
Recombinase Rad51 is a key protein in HR repair. Rad51 is recruited to DSBs and controls homology search and recombination, which permits cells to utilize genetic information contained in the sister chromatids to repair DNA damage in an error-free manner. Because I-SceI-induced HR repair assays showed that β-thujaplicin inhibits HR repair, we explored the mechanism responsible for this effect. We first examined whether β-thujaplicin affects the recruitment of Rad51 to DSBs. We treated cells with β-thujaplicin and induced DSBs by exposure to IR. Rad51 was recruited to DSBs, forming discrete nuclear foci in control cells. In cells treated with β-thujaplicin, Rad51 foci formation was significantly reduced (Fig. 6A–C). We then examined whether β-thujaplicin may induce DNA damage by staining two DNA DSBs markers 53BP1 and γ-H2AX. As shown in Fig. 6D–I, β-thujaplicin treatment did not induce accumulation of DNA DSBs in the absence of DNA damage, which is consistent with the apoptosis assay (Fig. 7C–D). These data suggest that β-thujaplicin inhibits HR through impaired recruitment of Rad51 to DSBs.
Fig. 7.
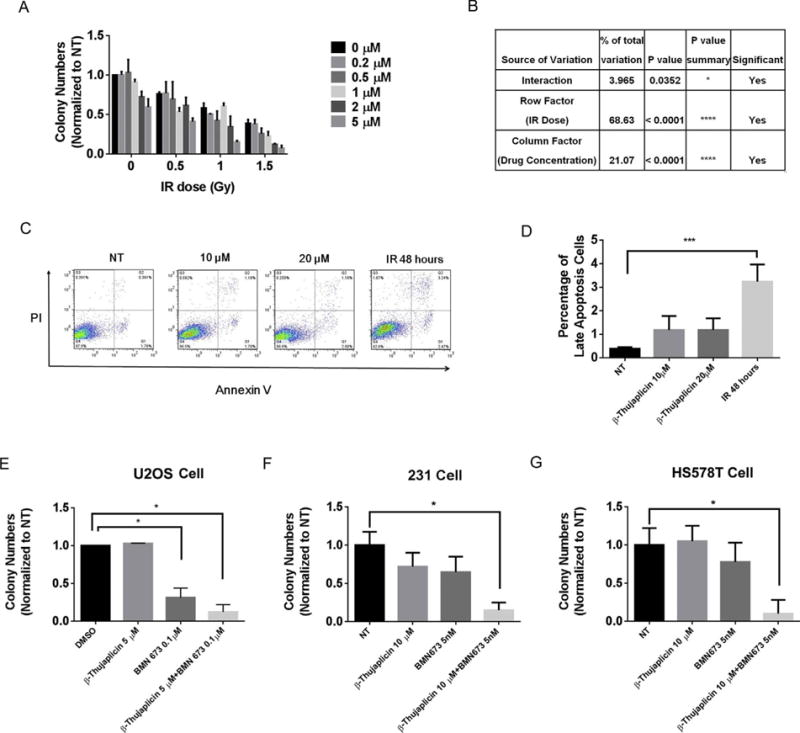
Effect of β-thujaplicin on radiosensitivity or combination of PARP inhibitor.
A. Survival of U2OS cells treated with DMSO or β-thujaplicin at the indicated concentration (0.2, 0.5, 1, 2, 5 μM) and either not exposed to IR or exposed to IR at the indicated doses (0.5, 1, 1.5 Gy).
B. Statistical analysis of impact of interaction between β-thujaplicin concentration and IR dose on cell survival. *P < 0.05, ****P < 0.0001.
C. The apoptotic profile of U2OS cells treated with DMSO or β-thujaplicin at the indicated doses for 24 h. Positive control cells were exposed to IR (10 Gy).
D. The quantitative data for apoptosis assay from three independent experiments shows β-thujaplicin at the indicated concentration (10, 20 μM) doesn’t induce significant cell apoptosis. ***P < 0.001.
E. Survival assay of U2OS cells treated with DMSO, β-thujaplicin (5 μM), BMN673 (0.1 μM), and the combination group.
F. Survival assay of breast cancer MDA-MB-231 cells treated with DMSO,β-thujaplicin (10 μM), BMN673 (5 nM),and the combination group.
