

Abstract
Increasing evidence suggests that Alzheimer's disease pathogenesis is not restricted to the neuronal compartment but strongly interacts with immunological mechanisms in the brain. Misfolded and aggregated proteins bind to pattern recognition receptors on micro- and astroglia and trigger an innate immune response, characterized by the release of inflammatory mediators, which contribute to disease progression and severity. Genome wide analysis suggests that several genes, which increase the risk for sporadic Alzheimer's disease en-code for factors that regulate glial clearance of misfolded proteins and the inflammatory reaction. External factors, including systemic inflammation and obesity are likely to interfere with the immunological processes of the brain and further promote disease progression. This re-view provides an overview on the current knowledge and focuses on the most recent and exciting findings. Modulation of risk factors and intervention with the described immune mechanisms are likely to lead to future preventive or therapeutic strategies for Alzheimer's disease.
Introduction
At first glance, realms of immunology and neurobiology could not be further apart. From a cellular perspective, the brain represents stasis whereas the immune system represents motion. These two perspectives come together as the importance of neurodegenerative disease is increasingly appreciated. Indeed, understanding and controlling their interactions may hold the keys to the prevention or delay of a majority of late onset CNS diseases. In Alzheimer's disease (AD) neuroinflammation, instead of being a mere bystander activated by emerging senile plaques and neurofibrillar tangles, contributes as much or more to the pathogenesis as do the plaques and tangles themselves 1. This is underlined by recent findings that genes for immune receptors, including TREM2 2 and CD33 3,4, are associated with AD. Analysis of clinical manifestations that precede AD, such as mild cognitive impairment (MCI), further argue for an early and substantial involvement of inflammation in the pathogenesis of the disease. Therefore, we give a current view on the neuroinflammatory landscape during AD including the cell types and mediators involved, the ways used to visualize neuroinflammation, as well as its clinical presentation and potential treatments.
Cellular Players
Microglia
Microglia, the resident phagocytes of the CNS, are ubiquitously distributed in the brain. They constantly survey their assigned brain regions using their highly motile processes for the presence of pathogens and cellular debris, and simultaneously providing factors that support tissue maintenance (Figure 1) 5. At the same time, microglia contribute to the protection and remodeling of synapses for proper maintenance and plasticity of neuronal circuits 6. To some extent, this action is mediated by the release of trophic factors including brain derived neurotrophic factor, which contributes to memory formation 7. Once activated by pathological triggers, like neuronal death or protein aggregates, microglia extend their processes to the site of injury, later start migrating to the lesion, and initiate an innate immune response (Figure 2 A,B). The perception of pathological triggers is mediated by receptors originally designed to recognize danger or pathogen associated molecular patterns (DAMPs/PAMPs). In AD, microglia are able to bind to soluble amyloid β (Aβ) oligomers and Aβ fibrils via receptors including class A scavenger receptor A1, CD36, CD14, α6β1 integrin, CD47 and toll like receptors (TLR2, TLR4, TLR6 and TLR9) 8–11, which is thought to be part of the inflammatory reaction in AD. The Aβ peptide derives from a larger precursor, the amyloid precursor protein, by subsequent cleavages of two membrane-bound proteases (for review see 12). The initial cleavage is mediated by a protease termed BACE1 (β-site APP cleaving enzyme 1) followed by an unconventional cleavage by the γ-secretase complex within the transmembrane region of APP resulting in differentially truncated C-termini, ranging from amino acid 37 to 42 13 (Figure 3). In particular, the 42 amino acid long form of Aβ has a strong tendency to form the aforementioned soluble oligomers and fibrill. The binding of Aβ with CD36, TLR4 and TLR6 results in activation of microglia which start to produce proinflammatory cytokines and chemokines (Figure 4) 10,14. In turn, genetic deletion of CD36, TLR4, or TLR6 in vitro reduces Aβ-induced cytokine production 10,14,15 and prevents intracellular amyloid accumulation and inflammasome activation 15.
Figure 1. Pathomechanistic sequale of immune activation.
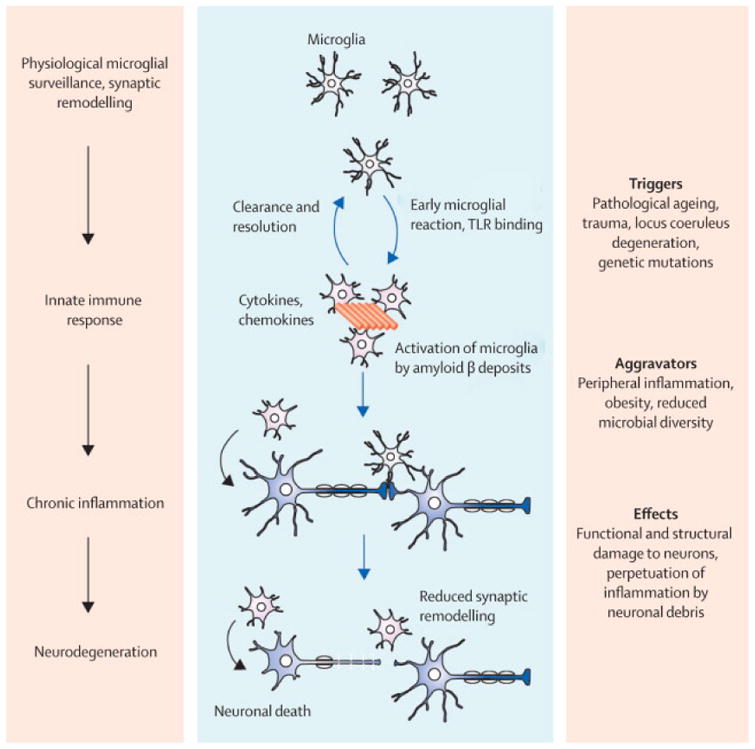
Physiological functions of microglia including tissue surveillance and synaptic remodelling are compromized when microglia sense pathological Aβ accumulations. Initially the acute inflammatory response is thought to aid the clearance and to restore tissue homeostasis. Triggering factors and aggravators promote the sustained exposure and immune activation which ultimately leads to chronic neuroinflammation. The perpetuation of microglial activation, persistent exposure to proinflammatory cytokines and process retraction, causes functional and structural changes which finally end in neuronal degeneration.
Figure 2. Micro- and astroglial changes in Alzheimer´s disease brain and APP/PS1 mice.
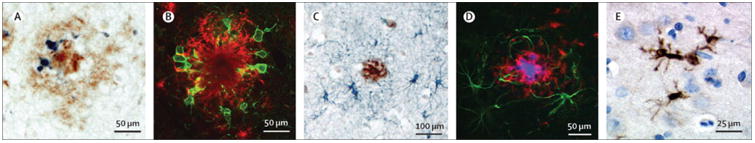
(A) CD11b positive microglia (blue) within a Aβ deposit (brown) in the parietal cortex of a human AD brain section (bar = 50 μm). (B) Activated, Iba1-positive microglia (green) at a Aβ plaque site (red) in a section of a APP/PS1 transgenic mouse (bar = 50 μm). (C) GFAP positive astrocytes (blue) surround the site of Aβ deposition (brown) in the parietal cortex of a human AD brain section (bar = 100 μm). (D) GFAP-positive astrocytes (green) at a Aβ plaque site (red) in a section of a APP/PS1 transgenic mouse (bar = 50 μm). (E) Interleukin-1β positive microglia (brown) in the frontal cortex of a human AD brain section (bar = 25 μm). Courtesy of Drs. Markus Kummer and Michael Heneka.
Figure 3. Amyloidogenic processing of the amyloid precursor protein (APP).
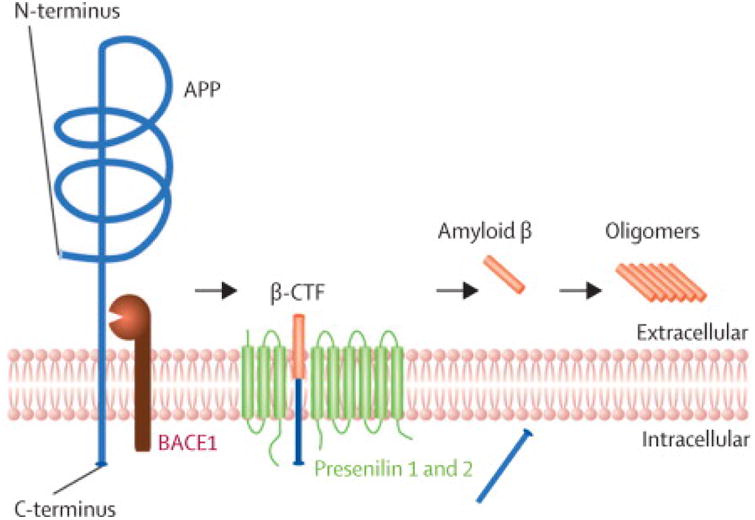
APP is processed by subsequent cleavages of two aspartate-proteases: the β-side cleaving enzyme 1 (BACE1) and the presenilin 1 and 2, which are part of the γ-secretase complex. BACE1 cleavage generates a C-terminal fragment (β-CTF) that is finally processed by presenilin 1 or 2 to liberate the amyloid β peptide, that is prone to aggregate into oligomers.
Figure 4. Impact of amyloid β on microglia.
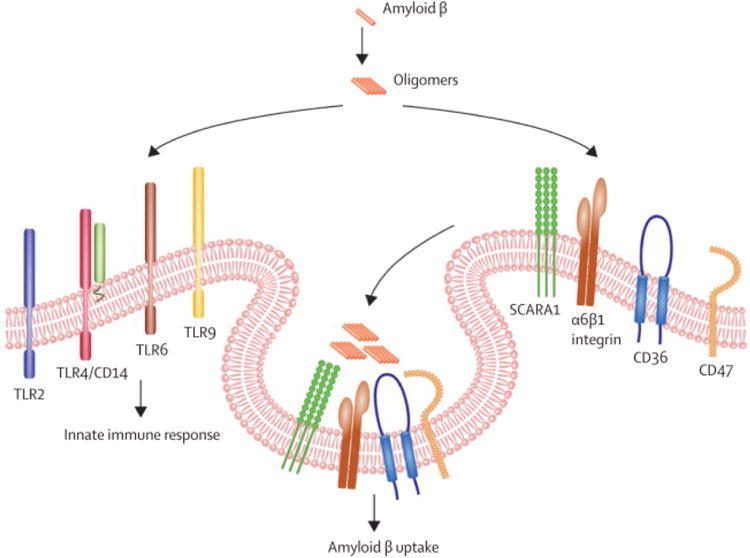
Aβ aggregates (oligomers) act on several TLR receptors on the microglial surface provoking reactions of the innate immune system. On the other side Aβ oligomers are internalized by microglia by the help of the the scavanger receptor A1 (SR-A1), α6β1-integrins, CD36 and CD47.
