

Abstract
Using progeny of a cross between Japanese soybean Enrei and Chinese soybean Peking, we developed a high-density linkage map and chromosomal segment substitution lines (CSSLs). The map consists of 2,177 markers with polymorphism information for 32 accessions and provides a detailed genetic framework for these markers. The marker order on the linkage map revealed close agreement with that on the chromosome-scale assembly, Wm82.a2.v1. The differences, especially on Chr. 5 and Chr. 11, in the present map provides information to identify regions in the genome assembly where additional information is required to resolve marker order and assign remaining scaffolds. To cover the entire soybean genome, we used 999 BC3F2 backcross plants and selected 103 CSSLs carrying chromosomal segments from Peking in the genetic background of Enrei. Using these low-genetic-complexity resources, we dissected variation in traits related to flowering, maturity and yield into approximately 50 reproducible quantitative trait loci (QTLs) and evaluated QTLs with small genetic effects as single genetic factors in a uniform genetic background. CSSLs developed in this study may be good starting material for removing the unfavourable characteristics of Peking during pre-breeding and for isolation of genes conferring disease and stress resistance that have not yet been characterized.
Keywords: glycine max, SSR, SNP, CSSL
1. Introduction
Soybean, Glycine max (L.) Merr., is the most important legume and is the fourth crop next to rice, wheat and maize in terms of world crop production. The estimated size of the soybean genome is 1.1 Gb.1 The genome sequence of the US cultivar Williams 82, Glyma0, became available on the Phytozome in January 2008. The first chromosome-scale assembly, Glyma1.01, became available in December 2008, and a new assembly, Wm82.a2.v1, was released in January 2014 (https://phytozome.jgi.doe.gov/pz/portal.html (1 November 2017, date last accessed)). According to the Phytozome web site, annotations of some genes have been improved on the basis of RNA-seq data and the number of unmapped scaffolds has been reduced by using new assembly methods and constructing high-density linkage maps. Two other soybean chromosome-scale assemblies are available from the NCBI web site (Glycine_max_v1.1 and Glycine_max_v2.0). However, gene names, numbers and genomic positions are not comparable among different assemblies and are sometimes very confusing to users. In the present study, we discuss only the Glyma1.01 and Wm82.a2.v1 assemblies at the Phytozome web site.
A reference genome sequence is a versatile tool with which to characterize the relationships between genes and agronomically important traits. However, additional genome information and experimental materials suitable for genetic characterization are also needed. New next-generation sequencing (NGS) technologies are expected to yield genomic sequences of a wide variety of soybean germplasms.2 In addition, the development of soybean mutant libraries and the identification of mutants related to agronomically important traits from their phenotypes and reverse-genetic approaches based on NGS are expected to provide new genetic resources.3 The efficient use of this information and experimental materials is necessary for further soybean breeding.
Recent re-sequencing information has enabled the development of molecular markers for soybean gene discovery and breeding. Among molecular markers, simple sequence repeats (SSRs) and microsatellites are useful tools because of their abundance, multi-allelic features, co-dominant inheritance, high variability and ease of analysis. SSR markers developed by previous studies4,5 have been widely used in soybean breeding programs worldwide and are available from SoyBase (https://www.soybase.org/ (1 November 2017, date last accessed)). A genetic linkage map is essential for soybean chromosome assembly. The reference sequence Glyma1.01 was assembled on the basis of a consensus linkage map,1 and Wm82.a2.v1 (Glyma2) on the basis of high-density linkage maps.6 However, chromosomal translocation and inversion in soybean have been reported with cytogenic analysis7 and karyotyping based on fluorescence in situ hybridization has also identified translocations and inversions in several soybean cultivars.8 Comparison of high-density linkage maps is expected to be deepen with our knowledge about chromosomal structure in soybean cultivars.
Soybean is an important source of traditional staple foods such as tofu, natto, miso and soy sauce in Japan. The unique cuisine, geographical and historical isolation of Japan probably shaped distinct agro-morphological characteristics of Japanese soybeans from those of continental soybeans. Since Enrei is a major cultivar (9% of total soybean cultivation area in Japan in 2014) with seeds of high quality for food processing, previous study9 sequenced the whole genome of Enrei as a representative Japanese cultivar. The sequencing average read coverage at a locus was 22.2×, and 1,659,041 SNPs and 344,418 insertions/deletions between the Enrei assembly and the reference sequence of Williams 82 were identified. One 4-coumaroyl-CoA-ligase gene (out of 10 genes in the William 82 reference genome), seven chalcone synthase genes (out of 24 genes), three chalcone isomerase genes (out of 16 genes), one flavonol synthase gene (out of 4 genes) and six dihydroflavonol 4-reductase genes (out of 10 genes), those predicted as anthocyanin and flavonoid biosynthesis genes in Williams 82 genome, were not found in the Enrei genome. Hence, the accumulation of genome and marker information for a wide range of soybean elite cultivars and the development of experimental resources to facilitate the evaluation of useful genes that differ from Williams 82 orthologs are necessary to utilize a wide range of the genetic diversity in soybean breeding.
Peking is a landrace that has been extensively used in a breeding programs because of its resistance to the soybean cyst nematode Heterodera glycines.10 In 1906, the accession was introduced from Beijing, China, into the USA by the US Department of Agriculture (USDA).11 Peking is also resistant to soybean mosaic virus,12 peanut mottle virus,13 bacterial blight,14 frogeye leaf spot (Cercospora sojina),15 soybean dwarf disease,16 soybean stem canker,17 reniform nematode Rotylenchulus reniformis,18Phytophthora stem and root rot19 and germinates well under wet conditions.20,21 However, the genes responsible for these traits, except for resistance to cyst nematode and phytophthora stem and root rot, remain uncharacterized.
Many quantitative trait loci (QTLs) related to important agronomic traits such as flowering time, plant height, maturity, seed weight, yield, seed nutrients, seed oil contents and seed protein are summarized in SoyBase. Isolation and characterization of genes for these traits are very important in facilitating MAS with DNA markers tightly linked to a locus or nucleotide polymorphism suitable for distinguishing functional alleles. However, the development of experimental material suitable for positional cloning is time consuming. Among such materials, ‘heterozygous inbred families22’ and ‘residual heterozygous lines23’ have been used to analyze QTLs as single Mendelian factors for fine mapping. In addition, series of near-isogenic lines (NILs), which have a common genetic background, are advantageous for QTL evaluation. First NIL library, which consisted of a series of NILs, currently referred to as chromosomal segment substitution lines (CSSLs), each having different chromosomal segments originating from a wild donor parent in the genetic background of cultivated tomato, was developed and used for identification of a yield-related QTL.24 CSSLs were developed in many plant species, including tomato,24 Arabidopsis,25 rice26), barley,27 peanut28 rye,29 lettuce30 and wild soybean.31 In soybean, one genetic locus from wild soybean increasing yield was also reported.32 Genomic sequences indicate that the genetic diversity of cultivated soybean is narrow compared with that of landraces and wild soybean (G. soja).33 Although CSSLs carrying segments of the wild soybean genome have been developed,31 the availability of CSSLs from out of Japan is limited, and the development of CSSLs using various combinations of soybean germplasms would provide novel breeding materials and increase genetic diversity.
In the present study, using progeny from a cross between the leading Japanese cultivar, Enrei, and the Chinese landrace Peking, we developed a high-density linkage map and CSSLs. Enrei is a representative Japanese cultivar with high quality for food processing, whereas Peking is an excellent Chinese germplasm resistant to various diseases and stresses. The genetic distance between these parents is much larger than that between Japanese landraces and Enrei but smaller than wild soybean and Enrei.34 In addition to re-sequencing information,9,35 genes controlling flowering time and growth habit have been characterized for these parents.36 Therefore, genomic resources developed in the present study may help to characterize agronomically important genes.
2. Materials and methods
2.1. Plant materials
A total of 32 Soybean lines including 16 Japanese breeding varieties, two USA breeding varieties, six Japanese landraces, four exotic landraces and four wild soybean accessions were used to identify SSR polymorphisms (Supplementary Table S1). A cross between Enrei [G. max; accession number in National Agriculture and Food Research Organization (NARO) Genebank, Japan: GmJMC025] as the female parent and Peking (GmWMC084) as the male parent was performed in 2005, and an F2 mapping population was developed. The F2 population (189 plants) and 20 plants of each parent were grown with an inter-row spacing of 80 cm and a hill spacing of 30 cm in the field at NARO in Tsukuba, Ibaraki, Japan (36°01'25.6”N 140°06'59.1”E). All experimental populations were evaluated at the same location. Seeds were sown on 30 May 2007. Images of the parent plants are shown in Fig. 1. The breeding scheme is shown in Supplementary Fig. S1.
Figure 1.

Seeds and mature plants of Enrei and Peking. The ruler is in centimeters.
2.2. Development of a backcross population and CSSLs
Enrei was used as the recurrent parent and Peking as the donor parent. Pollen from F1 plants was used to pollinate Enrei flowers before blooming, and >1,000 BC1F1 seeds were obtained in 2006. All BC1F1 seeds were sown, and 999 BC1F1 plants were crossed with Enrei to produce BC2F1 in 2007 and BC3F1 in 2008. BC3F2 seeds were obtained by self-pollinating each BC3F1 plant; the BC3F2 population (999 plants) was sown on 16 June 2010. Next year, CSSLs (103 lines) were selected from the BC3F2 population (as described below) and grown under natural day length in the same field. CSSLs were sown on 24 June 2011 (10 plants per line) and on 27 June 2012 (4–20 plants per line). Selection of CSSLs was based on the data obtained from 320 SSR markers that covered all chromosomes evenly. The proportion and length of donor chromosomal segments were calculated from genetic distances between DNA markers, and the positions of recombination breakpoints in all chromosomes were calculated in individual lines. Candidate lines with low proportions of donor chromosomal segments per chromosome were first selected, and then 103 CSSLs were selected so that the donor segments of 4–6 lines covered each chromosome.
2.3. Phenotypic evaluation
Agronomic traits of each plant in the F2, BC3F2 and CSSL populations were evaluated (Table 1). Days to first flowering (DFF) corresponded to the R1 stage.37 Days to flowering of the top raceme (DFT) corresponded to the R2 stage. Days to harvest (DH) corresponded to the R8 stage. Flowering period (FP = DFT − DFF), reproductive period (RP = DH − DFF) were calculated. Plant height (PH), number of pods (NP), one-hundred-seed weight (SWH) and total seed weight (TSW) were measured in each BC3F2 plant and CSSL population. Genetic variance (heritability) was calculated from the phenotypic variance of each population, F2 and BC3F2, with that of the parents as follows.
