

Abstract
The hypersensitive response (HR) triggered on Nicotiana edwardsonii by tobacco mosaic virus was studied using a modified viral genome that directed expression of the green fluorescent protein. Inoculated plants were initially incubated at 32°C to inhibit the N gene-mediated HR. Transfer to 20°C initiated the HR, and fluorescent infection foci were monitored for early HR-associated events. Membrane damage, which preceded visible cell collapse by more than 3 h, was accompanied by a transient restriction of the xylem within infection sites. Following cell collapse and the rapid desiccation of tissue undergoing the HR, isolated, infected cells were detected at the margin of necrotic lesions. These virus-infected cells were able to reinitiate infection on transfer to 32°C, however, if maintained at 20°C they eventually died. The results indicate that the tobacco mosaic virus-induced HR is a two-phase process with an early stage culminating in rapid cell collapse and tissue desiccation followed by a more extended period during which the remaining infected cells are eliminated.
The hypersensitive response (HR), induced following the infection of a resistant plant by an incompatible pathogen, is a well conserved defense mechanism used against viral, bacterial, fungal, and nematode pathogens. By definition, the HR is characterized as the rapid death of a limited number of cells in the vicinity of the invading pathogen that is often associated with a block on the progression of the infection (Goodman and Novacky, 1994). In many cases this response is manifested in the development of necrotic lesions in which cell collapse is followed by localized desiccation and browning of the affected cells. Triggering of the HR is usually a highly specific event, which depends on a matching specificity between a disease resistance gene in the plant and an avirulence gene in the pathogen, a concept referred to as gene-for-gene resistance (Flor, 1942).
Gene-for-gene interactions are generally interpreted in terms of a specific interaction between a resistance gene-encoded receptor and a pathogen-derived elicitor (Keen, 1990; Scofield et al., 1996). Recognition of the elicitor by the host receptor initiates a cascade of events, culminating in the collapse and death of host cells in the vicinity of the pathogen, and the limitation of further pathogen accumulation. Although cell death and the development of necrotic lesions represent the defining phenotype of the HR, the function of cell death as cause or consequence of resistance is less certain (Heath, 1999; Richael and Gilchrist, 1999). It is an intuitively appealing concept that the death of cells infected with an obligate biotrophic pathogen could provide an effective means of blocking accumulation of the pathogen and thus confer resistance. However, proof that cell death is causal in determining resistance is limited.
In fact there are several examples where gene-for-gene resistance occurs either in the absence of cell death or where cell death can be blocked without inhibiting the resistance to pathogen accumulation and spread. For example the potato gene Rx confers resistance to many strains of potato virus X (PVX) without the appearance of visible symptoms or necrotic lesion formation. However, when Rx is expressed in transgenic Nicotiana benthamiana, resistance to avirulent strains of PVX is associated with the development of necrotic local lesions. Thus in the PVX/Rx pathosystem, cell death and resistance are separate events, and although lesion formation can accompany Rx-mediated resistance to PVX it is not necessary for an effective host response (Bendahmane et al., 1999). In the case of the HR to tobacco mosaic virus (TMV) in tobacco carrying the N gene, oxygen has been shown to be necessary for necrotic lesion formation (Mittler et al., 1996). However, resistance to TMV is not compromised in a low-oxygen environment despite the absence of necrotic lesions (Mittler et al., 1996). Further evidence for the separation of cell death and resistance is provided by an Arabidopsis mutant, dnd1, that exhibits constitutively high levels of salicylic acid (SA) and is defective in HR cell death but that nevertheless retains a characteristic resistant phenotype when challenged with an avirulent bacterial pathogen (Yu et al., 1998). However, despite the exceptions listed above it is notable that hypersensitive cell death is very commonly associated with gene-for-gene resistance, and the fact that cell death per se can activate many defense-associated responses suggests that, in part, death may serve to strengthen the induction of host defenses (Dangl et al., 1996).
The interaction between TMV and tobacco plants carrying the N gene is one of the best studied of all viral pathosystems, and both the viral elicitor and the plant resistance gene product have been characterized (Padgett and Beachy, 1993; Whitham et al., 1994). In addition to the formation of necrotic lesions the TMV/N gene interaction displays characteristic hallmarks of the HR prior to the onset of cell death including the oxidative burst and loss of plasma membrane integrity (Weststeijn, 1978; Doke and Ohashi, 1988). One feature of the N gene-mediated response, which distinguishes it from many other HR pathosystems, is the temperature dependence of the host response with the HR and resistance occurring effectively only at temperatures below 27°C (Weststeijn, 1981). This feature of the N gene response allows plants to be inoculated and maintained at temperatures at which the resistance response is inoperative prior to lowering the temperature to initiate and synchronize the induction of the HR.
Despite a great many studies into the ultrastructural, physiological, and biochemical events associated with the N gene-mediated HR (Milne, 1966; Dunigan and Madlener, 1995; Mittler et al., 1996, 1997; Zhang and Klessig, 1998), an inherent difficulty in studying any virus-induced HR is that infection sites cannot be localized noninvasively prior to the onset of visible symptoms. To circumvent this problem and to allow the precise localization of virus-infected cells in relation to the development of the local lesion response, we investigated the induction of the N gene-mediated HR against TMV expressing the green fluorescent protein (GFP). Here we report our analysis of both the early and late phases of the N gene-mediated resistance process using a range of imaging techniques to correlate the localization of virus in relation to the host response.