G. Survival of breast cancer HS578T cells treated with DMSO, β-thujaplicin (10 μM), BMN673 (5 nM), and the combination group.
3.7. β-thujaplicin sensitizes cancer cells to radiation and PARP inhibitor
Patients with osteosarcoma often exhibit resistance to radiation therapy. As we observed an inhibitory effect of β-thujaplicin on HR repair, we next tested whether β-thujaplicin can sensitize osteosarcoma U2OS cells to radiation therapy. We used colony formation assay to determine whether β-thujaplicin reduced the survival of U2OS osteosarcoma cells after IR. To identify an optimal combination of β-thujaplicin concentration and IR doses, we treated cells with β-thujaplicin at a range of concentrations (0, 0.2, 0.5, 1, 2, 5 μM) in combination with IR at a range of doses (0, 0.5, 1, 1.5 Gy). Our results showed that β-thujaplicin radiosensitized cells in a β-thujaplicin dose-dependent manner. When β-thujaplicin was combined with IR treatment at different drug concentrations and IR doses, colony formation decreased significantly as the drug concentration and IR dose increased (Fig. 7A). Two-way analysis of variance showed that both drug concentration (column factor) and IR dose (row factor) significantly affected cell proliferation. More importantly, the interaction of these two factors remarkably affected cell proliferation. These data strongly supported the concept that β-thujaplicin has a synergistic effect with IR and sensitizes cancer cells to radiation therapy (Fig. 7B). And the result of the apoptosis assay also suggested that β-thujaplicin did not induce significant cell apoptosis at the various concentration (Fig. 7C–D).
Next we tested the combination treatment using β-thujaplicin and PARP inhibitor BMN673 on cell proliferation. We used colony formation assay to determine whether β-thujaplicin can sensitize cancer cells to PARP inhibitor BMN673. We treated U2OS cells with β-thujaplicin at the concentration of 5 μM in combination with BMN673 at the concentration of 0.1 μM. Our results showed β-thujaplicin and PARP combination treatment significantly reduced cell survival, while monotherapy showed very little effect (Fig. 7E). In addition, we also tested two difference cancer cell lines to confirm the synergy between β-thujaplicin and PARP inhibitor BMN673. We treated breast cancer MDA-AB-231 and HS578T cells with β-thujaplicin at the concentration of 10 μM in combination with BMN673 at the concentration of 5 nM. Our results showed that these cancer cells were more sensitive to β-thujaplicin and PARP inhibitor combination treatment compared to monotherapy (Fig. 7F–G).
4. Discussion
The purpose of our study is to identify natural compounds and bioactive compounds from high-throughput image-based drug screening that act as HR repair inhibitors and apply to be radiosensitizers. Indeed, we identified β-thujaplicin as a natural compound with potent HR repair inhibitor activity. We also found that β-thujaplicin can inhibit the recruitment of key HR repair proteins to DNA damage sites, which can cause persistent binding of RPA to ssDNA,leading to RPA phosphorylation. In addition, we found that β-thujaplicin can radiosensitize osteosarcoma U2OS cells, a cancer cell type normally insensitive to IR. Finally,β-thujaplicin shows a synthetical lethality effect with PARP inhibitor in different cancer types. Together, our results suggest that β-thujaplicin can potentially be used as a novel natural compound-based radiosensitizer in osteosarcoma.
From the high-throughput HR inhibitor screening, we focused on the natural product library, containing a variety of natural compounds, which have translational research potential. We selected β-thujaplicin as our top candidate as previously described in results part. To validate our screening data,we performed second validation screening assay for the top candidates. Moreover, given the fact that HR occurs only in S and G2 phases of the cell cycle, HR levels are exquisitely sensitive to fluctuations in cell cycle progression, to demonstrate that β-thujaplicin’s HR inhibition effect is not due to the G1 or S arrest previously shown in other studies [33,38], we conducted the assays previously used in our own publication [22] to exclude cell cycle effects particularly the shift to G1 phase on HR repair by synchronizing cells using contact inhibition. As shown in Fig. 4F, in the contact inhibition condition, β-thujaplicin treatment at different concentrations slightly increased G1 population,but no significant G1 arrest was observed compared to untreated cells. In addition, HR repair was dramatically reduced at 10 μM (Fig. 4D–E). These data indicated that β-thujaplicin doesn’t significantly induce G1 arrest at the specific concentrations in our study; its inhibitory effect on HR repair is not completely dependent on G1 arrest.