Table 1.
Description of investigated traits, broard sence heritability and number of QTLs detected in the present study
| Trait | Trait abbreviation | QTL abbreviation | Trait evaluation | Broad-sense heritability |
Number of QTLs detected in each population |
Number detected QTLs common between populationsa |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F2 | BC3F2 | CSSLs (2011) | CSSLs (2012) | F2 | BC3F2 | CSSL | F2 vs BC3F2 | F2 vs CSSL | BC3F2 vs CSSL | ||||
| Days to first flowering (R1) | DFF | qDFF | Days from sowing to first flowering | 97.6% | 92.0% | 77.9% | 89.0% | 7 | 6 | 7 | 6 | 7 | 6 |
| Days to flowering of top raceme (R2) | DFT | qDFT | Days from sowing to flowering of the top raceme | NA | 96.3% | 94.3% | 79.9% | Not evaluated | 6 | 6 | Not evaluated | Not evaluated | 6 |
| Flowering period (days from R1 to R2) | FP | qFP | R2−R1 | NA | 89.0% | 55.8% | 51.0% | Not evaluated | 2 | 2 | Not evaluated | Not evaluated | 2 |
| Days to harvest (R8) | DH | qDH | Days from sowing to harvest (maturity stage) | 89.7% | 35.9% | 94.5% | 61.8% | 5 | 9 | 7 | 5 | 5 | 7 |
| Reproductive period (days from R1 to harvest) | RP | qRP | Harvesting day–first flowering day | 74.9% | 5.2% | 85.3% | 48.4% | 2 | 6 | 4 | 2 | 2 | 4 |
| Plant height | PH | qPH | Distance from the primary leaf node to the base of the top raceme | 89.8% | 87.7% | 93.7% | 87.1% | 3 | 4 | 4 | 3 | 3 | 4 |
| Number of pods | NP | qNP | Number of harvested total pods | 90.6% | 76.5% | 81.9% | 66.9% | 2 | 8 | 5 | 2 | 2 | 5 |
| Seed weight per plant (total seed weight) | TSW | qTSW | Weight of harvested total seed | NA | 72.3% | 64.5% | 51.8% | 2 | 8 | 8 | 2 | 2 | 8 |
| Seed weight (100 seeds) | SWH | qSWH | Weight of 100 seeds | 60.3% | 63.3% | 63.6% | 71.2% | Not evaluated | 7 | 7 | Not evaluated | Not evaluated | 7 |
| Total | 21 | 56 | 50 | 20 | 21 | 49 | |||||||
aPartially confirmed QTLs are included.
2.4. Total genomic DNA extraction
DNA of parents and F2 plants was extracted from fresh leaves (3 g) by a Cetyltrimethylammonium Bromide (CTAB) protocol.38 DNA of cultivars and BC3F2 plants was isolated as described previously.39 DNA was quantified on a fluorescence microplate reader (ARVO; Perkin Elmer, Boston, MA, USA) according to the manufacturer‘s instructions and was adjusted to 50 and 20 ng/μl for SSR and SNP analyses, respectively.
2.5. SSR marker analysis
Marker design: SSR core motifs in Glyma1.01 and in large scaffolds (>100 kb) of Glyma0 were extracted in the read2Marker program with default parameters.40 Primer pairs to amplify the core motifs were designed in Primer3 software.41 Three types of primer pairs with different amplicon sizes (small, 80–210 bp; medium, 211–340 bp; large, 341–500 bp) were designed; the parameter settings were Opt_Tm = 60, Min_Tm = 53, Max_Tm = 70 and Max_Poly_X = 3. Sequences of the primer pairs were searched against Glyma1.01 to determine the number of binding sites, amplicon sizes and locations in Genome tester software42 with default parameters until a single amplicon was obtained (Supplementary Table S2). The SSR motifs that were consistent with BARC soybean potential SSR markers in SoyBase and EST-SSR markers43 are denoted in Supplementary Table S2.
Detection with fluorescently labelled primers: Primers were labelled with 6-FAM, HEX or NED fluorescent dyes. Multiplex PCR mixture (6 µl) contained DNA (10 ng/µl), 0.1 unit/µl of Taq DNA polymerase (Finnzymes), 1× Optimized Taq buffer, primer mix (0.02–0.2 µM each), 200 µM dNTPs and 1.6 M betaine. PCR was performed on a GeneAmp 9700 amplifier (Applied Biosystems, Foster City, USA) as follows: 1 cycle of initial denaturation at 95 °C for 2 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 2 min and extension at 72 °C for 1 min; and final extension at 72 °C for 3 min. The PCR product was diluted with water (1 : 10), and 1 µl was added to a mixture of Hi-Di formamide (10 µl) and GeneScan 400HD ROX size standard (0.2 µl). The sample was separated on an ABI3730 capillary sequencer (Applied Biosystems). PCR fragments detected simultaneously with three dyes were resolved in ABI GeneMapper v. 4.0 software (Applied Biosystems). To measure allelic size, relative fluorescent units and sizes (bp) of the highest stutter peaks of the 32 accessions were sorted by peak size rounded to integer numbers (Supplementary Table S3).
Detection with modified universal fluorescently labelled (UFL) primer: The UFL method44 was modified to attain multiplex amplification. Short polylinker sequences from the pBluescript vector were used as queries in blastn searches45 against Glyma1.01; the parameter settings were word_size = 7, evalue = 0.1 and perc_identity = 90. Three oligonucleotide tag sequences that did not show any hits in the soybean genome were used instead of the original M13 universal primer and were fluorescently labelled: 5′-CCACCGACGTGTCGCAC with 6-FAM, CCGTGCAGTCCGTCAGC with HEX and GGTGGCGACTCCTGGAG with NED (all dyes from Applied Biosystems). The concentrations of the oligonucleotide tag sequence (0.04 µM), 5′-tagged forward primer (0.04 µM) and unlabelled reverse primer (0.4 µM each) per marker were optimized to attain multiplex amplification. PCR conditions, detection of PCR fragments, genotyping and allele sizing were as described above.
Genotyping of the mapping population: PCR conditions were optimized to attain multiplex amplification for 12–18 fluorescently labelled SSR markers and 9–12 UFL SSR markers. Multiplex PCR mixture (3–5 µl) contained total DNA (50 ng/µl), 1× Multiplex PCR Mix (Qiagen), 1× Q solution and primer mix (described above). The following conditions were used: initial denaturation at 95 °C for 15 min; 18 cycles total of 94 °C for 30 s and 3 cycles each of 68, 66, 64, 62, 60 and 58 °C for 3 min; then 40 cycles of 30 s at 94 °C, 3 min at 55 °C and 1 min at 72 °C; and final extension at 72 °C for 10 min. The primer concentration for fluorescently labelled SSR markers can be reduced to 1/10. Genotyping conditions were as for SSR marker detection.
2.6. SNP marker analysis
Sequence-tagged sites containing SNP information developed46 were used as queries in blastn searches against Glyma1.01 with default parameters. Multiplex assays for 1,000 randomly selected SNPs distributed throughout the genome (Supplementary Table S3) were designed to amplify low-copy sequences in Sequenom Assay Design 3.1 software (Sequenom). The Sequenom MassARRAY system47 was used for SNP genotyping. Multiplex PCR followed by template-directed single base extension at each SNP site was conducted with a MassARRAY iPLEX Gold kit (Sequenom) following the manufacturer‘s protocol. The genotypes were determined in MassARRAY Typer 4.0 software (Sequenom).
2.7. Linkage map construction and QTL detection
The linkage maps of F2 and BC3F2 populations were constructed by using JoinMap v. 4.0 software.48 The logarithm of odds (LOD) threshold for grouping of DNA markers was 4.0. The marker order was determined using the maximum likelihood mapping algorithm. The recombination frequency was converted into genetic distance (cM) using the Haldane mapping function. Marker genotypes were examined by eye to check whether the pattern of marker segregation changed gradually at each round of map construction in order to ensure the correct marker order. When a discrepancy between the marker order and chromosome assembly was found, additional markers were integrated into the position to confirm the discrepancies. QTLs were analyzed by composite interval mapping implemented in the R/qtl package49 with threshold values (P < 0.1) that were calculated by the 1,000-permutation test for each trait. The detected peak positions were used for the ‘refineqtl’ and ‘fitqtl’ functions to estimate the maximum-likelihood position for each QTL model, the effects of each QTL and genetic variance. The phenotypic values of CSSLs and Enrei were evaluated in 2011 and 2012 using the Tukey-Kramer test in R v. 3.3.1 software50 with significance level, P < 0.01.
2.8. Comparison of the linkage map with the chromosome assembly
The positions of primer sequences of mapped markers on Glyma1.01 and Wm82.a2.v1 were estimated by similarity searches using Genome tester and/or blastn described above. The first base position of either forward or reverse primer sequences on the coordinates of the chromosome assembly and the size and number of expected PCR products are listed in Supplementary Table S3. When multiple polymorphic fragments are amplified by a primer set, we added the 0.1, 0.2, 0.3 suffixes after name of the marker mapped to different linkage groups. In the comparison of the linkage map with the chromosome assembly, we omitted markers being far from the expected position based on information of surrounding markers and then used only information of the marker position of chromosome assembly which marker designed in Glyma1.01 and Wm82.a2.v1. Thirty-one public SSR markers for which only information on primer sequences but difficult to locate precise position on chromosome assemblies were not included. The Marey map approach51 was used to visualize the corresponding positions between the linkage map and the genome assemblies. Genetic positions of the markers were interpolated using the cubic spline method with default parameters settings in MareyMap version 1.3.3.52
3. Results and discussion
3.1. DNA markers
SSR markers has been used in MAS because of multi allelic behavior, easy handling and low cost. Information on their polymorphism, genotyping quality and genetic positions is useful for marker selection. We identified 171,915 SSR loci (62,739 di-, 41, 696 tri-, 154 tetranucleotide and 67,326 compound loci) in the soybean reference sequence Glyma1.01.1 Because extended regions in the soybean genome are duplicated as a result of ancient polyploidization,1 novel primer pairs for 148,569 SSR loci were designed to amplify single PCR products to avoid analytical complexity (Supplementary Table S2). Polymorphisms of 2,235 SSR loci and that of previously developed 982 SNP markers46 were evaluated in 32 soybean germplasms (Supplementary Table S3). Sizes of amplified PCR product containing SSRs and nucleotides for SNPs, the number of PCR products, polymorphic information content and genetic positions estimated from the high-density linkage map are listed in Supplementary Tables S2 and S3.
