

Abstract
Background
Glioblastoma (GBM) is the most common primary malignant brain cancer, and is currently incurable. Chimeric antigen receptor (CAR) T cells have shown promise in GBM treatment. While we have shown that combinatorial targeting of 2 glioma antigens offsets antigen escape and enhances T-cell effector functions, the interpatient variability in surface antigen expression between patients hinders the clinical impact of targeting 2 antigen pairs. This study addresses targeting 3 antigens using a single CAR T-cell product for broader application.
Methods
We analyzed the surface expression of 3 targetable glioma antigens (human epidermal growth factor receptor 2 [HER2], interleukin-13 receptor subunit alpha-2 [IL13Rα2], and ephrin-A2 [EphA2]) in 15 primary GBM samples. Accordingly, we created a trivalent T-cell product armed with 3 CAR molecules specific for these validated targets encoded by a single universal (U) tricistronic transgene (UCAR T cells).
Results
Our data showed that co-targeting HER2, IL13Rα2, and EphA2 could overcome interpatient variability by a tendency to capture nearly 100% of tumor cells in most tumors tested in this cohort. UCAR T cells made from GBM patients’ blood uniformly expressed all 3 CAR molecules with distinct antigen specificity. UCAR T cells mediated robust immune synapses with tumor targets forming more polarized microtubule organizing centers and exhibited improved cytotoxicity and cytokine release over best monospecific and bispecific CAR T cells per patient tumor profile. Lastly, low doses of UCAR T cells controlled established autologous GBM patient derived xenografts (PDXs) and improved survival of treated animals.
Conclusion
UCAR T cells can overcome antigenic heterogeneity in GBM and lead to improved treatment outcomes.
Keywords: EphA2, glioblastoma, HER2, IL13Rα2, universal CAR T-cells
Glioblastoma (GBM), the most common primary malignant brain cancer, is currently incurable. Even with optimal multimodal therapy, to date the median survival rate is less than 15 months. GBM cells overexpress distinct targetable antigens on their surface,1–4 and a number of these antigens, such as human epidermal growth factor receptor 2 (HER2), interleukin-13 receptor subunit alpha-2 (IL13Rα2), ephrin-A2 (EphA2), and epidermal growth factor receptor variant III (EGFRvIII), have been successfully targeted using T cells that express chimeric antigen receptors (CARs). CARs are artificial molecules with an antigen-recognition extracellular domain, usually derived from an antibody, fused to an intracellular zeta (ζ)-signaling chain of the T-cell receptor.5 We have shown that autologous HER2-CAR T cells killed primary GBM and GBM stem cells and induced regression of patient-derived xenografts (PDXs).6 More recently, we reported a phase I clinical trial in which 17 pediatric and adult patients with progressive GBM were infused systemically with autologous HER2-CAR T cells, establishing a favorable safety profile and demonstrating promising objective clinical responses and prolonged survival.7
While CAR T cells have high curative potential, the complex biology of GBM necessitates tumor-specific refinements in preclinical models to achieve complete tumor elimination.8,9 Challenges to CAR T-cell therapy include a hostile tumor microenvironment that hinders immune response sustenance, tumor antigen heterogeneity that renders GBM resistant to monotherapy, and poor T-cell trafficking to GBM augmented by the effect of the blood–brain barrier.10 High tumor antigenic heterogeneity is of particular pertinence to GBM. Interpatient heterogeneity hinders the development of a universal monovalent CAR T cell for GBM, in the same sense as do cluster of differentiation (CD)19 CAR T cells to B-cell acute lymphoblastic leukemia. Intrapatient heterogeneity results in the survival of CAR-targeted antigen-deficient variants in patients infused with monovalent CARs.11–13 Recent data from our group have shown a clear advantage for simultaneous targeting of a second glioma antigen. T cells coexpressing either 2 distinct CAR molecules or a single bivalent CAR (also known as tandem CAR or TanCAR) offset antigen escape and exhibited enhanced functionality.11,12 However, specific antigen pairs varied between patients, such that the need for generating permutations of bivalent T-cell products will make the successful clinical translation of this approach challenging.
Therefore, we explored whether targeting a third antigen could overcome this interpatient antigenic variability and could be a more universally applicable approach for the majority of GBM patients. Therefore, the goal of this work was to determine the frequency of 3 targetable antigens to assess the feasibility of killing nearly 100% of tumor cells treated in a cohort of GBM patients.
Materials and Methods
Blood Donors, Primary Tumor Cells, and Cell Lines
Blood samples from healthy donors and GBM patients and primary tumor cells from GBM patients were obtained on a protocol approved by the Baylor College of Medicine and Houston Methodist Hospital institutional review boards. Tumor samples were processed aseptically and primary cell cultures were initiated using Dulbecco’s modified Eagle’s medium (DMEM) with 15% heat-inactivated fetal calf serum (FCS), 2 mM GlutaMAX-I, 1% insulin-transferrin-selenium-X supplement, and 1% penicillin-streptomycin mixture (Invitrogen). Cells were used within 7 days of plating or established as primary cell lines.