RESULTS
Time Course of Lesion Development
The development of a visible HR in plants carrying the N gene from Nicotiana glutinosa following infection with TMV has been described extensively (e.g. Holmes, 1929; Mittler et al., 1997). Although necrotic lesions are ultimately formed following infection with TMV, the timing of symptom development differs depending on the strain of virus as well as on the genotype and physiological status of the host. In order to correlate the onset of visible symptoms of the HR with other changes associated with the response, we initially monitored TMV.GFP-infected Nicotiana edwardsonii plants under both natural light and blue light (488 nm), following transfer of plants from 32°C to 20°C, a treatment that serves to trigger the HR response (Weststeijn, 1981). N. edwardsonii, which is a hybrid between N. glutinosa and Nicotiana clevelandii, carries the N gene from N. glutinosa and was used in this study due to its high susceptibility to virus infection (Christie, 1969). Figure 1 shows the appearance of infection foci at 11 (Fig. 1, A and B) and 15 (Fig. 1, E and F) h post-transfer (hpt). From the time of transfer until approximately 12 hpt no visible symptoms of the HR were seen under natural light. The first visible sign of lesion formation occurred from approximately 13 hpt onwards when tissue destined to undergo the HR developed a glassy appearance (Fig. 1E). With time, developing lesions became more distinct from the surrounding tissue, appearing more clearly necrotic and collapsed.
Figure 1.

TMV infection foci following induction of the HR. Images of the same TMV infection focus observed at 11 hpt (A–D) and 15 hpt (E–H) viewed using either transmitted light under the stereomicroscope (A and E) or blue light under the CLSM (B–D and F–H). The images in A through D show the infection focus prior to visible cell collapse, whereas images in E through H show the infection focus following the development of visible symptoms. The CLSM images in C and G are focused on the upper leaf epidermis. Images in D and H are focused on the palisade mesophyll. Scale bars = 1 mm (A, B, E, and F), = 0.1 mm (C, D, G, and H).
Examination of developing necrotic lesions under the confocal laser scanning microscope (CLSM) also showed distinct changes first occurring from around 13 hpt. The initial stages of lesion development correlated with loss of turgor in the epidermal cells and the collapse of the epidermal layer; Figure 1G (15 hpt), taken after epidermal cells had collapsed, shows both epidermal and mesophyll cells in the same confocal plane, whereas at 9 hpt the epidermis remained turgid (Fig. 1C). Subsequently, between 15 and 18 hpt, the underlying palisade cells also lost turgidity leading to the complete collapse of the infected tissue. The subcellular distribution of GFP within infected cells also changed after the onset of visible symptoms. Initially GFP was localized to the nuclei and cytoplasm of infected cells (Fig. 1D), however, from around 15 hpt the distribution of GFP was seen to change, often accumulating in the vacuole and in chloroplast of collapsing cells (Fig. 1H; data not shown).
Low-Temperature Scanning Electron Microscopy (LTSEM) of Developing Necrotic Lesions
Examination of infection foci under the LTSEM at the earliest appearance of visible symptoms approximately 13 hpt, confirmed the loss of turgor and collapse of the epidermis; only guard cells remained turgid while the surrounding epidermal cells appeared deflated (Fig. 2A). Even at this early time point in lesion development the boundary between turgid and collapsing cells is clearly delimited (Fig. 2A). At later time points this boundary became more prominent (Fig. 2B) and was coincident with the collapse of the mesophyll as observed under the CLSM. The complete collapse of the infected tissue was also marked by the loss of turgor in guard cells (Fig. 2B).
Figure 2.

LTSEM of TMV-induced HR lesions. A, Upper epidermis of TMV.GFP-infected N. edwardsonii leaves shortly after visible lesion development at 14 hpt. Turgid and collapsed epidermal cells can be clearly differentiated; at this time point guard cells retain turgor. B, A later stage of lesion development at 20 hpt with both epidermal and guard cells showing loss of turgor. Scale bar = 0.1 mm (A and B).
Analysis of Plasma Membrane Integrity before and during the Visible HR
A common feature of tissues undergoing the HR is the efflux of electrolytes that accompanies the loss in cell turgor. Electrolyte leakage, which reflects damage to cellular membranes (Goodman, 1968), has been shown to occur during the TMV-induced HR approximately 1 h before visible cell collapse (Weststeijn, 1978). To correlate spatial and temporal patterns of membrane damage to cells within infection foci undergoing the HR we used Evans blue, a dye that is excluded by membranes of living cells but diffuses into dead cells (Gaff and Okong ‘O-Ogola, 1971). Staining of detached leaves by vacuum infiltration with aqueous Evans blue was performed at hourly intervals after transfer of TMV.GFP-infected N. edwardsonii from 32°C to 20°C.
From 1 to 8 hpt, no specific labeling of infected cells was observed as determined by the lack of Evans blue staining of cells within the green fluorescent infection foci (Fig. 3, A and B). By 9 hpt a subset of the infection foci seen using violet light under a fluorescence stereo microscope showed labeling with Evans blue with noticeably stronger dye uptake in cells close to major veins (Fig. 3, C and D). At increasing times post-transfer, staining with Evans blue showed more intense labeling of the infection foci, and by 11 to 12 hpt, the majority of infection foci showed Evans blue labeling in most of the cells expressing GFP (data not shown).
Figure 3.

Cell death prior to the onset of visible tissue collapse. Stereomicroscope bright field (A, C, and E) and fluorescence (B, D, and F) micrographs showing TMV.GFP infection foci after staining with Evans blue. A and B, Eight hpt, scale bar = 1 cm (B); C and D, 9 hpt, scale bar = 0.5 cm (D); E and F, 14 hpt, scale bar = 0.5 cm (F).