3.2. High-density linkage map
A high-density linkage map was constructed by using a single F2 mapping population. Genomic SSR markers,4,5 EST-SSR markers43 and the new SSR markers described above were incorporated into the map to cover the entire genome. The map spans 2885.7 cM and contains 1,667 SSR and 510 SNP markers (Fig. 2 and Supplementary Fig. S2, left white bars); the average marker distance is 1.3 cM and maximum distance is 6.8 cM. Severe segregation distortion (P ≤ 0.001) was observed for the markers on Gm06 (115.5–118.4 cM, corresponding to 46.7–47.3 Mbp) and Gm11 (116.3–119.3 cM, 36.1–36.2 Mbp) (Fig. 2, blue lines). In these regions, the frequency of Peking alleles was less than that of Enrei alleles.
Figure 2.

A high-density genetic linkage map between Enrei and Peking (left) aligned with a physical map of Williams 82 (Glyma1.01) (right), with marker locations connected by black lines. The regions with suppressed recombination, shown on the same scale on the right, are connected with the linkage map by orange lines and may correspond to the pericentromeric regions. Red trapezoids indicate discrepancies between genetic and physical maps; blue bars indicate regions with severe segregation distortion. The discrepancies that have been resolved in the genome assembly Wm82.a2.v1 are indicated by red dot trapezoids.
We compared marker locations on the linkage map (Fig. 2, left white bars) with those on Glyma1.01 (Fig. 2, right black bars) and Wm82.a2.v1 assemblies (Supplementary Table S3). Because important flowering- and yield-related QTLs in the region of Gm11 (11.0–14.8 Mb) of Glyma1.01 assembly are absent on Chr. 11 of Wm82.a2.v1, we discussed based on the Glyma1.01 assembly. The corresponding region was found to be included in two unassigned scaffolds of Wm82.a2.v1 assembly, scaffold_21 and scaffold_32 (Supplementary Table S3). In total, 2,155 of 2,177 markers (99%) were anchored on Glyma1.01. The short-range order of most markers on the linkage map closely agreed with that of their physical positions in the genome assembly, but a wide range of regions differed at the distal ends of chromosomes. For example, reverse marker order on Gm05, Gm11, Gm13, Gm14 and Gm19 was observed at the distal ends of the corresponding linkage groups (Fig. 2, red rectangles). In particular, the orientation of the top of Gm13 (14.8–15.6 Mb) containing nucleolar organizer region (NOR)53 was reversed in our map. Similarly, by using fluorescent CentGm probes, the reverse orientation of this region was previously detected in a cytogenetic study of Peking.54 These large discrepancies have been resolved in Wm82.a2.v1,6 although some differences, especially on Chr. 05 (8.6–22.0 Mb) and Chr. 11 (11.1–28.0 Mb), remain. As for Chr. 11, the two unassigned scaffolds described above are not integrated into the two high density linkage maps,6 because no SNP marker to anchor the scaffolds is not available. In contrast, order of SNP markers in the region of Chr. 05 are consistent among the two maps, therefore, the discrepancy with information of the present map might reflect the genomic differences in different accessions. Thus, the differences in the present map provides information to identify regions in the genome assembly where additional information is required to resolve marker order and assign remaining scaffolds.
3.3. Relationships between physical and genetic distances
Integration of markers that have been used for MAS and QTL mapping by breeders and researchers into the high-density linkage map allowed us to determine relative genetic and physical relationships through marker positions. Corresponding position between the linkage map and the genome assembly was visualized by the Marey map approach.51 The corresponding physical distance to genetic distance was ∼360 kb/cM, assuming a genome size of ∼1.1 Gb, although positional biases (50 kb/cM–7 Mb/cM) were found. The ratio of physical to genetic distance varied considerably depending on the chromosomal region, and looked like a sigmoid curve. For example, the ratio in the middle regions of Gm06 (100–110 cM) and Gm11 (94–103 cM) was >10 times that in the other regions (Fig. 3). Such regions with highly suppressed recombination flanking the centromeres are termed the pericentromeres. In each chromosome, highly suppressed recombination between markers was observed in the pericentromere, but the extent of suppression differed among chromosomes (Fig. 2). A pair of markers in such regions would provide insufficient information on recombination for genetic mapping even if they are located physically far from each other in the reference sequence. Therefore, information on recombination frequency across the genome is useful for genome-wide association study and for marker choice for MAS and QTL mapping. We estimated the genetic positions of all markers from the Marey map (Supplementary Tables S2 and S3). The choice of markers based on their genetic positions rather than their physical positions in the reference sequences would reduce the cost of MAS for breeding and QTL mapping.
Figure 3.

Marey maps51 for chromosomes Gm06 and Gm11. Small dots indicate marker locations. Black boxes indicate the locations of the centromere repeat sequences. The recombination rate apparently decreases in the pericentromeric region. Arrow indicates the location of the E1 gene.
Surprisingly, low-recombination regions covered ∼555 Mb (∼60%) of the published soybean genome sequence and sometimes they formed patches in euchromatic regions (e.g. on Gm11 76.5–78.6 cM; Fig. 3). Interestingly, the distribution of low-recombination regions in all chromosomes coincided with the abundance of long terminal repeat retrotransposons reported in soybean.55 Gm07 and Gm16 had more than one apparent peak of suppressed recombination in addition to the pericentromeric regions.
3.4. QTLs in F2 population
High-density genetic linkage maps have made it possible to genetically dissect flowering time differences between parents, and thus to better understand the genetic basis for soybean flowering by comparing previously reported flowering- and/or maturity-related QTLs. Both parents belong to soybean maturity group IV56; DFF was 48.2 for Enrei and 60.7 for Peking. The F2 population showed transgressive segregation (Supplementary Fig. S3), and the broad-sense heritability of DFF was 75.5%. Seven QTLs (qDFF, Table 2) for DFF were identified. Both parents had alleles that accelerated flowering; Enrei had such alleles on Gm10, Gm12, Gm16 and Gm19 and Peking on Gm06, Gm11 and Gm13. Despite the high genetic complexity of the F2 population, almost all phenotypic variation in DFF was explained (91.4%) by the four major QTLs (on Gm06, Gm10, Gm12 and Gm19) and three minor QTLs (on Gm11, Gm13, Gm16). Only qDFF_Gm06 was located close to the low-recombination region. In F2 population, 19 out of 21 QTLs (expect for qDFF_Gm13 and qPH_Gm10) showed higher LOD score than 1% significant level threshold value. Surprisingly, most QTLs for other traits (PH, NP, SWH, DH and RP) were clustered with QTLs for DFF (Table 2), suggesting two possibilities that the latter have pleiotropic effects on other traits or genes for these traits are clustered together.
Table 2.
QTLs detected in F2 and BC3F2 populations
| QTLs detected in F2 populationa |
QTLs detected in BC3F2 populationa |
||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trait | QTL name | QTL positiona | Flanking marker 1 | Flanking marker 2 | Closest DNA marker | LOD | PVE (%) | Additive effect of Peking allele | s.d. | Dominance effect | s.d. | QTL positionb | Flanking marker 1 | Flanking marker 2 | Closest DNA marker | LOD | PVE (%) | Additive effect of Peking allele | s.d. | Dominance effect | s.d. | Confirmation of QTL with CSSLc | Confirmed QTLs between F2 and BC3 F2 population | Candidate genes or related QTLs | ID of related QTLs reported in Soybase | References | |
| DFF | qDFF_Gm19 | Gm19@118.9 | FT3-751 | C19-BARC- 018175-02533 | FT3-751 | 51.2** | 23.9 | 4.9 | 0.2 | 1.6 | 0.3 | Gm19@106.0 | Sat_286 | FT3-751 | FT3-751 | 174.8** | 29.5 | 3.3 | 0.1 | 0.5 | 0.2 | Confirmed | Confirmed | Dt1/E3 | Liu et al. 2009, Watanabe et al. 2009 | ||
| qDFF_Gm06 | Gm06@102.0 | sF3H | C06-BARC- 041345-07969 | C06-BARC- 041345-07969 | 57.5** | 29.6 | –5.5 | 0.2 | 2.6 | 0.4 | Gm06@106.4 | sF3H | FT1SSR9 | FT1SSR9 | 145.1** | 22.7 | –3.2 | 0.1 | 1.2 | 0.2 | Confirmed | Confirmed | E1 | Xia et al. 2012 | |||
| qDFF_Gm12 | Gm12@33.0 | C12-BARC- 015603-02006 | s024200450 | s024200450 | 36.3** | 13.6 | 3.7 | 0.2 | 0.3 | 0.4 | Gm12@49.0 | s024200450 | s023500372 | s024200450 | 74.5** | 9.8 | 2.0 | 0.1 | –0.1 | 0.2 | Confirmed | Confirmed | qDFF_Gm12 | First flower 25–2 | Kuroda et al. 2013 | ||