The U373-GBM cell line was purchased from the American Type Culture Collection and maintained in DMEM with 10% FCS, 2 mM GlutaMAX-I, 1.5 g/L sodium bicarbonate, 0.1 mMol/L nonessential amino acids, and 1.0 mMol/L sodium pyruvate. T cells were maintained in T-cell media (250 mL Roswell Park Memorial Institute medium–1640, 200 mL Click’s Medium with 10% FCS containing 2 mMol/L GlutaMAX-I). All cell lines were validated using short tandem repeat analysis before use from the Characterized Cell Line Core Facility of the MD Anderson Cancer Center, Houston, Texas.
Construction, Delivery, and Expression of the UCAR-encoding Transgene
The IL13Rα2 binding IL-13 mutein, HER2-specific single-chain variable fragment (scFv), FRP5, and EphA2-specific scFv, 4H5, were previously described.14,15 The trivalent transgene—each consisting of the scFv followed by a short hinge, the transmembrane and signaling domain of the costimulatory molecule CD28, and the ζ-signaling domain of the T-cell receptor—was assembled on Clone Manager (Sci-Ed Software) separated by 2A sequences with restriction enzyme sites at the ends for cloning. The expression optimized UCAR transgene was synthesized by GeneArt Gene Synthesis service (Thermo Fisher Scientific), cloned into the Gateway entry vector pDONR 221, sequence-verified, subcloned in frame into an SFG retroviral vector, and the construct confirmed using restriction digests. The 5ʹ-3ʹ as well as the 3ʹ-5ʹ sequences of the whole construct were confirmed using pyrosequencing (SeqWright DNA-Technology) with >97% homology with the optimized construct map.
To produce UCAR retroviral supernatant, human embryonic kidney 293T cells were co-transfected with the UCAR-encoding retroviral transfer plasmid, Peg-Pam-e plasmid encoding MoMLV gag-pol, and plasmid containing the sequence for RD114 envelope, using GeneJuice (EMD Biosciences) and supernatants collected 48 and 72 hours later. Anti-CD3 (OKT3)/anti-CD28–activated T cells were transduced with retroviral vectors as described in the Supplemental Methods.
Flow Cytometry
Surface staining of tumor cells was done using a goat anti-human IL13Rα2-allophycocyanin, a mouse anti-human HER2-phycoerythrin (PE), and a mouse anti-human EphA2–Alexa Fluor 488 (R&D Systems). Cell surface expressions of FRP5 (HER2 CAR), IL-13 mutein, and EphA2 CAR were detected separately using HER2 Fc, IL13Rα2.Fc (R&D Systems), and EphA2.GST (Thermo Fisher Scientific) chimeric proteins, respectively, followed by PE-conjugated goat anti-human Fc (Thermo Fisher Scientific) for HER2.Fc and IL13Rα2.Fc and anti–glutathione S-transferase (GST)-PE (Abcam) for EphA2.GST. After 30-minute incubation at 4°C in the dark, cells were washed with fluorescence activated cell sorting buffer (PBS containing 2% FBS and 0.1% sodium azide) and fixed in 0.5% paraformaldehyde for analysis. Analysis was done on Gallios (Beckman Coulter) or Accuri C6 (Becton Dickinson). Kaluza (Beckman Coulter) or FlowJo data analysis software was used for all analyses.
Confocal Imaging of CAR Synapse
For confocal microscopy, conjugates between CAR T cells and GBM cells were incubated for 45 minutes at 37°C and then fixed, permeabilized, and stained for F-actin (phalloidin), perforin, and α-tubulin. Conjugates were imaged as Z stacks of 0.2 micron thickness to cover the entire volume of the immunological synapse, determined individually for each conjugate, on a Zeiss Axio-Observer Z1 equipped with a Yokogawa CSU10 spinning disc, Zeiss 63X 1.43 NA objective, and Hamamatsu Orca-AG camera. Images were acquired and analyzed with Velocity software (PerkinElmer).
Analysis of T-Cell Cytokine Production
T cells were co-cultured with autologous GBM or U373 cells at equal ratios (1 × 105 cells), and levels of interferon (IFN)-γ and IL-2 were determined in conditioned-culture supernatants 24 hours post incubation using an enzyme-linked immunosorbent assay (ELISA) per manufacturer’s instructions (R&D Systems).
To assess T-cell activation upon encountering immobilized target, nontissue culture treated 24-well plates (BD Falcon) were kept overnight at 4°C with HER2.Fc (range 0–0.8 µg/mL), IL13Rα2.Fc (range 0–10 µg/mL), EphA2.Fc (range 0–10 µg/mL; R&D Systems), or an irrelevant target (monoclonal anti-idiotype 1A716; TriGem Titan) and a nonspecific T-cell receptor stimulant (OKT3). After T-cell incubation for 24 hours at 37°C, the supernatant was analyzed for IFN-γ and IL-2.
