

Abstract
Dendritic cells are the principal antigen presenting cells that are responsible for acquiring and transporting antigen from the peripheral tissue to the secondary lymphoid tissue. There they present it to T cells which ultimately initiate an antigen specific immune response. In vivo, the migration of dendritic cells (DCs) and T cell activation are intimately linked. However, ex vivo systems that facilitate integrated evaluation of DC chemotaxis and resulting T cell activation by migrated DCs are lacking. In this work, we have developed a microfabricated platform that integrates DC chemotaxis with T cell activation. The basic design of the microdevice includes two layers of PDMS, with the top layer comprising the chemotaxis compartment and the bottom layer containing a T cell compartment. In the chemotaxis compartment, the DCs are subjected to a chemokine gradient, and their migratory response is evaluated. In the T cell compartment, rapid DC-induced activation of T cells is evaluated by measuring the level of calcium in T cells. We demonstrate the efficacy of our approach by evaluating the integrated response of mature DCs, whereby the overall T cell activation response is governed both by the chemotaxis and the T cell activation potential of mature DCs relative to immature DCs. Our system provides a powerful platform for systematically probing various aspects of antigen induced immune responses – DC maturation, migration and T cell activation – in an integrated fashion.
Introduction
Dendritic cells are potent antigen presenting cells that provide a key functional link between the innate and the adaptive immune responses. Upon exposure to an antigen, dendritic cells (DCs) in the peripheral tissues undergo maturation and migrate to secondary lymphoid organs where they present the antigen to naïve T cells, thus initiating the adaptive immune response.1 This well orchestrated sequence of events forms the basis for antigen induced immune responses following pathogen infection2 and allergic contact dermatitis.3 Developing systems that enable ex vivo interrogation, as well as manipulation of these cellular events, in an integrated fashion will greatly enhance our understanding of various factors that influence antigen induced immune responses.
In recent years, microfluidic devices are gaining prominence for creating integrated platforms for biological applications. In the context of adaptive immune response induced by an antigen, microfluidic devices have been utilized to evaluate DC-T cell immunological synapses4 and T cell activation by antibody coated beads.5 These studies employed specialized microfabrication design or external electric field in combination with microfluidics, to bring T cells in contact with DCs or beads. Although these strategies facilitated analysis of T cell activation at a single cell level, they did not attempt to create a platform where other cellular events such as chemotaxis of DCs are integrated with DC-T cell immunological synapses. In comparison to somewhat limited studies that employed microdevices for examining DC-T cell immunological synapses, chemotaxis has been widely investigated in microfabricated devices.
As compared to the traditional methods such as the Boyden chamber (transwell) assay, microdevices enable more exquisite spatial and temporal control over the chemokine gradient6 and also permit time lapse imaging to monitor cell migration. A number of studies have focused on utilizing microdevices to examine migration behavior of adherent cells such as neutrophils,6-11 cancer cells,12 and endothelial cells.13 However, weakly adherent cells such as dendritic cells present additional challenges as flow introduced to apply a chemokine gradient should not disturb chemotactic migration of cells. Haessler et al.14 circumvented this issue by fabricating an agarose based device where dendritic cells were embedded in the ECM matrix in the cell chamber and further flow isolation was achieved by an agarose wall that separated the flow chamber from the cell chamber. Even though this approach facilitated evaluation of chemotaxis of dendritic cells, it is not readily amenable for integration with other steps such as T cell activation by migrated DCs. Another approach that allows the investigation of non-adherent cell chemotaxis, relies on trapping the cells in the microchannels comparable to the size of the cells and subjecting the front and back of the cells to a chemokine gradient.15 This device was utilized to evaluate migration behavior of neutrophils under mechanically constrained conditions. Another study employed a similar design to evaluate morphological changes associated with spontaneous migration of DCs.16 However, integration of other steps such as DC-T cell synapse with chemotaxis of DCs has not been realized.
In this work, we have developed a microfabricated platform that facilitates evaluation of chemotaxis of DCs and DC-T cell immunological synapses in an integrated fashion. Specifically, the microdevice consists of a chemotaxis compartment and a T cell compartment. Mature DCs are introduced into the chemotaxis compartment where they are exposed to a gradient of the chemokine, CCL 19. The DCs that undergo chemotaxis enter into the T cell compartment. Subsequently, T cells labeled with a calcium sensitive fluorescent dye are introduced in the T cell chamber and T cell activation is monitored by measuring intracellular calcium levels.