| qDFF_Gm10 | Gm10@116.0 | C10-BARC- 015003-01948 | C10-BARC- 015925-02017 | C10-BARC- 015925-02017 | 37.4** | 14.3 | 4.0 | 0.2 | 1.2 | 0.4 | Gm10@103.0 | Satt331 | FT2-300H01d | FT2-300H01d | 70.7** | 9.2 | 1.9 | 0.1 | 0.2 | 0.2 | Confirmed | Confirmed | E2 | Watanabe et al. 2011 | |||
| qDFF_Gm11 | Gm11@71.0 | s008000014-2 | T001111280m | T001111280m | 18.9** | 5.6 | –2.4 | 0.2 | 0.0 | 0.4 | Gm11@61.5 | GMES0766 | T001111280m | T001111280m | 38.7** | 4.7 | –1.2 | 0.1 | –0.2 | 0.2 | Confirmed(partially) | Confirmed | qDFF_Gm11 | First flower 8–4 | Yamanaka et al. 2001 | ||
| qDFF_Gm16 | Gm16@33.0 | C16-BARC- 020505-04644 | GMES0775 | GMES0775 | 10.3** | 2.7 | 1.7 | 0.2 | 0.8 | 0.4 | Gm16@68.5 | GMES4751 | s026800043 | GMES4751 | 16.9** | 1.9 | –0.9 | 0.1 | –0.2 | 0.2 | Confirmed | Confirmed | GmFT5a | Takeshima et al. 2016 | |||
| qDFF_Gm13 | Gm13@7.0 | s000702358-2 | s000701869-2 | s000701869-2 | 6.8* | 1.7 | –1.5 | 0.3 | –0.1 | 0.4 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Confirmed | - | qDFF_Gm13 | First flower 11–4 | Gai et al. 2007 | ||
| DFT | qDFT_Gm19_1d | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm19@102.0 | Sat_286 | FT3-751 | FT3-751 | 285.3** | 55.3 | 8.8 | 0.2 | 0.0 | 0.4 | Confirmed | NA | Dt1 | Liu et al. 2009 | ||
| qDFT_Gm19_2d | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm19@106.0 | Sat_286 | FT3-751 | FT3-751 | 193.8** | 37.9 | 4.8 | 0.2 | 2.8 | 0.3 | Confirmed | NA | E3 | Watanabe et al. 2009 | |||
| qDFT_Gm06 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm06@105.6 | Satt277 | sF3H | sF3H | 78.8** | 11.5 | –3.5 | 0.2 | 1.1 | 0.3 | Confirmed | NA | E1 | “ | |||
| qDFT_Gm10 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm10@103.0 | Satt331 | FT2-300H01d | FT2-300H01d | 74.5** | 10.7 | 3.1 | 0.2 | 0.1 | 0.3 | Confirmed | NA | E2 | “ | |||
| qDFT_Gm12 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm12@49.0 | s024200450 | s023500372 | s024200450 | 68.5** | 9.7 | 3.0 | 0.2 | –0.2 | 0.3 | Confirmed | NA | qDFF_Gm12 | “ | |||
| qDFT_Gm11 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm11@60.0 | GMES0766 | T001111280m | T001111280m | 35.3** | 4.6 | –2.1 | 0.2 | 0.4 | 0.3 | Confirmed | NA | qDFF_Gm11 | “ | |||
| FP | qFP_Gm19_1d | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm19@100.0 | Sat_286 | FT3-751 | FT3-751 | 242.4** | 67.4 | 5.4 | 0.1 | –0.2 | 0.3 | Confirmed(partially) | NA | Dt1 | “ | ||
| qFP_Gm19_2d | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm19@102.0 | Sat_286 | FT3-751 | FT3-751 | 131.9** | 45.8 | 2.7 | 0.2 | 2.6 | 0.3 | Confirmed(partially) | NA | E3 | “ | |||
| DH | qDH_Gm19 | Gm19@118.0 | Satt229 | FT3-751 | FT3-751 | 22.4** | 14.2 | 8.1 | 0.7 | 2.5 | 1.0 | Gm19@105.0 | Sat_286 | FT3-751 | FT3-751 | 39.7** | 12.1 | 6.1 | 0.5 | –0.6 | 0.9 | Confirmed | Confirmed | Dt1/E3 | “ | ||
| qDH_Gm06 | Gm06@102.0 | sF3H | C06-BARC- 041345-07969 | C06-BARC- 041345-07969 | 34.8** | 26.2 | –11.2 | 0.7 | 2.1 | 1.0 | Gm06@104.4 | Satt277 | sF3H | sF3H | 30.6** | 9.1 | –5.4 | 0.5 | 0.8 | 0.9 | Confirmed | Confirmed | E1 | “ | |||
| qDH_Gm11 | Gm11@71.0 | s008000014-2 | T001111280m | T001111280m | 29.9** | 21.1 | –10.0 | 0.7 | 0.0 | 1.0 | Gm11@41.0 | GMES0045 | Satt197 | Satt197 | 18.8** | 5.5 | –4.3 | 0.5 | 0.5 | 1.1 | Confirmed | Confirmed | qDFF_Gm11 | Reproductive stage length 8–1 | Komatsu et al. 2012 | ||
| qDH_Gm10 | Gm10@119.0 | FT2-300H01d | s005205820-2 | s005205820-2 | 17.6** | 10.5 | 7.3 | 0.8 | 1.9 | 1.0 | Gm10@103.0 | Satt331 | FT2-300H01d | FT2-300H01d | 13.3** | 3.8 | 3.4 | 0.5 | –0.4 | 0.9 | Confirmed | Confirmed | E2 | “ | |||
| qDH_Gm12 | Gm12@33.5 | s024200450 | s024200389 | s024200450 | 16.4** | 9.6 | 6.4 | 0.7 | 1.3 | 1.0 | Gm12@49.0 | s024200450 | s023500372 | s024200450 | 11.8** | 3.4 | 3.0 | 0.5 | 0.0 | 0.9 | Confirmed | Confirmed | qDFF_Gm12 | “ | |||
| qDH_Gm02 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm02@48.0 | Satt701 | Satt296 | Satt701 | 8.2** | 2.3 | 2.3 | 0.5 | 1.6 | 1.1 | –– | –– | qRP_Gm02 | Pod maturity 22–4 | Reinprecht et al. 2006 | ||
| qDH_Gm13 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm13@125.0 | Satt362 | s003004926-2 | Satt362 | 5.2** | 1.5 | –2.3 | 0.5 | 2.7 | 1.0 | Confirmed | –– | qRP_Gm13 | Novel | |||
| qDH_Gm17 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm17@46.0 | T001709449l | T001710954m | T001709449l | 5.0* | 1.4 | –2.5 | 0.6 | 1.3 | 1.0 | Confirmed | –– | qRP_Gm17 | Reproductive period 1–7 | Wang et al. 2015 (position is slightly different) | ||
| qDH_Gm01 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm01@43.8 | Satt221 | Satt370 | Satt221 | 4.6* | 1.3 | 2.2 | 0.5 | –2.2 | 0.9 | –– | –– | qRP_Gm01 | Pod maturity 1–2 | Keim et al. 1990B | ||
| RP | qRP_Gm11 | Gm11@70.0 | s008000014-2 | T001111280m | s008000014-2 | 16.4** | 27.4 | –7.0 | 0.7 | 0.1 | 1.1 | Gm11@38.0 | GMES0045 | Satt197 | GMES0045 | 12.3** | 4.6 | –3.4 | 0.5 | 0.2 | 1.1 | Confirmed | Confirmed | qDFF_Gm11 | Reproductive stage length 8–1 | Komatsu et al. 2012 | |
| qRP_Gm02 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm02@49.0 | Satt701 | Satt296 | Satt701 | 9.3** | 3.5 | 2.6 | 0.6 | 1.2 | 1.1 | –– | –– | qRP_Gm02 | “ | |||
| qRP_Gm13 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm13@124.0 | Satt362 | s003004926-2 | Satt362 | 7.5** | 2.8 | –2.6 | 0.5 | 2.8 | 1.0 | Confirmed | –– | qRP_Gm13 | Novel | |||
| qRP_Gm19 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm19@105.0 | Sat_286 | FT3-751 | FT3-751 | 7.2** | 2.7 | 2.7 | 0.5 | –1.1 | 0.9 | Confirmed | –– | Dt1/E3 | “ | |||
| qRP_Gm06 | Gm06@98.4 | Satt277 | C06-BARC- 014491-01562 | Satt277 | 10.9** | 16.9 | –5.4 | 0.7 | –0.9 | 1.1 | Gm06@104.4 | Satt277 | sF3H | sF3H | 6.3** | 2.3 | –2.2 | 0.5 | –0.1 | 0.9 | Confirmed | Confirmed | E1 | “ | |||
| qRP_Gm16 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm16@54.5 | Satt596 | s014300091-2 | Satt596 | 5.7** | 2.1 | 2.3 | 0.5 | –0.5 | 1.0 | –– | –– | GmFT5a | “ | |||
| qRP_Gm17 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm17@46.0 | T001709449l | T001710954m | T001709449l | 4.5** | 1.6 | –2.3 | 0.5 | 1.1 | 0.9 | Confirmed | –– | qRP_Gm17 | “ | |||
| qRP_Gm01 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm01@44.8 | Satt221 | Satt370 | Satt221 | 4.4** | 1.6 | 2.1 | 0.5 | –2.0 | 0.9 | –– | –– | qRP_Gm01 | “ | |||
| PH | qPH_Gm19_1d | Gm19@114.0 | Satt664 | C19-BARC- 030101-06809 | Satt664 | 30.4** | 42.7 | 15.5 | 1.1 | 0.5 | 1.6 | Gm19@101.0 | Sat_286 | FT3-751 | FT3-751 | 129.8** | 45.7 | 14.4 | 0.5 | –3.5 | 1.0 | Confirmed | Confirmed | Dt1 | “ | ||
| qPH_Gm19_2d | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm19@102.0 | Sat_286 | FT3-751 | FT3-751 | 78.7** | 27.6 | 7.0 | 0.6 | 7.2 | 1.1 | Confirmed | –– | E3 | “ | |||
| qPH_Gm10 | Gm10@117.2 | FT2-300H01d | s005205820-2 | FT2-300H01d | 6.3* | 6.4 | 5.8 | 1.1 | 4.1 | 1.6 | Gm10@102.0 | Satt331 | FT2-300H01d | FT2-300H01d | 17.4** | 5.3 | 4.0 | 0.5 | –0.4 | 0.9 | Confirmed | Confirmed | E2 | “ | |||
| qPH_Gm06 | Gm06@103.0 | s005700017 | s007100484 | s005700017 | 16.6** | 19.3 | –10.5 | 1.1 | 3.8 | 1.6 | Gm06@106.4 | sF3H | FT1SSR9 | FT1SSR9 | 15.9** | 4.8 | –3.8 | 0.5 | 1.1 | 0.9 | Confirmed | Confirmed | E1 | “ | |||
| NP | qNP_Gm19 | Gm19@116.0 | C19-BARC- 030101-06809 | Satt229 | C19-BARC- 030101-06809 | 15.0** | 26.4 | 84.4 | 9.7 | 17.0 | ## | Gm19@101.0 | Sat_286 | FT3-751 | FT3-751 | 148.2** | 39.3 | 80.0 | 3.5 | 30.0 | 6.8 | Confirmed | Confirmed | Dt1/E3 | “ | ||
| qNP_Gm12 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm12@50.0 | s024200450 | s023500372 | s024200450 | 33.9** | 6.8 | 31.6 | 3.0 | 6.7 | 6.0 | Confirmed | –– | qDFF_Gm12 | “ | |||
| qNP_Gm10 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm10@109.0 | FT2-300H01d | T001045537m | FT2-300H01d | 27.7** | 5.4 | 28.3 | 3.1 | 5.3 | 5.6 | –– | –– | E2 | “ | |||
| qNP_Gm06 | Gm06@103.0 | s005700017 | s007100484 | s005700017 | 11.5** | 19.3 | −73.0 | 9.5 | 9.2 | ## | Gm06@104.4 | Satt277 | sF3H | sF3H | 23.5** | 4.6 | –29.7 | 3.2 | 5.1 | 5.8 | Confirmed | Confirmed | E1 | “ | |||
| qNP_Gm11 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm11@61.5 | GMES0766 | T001111280m | T001111280m | 15.3** | 2.9 | –21.2 | 2.8 | 1.7 | 5.4 | Confirmed | –– | qDFF_Gm11 | “ | |||
| qNP_Gm16 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm16@69.5 | GMES4751 | s026800043 | s026800043 | 7.4* | 1.4 | –18.5 | 3.2 | 16.3 | 6.1 | –– | –– | GmFT5a | “ | |||
| TSW | qTSW_Gm19 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm19@102.0 | Sat_286 | FT3-751 | FT3-751 | 155.5** | 37.3 | 37.9 | 1.7 | 18.3 | 3.2 | –– | –– | Dt1/E3 | “ | ||