Cytotoxicity Assays
Cytolytic activity of T cells was assessed using a chromium-51 (51Cr) assay described in the Supplementary Methods as earlier.17
Orthotopic Xenogeneic SCID Mouse Model of GBM
All animal experiments were conducted according to protocol AN-3949 approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Recipient nonobese diabetic severe combined immunodeficient (NOD-SCID) mice (IcrTac-Prkdcscid), 4 groups of 5 mice each per PDX, were purchased from Taconic Biosciences. Patient derived GBM cell lines (transduced with enhanced green fluorescent protein [eGFP]. Firefly luciferase fusion gene) and autologous T cells were injected into the right caudate nucleus of mouse brain as described in the Supplementary Methods.
All animals with progressively growing xenografts were randomly assigned a condition of treatment with nontransduced (NT) T cells, best monospecific T cells (EphA2 for UPN001 and IL13Rα2 for UPN005), best bispecific CAR T cells (IL13Rα2 and EphA2 biCAR for UPN001 and UPN005), or UCAR T cells and received an intratumoral injection of 1 × 106 T cells on days 5 and 12 following tumor injection. Bioluminescence imaging was performed using the Xenogen IVIS in vivo imaging system as mentioned in the Supplementary Methods.
Immunofluorescence Imaging
Brains were collected from mice at the end of the experiment or upon death and embedded in optimal cutting temperature compound. Brain tumor xenografts (n = 3), from NT and UCAR treated mice, were sectioned to 7-µm frozen sections in a cryomicrotome. Sections were fixed with methanol/acetone, blocked with 5% horse serum, incubated overnight at 4°C with primary antibodies, mouse anti-HER2 (Abcam) in 1:10 dilution, rabbit anti-EphA2 (Cell Signaling) in 1:100 dilution, and goat anti-IL13Rα2 (R&D Systems) in 1:10 dilution. Slides were incubated for 1 hour at room temperature in secondary antibodies diluted at 1:200 (anti-mouse 488, anti-rabbit 568, and anti-goat 647, respectively; Invitrogen). Microscopy images of 4′,6′-diamidino-2-phenylindole (DAPI) counterstained slides were captured using a Zeiss Axioimager spinning disc confocal microscope at 40x magnification. Nucleus-tethered quantifications of cells expressing HER2, IL13Rα2, and EphA2 were performed on 20 high power fields collectively from 3 mice in each group using ImageJ software (National Institutes of Health).
Statistical Analysis
Data were summarized using descriptive statistics. Elliptical Venn diagrams were constructed using the Euler APE drawing tool (University of Kent Computing).18 Comparisons of percentage tumor coverage when targeting 1, 2, or 3 tumor antigens were made using the Wilcoxon signed rank test. For ELISA, cytotoxicity assays, and immunofluorescence quantifications, comparisons between groups were carried out using one-way ANOVA or t-test. P-values were adjusted for multiple comparisons using the Hommel method or Tukey’s test when appropriate. The Kaplan–Meier method was used to estimate survival curves, and the Gehan-Breslow-Wilcoxon test was used to compare the curves. GraphPad Prism 7 software and SAS 9.4 were used for statistical analysis. A P-value of less than 0.05 was considered significant.
Results
Interpatient Antigenic Variability in Primary GBM
Individual GBMs show substantial intercellular heterogeneity in the surface expression of glioma antigens,1–4,12,19,20 yet interpatient heterogeneity of antigen expression has not been well characterized.21,22 We thus used multicolor flow cytometry to study the simultaneous single-cell expression pattern of 3 targetable glioma-restricted antigens, HER2, IL13Rα2, and EphA2, in 15 primary GBM samples obtained from surgical resections (Fig. 1A and 1B; Supplementary Table S1). All 3 antigens were expressed in this cohort except in 2 patient lines, but the percentage of cells in each patient tumor and the percentage of cells included in various permutations of antigen pairs varied from patient to patient (Supplementary Table S2). In 6 of 15 patients (40%), targeting HER2 and EphA2 was shown to result in the highest percentage of tumor killing, while 7/15 (47%) patients were correlated to EphA2 and IL13Rα2, and 2/15 (13%) to HER2 and IL13Rα2. In order to assess superiority of trivalent CAR T-cell product for patients in the studied cohort, we analyzed percentage tumor coverage when targeting 1, 2, and 3 tumor antigens in each patient. It was observed that targeting all 3 antigens provided significant tumor coverage over 2 antigens (P = 0.0001), which had superior coverage over targeting a single antigen (P = 0.0001; Supplementary Table S3).
Fig. 1.

Antigen expression pattern of HER2, IL13Rα2, and EphA2 for 15 primary patient GBM samples. Patient tumor samples were co-stained for all 3 antigens, and ≥100000 primary GBM cells were simultaneously interrogated using flow cytometry. (A) Sample of flow cytometry histograms for patient UPN001. (B) Euler diagrams with ellipsis representing the percentage of cells in patient tumor expressing each antigen. Areas of overlap indicate percentage of cells expressing multiple antigens.