In the majority of lesions examined staining was initially more prominent around the margin of the lesion (data not shown). However, a pattern of concentric zones of Evans blue staining with both the periphery and the central core of the infection site showing strongest labeling was also frequently observed (Fig. 3, E and F).
Timing of Irreversible Cell Damage during the Onset of the HR
Although the uptake of Evans blue from approximately 9 hpt provided the first detectable evidence of the onset of the HR, the cellular processes leading to cell death were presumably established prior to detectable membrane damage. In order to identify the point at which irreversible cellular damage was occurring in the period leading up to cell death, further temperature shift experiments were conducted. Following an initial temperature shift from 32°C to 20°C, plants were maintained at the lower temperature for progressively longer periods before being returned to 32°C. After a further 24-h incubation at 32°C, inoculated leaves were examined for visible evidence of HR development and stained with Evans blue to determine whether infected cells had suffered membrane damage. The cumulative results of three experiments, summarized in Figure 4, demonstrated that irreversible progression to membrane damage began from approximately 5 hpt. However, irreversible commitment to HR lesion formation was not established until 10 hpt or later.
Figure 4.

Irreversible commitment to cell death and lesion formation. The percentage of infection foci, as determined by GFP fluorescence, showing staining with Evans blue (█) and the percentage of infection foci that developed visible HR lesion (□) after a temperature shift to 20°C for the indicated time followed by maintenance at 32°C for a further 24 h.
Symplastic Movement of Micro-Injected Dyes
The loss of cell turgor and the collapse of infected cells implies that symplastic continuity must be lost between cells in the developing lesion and healthy neighboring cells. Because intercellular virus movement is dependent on plasmodesmata, which provide a symplastic continuum between mesophyll and epidermal cells, we investigated whether the breakdown of symplastic continuity preceded or accompanied the visible collapse of cells at the onset of lesion development. Micro-injection of the low Mr, membrane-impermeant, dye sulforhodamine B was used to determine whether epidermal cells immediately adjacent to the viral infection front were symplastically coupled to infected cells. Of 29 injections into cells adjacent to non-collapsed infection foci performed between 10 and 14 hpt, 25 (86%) showed extensive movement of the fluorescent probe into adjacent infected and noninfected cells (Fig. 5A). In contrast, of 15 injections performed between 16 and 20 hpt into cells adjacent to collapsing infection foci dye movement into adjacent noninfected cells was seen, however, no labeling of infected cells was observed (Fig. 5B).
Figure 5.

Symplastic continuity between cells at the periphery of TMV infection foci. Movement of sulforhodamine B between epidermal cells 5 min after micro-injection of individual epidermal cells adjacent to TMV.GFP infection foci observed under the CLSM. A, TMV.GFP infection focus at 10 hpt; B, TMV.GFP infection focus at 16 hpt. The image has been false-colored to show sulforhodamine B in red and GFP in green. The injected cells are outlined and marked with an asterisk. Scale bars = 0.2 mm.
Apoplastic Transport of Texas Red Delivered by the Xylem
An obvious feature of the HR induced by TMV on N. edwardsonii, as well as in many other examples of the HR, is the rapid desiccation of the infected tissue, which follows the initial phase of cell collapse. In order to investigate xylem transport within infection sites, leaves were detached at successive times following transfer of plants from 32°C to 20°C and the leaf petioles immersed in a solution containing the fluorescent dye Texas Red. All vein classes became heavily labeled with dye that was taken up by the xylem, and the subsequent exit of dye into the apoplast led to the uptake of the fluorescent dye and its sequestration in the vacuoles of mesophyll cells neighboring the veins, giving a characteristically diffuse pattern of fluorescence labeling around the veins (Fig. 6B). For leaves labeled up to 10 hpt, xylem transport and uptake of Texas Red by mesophyll cells was identical in both virus-infected and -noninfected areas of leaves (Fig. 6, A and B). However, in leaves loaded with dye at 11 hpt the xylem transport pathway across infection foci was severely restricted, leading to the absence of Texas Red labeling specifically in cells within and surrounding infection foci (Fig. 6, C–E). This restriction of xylem transport through infection foci, which occurred up to 2 h before visible signs of tissue collapse, persisted for several hours during which time visible necrotic lesions developed. To determine whether the observed restriction in xylem transport could have resulted from reduced transpiration in the virus-infected tissue small sectors of noninfected N. edwardsonii, leaves were coated with vacuum grease on both the abaxial and adaxial surfaces. As shown in Figure 6F xylem within the non-transpiring, grease-coated areas of leaves still showed extensive labeling with Texas Red, and the treatment served only to reduce the escape of dye from the xylem. The restriction in xylem transport seen prior to the onset of the HR was not permanent, and by 17 hpt the transport pathway through the developing necrotic lesions was again open (Fig. 6G) and remained open for at least a further 24 h (data not shown). Although xylem transport of Texas Red through developing lesions resumed, the pattern of labeling observed with Texas Red confined to the xylem elements (Fig. 6, G and H) was clearly different from the distribution of Texas Red in noninfected areas of the leaf where sequestration of the dye from the apoplast into mesophyll cells was observed.
Figure 6.