| qTSW_Gm12 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm12@50.0 | s024200450 | s023500372 | s024200450 | 44.2** | 8.0 | 18.1 | 1.5 | 1.7 | 2.9 | Confirmed | –– | qDFF_Gm12 | “ | |||
| qTSW_Gm11 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm11@61.0 | GMES0766 | T001111280m | T001111280m | 36.2** | 6.5 | –16.3 | 1.4 | 1.4 | 2.6 | Confirmed | –– | qDFF_Gm11 | “ | |||
| qTSW_Gm06 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm06@105.4 | Satt277 | sF3H | sF3H | 35.7** | 6.4 | –17.4 | 1.5 | 3.2 | 2.6 | Confirmed | –– | E1 | “ | |||
| qTSW_Gm10 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm10@110.0 | FT2-300H01d | T001045537m | FT2-300H01d | 20.9** | 3.6 | 10.7 | 1.5 | 5.2 | 2.8 | Confirmed | –– | E2 | “ | |||
| qTSW_Gm16 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm16@68.5 | GMES4751 | s026800043 | GMES4751 | 8.7** | 1.5 | –9.7 | 1.5 | 9.4 | 2.9 | Confirmed | –– | GmFT5a | “ | |||
| qTSW_Gm20 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Gm20@99.0 | T002042675s | Sct_189 | T002042675s | 6.8** | 1.1 | –9.4 | 2.0 | 1.5 | 3.9 | Confirmed | –– | qTSW_Gm20 | Seed yield 15–15 | Kabelka et al. 2004 | ||
| SWH | qSWH_Gm08 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm08@46.0 | GMES1620 | AY262686B | AY262686B | 33.3** | 10.1 | –2.8 | 0.2 | 1.9 | 0.4 | Confirmed | –– | qSWH_Gm08 | Seed weight 4–5 | Maughan et al. 1996 (close to I locus) | |
| qSWH_Gm06 | Gm06@98.4 | Satt277 | C06-BARC- 014491-01562 | Satt277 | 8.9** | 17.1 | –2.4 | 0.4 | 1.7 | 0.5 | Gm06@106.4 | sF3H | FT1SSR9 | FT1SSR9 | 29.1** | 8.8 | –2.3 | 0.2 | 1.1 | 0.4 | Confirmed(partially) | Confirmed | E1 | “ | |||
| qSWH_Gm11 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm11@59.0 | GMES0766 | T001111280m | T001111280m | 22.3** | 6.6 | –1.8 | 0.2 | 0.0 | 0.4 | Confirmed | –– | qDFF_Gm11 | “ | |||
| qSWH_Gm17 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm17@51.0 | T001709449l | T001710954m | T001710954m | 15.1** | 4.4 | –2.0 | 0.3 | 0.5 | 0.5 | Confirmed | –– | qSWH_Gm17 | Seed yield 5-2 | Renyna and Sneller 2001 | ||
| qSWH_Gm19 | Gm19@118.9 | FT3-751 | C19-BARC- 018175-02533 | FT3-751 | 8.4** | 15.9 | 2.4 | 0.4 | 1.1 | 0.5 | Gm19@107.1 | Sat_286 | FT3-751 | FT3-751 | 10.7** | 3.1 | 1.1 | 0.2 | 0.1 | 0.3 | Confirmed | Confirmed | Dt1/E3 | “ | |||
| qSWH_Gm20 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm20@98.0 | T002042675s | Sct_189 | T002042675s | 6.6** | 1.9 | –1.5 | 0.3 | 1.1 | 0.5 | Confirmed(partially) | –– | qSWH_Gm20 | “ | |||
| qSWH_Gm13 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm13@16.0 | Satt146 | Satt252 | Satt252 | 6.6** | 1.9 | –1.1 | 0.2 | 0.0 | 0.5 | Confirmed(partially) | –– | qDFF_Gm13 | “ | |||
| qSWH_Gm02 | –– | –– | –– | –– | –– | –– | –– | –– | –– | –– | Gm02@22.0 | Sat_096 | Sat_351 | Sat_351 | 5.9** | 1.7 | –0.9 | 0.2 | –0.4 | 0.5 | Confirmed | –– | qSWH_Gm02 | “ | |||
NA, the trait was not evaluated in the F2 population; LOD, logarithm of odds of the presence of a QTL; PVE, percentage of variance explained by the QTL.
Sowing date of F2 population and BC3F3 population were 30 May 2007 and 24 June2011, respectively.
Indicated as chromosome name and position in cM separated by @.
‘Confirmed partially’ indicates that a singificant difference was observed between Enrei and a CSSL habouring several donor choromosomal fragments.
Effects of the QTLs corresponding to E3 and Dt1 loci were calculated separately.
Significant at P < 0.05 after 1,000 permutation; **Significant at P < 0.01 after 1,000 permutation.
Our high-density linkage map offers an opportunity to explore candidate genes for flowering time and related QTLs. For instance, the physical location of two major QTLs, qDFF_Gm19 and qDFF_Gm10, estimated from the Marey map was 47.6 Mb and 44.9 Mb, respectively. Genes responsible for E357 and E258 are located at ∼47.5 Mb on Gm19 and ∼44.7 Mb on Gm10, respectively. The estimated physical locations of the two QTLs differ from those of E2 and E3 by only ∼200 kb. Thus, the positions of SSR markers and interpolated genetic distance provide great potential to narrow down the boundaries of genomic regions that include candidate genes.
3.5. QTLs in BC3F2 population
Nine agronomic traits (Table 1) of 999 BC3F2 individuals were evaluated and the average values of each trait was compared with those of the recurrent parent (Enrei); the average value of each trait of BC3F2 were almost the same as those of Enrei (Supplementary Table S4, Fig. S4). Whereas the effects of Peking alleles increase the phenotypic variance observed in BC3F2. Among the genetic variances (heritability) of all traits, some traits such as DFF, DFT and PH were highly heritable, whereas heritability of yield-related traits, such as TSW and SWH, were intermediate, and that of DH and RP was very low (Table 1). Enrei tends to suffer from green stem disorder, caused by biotic and abiotic stresses59; this disorder, which delays harvesting, would broaden the range of the values of maturity-related traits of the recurrent parent and the BC3F2 population and decrease the heritability of these traits.
The linkage map was constructed for the BC3F2 population to perform QTL mapping for these nine traits. The genotype of each BC3F2 line was determined by using 320 SSR markers selected from the high-density genetic linkage map described above, so that they covered the whole genome (Supplementary Fig. S5). The ratios of Peking-homozygous (3.1%), heterozygous (6.3%) and Enrei-homozygous (90.6%) loci in the population coincided well with the theoretical ratios (3.3%, 6.0% and 89.1%, respectively). The map consisted of 20 linkage groups covering 2475.3 cM with an average interval of 8.3 cM, and showed a genetic order of DNA markers similar to that expected from the high-density linkage map of the F2 population.
In total, 56 QTLs scattered over 12 chromosomes were identified from the positions of LOD peaks for the nine traits. Six QTLs for DFF, six for DFT, two for FP, nine for DH, eight for RP, four for PH, six for NP, seven for TSW and eight for SWH showed significant LOD scores. Among them, 53 QTLs (expect for qDH_Gm01, qDH_Gm17 and qNP_Gm16) showed higher LOD score than 1% significant level of threshold value (Table 2). Six out of seven QTLs for DFF detected in the F2 population were reproducible in the BC3F2 population. As in the F2 population, many QTLs for other traits were clustered with QTLs for DFF, suggesting that QTLs for flowering time strongly affect other traits. However, additional QTLs for maturity and yield traits were separate from DFF QTLs in the BC3F2 population.
We previously characterized the sequences of the known flowering genes, E1–E4, in many soybean cultivars36; on this information, Enrei has E1, e2, e3 and E4 alleles, and Peking has E1, E2, E3 and E4 alleles. Therefore, we expected that only the E2 and E3 loci would segregate in the population; however, qDFF_Gm06 was identified in a region close to the E1 locus (Table 2). Allelic effects of qDFFs agreed well with the results of QTL analysis in the F2 population. In addition, two soybean orthologs of Arabidopsis Flowering locus T (FT), GmFT2a60 and GmFT5a61 delay flowering in cultivars adapted to northern Japan. The location of qDFF_Gm16 was very close to the GmFT5a locus, and the Peking allele delayed flowering.
A major QTL for DFT, qDFT_Gm19, was identified close to the Dt1 locus. The Peking allele significantly delayed flowering of the top raceme. The dominant allele of Dt1, which is a homolog of Arabidopsis TERMINAL FLOWER 1, controls indeterminate growth habit in soybean.62 The growth habit of Peking is closer to that of a semi-determinate isoline (Dt1, Dt2) than to that of a determinate isoline (dt1, dt2) in top-leaf and stem traits, but the Peking allele is controlled by dt1-t, which is allelic to dt1.63 Together with the semi-determinate growth of Peking, delaying flowering of the top raceme will increase both the number of nodes on the main stem and plant height. The corresponding traits, NP and PH, were controlled by QTLs detected near the Dt1 locus (Fig. 4). The genetic variance of DFT and PH explained by the Dt1 locus accounted up to 55.3% and 45.7%, respectively, of total variance (Table 2), indicating that the Dt1 locus strongly affects these traits.
Figure 4.

Whole-chromosome view of the graphical genotypes of the CSSLs and locations of QTLs identified in the BC3F2 population. Marker genotypes: black, Peking homozygous; white, Enrei homozygous; gray, heterozygous. QTL positions on the right side are in black.