Microglial Aβ clearance mechanisms
As a response to receptor ligation, microglia start to engulf Aβ fibrills by phagocytosis. As a consequence, these fibrils enter the endosomal/lysosomal pathway. In contrast to fibrillar Aβ, which is largely resistant to enzymatic degradation, soluble Aβ can be degraded by a variety of extracellular proteases 16. In the microglial context, two proteases, neprilysin and insulin degrading enzyme (IDE) are of major importance.
In sporadic cases of AD, inefficient clearance of Aβ has been identified as a major pathogenic pathway 17. It has been suggested, that increased cytokine levels are responsible for the insufficient microglial phagocytic capacity by downregulating Aβ phagocytosis receptors 18. Further support for the hypothesis that compromised microglial function comes from two recent studies identifying rare mutations, which convey a higher risk of AD: a rare mutation in the extracellular domain of TREM2 increases AD risk to a similar extent as apoEε4 2. TREM 2 is highly expressed by microglia 19,20 and mediates phagocytic clearance of neuronal debris 21. Although TREM2 is an orphan receptor so far, TREM2 binding activity (putative TREM2 ligand expression) is detected on reactive astrocytes surrounding amyloid plaques and on damaged neurons and oligodendrocytes 21. Likewise a SNP in the gene encoding the microglial surface receptor CD33 reduces Aβ phagocytosis by peripheral macrophages isolated from hetero- and homozygous mutation carriers. In addition, increased Aβ deposition as determined by Pittsburgh compound B (PiB)-PET was detected in the brain of these patients.
Microglial diversity
Microglial activation is a complex process that produces multiple phenotypes. Outside of the CNS, activated macrophages have been categorized as classical, proinflammatory (M1) phenotype associated with the expression of cytotoxic genes 22 and a non-inflammatory, alternative activation (M2) phenotype, based upon induction of specific proteins like arginase-1, FIZZ1, YM-1 and IGF-1 23,24. Classical M1 activation is characterized by elevated pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6, IL-12 and IL-18, and is accompanied by impaired phagocytic capacity 25. The M2 state is characterized by secretion of the anti-inflammatory cytokines IL-4, IL-10, IL-13 and transforming growth factor-beta (TGF-β), and elevated phagocytic capacity without production of toxic NO 26–28. A third phenotype may be a deactivated one associated with corticosteroids or TGF-β 29,30. The M1/M2 activation states represent the polar extremes of myloid cell activation. There is a large diversity and graduation of phenotypic states in peripheral monocyte-derived macrophages, in particular under conditions of chronic inflammation 31. It is therefore most likely that this applies to microglia as well. Microglia exhibit a diverse range of phenotypes reflective of their response to the local environment, including physical interaction with other cells and their physiological activity in the brain. Central to this matter is our limited ability, at present, to isolate or image subsets of unperturbed microglia in order to characterize their gene expression and mode of action as discriminated by physiological markers.
Microglia priming
In the aging CNS of mice, rats and primates, microglia show enhanced sensitivity to inflammatory stimuli (reviewed in Perry and Teeling, 2013), similar to that seen in microglia in brains with ongoing neurodegeneration. This phenomenon has been termed priming. Priming may be caused by microglial senescence and may represent a consequential element of the “time factor” for age-related neurodegenerative disease. On the transcriptomic level, endogenous ligands are downregulated during aging, whereas transcripts involved in host defense and neuroprotection are upregulated 20. It remains uncertain to what extent age-related microglial priming results from cell-autonomous cellular aging, as opposed to prolonged exposure to the aged neural environment. In physiologically-aged and senescence-accelerated mice, profound microglial priming was characterized by increased production of cytokines and reactive oxygen species as well as enhanced phagocytic capacity. This model provided proof of principle that environmental influences, such as neuronal aging, can drive microglial priming. Weighted gene correlation network analysis revealed a characteristic pattern of gene expression for microglia priming, featuring increased pattern recognition and interferon signaling genes. A similar gene expression network was also found in mouse models for age-related neurodegeneration, including APP/PS1 transgenic mice 33. As a further component, microglia may be primed by systemic inflammation in response to peripheral immune reaction.
Blood-derived mononuclear cells
The precise contribution of blood-derived mononuclear cells infiltrating the CNS in AD like innate immune responses of the brain remains unclear to date and the current knowledge is restricted to animal studies. Those have shown infiltration of peripheral mononuclear cells associated with amyloid plaques in mouse models 34. Further, ablation of CD11b+ cells in the APP/PS1 model of AD showed that peripheral mononuclear phagocytes play an important role in restricting Aβ plaques 34. Restricting the entry of blood-derived mononuclear cells into the brain, by deletion of the chemokine receptor CCR2 in the Tg2576 mouse model, led to increased plaque load 35, although the particular mononuclear cell type was not specified. However, most of these studies used bone marrow irradiation and subsequent transplantation with fluorescent, and therefore traceable, cells. Irradiation of whole animals is likely to cause damage to the blood brain barrier. A further study shielding the brain, thereby restricting irradiation to the rest of the body, did not find any cerebral infiltration by peripheral macrophages, but concluded that perivascular macrophages, protected by the shielding of the brain, were able to modulate Aβ deposition depending on the presence of CCR2 36. The involvement of perivascular macrophages has also been shown for the removal of Aβ in a mouse model of cerebral amyloid angiopathy 37. Nevertheless, recruitment of bone-marrow derived cells is almost absent in parabiosis mouse models, even 12 months after initiation 38. In this context it is interesting that ablation of microglia in APP/PS1 mice by HSV thymidine kinase/ganciclovir system did not change the amyloid pathology, although 95 % of microglia were lost and blood-derived monocytes were spared by the use of bone marrow-chimeric mice 39. This result argues against the participation of peripheral cells in the phagocytosis of amyloid plaques, albeit the observation time was only 2-4 weeks. Currently, these results speak against a significant contribution of blood-derived monocytes, but support the idea that at least perivascular macrophages have some impact on the removal of CNS Aβ depositions.
Modulating innate immunity
The emerging role of microglial activation for AD pathogenesis makes these cells a legitimate therapeutic target. However, a caveat is that in some circumstances microglial activation appears in both beneficial and detrimental guises. Part of the confusion is, that depending on the particular disease stage and particular model in which a brain region is affected, microglia may have different roles and effects. Upon exposure to a DAMP or PAMP, the acute microglial reaction aims at the removal of the recognized abnormality or pathology. In the case of AD, this type of inflammatory reaction is sterile, as it involves the very same receptors but no living pathogens. Under normal circumstances such a reaction quickly resolves pathology with an immediate benefit to the nearby environment. In AD however, several mechanisms including the ongoing formation of Aβ and positive feedback loops between inflammation and APP processing compromise cessation of inflammation. Instead, the further accumulation of Aβ, neuronal debris, and most likely further activating factors establish a chronic, non-resolving inflammation. The sustained exposure to Aβ itself, chemokines, cytokines, and other inflammatory mediators seems responsible for the persistent functional impairment of microglial cells observed at plaque sites 40,41. As an intracellular regulator of microglial function, the autophagy protein beclin 1 was found to be reduced in the brains of AD patients 42. Beclin 1 plays a role in retromer-mediated sorting of cellular components, including Trem2, APP, BACE 1 and CD36, in the endosomal-lsosomal pathway. Reduction of Beclin 1 in vitro and in vivo interferes with efficient phagocytosis resulting in decreased receptor recycling of CD36 and Trem2 42, but more receptors may be affected.
Plasticity of the microglial phenotype is of fundamental importance, since it has become clear that the resolution of inflammation involves conversion to an “alternative”, i.e. similar to M2, activation state associated with tissue repair, phagocytosis and anti-inflammatory actions. Turning microglia from villains to samaritans may be achieved by the modulation of proinflammatory signaling pathways such as the NLRP3 inflammasome. Modifying these pathways, however, implies that they are exclusively restricted to microglia and do not possess critical functions in other cell types. Pharmacologically, the transition to an “alternative” activation state could be achieved through the heterodimeric type II nuclear receptors, PPARγ:RXR, PPARδ:RXR, and LXR:RXR. Agonists of these receptors are robustly anti-inflammatory and stimulate phagocytosis, through induction of CD36 leading to increased microglial Aβ uptake 43. Another target is the RXR itself, which might have a positive effect on both LXR and PPARγ controlled genes. Recently, it was shown that agonism of RXR by bexarotene causes rapid reduction of soluble Aβ, plaque load, and behavioral deficits by apoE-dependent clearance of Aβ44. Nevertheless, this study could not be reproduced by others in its entirety 45–48. Although aberrant and ineffective activation of microglia has been relatively well documented for pre- and moderate-Alzheimer stage disease, but late stage effects are less understood. There is even some evidence of focal microglial senescence particularly surrounding neurofibrillary tangles 40.