Transport of Texas Red through the xylem of TMV.GFP infection foci. CLSM images of TMV.GFP-infected and control-noninfected tissue 30 min after labeling of detached leaves with the fluorescent dye Texas Red. A, TMV.GFP infection focus at 10 hpt with the corresponding image of Texas Red shown in B. C, An infection focus at 11 hpt is shown with the corresponding Texas Red image in D showing a restriction of dye movement into the infected area of the leaf. E, Low-magnification image of a TMV.GFP-infected leaf at 11 hpt with areas of Texas Red exclusion corresponding to the position of three TMV.GFP infection foci. F, Texas Red in the xylem of a leaf following prevention of transpiration from both upper and lower epidermis after localized application of vacuum grease (encircled area). G, Resumed transport of Texas Red through a TMV.GFP infection focus at 20 hpt. H, High-magnification image of a TMV.GFP infection site 20 hpt showing that the fluorescent dye is restricted to the xylem elements. Scale bars = 0.5 mm (A and B); 1 mm (C and D); 4 mm (E); 2 mm (F); 0.5 mm (G); 25 μm (H).
The Presence of Virus-Infected Cells at the Periphery of Necrotic Lesions
Analysis of developing HR lesions under the CLSM failed to show GFP in cells lying beyond the lesion periphery for at least 48 hpt (data not shown). However, from approximately 52 hpt onwards both isolated cells and small clusters of cells expressing the GFP were clearly visible at the margins of lesions (Fig. 7A). Occasionally fluorescent cells persisted at the lesion margin for up to 120 hpt after which time they were no longer detectable. The GFP containing cells seen at the periphery of HR lesions showed visible cytoplasmic streaming (data not shown), indicating that they were alive. To test whether these living, infected cells were able to reinitiate infection plants that had been transferred to 20°C for 72 h to induce necrotic lesion formation, then they were transferred back to 32°C. This change in temperature resulted in the resumption of TMV.GFP cell-to-cell movement, initiating from the green fluorescent cells at the lesion margin, and resulted in the development of secondary infection foci (Fig. 7, B and C).
Figure 7.
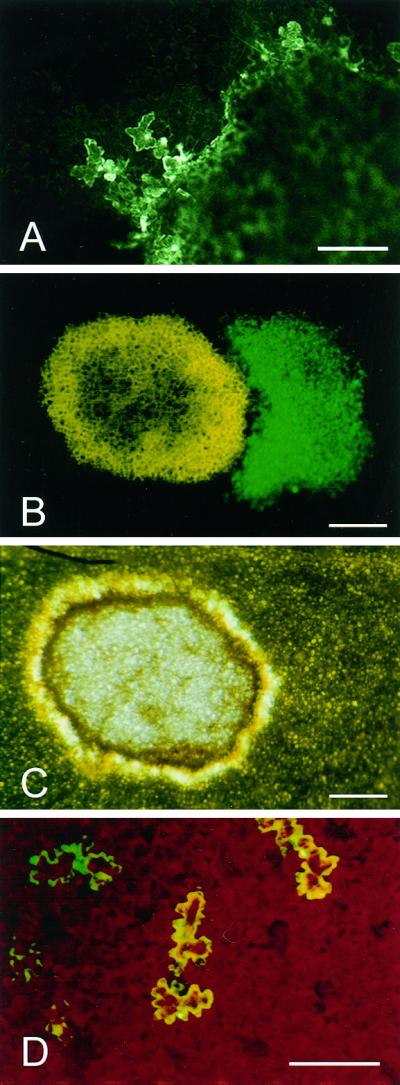
Survival of TMV-infected cells at the margin of necrotic lesions. A, CLSM image of TMV.GFP-infected epidermal cells adjacent to a collapsed necrotic lesion (right-hand side of image) at 52 hpt. B and C, Fluorescence and bright field images, respectively, of a necrotic lesion after a 65-h incubation at 20°C followed by a 96-h incubation at 34°C. The necrotic lesion shown in C appears as yellow autofluoresence in B and TMV.GFP-infected tissue appears green. D, Stereomicroscope fluorescence image showing individual epidermal cells infected with the cell-to-cell movement-deficient mutant, TMV.GFPΔMP, at 48 hpt. Scale bars = 0.2 mm (A and D) and 0.5 mm (B and C).
Single, Infected Cells Fail to Initiate the HR
The eventual disappearance of green fluorescent cells at the periphery of necrotic lesions when plants were kept at 20°C suggested that these infected cells had succumbed to the HR. The ability of single TMV-infected cells to mount an HR has not been described previously, and therefore we tested a mutant derivative of TMV.GFP that carried a frameshift mutation in the movement protein gene. Inoculation of this mutant, TMV.GFPΔMP, to N. edwardsonii at 32°C resulted in infections that were restricted to single epidermal cells, demonstrating that the mutant lacked the cell-to-cell transport function. To test whether isolated epidermal cells were capable of mounting an HR in response to TMV.GFP infection, N. edwardsonii plants infected with TMV.GFPΔMP were maintained at 32°C for 96 h prior to transfer to 20°C and monitored for the persistence of green fluorescence in epidermal cells. Leaves were detached from infected plants and examined under the CLSM to determine the number of infected epidermal cells immediately after transfer from high to low temperature. Of an initial population of 174 epidermal cells showing green fluorescence at 0 to 1 hpt, 150 cells were still detected at 48 hpt, and even at 120 hpt, 112 fluorescent epidermal cells were still evident (Fig. 7D).