The locations of QTLs for yield-related traits NP and TSW in the BC3F2 population were very similar to those of QTLs for DFF in the F2 population. The ratios of genetic variance explained by six flowering-time QTLs and the Dt1 locus were examined but the order of effects of qDFFs did not coincide with that of post-flowering including yield-related traits. For example, qRP_Gm11, detected at the same locus as qDFF_Gm11, had a larger additive effect on RP (−3.4 days) than on DFF (−1.2 days). Each QTL for DFF had a different effect on the phenotypic variance of other traits, likely reflecting functional differences between the underlying genes. Hence, dissection of these QTLs as single Mendelian factors would provide more precise information about their effects.
Four QTLs for SWH were identified on Gm02, Gm08, Gm17 and Gm20, where no QTLs for DFF were identified. None of the QTLs from small-seeded Peking increased SWH. The Peking allele of qSWH_Gm08 had the strongest effect of decreasing SWH (10% of phenotypic variance in the BC3F2 population explained). Because this QTL is close to Rhg4, an important locus for cyst nematode resistance of Peking,64 pre-breeding of an Rhg4 NIL to remove such an unfavourable Peking allele is important for breeding of large-seeded cultivars. Previous study65 identified a QTL (qSW17-1OA) with stable effects on seed weight in diverse environments over several years. The genetic position of qSWH_Gm17 is likely to be the same as that of qSW17-1OA and some other QTLs in previous study (Table 2). The locations of qSWH_Gm20 and qTSW_Gm20 were close to that of Ln, which controls leaflet shape and seed size.66 The gene responsible for Ln is homologous to Arabidopsis JAGGED.67 Although most Peking alleles had negative effects on yield-related traits in the Enrei genetic background, two QTLs related to late flowering (qDFF_Gm10, qDFF_Gm19) and one QTL extending the reproductive period (qRP_Gm19) associated with increased TSW in this genetic background (Table 2).
3.6. Development of CSSLs and confirmation of QTLs
The graphical genotypes of the selected 103 CSSLs are shown in Fig. 4. The average length (± standard deviation) of the donor chromosomal segment was 85.4 ± 42.1 cM for homozygous and 130.8 cM ± 59.6 cM for heterozygous alleles. A few donor segments other than the target segment remained in the genetic background of the recurrent parent; therefore, further backcrossing with MAS would be necessary to eliminate these extra segments. However, CSSLs harboring different donor chromosomal segments from Peking were still useful to dissect QTLs as single genetic factors and to evaluate the genetic effects of individual QTLs.
Two-year evaluations of nine agronomic traits using 103 CSSLs revealed that 50 of these lines had at least one trait that was significantly different from that of Enrei by the Tukey-Kramer test (P < 0.01). Most lines whose donor segments contained QTLs originating from Peking showed significantly different phenotypes. These differences agreed well with the effects of QTLs identified in the BC3F2 population (Supplementary Table S5). Of the 57 QTLs detected in the F2 and BC3F2 populations, 49 were also detected in CSSLs (Table 1). Therefore, we considered that majority of QTLs reported in this study are highly reliable. Interestingly, four CSSLs B0704, B0804, B0816 and B0879 had the same genotype as Enrei at all DFF QTLs but had a significantly (P <0.01) different DFF from Enrei (Supplementary Table S5). The results indicate that the effect of a novel QTL appeared in the simple genetic background of a CSSL but was undetectable in the BC3F2 population. In 2011 and 2012, 36 CSSLs showed significant differences in TSW from Enrei; 27 of them had QTLs for DFF, RP and SWH, but genetic factors in the remaining lines were not identified. Increasing the number of plants used for evaluation or crossing with Enrei would be necessary to uncover the cause of the increase in TSW and to use this QTL in soybean breeding programs.
We further evaluated the progeny of each line heterozygous for each of six flowering-time QTLs (E1, E2, E3, qDFF_Gm11, qDFF_Gm12 and GmFT5a) to evaluate whether the chromosomal segments from Peking contained flowering time QTLs. Progeny classification according to the genotypes of SSR markers near each QTL showed significant association between phenotypic values and QTL genotypes (data not shown). Representative graphical genotypes of two CSSLs harboring donor chromosomal segments that included qDFF_Gm11 (B0015 and B0676) and the effect of qDFF_Gm11 on the phenotype are shown in Fig. 5. Both lines differed significantly (P <0.01) from Enrei in RP (Fig. 5B). qDFF_Gm11 had the fifth strongest effect among seven DFF QTLs (Table 2), whereas qDFF_Gm11 had the strongest effect on RP among all RP QTLs. The decreased RP resulted in a clear difference in the maturity phenotype (Fig. 5C), and this QTL also affected TSW and SWH (Supplementary Table S4). Comparison of the effects of qDFF(qRP)_Gm11 on different traits suggests that the gene responsible for this QTL probably extends the pod-filling period. The results described above indicate that CSSLs developed in this study may help to dissect the genes underlying the detected QTLs into single genetic factors and provide breeding materials with the genetic background of an elite Japanese cultivar.
Figure 5.
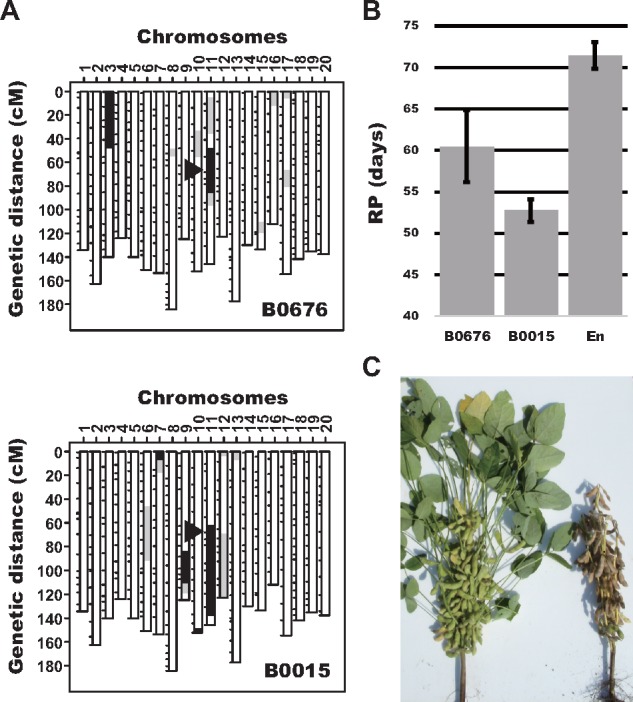
Confirmation of QTL effects on the reproductive period using CSSL lines. A, Graphical genotypes of the CSSLs B0676 and B0015. These lines have donor (Peking) chromosomal segments containing qDFF_Gm11, which affects the length of the reproductive period (arrowheads). B, Reproductive periods of the two CSSLs and the recurrent parent Enrei (En). C, Representative images of CSSL B0676 (right) in comparison with the recurrent parent Enrei (left). Early maturity in B0676 is caused by a chromosome segment that includes qDFF_Gm11 from Peking.
3.7. Novel flowering-related genes
We identified seven QTLs related to DFF in F2, BC3F2 populations and selected CSSLs (Table 2 and Supplementary Table S5). Among soybean maturity genes, the classical E1 locus has the strongest effect on flowering time.68 In response to photoperiod, a putative transcription factor encoded by E1 controls flowering time by regulating the expression of FT genes.69 The detection of qDFF_Gm06 close to E1 in the F2 and BC3F2 populations suggests that the E1 allele differs between Peking and Enrei. We assume that Peking has a recessive allele because of an early flowering effect of this QTL (Table 2). However, the coding region of Peking E1 is reported to be identical to that of Enrei E1.36 Thus, Peking may have a recessive allele different from the known one, because several different types of recessive alleles have been identified for soybean maturity genes, such as the E4 locus,70 and the promoter region of Peking has a high similarity to those of other cultivars with recessive e1 alleles.36 Alternatively, a novel gene affecting soybean flowering time and maturity may be located in this region. The qDFF_Gm06 was located in the pericentromeric region (Fig. 3). The other maturity gene E7 locus for flowering is located on the same chromosome and is genetically tightly linked to the E1 locus.71 Fine mapping would be needed to resolve these possibilities.
Genes responsible for qDFF_Gm11, qDFF_Gm12 and qDFF_Gm13 (and nearby QTLs for other traits) have not been characterized. QTLs corresponding to qDFF_Gm12 have been identified in two different populations derived from crosses between wild and cultivated soybeans.72 Satt442 (the closest DNA marker in the above study) was located close to the marker s024200450 in the present study. Previous study73 reported QTLs at positions similar to that of qDFF_Gm11. They identified a strong QTL, named qDfm1 (Duration from flowering to maturity), using RILs derived from a cross between Fukuyutaka and Ippon-sango in a low-latitude region of Japan (Kyushu, 32°52′N). Although the positions of qDFF_Gm11 and qDfm1 are close to each other on the linkage map, the effect of qDfm1 was not associated with DFF.73 The difference might be related to population size (192 F2 or 999 BC3F2 plants vs. 143 RILs) and a short photoperiod at low latitudes. In any case, both QTLs control RP under different photoperiod and temperature conditions (in the middle and southern parts of Japan). If the same gene is responsible for these QTLs, the Peking allele, which shortens RP, would be useful in wide latitudinal range. The evaluation of genetic interactions between E genes and qDFF_Gm11 by crossing CSSLs having different qDFF alleles would be important for controlling maturity.
3.8. Future characterization of useful traits in Peking
Peking was first identified as material resistant to the soybean cyst nematode H. glycines.74 Peking was introduced into the USA from Beijing, China, in 1906 (previous accession number: PI17852B; current USDA accession number: PI548402). However, other USDA accessions named Peking differ from PI548402 by RFLP analysis11: PI297543 (introduced from Hungary in 1964), PI438496 and PI438497 (from the Russian Federation in 1979). The Peking accession used in the present study revealed an SSR genotype patterns very similar to those of PI548402, but quite different from the patterns of PI438496A, PI438496B, PI438496C, PI438497 and two pure lines, PI548205 and PI548359, derived from Peking in the USDA germplasm collection, and from the pattern of JP28432 in NARO Genebank (data not shown).