Astroglia
Pathological responses of astrocytes are represented by reactive astrogliosis, a complex, multi-stage and pathology-specific reaction with remodeling of astrocytes generally aimed at neuroprotection and recovery of injured neural tissue 49,50. Next to activated microglia, hypertrophic reactive astrocytes accumulate around senile plaques and are commonly found in post-mortem human AD tissue 51 as well as in AD animal models 52 Glial cell activation may occur early in AD, even before Aβ deposition 53. Reactive astrocytes are characterized by increased expression of glial fibrillary acidic protein (GFAP) and signs of functional impairment 54, however astrocytes do not seem to lose their domain organization and there are no indications for scar formation (Figure 2 C,D). In animal models of AD the early response is represented by astroglial atrophy that may have far reaching consequences for synaptic connectivity since astrocytes are central for maintaining synaptic transmission thereby contributing to cognitive deficits 52,54–57. These signs of atrophy demonstrate clear spatio-temporal progression appearing first in the entorhinal cortex and affecting astrocytes located distantly from senile plaques in the later stages of AD. Similarly to microglia, astrocytes release cytokines, interleukins, nitric oxide, and other potentially cytotoxic molecules upon exposure to Aβ thereby exacerbating neuroinflammatory response. The importance of astroglial inflammation in AD has been recently investigated by AAV-driven suppression of the astrocytic reaction in APP/PS1 mice. Interfering with the calcineurin/ nuclear factor of activated T-cells signaling pathway revealed improved cognition, reduced astrogliosis, and lower Aβ levels 58. Astrocytes also have a potential role in internalizing and degrading Aβ in vivo 59. Astrocyte-mediated clearance of Aβ requires apoE 60, and astrocyte-dependent lipidation of ApoE increases the capability of microglia to clear Aβ 61,62. Furthermore, adult astrocytes up-regulate expression of extracellular Aβ-degrading proteases 63, such as neprilysin, “insulin degrading enzyme”, endothelin-converting enzyme-2, and angiotensin-converting enzyme-1 upon exposure to native Aβ deposits 64. These proteases and the impaired function and atrophy of astrocytes may contribute to reduced proteolytic clearance of Aβ. In addition to the above mentioned clearing pathways, astrocytes are involved in the clearance of soluble Aβ from the parenchyma by paravenous drainage 65. This pathway depends on the astrocytic water channel aquaporin-4, since deletion resulted in a strong decrease of clearance via this pathway.
Mediators and Modulators of Neuroinflammation
Cytokines
Microglia and astrocytes are arguably the major source of cytokines in AD. Cytokines contribute to nearly every aspect of neuroinflammation, including pro- and anti-inflammatory processes, bystander neuronal injury, chemoattraction and response of microglia to Aβ deposits. Microglial activation is both characterized and modulated by cytokines. Increase in Aβ in aging TgAPPsw and PSAPP transgenic mice is associated with increased pro-inflammatory cytokines including TNF-α, IL-6, IL-1α and granulocyte macrophage-colony stimulating factor (GM-CSF) 66. This observation suggests that pathological accumulation of Aβ is a key factor that drives neuroinflammatory responses in AD. In addition, exposure of microglia to pre-aggregated Aβ1-42 increases production of pro-inflammatory cytokines (i.e., pro-IL-1 β, IL-6, TNF-α), macrophage inflammatory peptide (MIP-1α) and macrophage colony-stimulating factor (M-CSF) 67. Furthermore, M-CSF levels in the plasma and CNS of AD patients are significantly elevated when compared to age-matched healthy controls or patients with mild cognitive impairment 68,69. Caspase-1 activation, which is required to maturate IL-1β from its inactive pro-forms is similarly elevated in the brains of patients suffering from MCI and AD 70. Consequently, high levels of the cardinal pro-inflammatory cytokine IL-1β are detected in microglial cells surrounding Aβ plaques in AD patient brains (Figure 2E) and cerebrospinal fluid (CSF). In vitro, IL-1β is released by activated microglia after stimulation with Aβ 71. IL-1β can, at least under certain circumstances, favor Aβ deposition by modulating APP expression and proteolysis 72. In addition to these cytokines, IL-12 and IL-23, well known from leukocytes, have been found to be produced by microglia in AD mouse models 73 and inhibition of IL-12/23 improves AD-like pathology 73,74, even so the regulation of IL-12 in human CSF is controversial 73,75.
There are multiple evidences suggesting that the pro-inflammatory enviroment present in the AD brain and in transgenic mouse models of cerebral amyloidosis assumes damaging proportions. For instance, risk for conversion from MCI to AD is higher in subjects with elevated CSF presence of the pro-inflammatory cytokine TNF-α and decreased antiinflammatory TGF-β levels 76. IL-1β, TNF-α and other cytokines may impair neuronal function even before leading to structural changes, as evidenced by suppression of long-term potentiation of synaptic transmission (LTP). Multiple interactions as well as elevated expression of additional cytokines/chemokines and innate immune receptors favor an M1-like activation state in AD. For example, in neuron-microglia co-cultures, the synergistic action of Aβ with either interferon-γ (IFN-γ) or CD40 ligand triggers TNF-α secretion and production of neurotoxic reactive oxygen species 77–79. In addition, the innate immune toll-like receptor 4 is responsible for elevated levels of TNF-α and MIP-1α in AD model mice 80.
Conversely, stimulation of some pro-inflammatory signaling pathways seems to be a beneficial approach in AD mouse models. Transgenic expression of IL-1β in APP/PS1 led to robust neuroinflammation and a reduction of amyloid plaque pathology 81,82. These findings implicate IL-1β expression in activating a “good” form of neuroinflammation in APP/PS1 mice. In another study, AAV-mediated expression of IFN-γ in the brains of the TgCRND8 mouse model demonstrated the ability of this pro-inflammatory cytokine to enhance clearance of amyloid plaques, alongside with a widespread increase astrogliosis and microgliosis 83. In addition, these mice exhibited decreased levels of soluble Aβ and Aβ plaque burden, without altered APP processing. Similar results were obtained using AAV-mediated expression of IL-6 and TNF-α 84,85. Using the opposite approach, expression of the anti-inflammatory cytokine IL-4 resulted in the exacerbation of Aβ deposition 86. These results suggest that certain “good” forms of pro-inflammatory microglial activation are potentially beneficial for reducing AD-like pathology in transgenic mouse models.
Chemokines
Chemokines in AD have been suggested to regulate microglial migration to areas of neuroinflammation, thereby enhancing local inflammation 87. In AD up-regulation of CCL2, CCR3 and CCR5 in reactive microglia has been reported 88,89, whereas CCL4 has been detected in reactive astrocytes near Aβ plaques 88. In vitro, Aβ causes the generation of CXCL8 (IL-8), CCL2, CCL3 and CCL4 in human macrophages and astrocytes 90, and microglia cultured from autopsies of AD revealed an increased expression of CXCL8, CCL2, and CCL3 after experimental exposure to Aβ 91. In AD mouse models a modulation of neuronal survival 92, plaque load 93, and cognition 94 by the CX3CR1/CX3CL1 system has been shown. Further, the receptors CCR5 95 and CCR2 35,96,97 can modulate the course of the disease by influencing microglial positioning and function.
Caspases
Caspases are a family of intracellular proteases, which are key mediators of apoptosis and inflammation. Of the inflammatory caspases, the catalytic activity of caspase-1 is tightly regulated by signal-dependent auto-activation within multiprotein complexes called “inflammasome”, that mediate caspase-1 autocatalytic activation and the subsequent cleavage of the precursors of IL-1β and IL-18 into bioactive cytokines 98,99. Aβ fibrils can activate NRLP3 inflammasomes via lysosomal damage in mouse microglia 100. Elevated levels of active caspase-1 are detected in the brains from AD patients and APP/PS1 mice. In addition, APP/PS1 mice deficient in NLRP3 or caspase-1 are largely protected from spatial memory impairment, loss of hippocampal synaptic plasticity, associated behavioral disturbances and other consequences associated with AD 70. Deficiency of NLRP3 or caspase 1 in APP/PS1 mice appeared to skew microglial cells from a pro-inflammatory ‘M1-like’ phenotype to a more neuroprotective ‘M2-like’ phenotype 70. Further, stimulation of microglia with various inflammogens, led to the orderly activation of the apoptotic caspase-8 and caspase-3/7. Activated caspase 3 modulates nuclear factor-κB activation via protein kinase Cδ and increases the production of neurotoxic pro-inflammatory mediators such as IL-1β, tumor necrosis factor-α and nitric oxide. Inhibition of these caspases hindered microglia activation and neurotoxicity 101. Incidentally these caspases were activated in microglia in AD subjects 102. Pharmacological interventions have been reported to successfully exert neuroprotective effects in AD mouse models 103,104.
Prostanoids and neuroprotectin D1
Prostanoids are derivatives of the arachidonic acid synthesized by the cyclooxygenase-1 and the inducible cyclooxygenase-2, both produced by microglia. In AD, the suppressive effect of cyclooxygenase-1 inhibition on glial activation, amyloid deposition and the expression of inflammatory marker, switching microglia to an alternative phenotype has recently been demonstrated in an AD mouse model 105. In addition, the proinflammatory prostaglandin PGE2, which binds to EP1-4 receptors, has been found to be elevated in patients with probable AD 106. EP1-3 receptors are expressed by microglia cells 107, but are also found in other cells of the brain, in particular neurons. In vitro, microglial EP2 receptors inhibit Aβ phagocytosis and enhance neurotoxic activities of microglia 108. This is complemented by the findings that deletion of the prostaglandin EP2 or EP3 receptor in AD mouse models decreased oxidative stress, neuroinflammation, Aβ-burden, and BACE expression 109–111.
Recently, the use of EP4 receptor agonist on microglia revealed suppression of inflammation and increased update of synthetic Aβ, whereas the deletion of the EP4 receptor in the APP/PS1 mouse model of AD increased the plaque burden and the production of proinflammatory cytokines like Il1-β and CCL3 112. Interestingly, the expression of the EP4 receptor was found to be decreased in the cortex of both MCI and AD patients 112, suggesting that this may contribute the inflammatory reaction in AD. However, the role of PGE2 in neurodegeneration is probably more complex due effects of PGE2 on other cell types like neurons.
The neuroprotective docosahexaenoic acid derivative 10R, 17S-DHA (neuroprotectin D1) is a major components of cell membranes 113, which has been found to be decreased in early stages of AD 114. Neuroprotectin D1 is an autocrine/paracrine mediator involved in the resolution response during early stages of neuroinflammation, that downregulates amyloidogenic processing of APP, switches off pro-inflammatory gene expression, and promotes neural cell survival. Moreover, anti-amyloidogenic processing by neuroprotectin D1 targets α- and β-secretases and PPARγ receptor activation. sAPPα, a peptide with neurotrophin-like activity, is an agonist for NPD1 synthesis and part also of a cycle to sustain generation of the lipid mediator 115.