Essential oils have been reported to exhibit anticancer effects against different types of human tumors [39]. β-thujaplicin (4-isopropyltropolone), isolated from the essential oil of the Japanese cypress, is a tropolone-based phenolic compound [40,41]. This compound has a safety profile and has been approved for use in both clinical practice and health care products [42–44]. It exerts a wide variety of biologic activities, including antimicrobial, antioxidant, and antitumor activities [45]. Additionally, It has also been reported to regulate tumor growth and metastasis, β-thujaplicin’s antitumor effects have been investigated in a variety of cancer models, including models of melanoma,breast cancer,lung cancer,colon cancer,prostate cancer, and oral cancer [33–35,38,46,47]. Some studies have shown that β-thujaplicin can inhibit cancer cell proliferation by inducing the p53-independent DNA damage response, S or G1 cell cycle arrest [33,34,48]. However,we found β-thujaplicin can inhibit HR repair through regulating the recruitment of RAD51 rather than inducing cell cycle arrest. Our study revealed that β-thujaplicin may work as a HR repair inhibitor and has a potential role acting as sensitizer to DNA damage-inducing therapy such as radiation and PARPi. This finding provides a rationale for developing a novel strategy for combination treatment in the near future.
For decades, DNA damage-inducing agents have been the mainstay of anticancer therapy such as chemotherapy and radiation therapy. Inhibition of DNA repair has also been investigated experimentally and clinically as a sensitizing approach to improve responses to radiation therapy. On the basis of our study, β-thujaplicin, also known as hinokitiol,has great potential to be a novel radiosensitizer and should be further explored. β-thujaplicin is an antimicrobial agent with an established good safety profile, it is known to be of fairly low cytotoxicity and teratogenic effects, and has minimal side effects in animal models [49,50]. Thus, β-thujaplicin has been widely used as a preservative in toothpastes, skin lotions, body soaps and other health care products [42–44]. Likewise, techniques have been established to improve the yield of β-thujaplicin production [51]. Therefore, we expect that β-thujaplicin and possibly similar natural products identified from our study will provide an important alternative approach to safely enhance the therapeutic effects of radiation by reducing DNA repair capacity in cancer cells. Furthermore, natural compounds cost less than targeted drugs that might be investigated for radiosensitizing effect, such as inhibitors of the DNA damage response kinases CHK1 and WEE1, thus, the exploration of natural compounds could significantly reduce the economic burden for cancer patients [25,26,52].
For our further study, firstly, the function of β-thujaplicin as a radiosensitizer will need to be explored in more cancer models. In fact, we plan to perform further in vivo and in vitro experiments to confirm the radiosensitization effects of β-thujaplicin. Secondly, it is our future interest to dissect how β-thujaplicin might regulate HR repair in mechanism. As described in Ononye S. N.’s study, β-thujaplicins was identified as a new chemotype of isozyme-selective histone deacetylase (HDAC) inhibitors [53]. The direct link between histone acetylation has been well-established in DNA repair and DNA damage response. It is possible that β-thujaplicin inhibits HR repair through regulating histone acetylation. Lastly, β-thujaplicin inhibits HR, which can potentially lead to major chromosomal instability. In addition, the therapeutic window of β-thujaplicin treatment is rather narrowed in our current testing conditions. Thus we propose to use β-thujaplicin target cancer cells with replication stress, which may cause a synthetic lethality. In our recent publication, we specifically reported that HR repair inhibitors targeting a key DNA resection enzyme DNA2 nuclease activity can potentially be used to target cells with replication stress, which requires HR repair capacity to overcome replication-associated DSBs [54]. It is our future interest to test whether the natural product such as β-thujaplicin containing HR repair inhibitory effect can have a similar therapeutic function targeting cancer cells with replication stress.
In summary, our findings from use of a functional DNA repair chemical screening assay indicate that the naturally occurring compound β-thujaplicin is a HR repair inhibitor that may serve as a novel radiosensitizer for cancer treatment.