DISCUSSION
Development of Visible Symptoms in Cells Undergoing the HR
The objective of this study was to correlate the onset and progression of the N gene-mediated HR with the presence of virus by using a GFP-tagged TMV genome. Using a temperature shift from 32°C to 20°C, to initiate and synchronize the host response (Weststeijn, 1981), the first macroscopic sign of turgor loss and lesion formation occurred at approximately 13 hpt. Cell collapse appeared to occur specifically in infected cells expressing levels of GFP that were detectable by fluorescence microscopy. The altered distribution of GFP, seen during the collapse of mesophyll cells, most likely results from damage to organelle membranes occurring in cells undergoing the HR (Goodman, 1968; Weststeijn, 1978). The maintenance of cell turgor by guard cells following the collapse of the epidermis, as seen under the LTSEM, may reflect the fact that at the time of inoculation the guard cells were symplastically isolated from surrounding cells and thus did not become infected. The lack of elicitor in the guard cells and/or their isolation from any symplastically transmitted signal(s) could explain their prolonged survival.
Membrane Damage and Cell Death Prior to the Onset of the Visible HR
The results obtained using Evans blue as a marker for dead and damaged cells showed clear evidence of cellular damage up to 4 h before collapse of the epidermis. As most of the detected staining with Evans blue was of mesophyll cells (data not shown), this demonstrates that significant damage to the plasma membrane of mesophyll cells was occurring at least 6 h before these cells finally lost turgor and collapsed. The initial association of Evans blue staining with cells close to vascular tissues is consistent with previous studies of other HR pathosystems (Hammond-Kosack et al., 1996) and may reflect a greater sensitivity of these cells to death-inducing stimuli.
Establishment of the Cell Death Program
At the time of the temperature shift, infected cells in which GFP is present must harbor the viral replication-associated protein that is the elicitor of the HR (Padgett and Beachy, 1993) and thus should be able to induce the host response. The delay of at least 5 h following the temperature shift before irreversible membrane damage is conditioned may in part be explained by a phase in which the component(s) of the N gene-mediated response pathway that is compromised by high temperature either recovers or is replaced. Although a commitment to membrane damage was established following periods of greater than 5 h at 20°C, the resulting cell death alone does not result in visible lesion formation, the latter requiring a further 5-h induction period. This result demonstrates that cell death and formation of a macroscopic lesion, although clearly associated, are not inexorably linked. Although we cannot exclude the possibility that a threshold number of damaged cells is necessary for the formation of visible lesions, other factors, for example tissue dehydration, may also be involved in the development of macroscopic lesions.
Dye Coupling within and around Infection Foci
The central role of plasmodesmata in the intercellular transport of viruses prompted us to investigate dye coupling between infected and noninfected cells both before and after the onset of visible symptoms. Prior to their collapse infected cells were symplastically connected to their noninfected neighbors and this continuity was only lost when cells began to collapse. This result demonstrates that, at least with respect to the transport of a low-Mr dye, symplastic continuity was maintained between infected and noninfected cells right up until the point when turgor loss and cell collapse began. Thus, plasmodesmatal closure does not appear to be important in the N gene-mediated restriction of TMV.
Xylem Closure Precedes Visible Lesion Formation
The finding that movement of a fluorescent tracer in the xylem within infection foci was restricted at least 1 h before the onset of visible cell collapse was unexpected and has not been described previously. Recent work has demonstrated that prenecrotic TMV infection foci show a reduced level of transpiration, apparently as a consequence of guard cell closure (Chaerle et al., 1999). However, reduced transpiration alone cannot explain the restricted xylem transport, as localized blocking of transpiration only affected unloading of the fluorescent tracer and did not block transport through the xylem. It is thus probable that some additional mechanism(s) is contributing to the observed restriction in xylem transport. Whatever the means by which xylem conductivity is restricted, the ensuing reduction in the water potential within the infection site could serve to increase water loss from cells with damaged cellular membranes and accelerate loss of cell turgor. Such a mechanism could explain the rapid desiccation of HR lesions (Fig. 6C) and could accelerate the cell death process, thereby reducing the likelihood of virus escape into neighboring cells.
Life at the Edge
A previous study demonstrated that following the formation of a visible HR in the TMV/N gene pathosystem a shift to high temperature (>30°C) results in the resumption of viral infection (Weststeijn, 1981). This observation has been interpreted as an indication that cell death is a consequence, rather than a cause, of resistance in this pathosystem. The results presented here clearly demonstrate that only a limited number of infected cells persist at the margin of necrotic lesions and that with time these cells are eliminated. That at the onset of the visible HR all cells expressing detectable levels of GFP collapse suggests that the infected cells at the edge of necrotic lesions, which are only seen at later time points, were only recently infected during the first phase of cell collapse. The low level of viral elicitor in newly infected cells is presumably insufficient to trigger cell death during the initial phase of lesion development. However, with time sufficient viral gene expression occurs to allow detection of GFP. The eventual demise of infected cells at the lesion edge, as determined by loss of GFP fluorescence, represents a second phase of the HR in which infected cells that escaped the first phase of lesion development are eliminated. The means by which the surviving infected cells are eventually incorporated into the necrotic lesion is uncertain but the fact that, under the conditions used here, necrotic lesions did not show any detectable increase in area after the initial phase of tissue collapse suggests that an efficient mechanism exists to selectively eliminate the few remaining infected cells. Furthermore, the fact that these cells can reinitiate the infection process following an increase in temperature suggests that infected cells at the lesion margin are not irreversibly modified from the outset of the HR.