Device design
In response to an antigen, dendritic cells migrate to the lymph node and present the antigen to the T cells. In order to mimic this process ex vivo, we have developed a microfabricated platform that facilitates integrated evaluation of DC chemotaxis and DC-T cell immunological synapses. The microdevice is comprised of a chemotaxis compartment connected to a T cell compartment as shown in Fig. 1. The outer region of the chemotaxis compartment consists of side channels through which medium with or without chemokine is pumped. These outer side channels are connected to the inner side channels via parallel transversal channels. The dimension of these transversal channels is such that they allow fluidic connection between the inner and outer side channels, but prevent passage of DCs. This design ensures that the cells remain confined within the inner region while fluid is actively pumped into the outer region. The middle region of the chemotaxis compartment is occupied by another set of parallel transversal channels, through which chemotaxis of DCs occur and are henceforth called the chemotaxis channels. The design and dimension of these chemotaxis channels is such that they facilitate mechanical confinement of DCs at the entrance of the constriction in the chemotaxis channels while loading the cells and also permit relatively fast retrieval of the cells that undergo chemotaxis across the channels. The chemotaxis channels are connected to the two inner side channels. One of the inner side channels is used for introducing the DCs into the chemotaxis compartment (cell loading channel) and connected to the DCs loading port, while the other channel (cell retrieval channel) is connected to the T cell compartment. The design also includes a waste channel connected to the cell retrieval channel as shown in Fig. 1. This channel is used for removing the excess cells that enter into the cell retrieval channel, while loading the cells into the chemotaxis compartment prior to chemotaxis. The flow from the cell retrieval channel is directed towards the T cell compartment or waste by actuating valves 1 and 2. Opening valve 1 and closing valve 2 directs flow towards the T cell compartment, while the flow is directed towards waste by closing valve 1 and opening valve 2. Finally, the T cell compartment is comprised of a circular trough structure in a level below the chemotaxis compartment, with the channels overhanging on top. This design ensures that DCs that undergo chemotaxis settle into the T cell compartment due to gravity without impeding the flow on top of the T cell compartment.
Fig. 1.
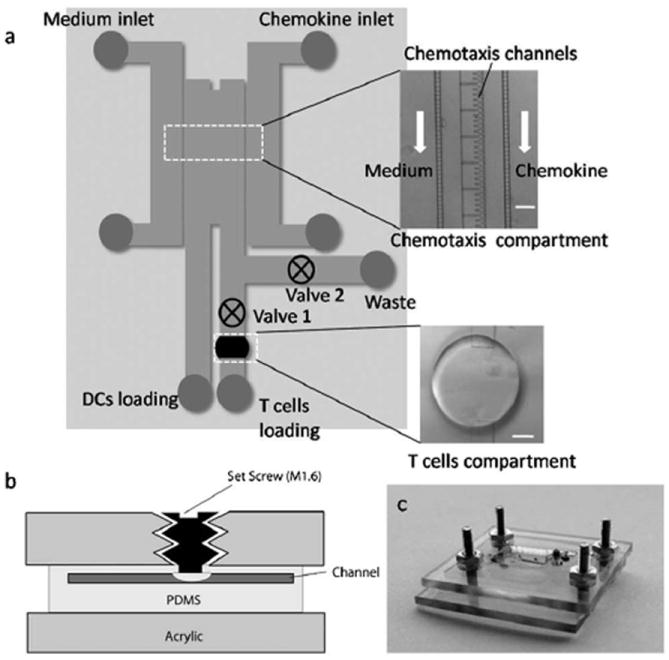
Schematic and images of microdevice. (a) Schematic of microdevice comprising of chemotaxis compartment connected to T cell compartment. The insets show micrographs of chemotaxis compartment and T cell compartment (scale bar: 100 μm). (b) Schematic illustrating basic operation of valve. (c) Image of the device mounted in the plastic manifold.
Materials and methods
Reagents
Advanced RPMI 1640 medium (Reduced serum RPMI), MEM Alpha medium with ribonucleosides, deoxyribonucleosides, and GlutaMax (MEM-α), glutamine, penicillin/streptomycin (P/S), 2-mercaptoethanol (2-Me), fetal bovine serum (FBS), and Fluo4 were purchased from Invitrogen technologies (Carlsbad, CA). Recombinant human GM-CSF, recombinant human tumor necrosis factor alpha (TNF-α), human transforming growth factor beta 1 (TGF-β1), recombinant human interleukin 6 (IL-6), recombinant human interleukin 1 beta (IL-1β), and recombinant human CCL19 were acquired from R&D systems (Minneapolis, MN). Prostaglandin E2 (PGE2) was purchased from Sigma-Aldrich (St. Louis, MO). MUTZ-3 cells were obtained from Massachusetts General Hospital (Boston, MA). 5637, a human renal carcinoma cell line, was obtained from American Type Culture Collection.