Complement system
The complement system is a major constituent of the innate immune system mostly involved in the defense against pathogens. Activation of the proteolytic complement cascade results in the opsonization and ultimately in the lysis of microorganisms. In the brain the major cells that contribute in the production of proteins of the complement system are microglia and to a minor extend astrocytes (reviewed in 116). In AD, activated complement factors products have been found to be associated with Aβ deposits 117. In addition, Aβ is able to activate the complement system in vitro via the so-called alternative pathway. The finding that variants of clusterin, or apolipoprotein J, as soluble inhibitor of the complement system and the complement receptor 1 (CR1), involved in processing and clearance of opsonized immune complexes and a regulator of C3 convertase activity, are associated with AD has further substantiated the importance of the complement system in AD 118,119.
Nitric oxide and reactive oxygen species
Next to their direct actions via surface receptors, cytokines stimulate inducible nitric oxide synthase (iNOS) in micro-and astroglia, producing high levels of NO that can be toxic to neurons. iNOS is upregulated in AD brains 120, and genetic knockout of iNOS is protective in mouse models of AD 121. Likewise, NADPH oxidase (PHOX) is highly expressed by microglia, upregulated in AD, and rapidly activated by inflammatory stimuli such as Aβ, resulting in hydrogen peroxide that further promotes microglial activation 122,123. Superoxide from PHOX reacts with iNOS-derived NO forming peroxynitrite 124. Increased expression of iNOS in AD has also been shown to introduce NO-caused posttranslational modifications 125, which include nitration, S-nitrosylation and dityrosine formation 125. Nitration of the Aβ peptide at tyrosine 10 has been recently shown to increase the propensity of Aβ to aggregate and has been identified in the core of the amyloid plaques 126. More compelling, this modified peptide was able to initiate plaque formation in APP/PS1 mice, suggesting a central role during the early phase of AD. Nitrated Aβ suppressed hippocampal LTP more effectively when compared to non-nitrated Aβ, indicating that this post-translational modification exerts functional as well as structural damage to the AD brain. There is evidence that oxidative stress supports the formation of this Aβ species 127. Other NO-mediated modifications that may relevant for AD have already been reported 128 and it is to be expected that there are more to follow.
Inflammatory changes of the neurovascular unit in AD
Numerous epidemiological, clinical and neuropathological studies have shown that vascular pathology represents the an important risk factor for the development of AD. Moreover, it has been shown that AD is associated with distinct inflammatory, functional and morphological alterations of cerebral blood vessels and perivascular glia and neurons. These early-onset and progressive changes, which are induced by combined effects of soluble Aβ oligomers and vascular Aβ deposits 129,130, ultimately lead to decreased cerebral blood flow and an impairment of functional hyperemia, i.e. the ability of local blood flow to increase in response to neuronal activation 131. Chronic cerebral hypoxia is further amplified by blood-bourne factors such as platelets, which are chronically activated in AD models and patients 132, ultimately resulting in micro-infarcts and neuronal injury. Moreover, the combination of mild hypoxia, inflammation of the neurovascular unit, and progressive Aβ accumulation in brain parenchyma, induces upregulation of the receptor for advanced glycation end products (RAGE), which mediates Aβ transport into the brain across the blood-brain barrier 133. Hypoxia also directly induces amyloidogenic APP processing through multiple pathways involving β-secretase, γ-secretase, neprilysin, and others 134. Taken together, chronic hypoxia in AD directly induces neuronal injury, but also amplifies neurodegeneration by inducing amyloidogenic pathways and decreasing brain clearance of Aβ.
Factors facilitating neuroinflammation
Aβ deposition alone may be sufficient to induce an inflammatory reaction which subsequently contributes to cognitive decline and development of AD. Given the possibility that Aβ deposition precedes “cognitive deficits” OR “clinical manifestation” by decades, one may speculate that exogenous or endogenous factors can modify the innate immune response mounted by Aβ exposed microglia. Thus environmentally modifiable AD risk factors including systemic inflammation, obesity and traumatic brain injury may impact risk through sustained neuroinflammatory drive.
Systemic inflammation
The development of sickness behavior 135 following a peripheral inflammatory challenge, such as an infection or an aseptic injury 136, demonstrates the communication of systemic inflammation to the brain. Sickness behavior in response to an acute event is usually self limited due to the presence of multiple regulatory mechanisms that dampen down the central inflammatory response to the peripheral challenge 137. However, it may be prolonged in response to chronic, low grade inflammation 138, possibly due to the lack of a anti-inflammatory response 139. Neuroinflammation in AD represents such a chronic reaction, since microglia may already be “primed”, and are therefore highly responsive to further insults causing a rapid switch to a damaging M1 phenotype 23,140. This microglial priming is likely to result from various activators including the chronic exposure to Aβ, neuronal debris and chronic vascular changes including cerebrovascular dysregulation and cerebral microinfarcts. This hypothesis is supported by animal studies showing an exaggerated inflammatory and oxidative stress response to peripheral stimuli in aged mice 141, increases in CNS IL-1β and neuronal apoptosis in the ME7 prion mouse following a peripheral LPS or poly-IC challenge 142–144. Another example are the consequences of osteoarthritis in the APP/PS1 / Col1-IL1βXAT mouse model resulting in accelerated neuroinflammation and Aβ pathology 145. In addition, clinical studies of AD showing increased cognitive decline and exacerbation of sickness behavior following acute and chronic systemic inflammation 146,147.
Obesity
Obesity is defined as a medical condition in which excess body fat has accumulated to the extent that it may have an adverse effect on health. Obesity increases the propensity to acquire more bacterial or viral infections and thus directly increases the likelihood for systemic inflammation 148,149. Moreover, the white fat tissue itself contains a high percentage of activated macrophages, which constantly secrete pro-inflammatory cytokines 150. Interestingly, midlife obesity has been identified as a risk factor for AD 151, which is partly caused by other AD risk factors, such as high cholesterol diet, reduced physical activity and a sedentary life style 152 . As a possible consequence of obesity, diabetes type II has been demonstrated to accelerate memory dysfunction and neuroinflammation in an AD mouse model 153. Obesity associated reduced gut microbial diversity was associated with increased levels of proinflammatory markers in the peripheral blood and thus could be viewed as factor contributing to AD risk 154. Together it seems possible that obesity increases the risk for AD by the systemic, chronic presence of proinflammatory cytokines.
Traumatic brain injury
Several studies have established traumatic brain injury (TBI) as a risk factor for the development of AD 155. Experimentally TBI aggravates learning and memory deficits and the deposition of Aβ in murine AD models 156,157. Experimental and human studies have shown that microglial activation can persist for months and years after TBI 158,159. This inflammatory reaction may initially be important for the phagocytic clearance of debris. However, a sustained cerebral inflammation may either directly or indirectly promote pathological development in AD. Certain cytokines implicated in TBI can potentially increase BACE-1 levels 160, thereby shifting the APP processing to the amyloidogenic generation of Aβ 161. The chronic release of cytokines may also decrease the capability of microglia to phagocyte and degrade Aβ or directly affect neuronal functions.
Locus coeruleus degeneration
Norepinephrine (NE), in addition to its role as a neurotransmitter, possesses potent anti-inflammatory, anti-oxidative, neurotrophic, and neuroprotective actions 162. The primary source of NE in the brain is the locus coeruleus (LC), located at the dorsal part of the brain stem. The LC neurons project throughout the brain, albeit the majority of terminals target the hippocampus and neocortex. NE released from LC projections acts on adrenergic receptors expressed on neurons, microglia and astrocytes 162. The number of cells in the LC as well as NE levels in the brain decrease during normal aging 163 although a much more pronounced cell loss occurs in AD 164. Experimental lesions of the LC in AD mouse models led to increased inflammation and neuronal damage, as well as increases in Aβ plaque burden 165,166. Thus, an early degeneration of the LC and the subsequent loss of NE-mediated innervation may substantially facilitate the inflammatory response to any stimulus including Aβ. Experimental loss of NE compromised microglial migration and Aβ phagocytosis in vivo, suggesting that the failure in the NE tone not only increases inflammation but also Aβ deposition. Increasing endogenous NE levels by use of selective NE re-uptake inhibitors 167 or α-2-adrenoceptor antagonists 165 or the NE precursor L-threo-DOPS reduces neuroinflammation and partly rescues microglial functions.
Analysis of immune activation
In vivo-laser scanning microscopy
Over the past decades, analyzing innate immunity of the brain was restricted to cell culture experiments and immunohistochemical detection of microglia. Little was known about the functional state of these cells in vivo. Recent methodological advances such as the generation of transgenic mice with eGFP-labeled microglia, cranial window implantation, and Aβ plaque-labeling with the fluorescent dye methoxy-XO4 have enabled longitudinal and live monitoring of the functional state of microglia in murine models of disease. This technique now permits the analysis of microglial phenotypes over time and in relation to the deposition of Aβ. These data revealed that both in control and plaque-bearing mice microglia migrate within the brain parenchyma with an average speed of 5-9 mm/month 168. Upon the formation of Aβ plaques the microglia rearranged their processes becoming polarized and moved their somata towards the plaque temporarily leaving the formerly surveyed area 168. Individual microglia migrated towards new or preexisting plaques within one-two days 168,169, similar to observations made upon acute laser-induced brain injury 170. In addition to Aβ deposits, microglia are also attracted to neurons that underwent Alzheimer's disease-associated elimination 92. Of note, microglial cells were recruited to neurons up to 7 days before their elimination and neuronal loss could be completely rescued by knockout of the receptor for chemokine fractalkine (CX3CR1) 92.