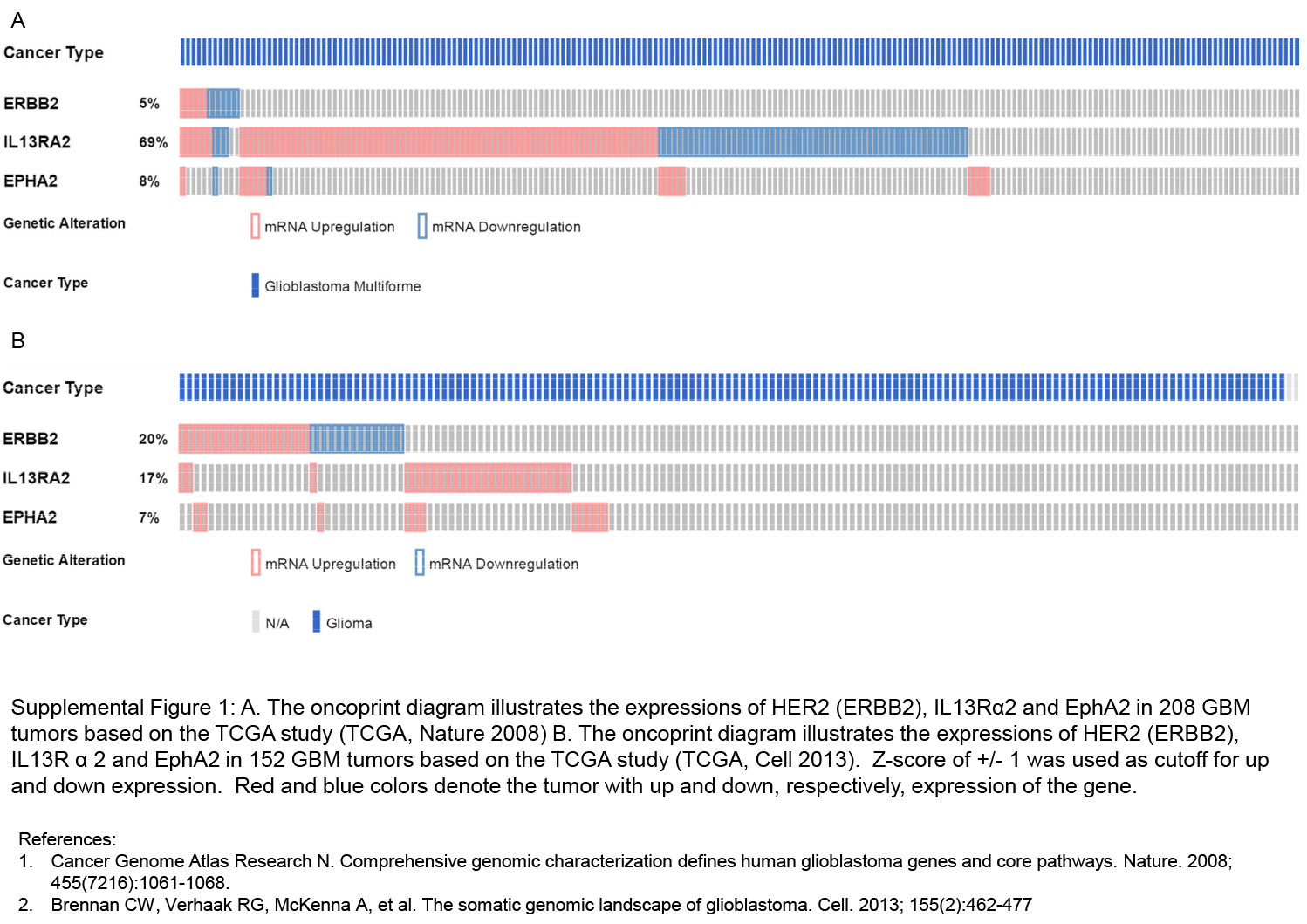
Further, RNA expression analyses were performed for these antigens (Supplementary Figure S1) on 2 large cohorts of GBM primary tumors (n = 206 and n = 152) from the glioblastoma datasets of The Cancer Genome Atlas via cBioPortal.23,24 In the Nature 2008 dataset, the oncoprint result showed that HER2 (Erb-B2 receptor tyrosine kinase 2 [ERBB2]), IL13Rα2, and EphA2 were differentially expressed in 5%, 69%, and 8% of the GBM patients (z-score = +/− 1). More importantly, 44% (n = 92/206) of the patients had at least one of the 3 genes upregulated in the tumors. In the Cell 2013 dataset, HER2 (ERBB2), IL13Rα2, and EphA2 were differentially expressed in 20%, 17%, and 7% of the GBM patients (z-score = +/− 1; Supplementary Fig. S1) with 32% (n = 48/152) of the patients having at least one of the 3 genes upregulated in the tumors.