DNA markers for the soybean cyst nematode resistance genes Rhg4 and Rhg1, based on functional SNPs in Peking, have been reported.64 Previous study75 isolated the Peking gene Rps1k, which promotes resistance to stem rot disease caused by Phytophthora sojae races 2 and 11.19 Peking has also been used in studies of interactions with nitrogen-fixing rhizobia (Rhizobium and Sinorhizobium species) owing to their host specificity and genetic control of symbiosis by a host gene.76 Peking carries rfg1, which controls effective nodulation of fast-growing rhizobia, and Rj4, which controls nodulation of slow-growing bradyrhizobia.77 The responsible genes were isolated for rfg178 and that for Rj4.79 Peking is also resistant to the reniform nematode Rotylenchulus reniformis.18
Peking has a Rsv4 gene for resistance to soybean mosaic virus and DNA markers have been developed for this gene.12 A recessive gene, rpv2, for resistance to peanut mottle virus has been reported in Peking.13 Peking is resistant to many isolates of the frogeye leaf spot pathogen, Cercospora sojina,15 which is controlled by a single dominant gene, RcsPeking, mapped near the SSR marker Satt244, and another resistance gene, Rcs3.80 Peking is tolerant to Pseudomonas syringae pv. glycinea race 6, which cause bacterial blight disease,14 and to soybean dwarf disease,16 soybean stem canker17 and corn earworm.81 Its seed-flooding tolerance at the germination stage has been explained by the structural characteristics of its seeds20,21 and is controlled by four QTLs.82 The success rate of somatic embryogenesis from immature embryo cultures of Peking is reportedly high,83 and six QTLs associated with somatic embryogenesis have been identified.84 Susceptibility of Peking to tumor formation by Agrobacterium tumefaciens is controlled by several genes,85 but these genes have not been isolated. The evaluation of CSSLs for resistance to these diseases and to stress would help to identify and characterize the resistance genes.
4. Conclusion
We developed a high-density linkage map and CSSLs carrying chromosomal segments from the Chinese soybean Peking in the background of the Japanese soybean Enrei. The map provides a detailed genetic framework within which to use molecular markers for breeding and to obtain a precise assembly of the genome sequence in the Japanese soybean genetic background. The marker order on the linkage map agreed well with the new genome assembly Wm82.a2.v1, but large differences were identified on Chrs. 05 and 11. These CSSLs are a unique resource that would be useful for evaluating minor QTLs as single genetic factors in a uniform genetic background. We demonstrated that many QTLs related to basic agronomic traits detected in the F2 and BC3F2 populations were reproducible in the CSSLs.
Peking has many useful genes for resistance to diseases, pests and stress, but most of these genes have not yet been characterized. CSSLs developed in this study would be a good resource for us in developing new cultivars harboring resistance genes and for gene isolation by positional cloning. To increase the genetic diversity of soybean for breeding, finding and confirmation of QTLs associated with agronomic traits using a mapping population derived from these lines would be necessary. CSSLs developed in this study would also be good starting materials for removing the unfavourable characteristics of Peking.
Supplementary Material
Acknowledgements
We thank Kazuhiro Yagasaki, Nagano Prefecture Vegetable and Ornamental Crops Experiment Station, for supplying soybean materials used in this study. We thank Koji Takahashi, Naohiro Yamada, Nobuhiko Oki, Kaori Hirata, Benitez Eduardo of Institute of Crop Science for their kind assistance for backcrossing. We also appreciate the technical support in field work from the staff of the National Institute of Agrobiological Sciences: T. Nobori, N. Karino, T. Ohmizu, T. Taguchi, Y. Tsubokura and K. Sugimoto. This work was supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation, DD-1010, SOY1002, SOY2003).
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at DNARES online.
References
- 1. Schmutz J., Cannon S.B., Schlueter J.. 2010, Genome sequence of the palaeopolyploid soybean, Nature, 463, 178–83. [DOI] [PubMed] [Google Scholar]
- 2. Zhou Z., Jiang Y., Wang Z., et al. 2015, Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean, Nat. Biotechnol., 33, 408–14. [DOI] [PubMed] [Google Scholar]
- 3. Tsuda M., Kaga A., Anai T., et al. 2015, Construction of a high-density mutant library in soybean and development of a mutant retrieval method using amplicon sequencing, BMC Genom., 16, 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cregan P.B., Jarvik T., Bush A.L., et al. 1999, An integrated genetic linkage map of the soybean genome, Crop Sci., 39, 1464–90. [Google Scholar]
- 5. Song Q., Jia G., Zhu Y., et al. 2010, Abundance of SSR Motifs and development of candidate polymorphic SSR Markers (BARCSOYSSR_1.0) in soybean, Crop Sci., 50, 1950. [Google Scholar]
- 6. Song Q.J., Jenkins J., Jia G.F., et al. 2016, Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1.01, BMC Genom., 17, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mahama A.A. 1999, Cytogenetic analysis of translocations in soybean, J. Heredity, 90, 648–53. [Google Scholar]
- 8. Findley S.D., Pappas A.L., Cui Y., Birchler J.A., Palmer R.G., Stacey G.. 2011, Fluorescence in situ hybridization-based karyotyping of soybean translocation lines, G3, 1, 117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimomura M., Kanamori H., Komatsu S., et al. 2015, The Glycine max cv. Enrei genome for improvement of Japanese soybean cultivars, Int. J. Genom., 2015, 358127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ross J., Brim C.A.. 1957, Resistance of soybean to the soybean-cyst nematode as determined by a double-row method, Plant Dis. Reporter, 41, 923–4. [Google Scholar]
- 11. Lorenzen L.L. 1994, Soybean cultivar development: a genome perspective. Ph.D. Dissertation, Iowa State University, Ames, IA. Available at: http://lib.dr.iastate.edu/cgi/viewcontent.cgi?article=11625&context=rtd (1 November 2017, date last accessed).
- 12. Hayes A.J., Ma G.R., Buss G.R., Maroof M.A.S.. 2000, Molecular marker mapping of Rsv4, a gene conferring resistance to all known strains of soybean mosaic virus, Crop Sci., 40, 1434–7. [Google Scholar]
- 13. Shipe E.R., Buss G.R., Tolin S.A.. 1979, 2nd gene for resistance to peanut mottle virus in soybeans, Crop Sci., 19, 656–8. [Google Scholar]
- 14. Staskawicz B.J., Dahlbeck D., Keen N.T.. 1984, Cloned avirulence gene of Pseudomonas syringae pv. glycinea determines race-specific incompatibility on Glycine max (L.) Merr, Proc. Natl. Acad. Sci. USA, 81, 6024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baker W.A., Weaver D.B., Qui J., Pace P.F.. 1999, Genetic analysis of frogeye leaf spot resistance in PI54610 and Peking soybean, Crop Sci., 39, 1021–5. [Google Scholar]
- 16. Tanimura Y., Tamada T.. 1976, Breeding for resistance to soybean dwarf disease 1. Resistance of soybean varieties to soybean dwarf disease, Bull. Hokkaido Prefect Agric. Exp. Stn., 35, 8–17. [Google Scholar]
- 17. Keeling B.L. 1982, A seedling test for resistance to soybean stem canker caused by Diaporthe phaseolorum var caulivora, Phytopathology, 72, 807–9. [Google Scholar]
- 18. Robbins R.T., Rakes L.. 1996, Resistance to the reniform nematode in selected soybean cultivars and germplasm lines, J. Nematol., 28, 612–5. [PMC free article] [PubMed] [Google Scholar]
- 19. Pacumbaba R.P., Sapra V.T., Prom L.K.. 1984, Effect of 2 commercial fungicides on incidence of Diaporthe phaseolorum var caulivora on susceptible soybean cultivars, Phytopathology, 74, 827. [Google Scholar]
- 20. Tian X.-H., Nakamura T., Kokubun M.. 2005, The role of seed structure and oxygen responsiveness in pre-germination flooding tolerance of soybean cultivars, Plant. Prod. Sci., 8, 157–65. [Google Scholar]
- 21. Muramatsu N., Kokubun M., Horigane A.. 2008, Relation of seed structures to soybean cultivar difference in pre-germination flooding tolerance, Plant. Prod. Sci., 11, 434–9. [Google Scholar]
- 22. Tuinstra M.R., Ejeta G., Goldsbrough P.B.. 1997, Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci, Theor. Appl. Genet., 95, 1005–11. [Google Scholar]
- 23. Yamanaka N., Watanabe S., Toda K., et al. 2005, Fine mapping of the FT1 locus for soybean flowering time using a residual heterozygous line derived from a recombinant inbred line, Theor. Appl. Genet., 110, 634–9. [DOI] [PubMed] [Google Scholar]
- 24. Eshed Y., Zamir D.. 1995, An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated qtl, Genetics, 141, 1147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koumproglou R., Wilkes T.M., Townson P., et al. 2002, STAIRS: a new genetic resource for functional genomic studies of Arabidopsis, Plant J. Cell Mol. Biol., 31, 355–64. [DOI] [PubMed] [Google Scholar]
- 26. Ali M.L., Sanchez P.L., Yu S., Lorieux M., Eizenga G.C.. 2010, Chromosome segment substitution lines: a powerful tool for the introgression of valuable genes from Oryza wild species into cultivated rice (O. sativa), Rice, 3, 218–34. [Google Scholar]
- 27. von Korff M., Wang H., Léon J., Pillen K.. 2004, Development of candidate introgression lines using an exotic barley accession (Hordeum vulgare ssp. spontaneum) as donor, Theor. Appl. Genet., 109, 1736–45. [DOI] [PubMed] [Google Scholar]
- 28. Fonceka D., Tossim H.-A., Rivallan R., et al. 2012, Construction of chromosome segment substitution lines in peanut (Arachis hypogaea L.) using a wild synthetic and QTL mapping for plant morphology, PLoS ONE, 7, e48642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Falke K.C., Sušić Z., Hackauf B., et al. 2008, Establishment of introgression libraries in hybrid rye (Secale cereale L.) from an Iranian primitive accession as a new tool for rye breeding and genomics, Theor. Appl. Genet., 117, 641–52. [DOI] [PubMed] [Google Scholar]
- 30. Jeuken M.J.W., Lindhout P.. 2004, The development of lettuce backcross inbred lines (BILs) for exploitation of the Lactuca saligna (wild lettuce) germplasm, Theor. Appl. Genet., 109, 394–401. [DOI] [PubMed] [Google Scholar]
- 31. Wang W., He Q., Yang H., Xiang S., Zhao T., Gai J.. 2012, Development of a chromosome segment substitution line population with wild soybean (Glycine soja Sieb. et Zucc.) as donor parent, Euphytica, 189, 293–307. [Google Scholar]