Induction of the HR Requires Multicellular Infections
TMV is unable to initiate cell death in isolated protoplasts of N gene tobacco, raising the possibility that intercellular communication is necessary in the N gene-mediated HR (Otsuki et al., 1972). The present data, showing that TMV.GFPΔMP did not trigger cell death, argues that it is not intercellular communication per se that is necessary for the HR and that the presence of elicitor, although necessary, is insufficient by itself to trigger N gene-mediated cell death. To explain this difference between multicellular and single-cell infection sites, it would seem likely that a further cue from neighboring cells is necessary to trigger death. A strong candidate for this intracellular signal is SA, which triggers defense gene activation and serves to prime a more rapid incompatible response (Mur et al., 1997). In transgenic tobacco expressing the SA degrading enzyme salicylate hydroxylase, necrotic lesions induced by TMV continue to propagate indefinitely indicating that SA, although neither sufficient nor necessary for cell death to occur, is nevertheless essential for timely host response and pathogen containment (Delaney et al., 1994; Mur et al., 1997). Furthermore, pretreatment of a TMV susceptible tobacco cultivar (genotype nn) with SA reduces the accumulation of both TMV RNAs and the viral coat protein (Chivasa et al., 1997), a factor which could serve to delay spread of virus from the living cells at the periphery of the necrotic lesions. Although SA possesses features that make it an attractive candidate for a diffusible signal, it is equally plausible that other signaling compounds, for example ethylene (Pontier et al., 1999) either alone or in concert, act to prime a heightened state of responsiveness in cells surrounding HR lesions.
A reduced rate of accumulation of virus is consistent with the delay of over 50 h between the initiation of necrotic lesion formation and the first detection of GFP in cells adjacent to HR lesions. This concept of a zone of heightened resistance surrounding TMV-induced lesions is consistent with earlier observations regarding the failure to initiate secondary necrotic lesions in a narrow zone surrounding established lesions following a second challenge with TMV (Ross, 1961). Recently it has been shown that senescence-related genes are selectively activated at the periphery of TMV-induced necrotic lesions, a response that could also contribute to the slowing of pathogen spread (Pontier et al., 1999).
The data presented here provide a number of novel insights into the TMV-induced HR. Using a GFP-tagged viral genome allowed an analysis of the earliest events during the onset of the HR due to the ability to precisely identify infected cells. In addition, because GFP can be imaged noninvasively, the changes occurring to cells and tissues undergoing the HR could be correlated with sites of infection. This allowed both the early and late phases of the N gene-mediated response to be examined in more detail than has previously been possible in studies of incompatible virus/host interactions. Significantly, the inability of single cells infected with the movement deficient mutant to initiate the HR indicates that elicitor alone is insufficient to trigger cell death and strengthens the case for a two-phase HR process (Pontier et al., 1999). In the early phase, rapid cell collapse and death, perhaps facilitated by a reduced local water potential, lead to the elimination of the majority of infected cells and the generation of a signal that diffuses into neighboring living cells. In the second phase of the response, infected cells that survived the first round of cell death due to low levels of the viral elicitor respond to the combination of newly synthesized elicitor and externally generated signal(s) by dying.
MATERIALS AND METHODS
Viral Inoculum and Plant Inoculation
The construct pTMV.GFP carries a cDNA encoding the GFP in the TMV vector cDNA p30B (Lacomme and Santa Cruz, 1999; Shivprasad et al., 1999). A derivative of pTMV.GFP, carrying a frameshift mutation in the movement protein gene, was prepared by digestion of pTMV.GFP with ApaI followed by the removal of overhanging 3′-DNA termini using T4 DNA polymerase to give pTMV.GFPΔMP. All plasmid manipulations were performed using standard techniques (Sambrook et al., 1989). Following linearization of pTMV.GFP with KpnI, in vitro run-off transcripts were synthesized as described previously (Santa Cruz et al., 1996). Transcription reaction products were inoculated directly to leaves of 8-week-old Nicotiana benthamiana plants by manual abrasion of aluminum oxide-dusted leaves. Inoculated plants were maintained in controlled environment chambers with a 16-h photoperiod (400 μEm−2 s−1) maintained at 32°C, 50% relative humidity. At 4 d post-inoculation, inoculated leaves were harvested and ground in 2 volumes (w/v) of 20 mm phosphate buffer, pH 7.2. Viral inoculum prepared from transcript inoculated plants is referred to by deletion of the prefix p from the plasmid name. Dilutions of the TMV.GFP inoculum prepared in the same buffer were used to determine a concentration of inoculum that produced an average of 10 to 20 infection foci per inoculated leaf; TMV.GFPΔMP inoculum was used directly without further dilution.
Plant Material
Nicotiana edwardsonii plants (Christie, 1969) were grown from seed in a heated glasshouse and used for experiments when they were between 7 and 8 weeks old. Infectious sap from TMV.GFP- and TMV.GFPΔMP-infected plants, diluted as described above, was used to inoculate aluminum oxide-dusted leaves. Inoculated plants were maintained in a growth cabinet at 32°C with a 16-h photoperiod (400 μEm−2 s−1, 50% relative humidity) for 48 to 60 h before transfer to a similar cabinet at 20°C to induce and synchronize the HR.
CLSM and Fluorescence Microscopy
Infected leaves were viewed using a Stereofluorescence microscope (MZFLIII, Leica, Deerfield, IL) with either bright field illumination or violet light for excitation of GFP (filter set GFP1: excitation 425/60 nm, barrier 480 nm) and photographed on Ektachrome EPT 160T or EES P1600X (Fig. 7D) film (Eastman-Kodak, Rochester, NY). Fluorescent infection sites and injections were also monitored using an MRC 1000 CLSM (Bio-Rad, Hercules, CA) equipped with a 25-mW krypton/argon laser. For GFP imaging, blue excitation at 488 nm with an emission filter of 522 DF 32 nm was used, and for both sulforhodamine B and Texas Red, green excitation at 568 nm with an emission filter of 605 DF 32 nm was used. False color was applied to CLSM images using Photoshop (Adobe Systems, Mountain View, CA).