A Single Tricistronic Transgene Encoding HER2, IL13Rα2, and EphA2 CAR Molecules Renders GBM Patients’ T Cells Trivalent
After concluding that a single T-cell product targeting these 3 glioma antigens could potentially overcome interpatient variability, we used a single tricistronic vector encoding 3 CAR molecules, specific for HER2, IL13Rα2, and EphA2 (Fig. 2A). The DNA construct successfully packaged 3 second-generation (CD28 ζ-signaling domain) CAR-encoding transgenes, and using a strategy specific for each individual CAR molecule, we detected CAR molecules in proportionate percentages on the surface of donor T cells (Fig. 2B) by flow cytometric analysis.
Fig. 2.

Expression of 3 separate CAR molecules simultaneously on the surface of T cells and their antigen specificity. (A) Plasmid map showing the single transgene encoding for 3 CAR molecules in tandem separated by viral 2A sequences. (B) Staining for 3 CAR molecules by flow cytometry in T cells for 2 separate donors. (C) IL-2 and IFN-gamma production by UCAR and NT T cells when exposed to plate-bound antigens HER2, IL13Rα2, and EphA2 measured by ELISA. ***P < 0.001. (D) Raji tumor force express model for humanized HER2 antigen (Raji-hHER2), IL13Rα2 antigen (Raji-hIL13Rα2), or EphA2 antigen (Raji-hEphA2) to test specificity of tumor killing by UCAR T cells. The reporter green fluorescent protein gene detection by flow cytometry is shown for each. (E) Four-hour 51Cr cytotoxicity assay demonstrating tumor killing of various CAR T-cell products with Raji cells that contain single tumor antigens and negative (Raji) and positive (U373) controls.
To test whether UCAR T-cells could distinctly recognize all 3 tumor antigens, IFN-γ and IL-2 release were measured in culture supernatants 24 hours after plate-bound stimulation with recombinant human (rh)HER2, rhIL13Rα2, or rhEphA2. UCAR T-cells released higher levels of IFN-γ and IL-2 compared with controls upon culture with all 3 individual plate-bound targets (Fig. 2C).
Further, we selected Raji cells, negative for all 3 antigens, as a platform to generate model tumor cells positive for single antigens. We used a lentiviral vector to generate IL13Rα2+ (Raji-IL13Rα2), HER2+ (Raji-HER2), or EphA2+ (Raji-EphA2) cell lines to test the antitumor activity of UCAR T cells against a cell platform expressing individual antigens (Fig. 2D).25 UCAR T cells showed significant killing of Raji-IL13Rα2, Raji-HER2, and Raji-EphA2 cells individually, while monovalent CAR T cells only recognized and killed the tumor cells expressing their respective target antigen (Fig. 2E). The glioblastoma line U373 expressing all 3 antigens (HER2: 77%; IL13Rα2: 80%, and EphA2: 81%) was also killed by UCAR T cells. We thus concluded that the UCAR is cytolytic to tumor cells expressing any of the target tumor antigens.
Visualization of the CAR Immune Synapse Reveals Improved Cytolytic Properties
The in vitro studies prompted us to examine the ultrastructural characteristics of the CAR immune synapse (CARIS) and the lytic machinery of UCAR T cells. F-actin polymerization at the CARIS, perforin containing lytic granule convergence to the microtubule organizing center (MTOC), and MTOC recruitment to the CARIS have been previously established as indicators of enhanced cytotoxic CAR synapse.11 U373 tumor cells were co-cultured with NT, HER2 CAR, and UCAR T cells and were stained for perforin, phalloidin, and α-tubulin to detect the MTOC (Fig. 3A). Compared with NT and HER2 CAR T cells, UCAR T cells had higher F-actin accumulation at the CARIS and significantly less distance on average between the synapse and the MTOC, indicating enhanced cytolytic potential of these CAR T cells (Fig. 3B). Increased F-actin polymerization at the synapse is one of the first steps for cytotoxic synapse formation and indicated greater cytolytic ability of these cells.11,26 The convergence of granules was not significantly different between UCAR and HER2 CAR T cells but was much higher for both than for NT cells. These findings collectively demonstrate that UCAR T cells have enhanced CARIS organization, which correlates with their enhanced cytolytic function.
Fig. 3.

Immune synapse (IS) imaging of CAR T-cell/tumor interface demonstrates enhanced cytolytic potential of UCAR T cells. (A) Conjugates of U373 and UCAR, HER2-CAR, and NT T cells stained and incubated with U373 cells. Co-stains performed for perforin (green), the microtubule organizing center (MTOC; α-tubulin, blue), and phalloidin (F-actin, red). Scale bar = 5 µm. (B) Distance of MTOC from IS measured for different combinations of T cells with U373.