Acknowledgments
We acknowledge the National Cancer Institute Developmental Therapeutics Program for providing the chemical screening compound plates. This work was supported in part by grants from the National Science Foundation of China (No. 81372852 and No. 81572961). This work was also supported by Cancer Center Support Grant CA016672 to The University of Texas MD Anderson Cancer Center and by Landon Foundation-American Association for Cancer Research (AACR) Innovator Award for Cancer Prevention Research 989258.
Footnotes
Disclosure of conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics 2014. CA: Cancer J Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 2.Feng H, Wang J, Chen W, Shan B, Guo Y, Xu J, Wang L, Guo P, Zhang Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J Bone Oncol. 2016;5:67–73. doi: 10.1016/j.jbo.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarz R, Bruland O, Cassoni A, Schomberg P, Bielack S. The role of radiotherapy in oseosarcoma. Cancer Treat Res. 2009;152:147–164. doi: 10.1007/978-1-4419-0284-9_7. [DOI] [PubMed] [Google Scholar]
- 4.Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treatment Rev. 2014;40:523–532. doi: 10.1016/j.ctrv.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Bielack SS, Hecker-Nolting S, Blattmann C, Kager L. Advances in the management of osteosarcoma. F1000Research. 2016;5:2767. doi: 10.12688/f1000research.9465.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Connell PP, Hellman S. Advances in radiotherapy and implications for the next century: a historical perspective. Cancer Res. 2009;69:383–392. doi: 10.1158/0008-5472.CAN-07-6871. [DOI] [PubMed] [Google Scholar]
- 8.Linam J, Yang LX. Recent developments in radiosensitization. Anticancer Res. 2015;35:2479–2485. [PubMed] [Google Scholar]
- 9.Liu ZG, Tang J, Chen Z, Zhang H, Wang H, Yang J, Zhang H. The novel mTORC1/2 dual inhibitor INK128 enhances radiosensitivity of breast cancer cell line MCF-7. Int J Oncol. 2016;49:1039–1045. doi: 10.3892/ijo.2016.3604. [DOI] [PubMed] [Google Scholar]
- 10.Chabot P, Hsia TC, Ryu JS, Gorbunova V, Belda-Iniesta C, Ball D, Kio E, Mehta M, Papp K, Qin Q, Qian J, Holen KD, Giranda V, Suh JH. Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: results of a randomized global, placebo-controlled study. J Neurooncol. 2017;131:105–115. doi: 10.1007/s11060-016-2275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhute VJ, Ma Y, Bao X, Palecek SP. The poly (ADP-Ribose) polymerase inhibitor veliparib and radiation cause significant cell line dependent metabolic changes in Breast cancer cells. Sci Rep. 2016;6:36061. doi: 10.1038/srep36061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cacan E, Spring AM, Kumari A, Greer SF, Garnett-Benson C. Combination treatment with sublethal ionizing radiation and the proteasome inhibitor, bortezomib enhances death-receptor mediated apoptosis and anti-tumor immune attack. Int J Mol Sci. 2015;16:30405–30421. doi: 10.3390/ijms161226238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiu HW, Lin SW, Lin LC, Hsu YH, Lin YF, Ho SY, Wu YH, Wang YJ. Synergistic antitumor effects of radiation and proteasome inhibitor treatment in pancreatic cancer through the induction of autophagy and the downregulation of TRAF6. Cancer Lett. 2015;365:229–239. doi: 10.1016/j.canlet.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Wang D, Qin Q, Jiang QJ, Wang DF. Bortezomib sensitizes esophageal squamous cancer cells to radiotherapy by suppressing the expression of HIF-1alpha and apoptosis proteins. J X-ray Sci Technol. 2016;24:639–646. doi: 10.3233/XST-160571. [DOI] [PubMed] [Google Scholar]
- 15.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, Youssoufian H, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 16.Blattmann C, Oertel S, Thiemann M, Dittmar A, Roth E, Kulozik AE, Ehemann V, Weichert W, Huber PE, Stenzinger A, Debus J. Histone deacetylase inhibition sensitizes osteosarcoma to heavy ion radiotherapy. Radiat Oncol. 2015;10:146. doi: 10.1186/s13014-015-0455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang X, Yuan F, Guo K. Repair of radiation damage of U2OS osteosarcoma cells is related to DNA-dependent protein kinase catalytic subunit (DNA-PKcs) activity. Mol Cell Biochem. 2014;390:51–59. doi: 10.1007/s11010-013-1955-5. [DOI] [PubMed] [Google Scholar]