Scanning Electron Microscopy
At various times after plants were shifted from 32°C to 20°C leaves were examined under long wavelength UV light using a Blak-ray hand-held lamp (UV Products, Upland, CA) to identify fluorescent infection foci. Areas of leaf carrying infection foci (5 × 5 mm) were excised with a scalpel and prepared for LTSEM as described by Glidewell et al. (1999). Briefly, frozen hydrated samples prepared by immersion in nitrogen slush (−210°C) were sputter coated with gold in an SP2000 Sputter Cryo System (Emscope Laboratories, Ashford, UK) before imaging under a T200 scanning electron microscope (JEOL, Tokyo) fitted with a macro stage. Photographs were taken at 10 kV using TM100 film (Kodak).
Evans Blue Staining
Detached leaves, completely submerged in a 0.1% (w/v) aqueous solution of Evans blue (Sigma, St. Louis, MN), were subjected to two 5-min cycles of vacuum followed by a 20-min maintenance under vacuum. The leaves were then washed by vacuum infiltration of phosphate-buffered saline plus 0.05% (v/v) Tween for 3 × 15 min.
Micro-Injection
Micro-injection of a 5 mm aqueous solution of sulforhodamine B (Sigma) was performed as described previously using a modified pressure probe to prevent vacuolar rupture during impalement (Oparka et al., 1990).
Labeling of Xylem with Texas Red
The xylem network was labeled by detaching inoculated leaves and immersing the cut petiole in a solution containing 20 μg mL−1 Texas Red (Molecular Probes, Eugene, OR) for 30 min prior to imaging. Analysis of dye transport through non-transpiring tissue was performed using leaves that had been treated 16 h prior to labeling by the application of vacuum grease (Dow Corning, Munich) to circular (5.5-mm diameter) opposed areas of the adaxial and abaxial epidermis.
ACKNOWLEDGMENT
We are grateful to Bill Dawson for providing the TMV vector cDNA plasmid p30B.
Footnotes
The Scottish Crop Research Institute is grant-aided by the Scottish Executive Rural Affairs Department.
LITERATURE CITED
- Bendahmane A, Kanyuka K, Baulcombe DC. The Rx gene from potato confers separate virus resistance and cell death responses. Plant Cell. 1999;11:781–791. doi: 10.1105/tpc.11.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaerle L, Van Caeneghem W, Messens E, Lambers L, Van Montagu M, Van Der Straeten D. Presymptomatic visualization of plant-virus interactions by thermography. Nat Biotech. 1999;17:813–816. doi: 10.1038/11765. [DOI] [PubMed] [Google Scholar]
- Chivasa S, Murphy AM, Naylor M, Carr JP. Salicylic acid interferes with tobacco mosaic virus replication through a novel salicylhydroxamic acid-sensitive mechanism. Plant Cell. 1997;9:547–557. doi: 10.1105/tpc.9.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SR. Nicotiana hybrid developed as a host for plant viruses. Plant Dis Rep. 1969;53:939–941. [Google Scholar]
- Dangl JL, Dietrich RA, Richberg MH. Death don’t have no mercy: cell death programs in plant-microbe interactions. Plant Cell. 1996;8:1793–1807. doi: 10.1105/tpc.8.10.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney TP, Ukness S, Vernooij B, Friedrich L, Weymann K, Negrotto D, Gaffney T, Gut-Rella M, Kessmann H, Ward E, Ryals J. A central role of salicylic acid in plant disease resistance. Science. 1994;266:1247–1250. doi: 10.1126/science.266.5188.1247. [DOI] [PubMed] [Google Scholar]
- Doke N, Ohashi Y. Involvement of an O2− generating system in the induction of necrotic lesions on tobacco leaves infected with tobacco mosaic virus. Physiol Mol Plant Biol. 1988;32:163–175. [Google Scholar]
- Dunigan DD, Madlener JC. Serine/threonine protein phosphatase is required for tobacco mosaic virus-mediated programmed cell death. Virology. 1995;207:460–466. doi: 10.1006/viro.1995.1105. [DOI] [PubMed] [Google Scholar]
- Flor HH. Inheritance of pathogenicity of Melampsora lini. Phytopathology. 1942;32:653–669. [Google Scholar]
- Gaff DF, Okong ‘O-Ogola O. The use of nonpermeating pigments for testing the survival of cells. J Exp Bot. 1971;22:756–758. [Google Scholar]
- Glidewell SM, Williamson B, Duncan GH, Chudek JA, Hunter G. The development of blackcurrant fruit from flower to maturity: a comparative study by 3D nuclear magnetic resonance (NMR) micro-imaging and conventional histology. New Phytol. 1999;141:85–98. [Google Scholar]
- Goodman RN. The hypersensitive reaction in tobacco: a reflection of changes in host cell permeability. Phytopathology. 1968;58:872–873. [Google Scholar]
- Goodman RN, Novacky AJ. The Hypersensitive Reaction in Plants to Pathogens: A Resistance Phenomenon. St. Paul: APS Press; 1994. [Google Scholar]
- Hammond-Kosack KE, Silverman P, Raskin I, Jones JDG. Race-specific elicitors of Cladosporium fulvum induce changes in cell morphology and the synthesis of ethylene and salicylic acid in tomato plants carrying the corresponding Cf disease resistance gene. Plant Physiol. 1996;110:1381–1394. doi: 10.1104/pp.110.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath MC. The enigmatic hypersensitive response: induction execution and role. Physiol Mol Plant Pathol. 1999;55:1–3. [Google Scholar]