UCAR T Cells Exhibit Improved In Vitro Effector Functions Against Autologous Primary Tumor Cells
We hypothesized that UCAR T cells would have improved activation and antitumor efficacy and tested UCAR T-cell effector function in a clinically relevant autologous system using matched patient tumor and T cells. The autologous UCAR T cells produced from UPN001 had 81%, 82%, and 77%, while UPN003 had 83%, 82%, and 79% of HER2, IL13Rα2, and EphA2 CAR expression, respectively (Fig. 4A). UPN001 T cells had 59% and 36%, UPN003 had 44% and 53% of CD8+ T cells and CD4+ T cells, respectively (Fig. 4B). The UCAR T cells did not show more exhaustion during transduction and propagation when compared with respective NT T cells (Supplementary Figure S2). The in vitro doubling times of the lines UPN001, UPN003, UPN005, and UPN007 were 46, 37, 45, and 61 hours, respectively (Supplementary Figure S3). We compared monovalent (CAR T cells) and bivalent (biCAR) T-cell products to UCAR T cells in an autologous setup for the GBM patients UPN001, UPN003, UPN005, and UPN007. IFN-γ secretion after 24 hours was significantly higher with UCAR T cells over other CAR T cells in 3 of 4 patients (one-way ANOVA; P-value < 0.05; Fig. 4C). A similar trend was observed for IL-2 release (Fig. 4D).
Fig. 4.

In vitro activity of UCAR, biCAR, single CAR, and NT T cells in autologous GBM patient models. (A) Staining for 3 CAR molecules by flow cytometry in T cells for UPN001 and UPN003. (B) CD4/CD8 distribution of autologous T cells after transduction and expansion. (C) IFN-gamma production and (D) IL-2 production by UCAR, biCAR, single CAR, and NT T cells derived from patient peripheral blood mononuclear cells (PBMCs) when exposed to patient primary tumor samples measured by ELISA. (E) Four-hour 51Cr cytotoxicity assay demonstrating tumor killing of UCAR, biCAR, single CAR, and NT T cells derived from patient PBMCs when exposed to patient primary tumor samples. *P < 0.05, **P < 0.01, and ***P < 0.001.
To assess the cytolytic activity in this autologous system, a 51Cr cytotoxicity assay was performed (Fig. 4E). Cytotoxicity was higher with biCAR T cells than single CAR T cells, while UCAR T cells consistently showed more killing compared with biCAR T cells in all 4 patients. Collectively, these ex vivo results indicate that UCAR T cells have enhanced in vitro antiglioma functionality against autologous GBM tumor cells.
Adoptive Transfer of UCAR T Cells Improves Elimination of Established Autologous Orthotopic PDXs
We established orthotopic PDXs from 2 patients (UPN001 and UPN005), modified with an eGFP.Firefly luciferase fusion reporter gene, to assess the in vivo anti-GBM functionality of UCAR T cells. In order to dissect the difference in efficacy between cellular products, we performed a “stress test” experiment wherein relatively large GBM PDXs (cellular doubling time ~45 h) were treated with 1 × 106 autologous T cells on days 5 and 12 post tumor injection (Fig. 5A and 5B). Tumors derived from patients UPN001 and UPN005 grew exponentially in NT T-cell injected animals. NT T-cell injection did not alter the growth pattern. Mice treated with single CAR T cells targeting the most prevalent antigens in these tumors showed only transient antitumor responses after T-cell injections. Mice with UPN005 PDX tumors treated with biCAR T cells also demonstrated transient antitumor responses. In contrast, antitumor responses to UCAR T cells were sustained and all treated mice were alive >60 days for UPN001, and 4 of 5 treated mice were alive >60 days for UPN005. The UCAR-treated mice had significantly better survival compared with biCAR T-cells and single CAR T-cells treated mice in PDX UPN005 (P = 0.0204 and P = 0.02, respectively, Gehan-Breslow-Wilcoxon test), while the UCAR-treated mice had significantly better survival compared with single CAR T-cells treated mice in PDX UPN001 (P = 0.0174, Gehan-Breslow-Wilcoxon test). Collectively, UCAR T cells mediated significantly higher and more sustained antitumor effects compared with the other products and induced a significantly longer survival in treated animals (Fig. 5C).
Fig. 5.

In vivo experiments demonstrate superior antitumor activity and survival in UCAR-treated mice in an autologous tumor model; 2.5 × 105 patient GBM cells were stereotactically injected into the right caudate nucleus of SCID mice. On days 5 and 12 after tumor cell injection (indicated by arrows), mice received an intratumoral injection of 3 × 106 autologous best single CAR (IL13Rα2), best biCAR (IL13Rα2 and EphA2), UCAR T cells, or NT T cells normalized for transduction efficiency. (A) Bioluminescence imaging to monitor tumor size for mice injected with 2 patient primary tumor samples and followed by treatment with UCAR, biCAR, single CAR, and NT T cells derived from the same patient’s peripheral blood mononuclear cells. Median bioluminescence (solid line) and individual mouse data (dashed lines) are shown for mice in each group (n = 5 for each group). (B) Representative images of the bioluminescence imaging to monitor tumor size. (C) Kaplan–Meier curves for the in vivo experiments followed to 60 days post tumor injection.