Imaging inflammation in animal models
There are various molecular imaging techniques used in experimental and clinical settings to study the temporal and spatial relationship of inflammatory changes associated with neurodegeneration. Since microglial respond very fast to lesions, these cells are good candidates for diagnostic markers of disease progression 171. Therefore, several microglial cell surface and mitochondrial receptors were used for the development of in vivo imaging ligands. One of these targets is the translocator protein 18 kD (TSPO) 172, a protein of the outer mitochondrial membrane that has been found to be increasingly expressed under conditions of neuronflammation. Binding of the radiolabeled ligands 11C-(R)-PK11195 and 18F-DPA-714 to TSPO can be visualized using PET and SPECT 173. In AD mouse models progressive binding of PK11195 with age has been described 174,175. This binding could only reliably detected in APP/PS1 mice at a late age, when microglial and astroglial activation are already massive by immunohistological means 176. Binding of both 3H-(R)-PK11195 and 3H-DPA-713 could be decreased by the PPARγ-agonists pioglitazone and ciglitazone in the TASTPM mouse model of AD 175 have been shown to have anti-inflammatory and Aβ lowering effects in AD mouse models 177. The development of new TSPO ligands and other probes with improved bio-availability, decreased non-specific uptake, and higher specific binding 178,179, may permit the detection of activated microglia at an even earlier disease stage.
Imaging inflammation in humans
Similarly to the studies on animals, increased microglial activation has been detected in AD patients using the TSPO ligand 11C-PK11195. Binding potentials have been reported to be increased up to 50% in association cortex 180. Uptake of another TSPO ligand 11C-DAA1106 was reported to be elevated by up to 33% in AD 181. The cortical distribution of 11C-PK11195 binding parallels that of amyloid deposition, as detected by the thioflavin analogue 11C-PIB 182. Levels of cortical 11C-PK11195 signals in AD also correlate with cognitive impairment rated with the mini-mental state examination (MMSE) 183,184, suggesting that cortical microglial activation is detrimental to cognitive function in AD. In addition, a correlation of neuroinflammation and the severity of AD has been reported for the TSPO marker 11C-PBR28 185. In MCI patients, 11C-PK11195 PET detected inflammation in 40% of amnestic cases 186, although another study using the same tracer failed to detect microglial activation. A group of seven MCI cases showed a significant 27% mean increase in 11C-DAA1106 uptake across brain regions 181. Five of these seven MCI subjects, with 11C-DAA1106 uptake more than 0.5 standard deviations above the mean in controls, progressed to frank dementia over a two year follow up period.
Characterization and monitoring neuroinflammation in AD
While emerging evidence suggests that inflammation plays a causal role in AD pathogenesis, the detection of inflammatory markers has not yet been established as valuable tool for the diagnosis or monitoring of AD. Nevertheless, novel data from a recent study of postmortem late-onset AD brains by gene-expression analysis highlighted an immune and microglia network, which is dominated by genes implicated in phagocytosis 187. These data along with the new analysis of inflammation related biomarkers in the CSF, peripheral blood or directly in the brain by imaging, will be the focus of future studies. An important aspect of this search will be the discovery of inflammatory biomarkers that identify prodromal AD stages.
Detection of neuroinflammatory markers in the CSF
Over the last 25 years numerous studies have investigated levels of pro- and anti-inflammatory cytokines in the CSF of MCI and AD patients. The results of these studies are often controversial 188. Yet, there are indications that the time point of sampling – in other words, the stage of the disease – is a critical factor for investigation. Some studies highlight elevated CSF cytokines as risk factors for MCI to AD conversion or as correlates of the speed of cognitive decline and disease progression 76,189. As with imaging consortia, to overcome inter-individual variances and to obtain a more definite description of cytokine regulation and function in AD, a high degree of method harmonization and patients characterization, together with longitudinal sampling over years seems essential to leave the stage of cross-sectional descriptions.
Systemic biomarkers
A growing number of studies point to a sophisticated interaction between the systemic environment and the brain. Thus, systemic immune cells and secreted signaling proteins communicate with the brain and have been associated, not only with neuroinflammation, but neurodegenerative processes in general 190. While some of these interactions may involve cells entering the nervous tissue, it is likely that many more are mediated by soluble signaling molecules, like blood-borne factors, present in the systemic environment. These factors can inhibit or promote adult neurogenesis in an age-dependent fashion or restore regeneration of the aging brain in mice 191,192. In order to identify such factors, scientists have tried to discover molecular or cellular changes in blood associated with neurodegenerative diseases (reviewed in 193. Various proteomic methods have been used to identify blood-based biomarkers. Typical biomarkers have been cytokines and trophic factors such as brain-derived neurotrophic factor (BDNF) 194–197. Using multiplex ELISA in plasma from AD patients protein signatures were described which may be specific to prodromal stages of the disease 198, or which characterize patients who progress from a prodromal stage to AD 199. Other signatures appear to correlate with APOE 200 or with pathological changes such as Aβ and tau protein levels in CSF of AD patients 201. Close to 200 “communicome” proteins were measured in plasma samples from patients participating in the AD neuroimaging initiative yielding protein signatures that correlated with patients who converted from mild cognitive impairment to AD 202.
Clinical trials and epidemiological findings
Anti-inflammatory drugs
Although NSAID epidemiology and clinical trial results have produced some healthy skepticism about apparent stage-dependent outcomes, the normal physiological cytokine regulation of glial activation and microglial phenotypes is highly context and stage-dependent 5. Contextual factors modulating glial “activated” phenotypes include immune-modulatory apoE genotype and newer AD genes. Normal aging is also associated with chronic glial activation 203 and more focal stage-dependent injury-induced factors. Context-dependent responses should be expected for NSAIDs that act as cyclooxygenase (COX) inhibitors to reduce levels of prostaglandin products, notably PGE2, that act through EP receptors 1-4 to produce very different outcomes. For example, EP2 activation engages predominantly pro-inflammatory neurotoxic pathways and downregulates Aβ phagocytosis while EP4 ligands can produce anti-inflammatory and neuroprotective effects 204,205. Most significantly these receptors play a significant role in promoting the resolution of chronic neuroinflammation 206. Thus, it is understandable if conventional cyclooxygenase-inhibiting NSAIDs can block incipient inflammation-driven AD pathogenesis at early stages. They can also have adverse effects with advanced disease by potentially limiting resolution and interfering with phagocytic clearance of Aβ and extracellular tau aggregates.
Stage-dependent efficacy has also been hypothesized for anti-Aβ immunotherapy that stimulates microglial phagocytosis of Aβ and potential benefits may only be observed with early intervention. It is plausible to argue that aging, Aβ- or injury-induced inflammation may initiate tauopathy to drive neurodegeneration and downstream clinical decline. Thus, possible explanations for the lack of success with immunotherapy or anti-inflammatory therapy with more established dementia include a likely failure to halt the spread of fully established and seeded tauopathy and failure to rescue deficits driven by neuron loss. Whatever the explanations for past NSAID trial failures are, based on compelling new genetic evidence for a causal role for innate immunity in AD risk, new trials with both longer and earlier interventions and alternative approaches to favorably modulate neuroinflammation are warranted.
Interventional anti-inflammatory trials
Randomized placebo controlled trials (RPCT's) with NSAID's in AD evidence of success. Early trials with indomethacin hinting at reduced cognitive decline 207 were not replicated 208 and large scale trials with other NSAID's appeared unsuccessful 209,210. RPCT's with other anti-inflammatory agents including prednisone 211, hydroxychloroquine 212; simvastatin 213; atorvastatin 214,215; aspirin 216 and rosiglitazone 217 also failed to show significant changes in primary cognitive outcomes in subjects with AD dementia or prodromal symptoms. Although a large RPCT prevent study of the NSAIDs naproxen and celecoxib initially reported a detrimental effect for both 218, a longer term follow up of these subjects suggested that timing and choice of specific NSAID might be key 219. Thus, the early detrimental effects were largely attributable to a small group of subjects with early cognitive impairment and, in keeping with epidemiological studies, naproxen appeared thereafter to be protective in subjects who had been asymptomatic at baseline 219.
Immunization in AD mouse models and humans
In some instances (but not all) the characterization into M1 versus M2 phenotypes has been observed in response to specific conditions and cytokine exposures. With aging, Aβ-depositing mice increase in the expression of alternative activation state genes and deactivation state genes at the expense of classical activation state genes 220, however see also 221. When these mouse models are treated with antibodies, there is a shift in the activation state over the time with treatment. Initial studies 222 noted reciprocal changes in markers such as kinases, with MAP kinase p38 declining and MAP kinase 44/42 increasing over the duration of antibody treatment. Subsequent work using mRNA markers to distinguish the M1 versus M2 phenotype identified a transition from an initially elevated M2 phenotype before treatment to an M1 phenotype after antibody treatment 220. Similar shifts in microglial activation states appear associated with vaccination studies in humans with Alzheimer's disease. In comparison to other AD tissues, tissue from vaccinated patients have reduced staining for a number of microglial markers including the scavenger receptor A and Fcγ receptors, as was the amount of deposited Aβ 223. In cases coming to autopsy within 2 years of the vaccination, these cases also showed increased levels of microhemorrhage and vascular amyloid deposits, plus the appearance of phagocytic microglia 224. Similar outcomes were previously observed in aged mice treated with antibodies 225. In the phase 2 and phase 3 trials with the antibody bapineuzimab, events referred to as Aβ related imaging abnormalities (ARIA) were found on MRI scans 226. To some extent, these abnormalities in mouse models could be diminished by reducing antibody affinity for Fcγ receptors via deglycosylation 227, implying that the microglial activation caused by the antibody-antigen interaction may play a role in these vascular responses to immunotherapy.
Summary and outlook
The brain is no longer perceived as an immune privileged organ: we are in need of integrating advances in immunology into pathogenesis of diverse neurodegenerative conditions. The ligand-receptor interactions in the CNS micro-environment that keep microglia under tight control in the healthy brain are perturbed in chronic neurodegenerative disease but “the when” and “how” remain to be worked out. The notion of “activated microglia” is clearly useful shorthand but has hindered advances in the recognition of the diversity of the microglia phenotype and the extraordinary plasticity of these cells. The impact of systemic comorbidities and the associated systemic inflammation as well as ageing as a major risk factor, must be considered. The innate immune cells of the brain rapidly respond to systemic events and these responses are exaggerated in the ageing and diseased brain. The limited tools that allow us to assay the different states of microglia activation in vivo compound the difficulty of understanding the role of neuroinflammation in the human CNS. This is clearly an area with important unmet needs; better ligands for PET or other imaging modalities are vital. Clearly, the recognition that modifying the immune system contributes to the pathogenesis of chronic neurodegenerative diseases offers many potential routes to delaying the onset or progression.