Generating mature and immature dendritic cells
5637 cells were cultured in RPMI medium supplemented with 10% FBS, 4 mM glutamine, and 1% P/S. After the cells reached confluence, the cells were exposed to the fresh medium for 42–43 h. Next, the medium was collected and spun down to remove debris. The supernatant was kept frozen at −80 °C prior to use as the conditioned medium (CM) for preparing the medium to culture MUTZ-3 cells.17
MUTZ-3 cells were routinely cultured at a density of 105 cells ml−1 in the MEM-α medium supplemented with 20% FBS, 10% CM, 1% P/S and 50 μM 2-Me. MUTZ-3 cells were differentiated towards the dendritic cells lineage by exposing the cells to a cytokine cocktail for 7 days according to a slight variation of the procedure described in the literature.18 On day 0, the cells were differentiated at a density of 105 cells ml−1 in the MEM-α medium supplemented with 20% FBS, 1% P/S, 50 μM 2-Me and a cytokine cocktail of 100 ng ml−1 GM-CSF, 2.5 ng ml−1 TNF-α, and 10 ng ml−1 TGF-β1. On day 2 and day 5 of differentiation, the cells were exposed to the fresh medium supplemented with a cytokine cocktail of 50 ng ml−1 GM-CSF, 1.25 ng ml−1 TNF-α, and 5 ng ml−1 TGF-β1. On day 7, the cells were divided into two groups for either generating mature dendritic cells (mDCs) or immature dendritic cells (iDCs). In order to generate mDCs, the differentiated cells (2 × 105 cells ml−1) were exposed for 2 days to the MEM-α medium supplemented with 20% FBS, 1% P/S, 50 μM 2-Me and a cytokine cocktail of 100 ng ml−1 IL-6, 50 ng ml−1 TNF-α, 25 ng ml−1 IL-1β, and 1 mg ml−1 PGE2. The iDCs were obtained by treating the differentiated cells for 2 days with the same medium as above except without any cytokines. Treatment with the cytokine cocktail resulted in up-regulation of the DC maturation markers such as CD 86 (data not shown), which is consistent with prior reports.19 On day 9, the mDCs or iDCs were suspended at a density of 4–6 × 106 cells ml−1 in the fresh MEM-α medium supplemented with 20% FBS, 1% P/S, 50 μM 2-Me. Henceforth in the manuscript, the medium will refer to MEM-α medium supplemented with 20% FBS, 1% P/S, 50 μM 2-Me unless otherwise noted.
Isolation and labeling of T cells
Whole blood was acquired from the Blood Center of New Jersey. Peripheral blood mononuclear cells (PBMC) were isolated using the Ficoll-Paque (d = 1.077 kg L−1, GE Healthcare, Piscataway, NJ) density centrifugation protocol provided by the manufacturer. T cells were isolated from the PBMC fraction by positive selection using CD3 microbeads and a magnetic cell separator (Miltenyi Biotec Inc., Auburn, CA), following the manufacturer’s instructions. The T cells were frozen in liquid nitrogen and thawed prior to use. The T cells were suspended at a density of 1–2 × 106 cells ml−1 in the medium supplemented with 25 mM HEPES. The cells were labeled with the fluo4 dye diluted to a final concentration of 1 μM by incubating the cells at 37 °C for 20 min, and then washing twice to remove the remaining dye in the medium. Prior to loading into the microdevice, the labeled T cells were suspended at a concentration of 10 × 106 cells ml−1 in the fresh medium, which was supplemented with 25 mM HEPES.
Device fabrication
The microdevice was fabricated in PDMS using standard soft lithography and micromolding techniques. The masks were designed using the AutoCAD software and printed out on chrome (Fineline Imaging, Colorado Springs, CO). The device comprised of two layers of PDMS, which were molded separately using the two masters. The top layer of PDMS consisted of the chemotaxis compartment and the flow channels, while the bottom layer contained the T cell compartment. The master for the top layer was prepared by photo-lithographic patterning of SU8 on a silicon wafer to three different thicknesses − 40 μm for flow channels, 18 μm for chemotaxis channels, and 3 μm for flow reducing channels. The three-layered structure was made by successively spinning, patterning, and baking different formulations of SU8 (Su8 2002, Su8 2010, Su8 2025) starting from the lowest to the highest thickness, according to the manufacturer’s instructions. Finally, the master was hard baked at 150 °C for 20 min. Similarly, the master for the bottom layer was prepared using Su8 (SU8 2075) processed to a thickness of 225 μm on a silicon wafer.
The two masters were silanized to prevent PDMS sticking, by placing them in a desiccator with 10μl of Trichloro(1,1,2,2-perfluorooctyl) silane for 45 min. Meanwhile a 10 : 1 mixture of PDMS pre-polymer (Sylgard 184) and curing agent was prepared and degassed, and poured on top of the wafers. The wafers were baked in an oven for two hours at 65 °C to polymerize the PDMS. The molded PDMS layers were then cored with a biopsy punch (Harris Unicore™ from Ted Pella Inc.) to make access holes, cleaned with scotch tape, and sonicated in ethanol for 15 min to remove particulates. An optional 10 min dip in HCl was employed if the PDMS layers were not freshly prepared. The layers were then rinsed with DI Water, dried, and exposed to 50 W oxygen plasma (60% O2/air mixture) for 30 s to activate the PDMS surfaces. The activated layers were immediately aligned under a microscope, and brought into conformal contact for bonding. The bonded devices were then placed on a hot plate at 85 °C for 20 min, and gradually allowed to reach the room temperature.