- 18.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 19.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan MR, Hsieh HJ, Dai H, Hung WC, Li K, Peng G, Lin SY. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012;287:6764–6772. doi: 10.1074/jbc.M111.287037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, Lee JS, Hennessy B, Hanash S, Mills GB, Lin SY. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955–960. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 24.Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol Ther. 2014;142:258–269. doi: 10.1016/j.pharmthera.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, Canman CE, Normolle DP, Zabludoff SD, Maybaum J, Lawrence TS. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, Venook AP, Loechner S, Rosen LS, Shanahan F, Parry D, Shumway S, Grabowsky JA, Freshwater T, Sorge C, Kang SP, Isaacs R, Munster PN. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060–1066. doi: 10.1200/JCO.2014.57.5027. [DOI] [PubMed] [Google Scholar]
- 27.Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genomewide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomoda K, Tam YT, Cho H, Buehler D, Kozak KR, Kwon GS. Triolimus a multidrug loaded polymeric micelle containing paclitaxel, 17-AAG, and rapamycin as a novel radiosensitizer. Macromol Biosci. 2017;17 doi: 10.1002/mabi.201600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng G, Yim EK, Dai H, Jackson AP, Burgt I, Pan MR, Hu R, Li K, Lin SY. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat Cell Biol. 2009;11:865–872. doi: 10.1038/ncb1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Shen J, Yin Y, Peng Y, Wang L, Hsieh HJ, Shen Q, Brown PH, Tao K, Uray IP, Peng G. Identifying cell cycle modulators that selectively target ARID1A deficiency using high-throughput image-based screening. SLAS Discov. 2017;22:813–826. doi: 10.1177/2472555217698942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, Uray IP, Wu X, Brown PH, Shen X, Mills GB, Peng G. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5:752–767. doi: 10.1158/2159-8290.CD-14-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko J, Bao C, Park HC, Kim M, Choi HK, Kim YS, Lee HJ. beta-Thujaplicin modulates estrogen receptor signaling and inhibits proliferation of human breast cancer cells. Biosci Biotechnol Biochem. 2015;79:1011–1017. doi: 10.1080/09168451.2015.1008978. [DOI] [PubMed] [Google Scholar]
- 34.Li LH, Wu P, Lee JY, Li PR, Hsieh WY, Ho CC, Ho CL, Chen WJ, Wang CC, Yen MY, Yang SM, Chen HW. Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS One. 2014;9:e104203. doi: 10.1371/journal.pone.0104203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shih YH, Chang KW, Hsia SM, Yu CC, Fuh LJ, Chi TY, Shieh TM. In vitro antimicrobial and anticancer potential of hinokitiol against oral pathogens and oral cancer cell lines. Microbiol Res. 2013;168:254–262. doi: 10.1016/j.micres.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Godin SK, Sullivan MR, Bernstein KA. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol. 2016;94:407–418. doi: 10.1139/bcb-2016-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prado F. Homologous recombination maintenance of genome integrity during DNA damage tolerance. Mol Cell Oncol. 2014;1:e957039. doi: 10.4161/23723548.2014.957039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Yamauchi H. p27-Associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of Cdk2 function. Cancer Lett. 2009;286:240–249. doi: 10.1016/j.canlet.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 39.Bhalla Y, Gupta VK, Jaitak V. Anticancer activity of essential oils: a review. J Sci Food Agric. 2013;93:3643–3653. doi: 10.1002/jsfa.6267. [DOI] [PubMed] [Google Scholar]