- 32. Concibido V.C., La Vallee B., McLaird P., et al. 2003, Introgression of a quantitative trait locus for yield from Glycine soja into commercial soybean cultivars, Theor. Appl. Genet., 106, 575–82. [DOI] [PubMed] [Google Scholar]
- 33. Li Y.H., Zhou G.Y., Ma J.X., et al. 2014, De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits, Nat. Biotechnol., 32, 1045–52. [DOI] [PubMed] [Google Scholar]
- 34. Kaga A., Shimizu T., Watanabe S., et al. 2012, Evaluation of soybean germplasm conserved in NIAS genebank and development of mini core collections, Breed. Sci., 61, 566–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook D.E., Bayless A.M., Wang K., et al. 2014, Distinct copy number, coding sequence, and locus methylation patterns underlie Rhg1-mediated soybean resistance to soybean cyst nematode, Plant Physiol., 165, 630–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsubokura Y., Watanabe S., Xia Z., et al. 2014, Natural variation in the genes responsible for maturity loci E1, E2, E3 and E4 in soybean, Ann. Botany, 113, 429–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fehr W.R., Caviness C.E., Burmood D.T., Pennington J.S.. 1971, Stage of development descriptions for soybeans, Glycine max (L.) Merrill, Crop Sci., 11, 929. [Google Scholar]
- 38. Kurata N., Nagamura Y., Yamamoto K., et al. 1994, A 300 kilobase interval genetic map of rice including 883 expressed sequences, Nat. Genet., 8, 365–72. [DOI] [PubMed] [Google Scholar]
- 39. Khosla S., Augustus M., Brahmachari V.. 1999, Sex-specific organisation of middle repetitive DNA sequences in the mealybug Planococcus lilacinus, Nucleic Acids Res., 27, 3745–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukuoka H., Nunome T., Minamiyama Y., Kono I., Namiki N., Kojima A.. 2005, Read2Marker: a data processing tool for microsatellite marker development from a large data set, Biotechnology, 39, 472–6. [DOI] [PubMed] [Google Scholar]
- 41. Rozen S., Skaletsky H.. 2000, Primer3 on the WWW for general users and for biologist programmers, Methods Mol. Biol., 132, 365–86. [DOI] [PubMed] [Google Scholar]
- 42. Andreson R., Reppo E., Kaplinski L., Remm M.. 2006, GENOMEMASKER package for designing unique genomic PCR primers, BMC Bioinform., 7, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hisano H., Sato S., Isobe S., et al. 2007, Characterization of the soybean genome using EST-derived microsatellite markers, DNA Res., 14, 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schuelke M. 2000, An economic method for the fluorescent labeling of PCR fragments, Nat. Biotechnol., 18, 233–4. [DOI] [PubMed] [Google Scholar]
- 45. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Miller W., Lipman D.J.. 1997, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi I.Y., Hyten D.L., Matukumalli L.K., et al. 2007, A soybean transcript map: gene distribution, haplotype and single-nucleotide polymorphism analysis, Genetics, 176, 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oeth P., del Mistro G., Marnellos G., Shi T., van den Boom D.. 2009, Qualitative and quantitative genotyping using single base primer extension coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MassARRAY), Methods Mol. Biol., 578, 307–43. [DOI] [PubMed] [Google Scholar]
- 48. Van Ooijen J.W., Voorrips R.E.. 2001, JoinMap® Version 4.0: Software for the Calculation of Genetic Linkage Maps. Wageningen: Plant Research International.
- 49. Broman K.W., Wu H., Sen S., Churchill G.A.. 2003, R/qtl: QTL mapping in experimental crosses, Bioinformatics, 19, 889–90. [DOI] [PubMed] [Google Scholar]
- 50. R Development Core Team. 2008, R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/ (1 November 2017, date last accessed).
- 51. Chakravarti A. 1991, A graphical representation of genetic and physical maps: the Marey map, Genomics, 11, 219–22. [DOI] [PubMed] [Google Scholar]
- 52. Rezvoy C., Charif D., Gueguen L., Marais G.A.. 2007, MareyMap: an R-based tool with graphical interface for estimating recombination rates, Bioinformatics, 23, 2188–9. [DOI] [PubMed] [Google Scholar]
- 53. Yang K., Jeong S.C.. 2008, Genetic linkage map of the nucleolus organizer region in the soybean, Genetics, 178, 605–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Findley S.D., Cannon S., Varala K., et al. 2010, A Fluorescence in situ hybridization system for karyotyping soybean, Genetics, 185, 727–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Du J., Grant D., Tian Z., et al. 2010, SoyTEdb: a comprehensive database of transposable elements in the soybean genome, BMC Genom., 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nelson R.L., Amdor P.J., Orf J.H., et al. 1987, Evaluation of the USDA soybean germplasm collection: maturity groups 000 to IV (PI 273.483 to PI 427.107), T. Bull. USDA, 1718, 44–123. [Google Scholar]
- 57. Watanabe S., Hideshima R., Xia Z., et al. 2009, Map-based cloning of the gene associated with the soybean maturity locus E3, Genetics, 182, 1251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watanabe S., Xia Z., Hideshima R., et al. 2011, A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering, Genetics, 188, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hill C.B., Hartman G.L., Esgar R., Hobbs H.A.. 2006, Field evaluation of green stem disorder in soybean cultivars, Crop Sci., 46, 879–85. [Google Scholar]
- 60. Zhao C., Takeshima R., Zhu J., et al. 2016, A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog, BMC Plant Biol., 16, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takeshima R., Hayashi T., Zhu J., et al. 2016, A soybean quantitative trait locus that promotes flowering under long days is identified as FT5a, a FLOWERING LOCUS T ortholog, Exbotj, 67, 5247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu B., Watanabe S., Uchiyama T., et al. 2010, The soybean stem growth habit gene Dt1 is an ortholog of Arabidopsis TERMINAL FLOWER1, Plant Physiol., 153, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thompson J.A., Bernard R.L., Nelson R.L.. 1997, A third allele at the soybean dt1 locus, Crop Sci., 37, 757–62. [Google Scholar]
- 64. Shi Z., Liu S.M., Noe J., Arelli P., Meksem K., Li Z.L.. 2015, SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance, BMC Genom., 16, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kato S., Sayama T., Fujii K., et al. 2014, A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds, Theor. Appl. Genet., 127, 1365–74. [DOI] [PubMed] [Google Scholar]
- 66. Mandl F.A., Buss G.R.. 1981, Comparison of narrow and broad leaflet isolines of soybean, Crop Sci., 21, 25–7. [Google Scholar]
- 67. Jeong N., Suh S.J., Kim M.H., et al. 2012, Ln is a hey regulator of leaflet shape and number of seeds per pod in soybean, Plant Cell, 24, 4807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamanaka N., Ninomiya S., Hoshi M., et al. 2001, An informative linkage map of soybean reveals QTLs for flowering time, leaflet morphology and regions of segregation distortion, DNA Res., 8, 61–72. [DOI] [PubMed] [Google Scholar]
- 69. Xia Z., Watanabe S., Yamada T., et al. 2012, Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering, Proc. Natl. Acad. Sci. USA, 109, E2155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xu M.L., Xu Z.H., Liu B.H., et al. 2013, Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean, BMC Plant Biol., 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cober E.R., Voldeng H.D.. 2001, A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T, Crop Sci., 41, 698–701. [Google Scholar]
- 72. Kuroda Y., Kaga A., Tomooka N., et al. 2013, QTL affecting fitness of hybrids between wild and cultivated soybeans in experimental fields, Ecol. Evol., 3, 2150–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Komatsu K., Hwang T.Y., Takahashi M., et al. 2012, Identification of QTL controlling post-flowering period in soybean, Breed. Sci., 61, 646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ross J.P., Brim C.A.. 1957, Resistance of soybeans to the soybean cyst nematode as determined by a double-row method, Plant Dis. Rep., 41, 923–4. [Google Scholar]
- 75. Gao H., Bhattacharyya M.K.. 2008, The soybean-Phytophthora resistance locus Rps1-k encompasses coiled coil-nucleotide binding-leucine rich repeat-like genes and repetitive sequences, BMC Plant Biol., 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boboye B., Nyakatura G., Rosenthal A., Perret X., Broughton W.J., Boller T.. 2009, Sequence analysis of a DNA fragment from Sinorhizobium fredii USDA257 which extends the nitrogen fixation host range of Rhizobium species NGR234 to soybean, Glycine max (L.) Merr cultivar Peking, Symbiosis, 48, 110–9. [Google Scholar]
- 77. Devine T.E., O'Neill J.J.. 1989, Genetic allelism of nodulation response genes Rj1, Rj2, and Rj4 in soybean, Crop Sci., 29, 1347–50. [Google Scholar]
- 78. Yang S.M., Tang F., Gao M.Q., Krishnan H.B., Zhu H.Y.. 2010, R gene-controlled host specificity in the legume-rhizobia symbiosis, Proc. Natl. Acad. Sci. USA, 107, 18735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tang F., Yang S.M., Liu J.G., Zhu H.Y.. 2016, Rj4, a gene controlling nodulation specificity in soybeans, encodes a thaumatin-like protein but not the one previously reported, Plant Physiol., 170, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang W., Weaver D.B., Nielsen B.L., Qiu J.. 2001, Molecular mapping of a new gene for resistance to frogeye leaf spot of soya bean in Peking, Plant Breed., 120, 73–8. [Google Scholar]
- 81. Joshi J.M. 1980, Research Notes: evaluation of soybean germplasm for resistance to corn earworm–IV, Soybean Genet. Newslett., 7, 26. [Google Scholar]
- 82. Sayama T., Nakazaki T., Ishikawa G., et al. 2009, QTL analysis of seed-flooding tolerance in soybean (Glycine max [L.] Merr.), Plant Sci., 176, 514–21. [DOI] [PubMed] [Google Scholar]
- 83. Bailey M., Boerma H., Parrott W.. 1993, Genotype effects on proliferative embryogenesis and plant regeneration of soybean, In Vitro Cell. Dev. Biol.–Plant, 29, 102–8. [Google Scholar]
- 84. Song X., Han Y., Teng W., Sun G., Li W.. 2010, Identification of QTL underlying somatic embryogenesis capacity of immature embryos in soybean (Glycine max (L.) Merr.), Plant Cell Rep., 29, 125–31. [DOI] [PubMed] [Google Scholar]
- 85. Mauro A.O., Pfeiffer T.W., Collins G.B.. 1995, Inheritance of soybean susceptibility to Agrobacterium tumefaciens and its relationship to transformation, Crop Sci., 35, 1152–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.