Valve design and operation
The valve principle is similar to the screw valves already described in the literature.20 In our design, the setscrews were mounted in the custom designed plastic manifold (acrylic) as shown in the Fig. 1b. For valve operation, the PDMS device was mounted in the plastic manifold such that the setscrew aligned with the channel and upon tightening the screw, it applied pressure on PDMS, which resulted in closure of the channel. As we were not utilizing the valve for controlling the flow rate and were primarily interested in on and off operation, we only conducted basic characterization to ensure that under tightened condition, the setscrew was able to stop the flow in the channel and upon loosening the flow was restored.
Chemotaxis of DCs
The microdevices were primed using the medium. In some cases, the devices were left for 5 min in a desiccator operating under vacuum to remove the bubbles. The channels were continuously flushed with the medium. The primed devices were then placed in a custom designed plastic manifold. The manifold contained strategically placed setscrews, which aligned with the flow channels in the device, serving as valves 1 and 2 as shown in the Fig. 1. The device with the plastic manifold was mounted on a microscope and the two inlets in the outer side channels were connected to the two syringes containing medium with or without CCL19 (500 ng ml−1) that were mounted on a dual syringe pump (Harvard Apparatus (Holliston, MA)). The valve 1 connecting to the T cell compartment was kept open, while the valve 2 was closed. The gradient was set up by operating the dual syringe pump at a flow rate of 0.4–0.5 μl min−1.
The DCs were introduced from the DC loading port using a pipette tip that was filled with the cell suspension and the head was adjusted to 2 cm using additional medium. During the cell loading process, the syringe pump was turned off. Under these conditions, the cells started flowing into the chemotaxis compartment with the cells starting to get lodged at the entrance of the constriction in the chemotaxis channels. After several minutes, when most of the chemotaxis channels were occupied by the cells, the pipette tip was removed and then the flow in the outer side channels was started using the syringe pump. The movement of the DCs was recorded for 1.5–2 h by time lapse imaging of multiple locations using a computer controlled automated stage.
T cell activation by DCs
The devices were primed with the medium supplemented with 25 mM HEPES and mounted in the plastic manifold that could hold two devices. During these experiments, only the T cell compartment and surrounding channels were utilized. The mDCs or iDCs were introduced into the T cell compartments using a pipette tip placed at the waste ports. The head in the pipette tip was adjusted such that the DCs reaching the T cell compartment settled at the bottom of the T cell compartment and did not exit the T cell compartment. After loading similar numbers of mDCs and iDCs (based on the number of mDCs that migrated into the T cell compartment as described below in the next section), the pipette tips were removed and the waste and the T cell inlet ports were cleaned. Next, the T cells labeled with the fluorescent dye were introduced into the T cell compartments from the T cell inlet port using a pipette tip. In this case also, the head in the pipette tip was adjusted to facilitate settling of the T cells within the T cell compartments. Once a uniform distribution of T cells was achieved in the T cell compartments, the valve 1 was closed, the devices were mounted on a microscope and the images of the T cell compartment were acquired for 45 min.
Integrated evaluation of DC chemotaxis and T cell activation
For the integrated evaluation, the two devices were primed and mounted on a manifold as described above in the T cell activation section. In one device, mDCs were subjected to a gradient of the chemokine CCL19 and the migrated mDCs were collected in the T cell compartment. For loading of mDCs and the application of the CCL19 gradient in the chemotaxis compartment, similar steps were followed as described above in the chemotaxis section with a few modifications. In this case, during loading of the mDCs, the valve 1 was closed and the valve 2 was open. This prevented the mDCs that entered into the cell retrieval channel during loading, from entering into the T cell compartment. After removal of the pipette tip from the DC loading port and washing off residual mDCs from the cell retrieval channel into the waste, the valve 1 was opened and the valve 2 was closed. Next, the syringe pump was turned on to start the flow in the outer side channels. This also resulted in flow being directed in cell retrieval channel towards the T cell compartment. Thus only those mDCs that fully migrated across the chemotaxis channels, in response to a gradient of CCL19, were transported to the T cell compartment via flow in the cell retrieval channel. After 2 h of chemotaxis, the syringe pump was stopped and the T cell port was washed to remove the chemokine, CCL19. Next, in order to remove the chemokine, the T cell compartment was gently washed using a pipette tip mounted in the T cell port. This washing step also resulted in the distribution of mDCs that had accumulated under the outlet of the T cell compartment (Fig. 5b). Based on the number of mDCs that had migrated into the T cell compartment of the first device, a similar number of mDCs were directly introduced, from the waste port, into the T cell compartment of the second device. The rest of the procedure for inducing T cell activation was similar to the protocol already described above in the preceding section (T cell activation by DCs).
Fig. 5.