- 40.Erdtman H, Gripenberg J. Antibiotic substances from the heart wood of Thuja plicata Don. Nature. 1948;161:719. doi: 10.1038/161719a0. [DOI] [PubMed] [Google Scholar]
- 41.Nakano H, Ikenaga S, Aizu T, Kaneko T, Matsuzaki Y, Tsuchida S, Hanada K, Arima Y. Human metallothionein gene expression is upregulated by beta-thujaplicin: possible involvement of protein kinase C and reactive oxygen species. Biol Pharm Bull. 2006;29:55–59. doi: 10.1248/bpb.29.55. [DOI] [PubMed] [Google Scholar]
- 42.Higashi Y, Fujii Y. Determination of hinokitiol in skin lotion by high-performance liquid chromatography-ultraviolet detection after precolumn derivatization with 4-fluoro-7-nitro-2,1,3-benzoxadiazole. J Cosmet Sci. 2013;64:381–389. [PubMed] [Google Scholar]
- 43.Iha K, Suzuki N, Yoneda M, Takeshita T, Hirofuji T. Effect of mouth cleaning with hinokitiol-containing gel on oral malodor: a randomized, open-label pilot study. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:433–439. doi: 10.1016/j.oooo.2013.05.021. [DOI] [PubMed] [Google Scholar]
- 44.Higashi Y, Sakata M, Fujii Y. High-performance liquid chromatography with dualwavelength ultraviolet detection for measurement of hinokitiol in personal care products. J Cosmet Sci. 2009;60:519–525. [PubMed] [Google Scholar]
- 45.Morita Y, Matsumura E, Okabe T, Fukui T, Ohe T, Ishida N, Inamori Y. Biological activity of beta-dolabrin, gamma-thujaplicin, and 4-acetyltropolone hinokitiol-related compounds. Biol Pharm Bull. 2004;27:1666–1669. doi: 10.1248/bpb.27.1666. [DOI] [PubMed] [Google Scholar]
- 46.Seo JS, Choi YH, Moon JW, Kim HS, Park SH. Hinokitiol induces DNA demethylation via DNMT1 and UHRF1 inhibition in colon cancer cells. BMC Cell Biol. 2017;18:14. doi: 10.1186/s12860-017-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shih YH, Chang KW, Hsia SM, Yu CC, Fuh LJ, Chi TY, Shieh TM. In vitro antimicrobial and anticancer potential of hinokitiol against oral pathogens and oral cancer cell lines. Microbiol Res. 2013;168:254–262. doi: 10.1016/j.micres.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Lee YS, Choi KM, Kim W, Jeon YS, Lee YM, Hong JT, Yun YP, Yoo HS. Hinokitiol inhibits cell growth through induction of S-phase arrest and apoptosis in human colon cancer cells and suppresses tumor growth in a mouse xenograft experiment. J Nat Prod. 2013;76:2195–2202. doi: 10.1021/np4005135. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki H, Ueda T, Juranek I, Yamamoto S, Katoh T, Node M, Suzuki T. Hinokitiol, a selective inhibitor of the platelet-type isozyme of arachidonate 12-lipoxygenase. Biochem Biophys Res Commun. 2000;275:885–889. doi: 10.1006/bbrc.2000.3390. [DOI] [PubMed] [Google Scholar]
- 50.Cho YM, Hasumura M, Takami S, Imai T, Hirose M, Ogawa K, Nishikawa A. A 13-week subchronic toxicity study of hinokitiol administered in the diet to F344 rats. Food Chem Toxicol. 2011;49:1782–1786. doi: 10.1016/j.fct.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 51.Kwon S, Shimoda K, Hamada H, Ishihara K, Masuoka N, Hamada H. High production of beta-thujaplicin glycosides by immobilized plant cells of Nicotiana tabacum. Acta Biol Hung. 2008;59:347–355. doi: 10.1556/ABiol.59.2008.3.8. [DOI] [PubMed] [Google Scholar]
- 52.Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. ABBV Cell Cycle. 2013;12:3159–3164. doi: 10.4161/cc.26062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ononye SN, VanHeyst MD, Oblak EZ, Zhou W, Ammar M, Anderson AC, Wright DL. Tropolones as lead-like natural products: the development of potent and selective histone deacetylase inhibitors. ACS Med Chem Lett. 2013;4:757–761. doi: 10.1021/ml400158k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar S, Peng X, Daley J, Yang L, Shen J, Nguyen N, Bae G, Niu H, Peng Y, Hsieh HJ, Wang L, Rao C, Stephan CC, Sung P, Ira G, Peng G. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis. 2017;6:e319. doi: 10.1038/oncsis.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]