Table 1. Selected clinicals trials to date.
| Drug | Treatment | Outcome | Patients | |
|---|---|---|---|---|
| Rogers et al, 1993207 | Indomethacin | 6 months | Positive effects after marked attrition | 41 AD |
| De Jong et al, 2008208 | Indomethacin | 1 year | Neutral to positive effects after marked attrition | 51 mild to moderate AD |
| Aisen et al, 2000211 | Prednisone | 1 year | Neutral to negative effects | 138 AD |
| Aisen et al, 2003209 | Naproxen, rofecoxib | 1 year | Neutral to negative effects | 351 mild-to-moderate AD |
| Aisen et al, 2002228 | Nimesulide | 3 months | Neutral effects | 40 AD |
| Reines et al, 2004210 | Rofecoxib | 1 year | Neutral to negative effects | 692 mild-to-moderate AD |
| Thal et al, 2005229 | Rofecoxib | 3.5 years, preventive | Neutral to negative effects on conversion to AD | 1457 mild cognitive impairment |
| Van Gool et al, 2001212 | Hydroxychloro quine | 18 months | Neutral effects | 168 mild AD |
| ADAPT Research | Celecoxib | 2 years, preventive | Neutral to negative effects | 2528 normal with family |
| Research Group, 2007, 2008218,230 | effects | with family history of AD | ||
| ADAPT Research Group, 2007, 2008218,230 | Naproxen | 2 years, preventive | Neutral to negative effects | 2528 normal wit family history of AD |
| Simons et al, 2002213 | Simvastatin | 26 weeks | No effects on CSF Aβ | 44 AD |
| Sparks et al, 2005214 | Atorvastin | 72 weeks | Neutral effects | 640 mild to moderate AD |
| Feldman et al, 2010215 | Atorvastatin | 1 year | Neutral to positive effects | 67 mild AD |
| Gold et al, 2007217 | Rosiglitazone | 24 weeks | Neutral effects | 693 mild to moderate AD |
| Breitner et al, 2011219 | Naproxen | 4 years, preventive | Neutral to positive effects | 2528 normal with family history of AD |
| Breitner et al, 2011219 | Celecoxib | 4 years, preventive | neutral to negative effects | 2528 normal with family history of AD |
We searched PubMed for randomised controlled trials published in English. The table provides a list of trial results that have had a notable influence on the field, in our view, and is not an exhaustive list of studies. Priority was given to trials with sufficient numbers of participants, definition of clinical outcomes, and specification of design methodology to allow firm conclusions to be drawn (including inference of uncertainty). Two older trials were included because of their influence on later work, despite the fact that they failed to meet the foregoing criteria.
Possible future directions to advance the field.
- Development of new animal models that reflect multiple facets of AD and are not restricted to the transgenic expression of human mutations of familial Alzheimer's disease.
- New models should exceed beta amyloid or tau pathology and include aspects of multi-neurotransmitter loss, disease spreading and late onset of disease. Ideally these models would also show Alzheimer-like vascular pathology, synaptic destruction and neuronal loss. Future models and experiments should take disease modifiers such as systemic inflammation, insulin resistance, brain trauma, nutritional states, physical inactivity and obesity into account.
- Biomarker identification of the inflammatory component of Alzheimer's disease.
- New biomarkers may be developed for disease diagnosis, but also for the monitoring of preventive and therapeutic strategies. Such biomarkers could be blood, CSF or imaging based and allow to distinguish acute from chronic neuroinflammation.
- Definition of pathologies and periods of neuroinflammation during the course of Alzheimer's disease.
- Understanding of the individual contributions of microglia, macrophages, astrocytes, neurons and endothelial cells to neuroinflammation. This will provide information which inflammatory processes are good, which bad and which are irrelevant for disease pathogenesis at different stages of Alzheimer's disease.
- Exploit the influence of mutations associated with immune function, epigenetics and the microbiome on neuroinflammation in Alzheimer's disease.
- Recent discoveries which suggest a direct immune-related modification of the onset, progress and phenotype of Alzheimer's disease including SNPs in immune associated genes, epigenetic immune regulation and the influence of the microbiome on innate immunity should be taken into account.
- Observational and preventive clinical trials, which inhibit detrimental but foster benefical immunity during Alzheimer's disease.
- Observational trials should closely monitor the clinical course of patients carrying SNPs in immune-related genes, which have been associated with the risk to develop Alzheimer's disease. Clinical trials could include preventive trials, which inhibit detrimental aspects of neuroinflammation prior to any cognitive decline, relevant to NSAID epidemiology.
Acknowledgments
The authors of this review were participants of the 3rd Venusberg Meeting on Neuroinflammation taking place February 28th to March 2nd 2013 in Bonn, Germany.
Footnotes
Contributors: All authors provided sections of text covering their area of expertise and participated in the proofreading and in the discussion. MTH and MPK wrote the manuscript. MTH and MPK drafted the figures. MPK did the artwork.
Declaration of interest: GEL has a patent on RXR agonist in AD pending. AH reports grants from Boehringer Ingelheim Pharma GmbH & Co. KG. CV reports grants from Pfizer, outside the submitted work. JK reports grants from Baxter, personal fees from Medeia Therapeutics Ltd, outside the submitted work. KY reports grants from MEXT, Japan, during the conduct of the study. GMC reports grants from Veterans Affairs, grants from NIH, during the conduct of the study. SAF reports grants from Veterans Affairs, grants from NIH, grants from NIH, during the conduct of the study. In addition, SAF has a patent Curcumin Formulation 60,779,817 with royalties paid. RR reports grants from National MS Society, during the conduct of the study. MTH and MPK have a patent on nitration of amyloid β peptides pending.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradshaw EM, Chibnik LB, Keenan BT, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–50. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 6.Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PLoS ONE. 2013;8:e56293. doi: 10.1371/journal.pone.0056293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–65. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 10.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Walter S, Stagi M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–89. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 12.Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 13.Kummer MP, Heneka MT. Truncated and modified amyloid-beta species. Alzheimer's Research & Therapy. 2014;6:28. doi: 10.1186/alzrt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–66. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating the intracellular nucleation from soluble to particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–20. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117:949–60. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased Clearance of CNS β-Amyloid in Alzheimer's Disease. Science. 2010;330:1774–1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank S, Burbach GJ, Bonin M, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56:1438–47. doi: 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- 20.Hickman SE, Kingery ND, Ohsumi TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh CL, Koike M, Spusta SC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–56. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sierra-Filardi E, Puig-Kröger A, Blanco FJ, et al. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood. 2011;117:5092–101. doi: 10.1182/blood-2010-09-306993. [DOI] [PubMed] [Google Scholar]
- 23.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 26.Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen-presenting cells. Immunity. 1999;10:137–42. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- 28.Zelcer N, Khanlou N, Clare R, et al. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver × receptors. Proc Natl Acad Sci USA. 2007;104:10601–6. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–91. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 30.Town T, Laouar Y, Pittenger C, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–7. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue J, Schmidt SV, Sander J, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity. 2014;40:274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–12. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orre M, Kamphuis W, Dooves S, et al. Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain. 2013;136:1415–31. doi: 10.1093/brain/awt083. [DOI] [PubMed] [Google Scholar]
- 34.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 35.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 36.Mildner A, Schlevogt B, Kierdorf K, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–71. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci USA. 2009;106:1261–6. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grathwohl SA, Kälin RE, Bolmont T, et al. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–3. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–85. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucin KM, O'Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron. 2013;79:873–86. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARv/RXRa-induced and CD36-mediated microglial amyloid-(3 phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cramer PE, Cirrito JR, Wesson DW, et al. ApoE-directed therapeutics rapidly clear (3-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitz NF, Cronican AA, Lefterov I, Koldamova R. Comment on ApoE-directed therapeutics rapidly clear (3-amyloid and reverse deficits in AD mouse models'. Science. 2013;340:924–c. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price AR, Xu G, Siemienski ZB, et al. Comment on ‘ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models’. Science. 2013;340:924–d. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- 47.Tesseur I, Lo AC, Roberfroid A, et al. Comment on ‘ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models’. Science. 2013;340:924–e. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- 48.Veeraraghavalu K, Zhang C, Miller S, et al. Comment on ‘ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models’. Science. 2013;340:924–f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- 49.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–8. doi: 10.1016/j.expneurol.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia. 2010;58:831–8. doi: 10.1002/glia.20967. [DOI] [PubMed] [Google Scholar]
- 53.Kummer MP, Hammerschmidt T, Martinez A, et al. Ear2 Deletion Causes Early Memory and Learning Deficits in APP/PS1 Mice. J Neurosci. 2014;34:8845–54. doi: 10.1523/JNEUROSCI.4027-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer's disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener. 2011;6:55. doi: 10.1186/1750-1326-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeh CY, Vadhwana B, Verkhratsky A, Rodríguez JJ. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer's disease. ASN Neuro. 2011;3:271–9. doi: 10.1042/AN20110025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kulijewicz-Nawrot M, Verkhratsky A, Chvátal A, Syková E, Rodríguez JJ. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer's disease. J Anat. 2012;221:252–62. doi: 10.1111/j.1469-7580.2012.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beauquis J, Pavía P, Pomilio C, et al. Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer's disease. Exp Neurol. 2013;239:28–37. doi: 10.1016/j.expneurol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 58.Furman JL, Sama DM, Gant JC, et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J Neurosci. 2012;32:16129–40. doi: 10.1523/JNEUROSCI.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wyss-Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 60.Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–26. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 61.Jiang Q, Lee CYD, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–93. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terwel D, Steffensen KR, Verghese PB, et al. Critical role of astroglial apolipoprotein E and liver × receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31:7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saido T, Leissring MA. Proteolytic degradation of amyloid β-protein. Cold Spring Harb Perspect Med. 2012;2:a006379. doi: 10.1101/cshperspect.a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M. Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia. 2011;59:1643–57. doi: 10.1002/glia.21212. [DOI] [PubMed] [Google Scholar]