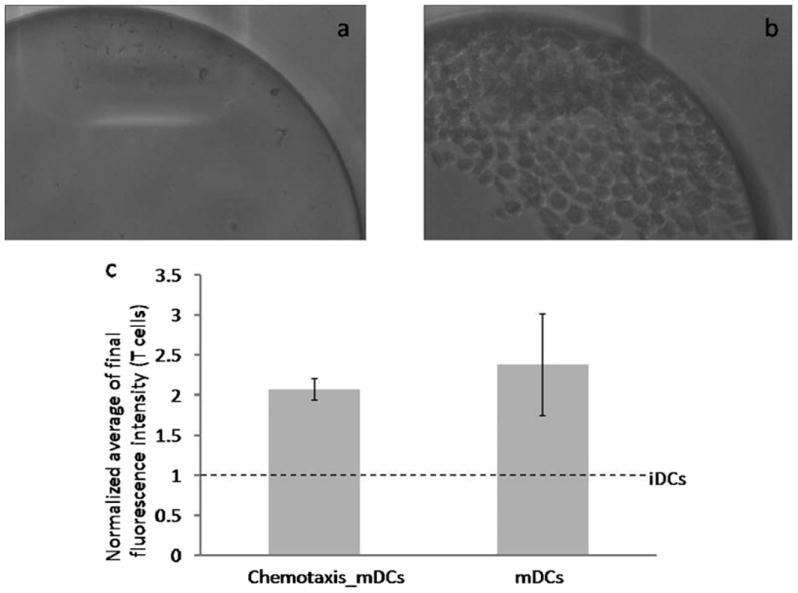
Integrated evaluation of DC chemotaxis and T cell activation. (a) Micrograph of T cell compartment before start of chemotaxis. (b) Micrograph of T cell compartment at the end of chemotaxis. (c) Normalized average of the final fluorescence intensity of T cells interacting with mDCs that migrated into the T cell compartment in response to a gradient of the chemokine CCL19 vs. mDCs not subjected to chemotaxis. The response was normalized with respect to iDCs that were directly introduced into the T cell compartment and interacted with the T cells.
Image capture
Images were captured using an Olympus (Center Valley, PA) IX81 inverted microscope equipped with XM10 digital camera and automated xyz stage. The fluorophores were excited using EXFO X-Cite 120 fluorescent light source. For both FITC-dextran and fluo4, fluorescence images were acquired using a GFP filter set. Olympus cellSens Dimension software was used for acquiring and saving the images before further image processing.
Image analysis for estimating migration velocity of DCs
Chemotaxis velocity was measured by tracking the movement of the front of the cell across the constriction in the chemotaxis channels. The choice of this metric was based on the consideration that even if a cell does not move all the way through the constriction of the chemotaxis channel, movement of the front of a cell is indicative of the migratory behavior. For the purpose of the measurement, cells, which were stuck in the constriction, with more than 50% of the cell body sticking out towards the chemokine at the initial time point, were discarded for the purpose of velocity estimation. The measurement was done manually for each cell, with t = 0 defined as the time when a cell was at the entrance of constriction, and measuring the distance moved thereafter, to calculate the velocity.
Image analysis for measuring T cell activation
The fluorescence images were analyzed using an in-house written MATLAB routine. The images were analyzed to extract fluorescence of T cells from the background. We first implemented a Sobel operator to detect rough borders of the T cells, and then manipulated the borders to create a mask that primarily covered the T cell areas. We then used a morphological reconstruction algorithm based on the original image and the masked image, to isolate the T cells from the background. Finally, we removed additional artifacts by size filtering and broke up adjoining cell borders using the watershed algorithm. The fluorescence of a T cell was estimated by averaging the pixel intensities inside a T cell, which was used to calculate the average of the fluorescence intensity of T cells. The fluorescence intensity distribution was determined by applying a normal distribution function to the fluorescence intensities of the T cells.
Results
Gradient characterization
In order to evaluate the gradient forming capability of our device, PBS with or without a fluorescent tracer was pumped through the outer side channels. The fluorescence tracer employed was FITC labeled dextran (10 kDa), which is of similar molecular weight as CCL 19 (8.8 kDa). The flow in the outer side channels generates reduced flow in the cell retrieval channel and the cell loading channel that leads to formation of concentration gradient across the chemotaxis channels as depicted in the fluorescence intensity profile across the cross section of the chemotaxis compartment (Fig. 2). The fluorescence intensity across the cross section (white line in Fig. 2a) was quantified (Image J software, NIH) and normalized based on the level of fluorescence in the side channels containing PBS with (100%) or without (0%) the tracer. Fig. 2b illustrates that across the cross section, the fluorescence intensity level increases from the cell loading channel towards the cell retrieval channel with the sharpest change occurring at the interface of the chemotaxis channel and the cell retrieval channel. These results indicate that our design can be used to apply a gradient of chemokine across the chemotaxis channels.
Fig. 2.