Further, we characterized the expression of glioma antigens HER2, EphA2, and IL13Rα2 on mice explants of PDXs UPN001 and UPN005 post treatment (Fig. 6A). In a control group treated with NT T cells, we found that the growing tumor xenografts had detectable levels of all 3 antigens. Post UCAR T-cell treatment, the recurrent tumors had expression of the 3 antigens, albeit at a lower level, except IL13Rα2 in UPN001 (Fig. 6B). The results show significant antigen loss (P < 0.01 to P < 0.001) in both PDXs in HER2 and EphA2, and in IL13Rα2 in UPN005 on recurrent tumors post treatment with UCAR T cells.
Fig. 6.

(A) Characterization of UPN001 and UPN005 xenografts post treatment from frozen mouse brain sections. GBM tumor xenografts of mice (N = 3) from the NT and UCAR T-cell treated groups were characterized post treatment using immunofluorescence staining for HER2 (green), EphA2 (red), and IL13Rα2 (purple), DAPI (blue), at 40x magnification. Representative images shown. Scale bar = 20 µm. (B) Dot plots representing HER2, EphA2, and IL13Rα2 positively stained cells in brain sections of mice quantified collectively from 20 high power fields in each group using ImageJ. **P < 0.01 and ***P < 0.001.
Discussion
Genetic and epigenetic interrogation of GBM reveals spatial heterogeneity. Despite this known intratumor heterogeneity, interpatient heterogeneity of antigen expression in GBM has not been well characterized.21,22 Here, we demonstrate a heterogeneous antigenic landscape in primary glioblastoma lines of 15 GBM patient samples. As varying expression patterns in areas of GBM could render some tumor cells resistant to targeted therapies, we created a multispecific yet targeted approach to parallel this intricate hierarchy of antigenic expression by targeting HER2, IL13Rα2, and EphA2. We demonstrated that trivalent UCAR T cells were able to approach killing 100% of tumor cells in nearly all patients modeled. UCAR T cells can overcome this intrapatient and interpatient antigenic variability in GBM and mediate enhanced antiglioma effector functions compared with mono- and bivalent CAR T-cell products.
In previous studies, we created and tested a clinically relevant bivalent T-cell product12; however, our antigenic profiling in a larger patient cohort (n = 15) showed that the odds of capturing the bulk of tumor cells by targeting 3 glioma antigens simultaneously were superior to any bivalent combination. Further, the degree of variability in the optimal bivalent combinations between patients justifies targeting all 3 glioma antigens with one immunotherapeutic product. While one bivalent combination (EphA2 and IL13Rα2) resulted in a higher median percentage of cells targeted for the majority, there was no universally favorable combination for the whole cohort. Thus, we reasoned that a trivalent product could lead to better tumor cell killing by extending the reach of effectors to the majority of tumor cells in all patients, broadening its therapeutic reach. Indeed, UCAR T cells showed superiority in autologous in vitro and in vivo experiments.
Our group and others have explored strategies to co-deliver multiple transgenes using retroviral constructs expressing 2A sequences.27,28 We demonstrated that these UCAR T cells can be efficiently generated and multiple CARs with distinct specificity can be comparably transduced by retroviral transduction using viral 2A sequences and consistently expressed without compromising T-cell activation, proliferation potential, or antitumor activity. Additionally, the trivalent activation and proliferation as demonstrated by surface staining and autologous in vitro models confirm that the 3 CAR molecules were expressed in their entirety in a proportionate and functional form.
Several attributes could explain the superiority of UCAR T cells to their bivalent CAR and monovalent CAR counterparts, including potentially enhanced signaling, engagement of a larger domain of GBM cells, and/or their ability to mediate a robust immune synapse.11 Indeed, our super-resolution imaging results indicated better organization of the killing machinery embodied in the proximity of the MTOC to the T-cell contact point with the tumor and better perforin clustering.26 Such arrangement indicates that the subsynaptic structure in the UCAR T cell is better poised for target cell killing. This could explain, at least in part, their superior activity against autologous GBM.
While this product can move to clinical trials, the optimization of a multispecific T-cell product for GBM may include modifications to enhance efficacy and decrease the likelihood of adverse effects where systemic administration is intended. These could include modifying or adding signaling domains to promote persistence of T cells or induce favorable phenotypes,29,30 adding a safety mechanism in the event of severe adverse events,31,32 or including or exchanging candidate CAR molecules for other known GBM tumor antigens, such as EGFRvIII.33
A recently completed pilot study showed feasibility, tolerability, and some evidence of efficacy of a trivalent peptide based vaccine targeting 3 validated surface-expressed glioma antigens—EphA2, IL13Rα2, and survivin—in pediatric malignant gliomas.34 The choice of target antigens was based on heterogeneous expression of these antigens in a previous study19 with development of a product that targeted tumor cells in nearly all patients with pediatric malignant gliomas. This product is effective only in human leukocyte antigen (HLA)-A2 positive patients, while a multivalent CAR T-cell approach is not limited by HLA restriction and thus may have broader applicability in this patient population.