- 65.Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation. 2005;2:9. doi: 10.1186/1742-2094-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lue LF, Rydel R, Brigham EF, et al. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–9. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 68.Laske C, Stransky E, Hoffmann N, et al. Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer's disease. Curr Alzheimer Res. 2010;7:409–14. doi: 10.2174/156720510791383813. [DOI] [PubMed] [Google Scholar]
- 69.Hirsch-Reinshagen V, Maia LF, Burgess BL, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280:43243–56. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- 70.Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akama KT, Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem. 2000;275:7918–24. doi: 10.1074/jbc.275.11.7918. [DOI] [PubMed] [Google Scholar]
- 72.Mrak RE, Sheng JG, Griffin WS. Glial cytokines in Alzheimer's disease: review and pathogenic implications. Hum Pathol. 1995;26:816–23. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vom Berg J, Prokop S, Miller KR, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–9. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 74.Tan MS, Yu JT, Jiang T, Zhu XC, Guan HS, Tan L. IL12/23 p40 inhibition ameliorates Alzheimer's disease-associated neuropathology and spatial memory in SAMP8 mice. J Alzheimers Dis. 2014;38:633–46. doi: 10.3233/JAD-131148. [DOI] [PubMed] [Google Scholar]
- 75.Rentzos M, Paraskevas GP, Kapaki E, et al. Interleukin-12 is reduced in cerebrospinal fluid of patients with Alzheimer's disease and frontotemporal dementia. J Neurol Sci. 2006;249:110–4. doi: 10.1016/j.jns.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 76.Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatr. 2003;74:1200–5. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–50. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 78.Tan J, Town T, Paris D, et al. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–5. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 79.Tan J, Town T, Crawford F, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–93. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 80.Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi KI. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghosh S, Wu MD, Shaftel SS, et al. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci. 2013;33:5053–64. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chakrabarty P, Ceballos-Diaz C, Beccard A, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184:5333–43. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chakrabarty P, Jansen-West K, Beccard A, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24:548–59. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chakrabarty P, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. doi: 10.1186/1750-1326-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chakrabarty P, Tianbai L, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener. 2012;7:36. doi: 10.1186/1750-1326-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. 1998;153:31–7. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–8. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 90.Smits HA, Rijsmus A, van Loon JH, et al. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–8. doi: 10.1016/s0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- 91.Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–56. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 92.Fuhrmann M, Bittner T, Jung CKE, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–3. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee S, Varvel NH, Konerth ME, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–62. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cho SH, Sun B, Zhou Y, et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–22. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee YK, Kwak DH, Oh KW, et al. CCR5 deficiency induces astrocyte activation, Abeta deposit and impaired memory function. Neurobiol Learn Mem. 2009;92:356–63. doi: 10.1016/j.nlm.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 96.Kiyota T, Yamamoto M, Xiong H, et al. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS ONE. 2009;4:e6197. doi: 10.1371/journal.pone.0006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Semple BD, Frugier T, Morganti-Kossmann MC. CCL2 modulates cytokine production in cultured mouse astrocytes. J Neuroinflammation. 2010;7:67. doi: 10.1186/1742-2094-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 99.Van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LAB. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011;32:110–6. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 100.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fricker M, Vilalta A, Tolkovsky AM, Brown GC. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J Biol Chem. 2013;288:9145–52. doi: 10.1074/jbc.M112.427880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Burguillos MA, Deierborg T, Kavanagh E, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–24. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- 103.Rohn TT, Kokoulina P, Eaton CR, Poon WW. Caspase activation in transgenic mice with Alzheimer-like pathology: results from a pilot study utilizing the caspase inhibitor, Q-VD-OPh. Int J Clin Exp Med. 2009;2:300–8. [PMC free article] [PubMed] [Google Scholar]
- 104.Biscaro B, Lindvall O, Tesco G, Ekdahl CT, Nitsch RM. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer's disease. Neurodegener Dis. 2012;9:187–98. doi: 10.1159/000330363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Choi SH, Aid S, Caracciolo L, et al. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer's disease. J Neurochem. 2013;124:59–68. doi: 10.1111/jnc.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Montine TJ, Sidell KR, Crews BC, et al. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53:1495–1495. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- 107.Slawik H, Volk B, Fiebich B, Hüll M. Microglial expression of prostaglandin EP3 receptor in excitotoxic lesions in the rat striatum. Neurochem Int. 2004;45:653–60. doi: 10.1016/j.neuint.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 108.Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 as a new target to increase amyloid beta phagocytosis and decrease amyloid beta-induced damage to neurons. Brain Pathol. 2005;15:134–8. doi: 10.1111/j.1750-3639.2005.tb00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liang X, Wang Q, Hand T, et al. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci. 2005;25:10180–7. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi J, Wang Q, Johansson JU, et al. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:788–98. doi: 10.1002/ana.23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiang Z, Ho L, Yemul S, et al. Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer's disease neuropathology. Gene Expr. 2002;10:271–8. doi: 10.3727/000000002783992352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Woodling NS, Wang Q, Priyam PG, et al. Suppression of Alzheimer-associated inflammation by microglial prostaglandin-E2 EP4 receptor signaling. J Neurosci. 2014;34:5882–94. doi: 10.1523/JNEUROSCI.0410-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bazan NG, Molina MF, Gordon WC. Docosahexaenoic acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer's, and other neurodegenerative diseases. Annu Rev Nutr. 2011;31:321–51. doi: 10.1146/annurev.nutr.012809.104635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lukiw WJ, Cui JG, Marcheselli VL, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–83. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao Y, Calon F, Julien C, et al. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer's disease models. PLoS ONE. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48:1592–603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Strohmeyer R, Ramirez M, Cole GJ, Mueller K, Rogers J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer's disease brain. J Neuroimmunol. 2002;131:135–46. doi: 10.1016/s0165-5728(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 118.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 119.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vodovotz Y, Lucia MS, Flanders KC, et al. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. 1996;184:1425–33. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nathan C, Calingasan N, Nezezon J, et al. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–9. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J Neuroinflammation. 2006;3:24. doi: 10.1186/1742-2094-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation. 2005;2:20. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Butterfield DA, Reed TT, Perluigi M, et al. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer's disease. Brain Res. 2007;1148:243–8. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kummer MP, Hermes M, Delekarte A, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–44. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 127.Thiabaud G, Pizzocaro S, Garcia-Serres R, Latour JM, Monzani E, Casella L. Heme Binding Induces Dimerization and Nitration of Truncated β-Amyloid Peptide Aβ16 Under Oxidative Stress. Angew Chem Int Ed Engl. 2013 doi: 10.1002/anie.201302989. published online June 20. [DOI] [PubMed] [Google Scholar]
- 128.Cho DH, Nakamura T, Fang J, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–5. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 130.Shin HK, Jones PB, Garcia-Alloza M, et al. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130:2310–9. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- 131.Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–97. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 132.Jarre A, Gowert NS, Donner L, et al. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer's disease. Cell Signal. 2014;26:2040–50. doi: 10.1016/j.cellsig.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 133.Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 134.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–38. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–37. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 136.Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–39. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 138.Undén AL, Andréasson A, Elofsson S, et al. Inflammatory cytokines, behaviour and age as determinants of self-rated health in women. Clin Sci. 2007;112:363–73. doi: 10.1042/CS20060128. [DOI] [PubMed] [Google Scholar]
- 139.Maitra U, Deng H, Glaros T, et al. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol. 2012;189:1014–23. doi: 10.4049/jimmunol.1200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pace JL, Russell SW, Torres BA, Johnson HM, Gray PW. Recombinant mouse gamma interferon induces the priming step in macrophage activation for tumor cell killing. J Immunol. 1983;130:2011–3. [PubMed] [Google Scholar]
- 141.Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am. 2009;29:321–37. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 142.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–84. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cunningham C, Campion S, Lunnon K, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–12. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kyrkanides S, Tallents RH, Miller JH, et al. Osteoarthritis accelerates and exacerbates Alzheimer's disease pathology in mice. Journal of Neuroinflammation. 2011;8:112. doi: 10.1186/1742-2094-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77:212–8. doi: 10.1212/WNL.0b013e318225ae07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Almond MH, Edwards MR, Barclay WS, Johnston SL. Obesity and susceptibility to severe outcomes following respiratory viral infection. Thorax. 2013;68:684–6. doi: 10.1136/thoraxjnl-2012-203009. [DOI] [PubMed] [Google Scholar]
- 149.Chidiac C. Pneumococcal infections and adult with risk factors. Med Mal Infect. 2012;42:517–24. doi: 10.1016/j.medmal.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 150.Bastard JP, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- 151.Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–64. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- 152.Brown BM, Peiffer JJ, Martins RN. Multiple effects of physical activity on molecular and cognitive signs of brain aging: can exercise slow neurodegeneration and delay Alzheimer's disease? Mol Psychiatry. 2013;18:864–74. doi: 10.1038/mp.2012.162. [DOI] [PubMed] [Google Scholar]
- 153.Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA. 2010;107:7036–41. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–6. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 155.Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer's disease. Neurosci Biobehav Rev. 2012;36:1376–81. doi: 10.1016/j.neubiorev.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 156.Brody DL, Holtzman DM. Morris water maze search strategy analysis in PDAPP mice before and after experimental traumatic brain injury. Exp Neurol. 2006;197:330–40. doi: 10.1016/j.expneurol.2005.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tajiri N, Kellogg SL, Shimizu T, Arendash GW, Borlongan CV. Traumatic brain injury precipitates cognitive impairment and extracellular aβ aggregation in Alzheimer's disease transgenic mice. PLoS ONE. 2013;8:e78851. doi: 10.1371/journal.pone.0078851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Koshinaga M, Katayama Y, Fukushima M, Oshima H, Suma T, Takahata T. Rapid and widespread microglial activation induced by traumatic brain injury in rat brain slices. J Neurotrauma. 2000;17:185–92. doi: 10.1089/neu.2000.17.185. [DOI] [PubMed] [Google Scholar]