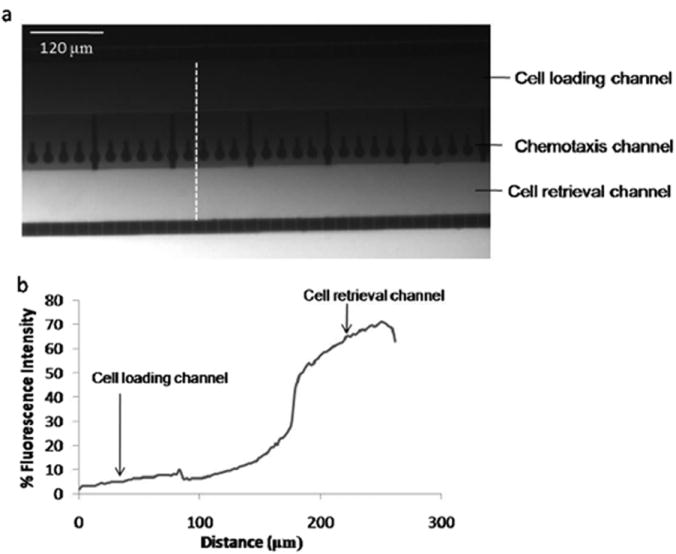
Gradient formation in the chemotaxis compartment. (a) Fluorescence micrograph illustrating concentration gradient across the chemotaxis compartment. PBS with or without FITC dextran was continuously flushed through the outer side channels using the syringe pump. (b) Quantified fluorescence intensity across the cross section (white line in Fig 2a) of the chemotaxis compartment. The intensity was normalized based on the fluorescence level in the outer side channels containing PBS with (100%) or without (0%) FITC dextran.
Chemotactic response of dendritic cells
The migratory response of dendritic cells was evaluated by subjecting the cells to a gradient of the chemokine, CCL 19. Furthermore, the gradient was formed by employing CCL 19 at a concentration of 500 ng ml−1. The gradient profile (Fig. 2b) indicates that the concentration of chemokine in the cell retrieval channel, adjacent to the chemotaxis channels, drops by ~50% of the bulk concentration in the chemokine side channel. Thus, the concentration of CCL 19 in the cell retrieval channel adjacent to the chemotaxis channels goes down to ~250 ng ml−1, which is what has been previously used for investigating migratory response of DCs.21,22 Fig. 3a shows the time lapse micrographs depicting the chemotactic response of mDC. At time t = 0, the mDC resides at the entrance of the constriction of the chemotaxis channel. Upon encountering a gradient of CCL 19, the cells undergo a shape change from circular to an elongated morphology as they traverse across the chemotaxis channels (Fig. 3a). This type of shape change during chemotaxis is reminiscent of the amoeboid like movement typically observed for dendritic cells. By the time t = 50 min, the mDC no longer appears in the image, as it has completely migrated across the chemotaxis channel and exited into the cell retrieval channel. To quantitatively estimate the differential migratory response of mature and immature dendritic cells, we evaluated velocity profiles of the cells across the chemotaxis channels. The velocity was measured by tracking the movement of the front of the cells initially lodged at the entrance of the constriction of the chemotaxis channels. Fig. 3b illustrates that ~35% mDCs displayed migratory movement. We also observed that a number of mDCs completely migrated across the chemotaxis channels. By contrast, in the case of iDCs a very small number of cells (<3%) displayed any movement, with none of the cells fully migrating across the chemotaxis channels. For mDCs that were exposed to the control medium on both sides of the chemotaxis channels (no chemokine gradient), ~16% of cells (Fig. S1, ESI†) showed slight movement, however none of the cells was able to fully migrate across the chemotaxis channels. These results indicate that only mDCs, upon encountering a gradient of the chemokine CCL 19, are able to completely migrate across the chemotaxis channels.
Fig. 3.
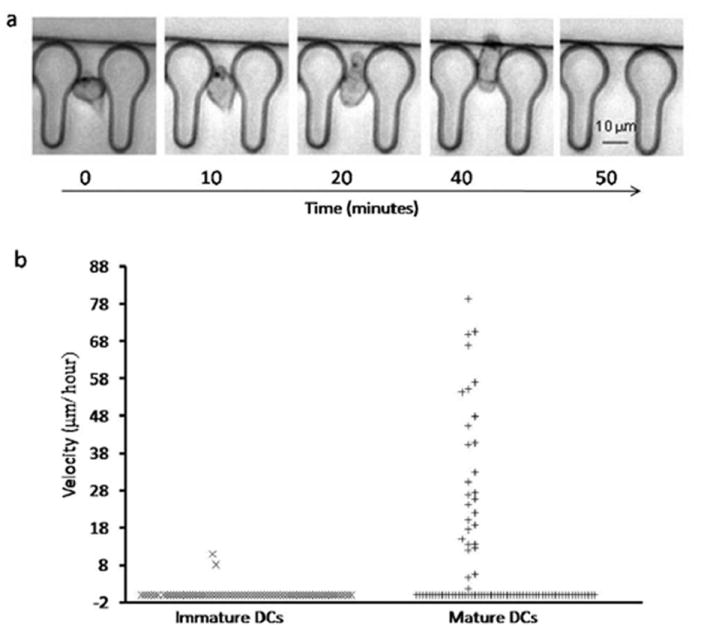
Chemotaxis response of mDCs and iDCs. (a) Time lapse images depicting representative migration of mDC across the chemotaxis channel subjected to a gradient of CCL19. (b) Velocity distribution of mDCs vs. iDCs subjected to a gradient of CCL19.