- Holmes FO. Local lesions in tobacco mosaic. Bot Gaz. 1929;87:39–55. [Google Scholar]
- Keen NT. Gene-for-gene complementarity in plant pathogen interactions. Annu Rev Genet. 1990;24:447–463. doi: 10.1146/annurev.ge.24.120190.002311. [DOI] [PubMed] [Google Scholar]
- Lacomme C, Santa Cruz S. Bax-induced cell death in tobacco is similar to the hypersensitive response. Proc Natl Acad Sci USA. 1999;96:7956–7961. doi: 10.1073/pnas.96.14.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RG. Electron microscopy of tobacco mosaic virus in leaves of Nicotiana glutinosa. Virology. 1966;28:527–532. doi: 10.1016/0042-6822(66)90237-6. [DOI] [PubMed] [Google Scholar]
- Mittler R, Lee S, Lam E. Pathogen-induced cell death in tobacco. J Cell Sci. 1997;110:1333–1344. doi: 10.1242/jcs.110.11.1333. [DOI] [PubMed] [Google Scholar]
- Mittler R, Shulaev V, Seskar M, Lam E. Inhibition of programmed cell death in tobacco plants during a pathogen-induced hypersensitive response at low oxygen pressure. Plant Cell. 1996;8:1991–2001. doi: 10.1105/tpc.8.11.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mur LAJ, Bi Y, Darby RM, Firek S, Draper J. Compromising early salicylic acid accumulation delays the hypersensitive response and increases viral dispersal during lesion establishment in TMV-infected tobacco. Plant J. 1997;12:1113–1126. doi: 10.1046/j.1365-313x.1997.12051113.x. [DOI] [PubMed] [Google Scholar]
- Oparka KJ, Murphy R, Derrick PM, Prior DAM, Smith JAC. Modification of the pressure probe technique permits controlled intracellular microinjection of fluorescent probes. J Cell Sci. 1990;99:557–563. [Google Scholar]
- Otsuki Y, Shimomura T, Takebe I. Tobacco mosaic virus multiplication and expression of the N gene in necrotic responding tobacco varieties. Virology. 1972;50:45–50. doi: 10.1016/0042-6822(72)90344-3. [DOI] [PubMed] [Google Scholar]
- Padgett HS, Beachy RN. Analysis of a tobacco mosaic virus strain capable of overcoming N gene-mediated resistance. Plant Cell. 1993;5:577–586. doi: 10.1105/tpc.5.5.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontier D, Gan S, Amasino RM, Roby D, Lam E. Markers for hypersensitive response and senescence show distinct patterns of expression. Plant Mol Biol. 1999;39:1243–1255. doi: 10.1023/a:1006133311402. [DOI] [PubMed] [Google Scholar]
- Richael C, Gilchrist D. The hypersensitive response: a case of hold or fold. Physiol Mol Plant Pathol. 1999;55:5–12. [Google Scholar]
- Ross FA. Localized acquired resistance to plant virus infection in hypersensitive hosts. Virology. 1961;14:329–339. doi: 10.1016/0042-6822(61)90318-x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Santa Cruz S, Chapman S, Roberts AG, Roberts IM, Prior DAM, Oparka KJ. Assembly and movement of a plant virus carrying a green fluorescent overcoat. Proc Natl Acad Sci USA. 1996;93:6286–6290. doi: 10.1073/pnas.93.13.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield SR, Tobias CM, Rathjen JP, Chang JH, Lavelle DT, Michelmore RW, Staskawicz BJ. Molecular basis of gene-for-gene specificity in bacterial speck disease of tomato. Science. 1996;274:2063–2065. doi: 10.1126/science.274.5295.2063. [DOI] [PubMed] [Google Scholar]
- Shivprasad S, Pogue GP, Lewandowski DJ, Hidalgo J, Donson J, Grill LK, Dawson WO. Heterologous sequences greatly affect foreign gene expression in tobacco mosaic virus-based vectors. Virology. 1999;255:312–323. doi: 10.1006/viro.1998.9579. [DOI] [PubMed] [Google Scholar]
- Weststeijn EA. Permeability changes in the hypersensitive reaction of Nicotiana tabacum cv. Xanthi nc. after infection with tobacco mosaic virus. Physiol Plant Pathol. 1978;13:253–258. [Google Scholar]
- Weststeijn EA. Lesion growth and virus localization in leaves of Nicotiana tabacum cv. Xanthi nc. after inoculation with tobacco mosaic virus and incubation alternately at 22°C and 32°C. Physiol Plant Pathol. 1981;18:357–368. [Google Scholar]
- Whitham S, Dinesh-Kumar SP, Choi D, Corr C, Baker B. The product of the tobacco mosaic virus resistance gene N: similarity to Toll and the interleukin-1 receptor. Cell. 1994;78:1101–1115. doi: 10.1016/0092-8674(94)90283-6. [DOI] [PubMed] [Google Scholar]
- Yu I, Parker J, Bent AF. Gene-for-gene disease resistance without the hypersensitive response in Arabidopsis dnd1 mutant. Proc Natl Acad Sci USA. 1998;95:7819–7824. doi: 10.1073/pnas.95.13.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Klessig DF. Resistance gene N-mediated de novo synthesis and activation of a tobacco mitogen-activated protein kinase by tobacco mosaic virus infection. Proc Natl Acad Sci USA. 1998;95:7433–7438. doi: 10.1073/pnas.95.13.7433. [DOI] [PMC free article] [PubMed] [Google Scholar]