- 159.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 160.Sastre M, Dewachter I, Landreth GE, et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Corrigan F, Pham CLL, Vink R, et al. The neuroprotective domains of the amyloid precursor protein, in traumatic brain injury, are located in the two growth factor domains. Brain Res. 2011;1378:137–43. doi: 10.1016/j.brainres.2010.12.077. [DOI] [PubMed] [Google Scholar]
- 162.O'Donnell J, Zeppenfeld D, McConnell E, Pena S, Nedergaard M. Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem Res. 2012;37:2496–512. doi: 10.1007/s11064-012-0818-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 164.Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–41. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 165.Kalinin S, Gavrilyuk V, Polak PE, et al. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28:1206–14. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 166.Heneka MT, Ramanathan M, Jacobs AH, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–54. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.O'Sullivan JB, Ryan KM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors inhibit expression of chemokines IP-10 and RANTES and cell adhesion molecules VCAM-1 and ICAM-1 in the CNS following a systemic inflammatory challenge. J Neuroimmunol. 2010;220:34–42. doi: 10.1016/j.jneuroim.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 168.Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 170.Kim JV, Dustin ML. Innate response to focal necrotic injury inside the blood-brain barrier. J Immunol. 2006;177:5269–77. doi: 10.4049/jimmunol.177.8.5269. [DOI] [PubMed] [Google Scholar]
- 171.Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 172.Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–88. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 173.Jacobs AH, Tavitian B INMiND consortium. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32:1393–415. doi: 10.1038/jcbfm.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Venneti S, Lopresti BJ, Wang G, et al. PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009;30:1217–26. doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Roberts JC, Friel SL, Roman S, et al. Autoradiographical imaging of PPARgamma agonist effects on PBR/TSPO binding in TASTPM mice. Exp Neurol. 2009;216:459–70. doi: 10.1016/j.expneurol.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 176.Rapic S, Backes H, Viel T, et al. Imaging microglial activation and glucose consumption in a mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:351–4. doi: 10.1016/j.neurobiolaging.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 177.Heneka MT, Reyes-Irisarri E, Hüll M, Kummer MP. Impact and Therapeutic Potential of PPARs in Alzheimer's Disease. Curr Neuropharmacol. 2011;9:643–50. doi: 10.2174/157015911798376325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chauveau F, Boutin H, Van Camp N, et al. In vivo imaging of neuroinflammation in the rodent brain with [11C]SSR180575, a novel indoleacetamide radioligand of the translocator protein (18 kDa) Eur J Nucl Med Mol Imaging. 2011;38:509–14. doi: 10.1007/s00259-010-1628-5. [DOI] [PubMed] [Google Scholar]
- 179.Chauveau F, Van Camp N, Dollé F, et al. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50:468–76. doi: 10.2967/jnumed.108.058669. [DOI] [PubMed] [Google Scholar]
- 180.Schuitemaker A, Kropholler MA, Boellaard R, et al. Microglial activation in Alzheimer's disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol Aging. 2013;34:128–36. doi: 10.1016/j.neurobiolaging.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 181.Yasuno F, Ota M, Kosaka J, et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer's disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiatry. 2008;64:835–41. doi: 10.1016/j.biopsych.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 182.Edison P, Archer H, Hinz R, et al. Relationship between the distribution of microglial activation and amyloid deposition in Alzheimer's disease: An 11C-PK11195 and 11C-PIB PET study. J Neurol Neurosurg Psychiatry. 2007;78:219–219. [Google Scholar]
- 183.Yokokura M, Mori N, Yagi S, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2011;38:343–51. doi: 10.1007/s00259-010-1612-0. [DOI] [PubMed] [Google Scholar]
- 184.Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–8. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 185.Kreisl WC, Lyoo CH, McGwier M, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer's disease. Brain. 2013;136:2228–38. doi: 10.1093/brain/awt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Okello A, Edison P, Archer HA, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shi H, Belbin O, Medway C, et al. Genetic variants influencing human aging from late-onset Alzheimer's disease (LOAD) genome-wide association studies (GWAS) Neurobiol Aging. 2012;33:1849.e5–18. doi: 10.1016/j.neurobiolaging.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer's Disease: a Comparative Overview. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8657-1. published online Feb 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Galimberti D, Fenoglio C, Scarpini E. Inflammation in neurodegenerative disorders: friend or foe? Curr Aging Sci. 2008;1:30–41. doi: 10.2174/1874609810801010030. [DOI] [PubMed] [Google Scholar]
- 190.Czirr E, Wyss-Coray T. The immunology of neurodegeneration. J Clin Invest. 2012;122:1156–63. doi: 10.1172/JCI58656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ruckh JM, Zhao JW, Shadrach JL, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10:96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Villeda S, Wyss-Coray T. Microglia--a wrench in the running wheel? Neuron. 2008;59:527–9. doi: 10.1016/j.neuron.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 193.Lista S, Faltraco F, Hampel H. Biological and methodical challenges of blood-based proteomics in the field of neurological research. Prog Neurobiol. 2013;101-102:18–34. doi: 10.1016/j.pneurobio.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 194.Green MJ, Matheson SL, Shepherd A, Weickert CS, Carr VJ. Brain-derived neurotrophic factor levels in schizophrenia: a systematic review with meta-analysis. Mol Psychiatry. 2011;16:960–72. doi: 10.1038/mp.2010.88. [DOI] [PubMed] [Google Scholar]
- 195.Kurita M, Nishino S, Kato M, Numata Y, Sato T. Plasma brain-derived neurotrophic factor levels predict the clinical outcome of depression treatment in a naturalistic study. PLoS ONE. 2012;7:e39212. doi: 10.1371/journal.pone.0039212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lee BH, Kim H, Park SH, Kim YK. Decreased plasma BDNF level in depressive patients. J Affect Disord. 2007;101:239–44. doi: 10.1016/j.jad.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 197.Martinotti G, Di Iorio G, Marini S, Ricci V, De Berardis D, Di Giannantonio M. Nerve growth factor and brain-derived neurotrophic factor concentrations in schizophrenia: a review. J Biol Regul Homeost Agents. 2012;26:347–56. [PubMed] [Google Scholar]
- 198.Hu WT, Holtzman DM, Fagan AM, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;79:897–905. doi: 10.1212/WNL.0b013e318266fa70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 200.Soares HD, Potter WZ, Pickering E, et al. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Arch Neurol. 2012;69:1310–7. doi: 10.1001/archneurol.2012.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Britschgi M, Rufibach K, Huang SLB, et al. Modeling of pathological traits in Alzheimer's disease based on systemic extracellular signaling proteome. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.008862. M111.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Johnstone D, Milward EA, Berretta R, Moscato P Alzheimer's Disease Neuroimaging Initiative. Multivariate protein signatures of pre-clinical Alzheimer's disease in the Alzheimer's disease neuroimaging initiative (ADNI) plasma proteome dataset. PLoS ONE. 2012;7:e34341. doi: 10.1371/journal.pone.0034341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cribbs DH, Berchtold NC, Perreau V, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010;184:7207–18. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tang EHC, Libby P, Vanhoutte PM, Xu A. Anti-inflammation therapy by activation of prostaglandin EP4 receptor in cardiovascular and other inflammatory diseases. J Cardiovasc Pharmacol. 2012;59:116–23. doi: 10.1097/FJC.0b013e3182244a12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brenneis C, Coste O, Altenrath K, et al. Anti-inflammatory role of microsomal prostaglandin E synthase-1 in a model of neuroinflammation. J Biol Chem. 2011;286:2331–42. doi: 10.1074/jbc.M110.157362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rogers J, Kirby LC, Hempelman SR, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–11. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 208.De Jong D, Jansen R, Hoefnagels W, et al. No effect of one-year treatment with indomethacin on Alzheimer's disease progression: a randomized controlled trial. PLoS ONE. 2008;3:e1475. doi: 10.1371/journal.pone.0001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 210.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 211.Aisen PS, Davis KL, Berg JD, et al. A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000;54:588–93. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 212.Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ, Scheltens PK. Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001;358:455–60. doi: 10.1016/s0140-6736(01)05623-9. [DOI] [PubMed] [Google Scholar]
- 213.Simons M, Schwärzler F, Lütjohann D, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52:346–50. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 214.Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62:753–7. doi: 10.1001/archneur.62.5.753. [DOI] [PubMed] [Google Scholar]
- 215.Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74:956–64. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- 216.AD2000 Collaborative Group. Bentham P, Gray R, et al. Aspirin in Alzheimer's disease (AD2000): a randomised open-label trial. Lancet Neurol. 2008;7:41–9. doi: 10.1016/S1474-4422(07)70293-4. [DOI] [PubMed] [Google Scholar]
- 217.Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30:131–46. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.ADAPT Research Group. Martin BK, Szekely C, et al. Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Breitner JC, Baker LD, Montine TJ, et al. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7:402–11. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wilcock DM, Zhao Q, Morgan D, et al. Diverse inflammatory responses in transgenic mouse models of Alzheimer's disease and the effect of immunotherapy on these responses. ASN Neuro. 2011;3:249–58. doi: 10.1042/AN20110018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Jimenez S, Baglietto-Vargas D, Caballero C, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–61. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–53. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- 223.Zotova E, Bharambe V, Cheaveau M, et al. Inflammatory components in human Alzheimer's disease and after active amyloid-β42 immunization. Brain. 2013 doi: 10.1093/brain/awt210. published online Aug 13. [DOI] [PubMed] [Google Scholar]
- 224.Boche D, Zotova E, Weller RO, et al. Consequence of Aβ immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- 225.Wilcock DM, Rojiani A, Rosenthal A, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241–9. doi: 10.1016/S1474-4422(12)70015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wilcock DM, Alamed J, Gottschall PE, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Aisen PS, Schmeidler J, Pasinetti GM. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. 2002;58:1050–4. doi: 10.1212/wnl.58.7.1050. [DOI] [PubMed] [Google Scholar]
- 229.Thal LJ, Ferris SH, Kirby L, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30:1204–15. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 230.ADAPT Research Group. Lyketsos CG, Breitner JCS, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007;68:1800–8. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
Box 1. Search strategy and selection criteria.
We searched PubMed for journal articles that were published between January, 2009 to October, 2014 restricted by the terms “neuroinflammation” or “inflammation” and “Alzheimer” and included those judged most relevant to the field. We also selected papers with groundbreaking findings that led to recent research.