T cell activation by DCs in the T cell chamber
Dendritic cells form immunological synapses and activate the T cells in the lymph node as part of the process for initiating the adaptive immune response induced by an antigen. In order to compare the T cell activation potential of mDCs and iDCs, the T cell compartment of the device was loaded with mDCs or iDCs and subsequently T cells loaded with calcium sensitive dye were introduced. Fig. 4a shows the fluorescence image of the T cell compartment loaded with mDCs and fluorescently labeled T cells. The upper panel of the inset shows the time lapse images depicting a temporal increase in the fluorescence of two T cells forming contact with mDC as indicated in the lower panel. The increase in fluorescence is indicative of the enhancement in the level of intracellular calcium in T cells. An increase in intracellular calcium occurs following the engagement of the T cell receptor and is considered one of the early markers of T cell activation.23 Fig. 4b and 4c show the distribution and average of the final fluorescence intensity of T cells interacting with the mDCs or iDCs. The plots indicate that the T cells interacting with mDCs exhibit a higher level of final fluorescence intensity as compared to those interacting with iDCs. These results indicate that the mDCs induce greater activation of T cells as compared to iDCs.
Fig. 4.
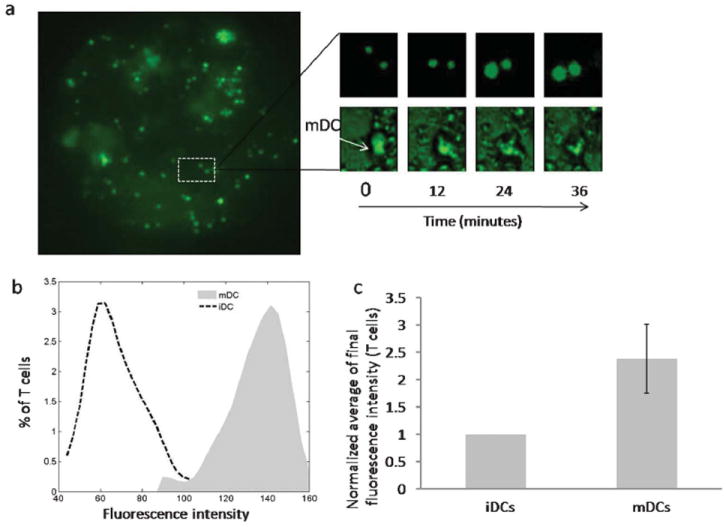
T cell activation by DCs in the T cell compartment. (a) Fluorescence image of the T cell compartment loaded with mDCs and fluorescently labeled T cells. The top inset shows time lapse images depicting temporal increase in the fluorescence of T cells. The bottom inset shows corresponding time lapse images depicting location of the mDC. (b) Final fluorescence intensity distribution of T cells interacting with mDCs or iDCs in the T cell compartment. (c) Normalized average of the final fluorescence intensity of T cells interacting with mDCs or iDCs.
Integrated analysis of DC chemotaxis and DC-T cell interaction
The dendritic cells have to migrate from the peripheral tissue to lymph node in order to present the antigen to the T cells and initiate the adaptive immune response. The overall T cell activation response in lymph node depends on both the ability of DCs to migrate to lymph node as well as the T cell activation potential. Having already established that only mDCs were able to fully migrate across the chemotaxis channels, we conducted integrated evaluation of mDCs. Fig. 5a and 5b shows the images of the T cell compartment before and after chemotaxis of mDCs. Fig. 5b illustrates robust trapping of mDCs that fully migrated across the chemotaxis channels. Next, we evaluated the T cell activation potential of migrated mDCs by introducing the T cells into the T cell compartment. Fig. 5c demonstrates that the T cell activation by the migrated mDCs was similar to that of mDCs that were not subjected to chemotaxis. These results indicate that the DCs, while traversing across the chemotaxis channels, do not sustain any kind of injury that precludes their ability to form immunological synapses and activate the T cells. However, iDCs due to their inability to migrate in response to a gradient of the chemokine CCL19 did not reach the T cell compartment and failed to activate the T cells. Thus, in the integrated device, the overall T cell activation response is controlled by the number of DCs that become available for interacting with the T cells. In our study, iDCs presented an extreme case where their inability to migrate resulted in the amplification factor to be zero and hence no T cell activation. These results establish that our system enables integrated evaluation whereby the overall T cell activation response depends on both the chemotaxis response of the DCs and their capability to activate the T cells.