This proof of concept could lead to applications in other diseases. While CAR therapy has shown lasting remissions in patients with relapsed acute lymphoblastic leukemia (ALL) and chronic lymphoblastic leukemia, failure of CD19 CAR T-cell therapy for ALL has been associated with development of CD19-negative tumor clones that may represent escape variants.35 The probability of antigen escape may be reduced if T cells are engineered to recognize multiple disease-specific B-cell antigens—such as CD22, CD20, or receptor tyrosine kinase-like orphan receptor 1 (ROR1)—in addition to CD19.13,36 In a wide spectrum of cancers, multiplex targeting of tumor antigens with T cells represents a platform to impact outcomes by targeting tumor tissue specifically while preventing escape variants. To our knowledge this is the first product successfully targeting 3 antigens with promise of broad spectrum activity to impact patient outcomes.
In summary, we have shown in a cohort of GBM patients’ tumors that simultaneous targeting of 3 glioma antigens with autologous trivalent CAR T cells, developed using a universal tricistronic construct, could overcome interpatient antigen variability. UCAR T cells exhibited enhanced antiglioma activity and better tumor control in 2 established, autologous orthotopic PDX mouse models.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was funded by the Childhood Brain Tumor Foundation (CBTF) Astrocytoma Grant, Alex’s Lemonade Stand Pediatric Cancer Foundation (ALSF), Alliance for Cancer Gene Therapy (ACGT, Inc), R01AI067946 (to J.S.O.), and Stand Up to Cancer–St Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113). Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. K.F. and T.T.B. were supported by NIH grants T32HL092332 (to Dr Helen Heslop) and by T32GM088129 from the National Institute of General Medical Sciences (NIGMS). This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or NIH.
Conflict of interest statement.
K.B., T.T.B., M.H., M.M., J.S.O., and N.A. have patent applications in the field of T-cell and gene-modified T-cell therapy for cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Ms Catherine Gillespie for the professional editing of the manuscript. We also thank Dr Stephen Gottschalk for providing the lentiviral vectors encoding IL13Rα2 and EphA2 used to generate the transgenic Raji cell lines.
References
- 1. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3(10):541–551. [DOI] [PubMed] [Google Scholar]
- 2. Zhang JG, Eguchi J, Kruse CA et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. 2007;13(2 Pt 1):566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67(17):7983–7986. [DOI] [PubMed] [Google Scholar]
- 4. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980–4986. [DOI] [PubMed] [Google Scholar]
- 5. Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. 2001;248(1–2):67–76. [DOI] [PubMed] [Google Scholar]
- 6. Ahmed N, Salsman VS, Kew Y et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmed N, Brawley V, Hegde M et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garfall AL, Maus MV, Hwang WT et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373(11):1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown CE, Alizadeh D, Starr R et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sengupta S, Mao G, Gokaslan ZS, Sampath P. Chimeric antigen receptors for treatment of glioblastoma: a practical review of challenges and ways to overcome them. Cancer Gene Ther. 2017;24(3):121–129. [DOI] [PubMed] [Google Scholar]
- 11. Hegde M, Mukherjee M, Grada Z et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hegde M, Corder A, Chow KK et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grada Z, Hegde M, Byrd T et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chow KK, Naik S, Kakarla S et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azemar M, Djahansouzi S, Jäger E et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res Treat. 2003;82(3):155–164. [DOI] [PubMed] [Google Scholar]
- 16. Yvon E, Del Vecchio M, Savoldo B et al. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15(18):5852–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gottschalk S, Edwards OL, Sili U et al. Generating CTLs against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood. 2003;101(5):1905–1912. [DOI] [PubMed] [Google Scholar]
- 18. Micallef L, Rodgers P. Euler APE: drawing area-proportional 3-venn diagrams using ellipses. PLoS One. 2014;9(7):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88(3):245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin Cancer Res. 2008;14(1):199–208. [DOI] [PubMed] [Google Scholar]
- 21. Kumar A, Boyle EA, Tokita M et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014;15(12):530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aubry M, de Tayrac M, Etcheverry A et al. From the core to beyond the margin: a genomic picture of glioblastoma intratumor heterogeneity. Oncotarget. 2015;6(14):12094–12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brennan CW, Verhaak RG, McKenna A et al. ; TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krebs S, Chow KK, Yi Z et al. T cells redirected to interleukin-13Rα2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy. 2014;16(8):1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell. 2010;21(13):2241–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quintarelli C, Vera JF, Savoldo B et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110(8):2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szymczak-Workman AL, Vignali KM, Vignali DA. Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb Protoc. 2012;2012(2):199–204. [DOI] [PubMed] [Google Scholar]
- 29. Long AH, Haso WM, Shern JF et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawalekar OU, O’Connor RS, Fraietta JA et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. [DOI] [PubMed] [Google Scholar]
- 31. Casucci M, Bondanza A. Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes. J Cancer. 2011;2:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Stasi A, Tey SK, Dotti G et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson LA, Scholler J, Ohkuri T et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015; 7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollack IF, Jakacki RI, Butterfield LH et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol. 2014;32(19):2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sotillo E, Barrett DM, Black KL et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.