Discussion and conclusion
Dendritic cells are the principal cells responsible for transporting the antigen from the peripheral tissue to the lymph node, where T cell activation occurs as part of the process of initiating antigen specific immune responses. Ex vivo, prior studies have focused separately on evaluating individual steps such as chemotaxis of DCs and DC-T cell immunological synapses. However, in vivo these two steps are intimately linked. In this work, we have developed a microfabricated platform that enables integrated evaluation of DC chemotaxis and DC-T cell immunological synapse activation. We demonstrated the efficacy of this microdevice by evaluating integrated responses of mDCs.
Our microdevice is comprised of a chemotaxis compartment integrated to a T cell compartment. The chemotaxis compartment design exploited a few features presented in the “polar stimulation device” previously described in the literature.15 The design included chemotaxis channels that facilitated loading of non-adherent DCs under mechanically constrained conditions. However, our design also included shallow transversal channels that served to reduce the flow in the cell loading and cell retrieval channels next to the chemotaxis channels. These shallow channels also facilitated exclusion of cells from the outer side channels through which media with or without chemokine were pumped. A similar design that employed shallow transversal channels has been used in the past to reduce shear stress in the cell culture chamber while subjecting the adherent cells to a gradient of soluble factor.10,13 Thus, by combining chemotaxis channels that permit mechanical trapping of DCs with the shallow transversal channel scheme, our design of chemotaxis compartment enabled optimal loading, monitoring of migration, and retrieval of DCs that underwent chemotaxis under non-adherent conditions. This design was also compatible with integrating the T cell compartment downstream of the chemotaxis compartment. The T cell compartment was realized by creating a trough structure that facilitated retention and washing of the DCs, and subsequent introduction of the T cells. A similar design has been used in a prior study to capture and wash cells while conducting various cell based assays on chip.24 In our studies, this design was successfully exploited to capture migrated DCs and evaluate DC-T cell immunological synapse formation and activation of T cells.
The migration of DCs towards the lymph node is largely believed to be regulated by the chemokines, CCL19 and CCL21,25 which are cognate ligands for the receptor CCR7. The maturation of DCs is accompanied by an increase in the expression of the receptor CCR7 resulting in mDCs migrating in response to a gradient of CCL19.26 We obtained similar results whereby exposure to a gradient of CCL19 resulted in a number of mDCs migrating across the chemotaxis channels and entering into the T cell compartment. The activation of T cells upon contacting DCs is influenced by various factors that include interactions mediated by co-stimulatory and adhesion molecules on DCs as well as interaction of peptide-MHC complex on DCs with T cell receptor.27 In our studies, we observed that mDCs, as compared to iDCs, induced stronger T cell activation responses. This is consistent with literature evidence where cytokine induced maturation of DCs is associated with an increase in the expression of co-stimulatory molecules such as CD 86 and enhanced T cell response.19
In the lymph node, it is plausible that the frequency of DC-T cell contact increases with a greater number of migrated DCs reaching the lymph node. Thus, it is possible that the overall T cell activation in the lymph node is regulated both by the number of DCs migrating and their T cell activation potential. In our integrated system, mDCs induced T cell activation by exploiting their ability to migrate across the chemotaxis channels and subsequently activate the T cells. By contrast, iDCs inability to migrate across the chemotaxis channels precluded their ability to activate the T cells. Thus, in our studies, chemotaxis of DCs served as a critical rate limiting step in regulating the final T cell interaction. This is reminiscent of the in vivo condition where chemotaxis of DCs from the peripheral tissues to lymph nodes enables co-localization of the DCs with the T cells, which ultimately regulates their interaction. Since we employed only one type of maturation stimulus, our studies did not attempt to investigate if the choice of maturation stimulus can affect the overall T cell activation by differentially influencing the number of DCs migrating into the T cell chamber and their T cell activation potential. This could be of particular interest in DC based vaccination strategies geared towards cancer, where maturation stimulus needs to be carefully considered in order to achieve optimal DC migration and corresponding T cell activation responses.28
In our studies, DCs were generated from the precursor cell line MUTZ-3, which was originally derived from the peripheral blood of a patient suffering from acute myelomonocytic leukaemia.29 However, our approach should be expandible to human DCs derived from other sources such as monocytes isolated from the peripheral blood.30 Since we are already using the T cells isolated from peripheral blood, this will facilitate evaluation of syngeneic T cell activation responses where both the DCs and the T cells are derived from the same donor and thus provide a more physiologically relevant system. We evaluated the T cell activation by measuring calcium influx, which is an indicator of the initial T cell activation and also believed to play an important role in governing the final fate of the T cells.31 We did not attempt to evaluate long term T cell activation responses such as proliferation of T cells, which require culturing of cells for long duration (2–5 days). However, our design of the T cell compartment is compatible with long term culture of T cells whereby nutrients can be continuously supplied by replenishing the medium on top of the T cell compartment, while cells reside at the bottom of the compartment.
In conclusion, we have developed a microfabricated system that facilitates integrated evaluation of chemotaxis and T cell activation, which are critical steps involved in antigen transport, and presentation by the DCs. This can be a powerful platform for systematically probing various aspects of antigen induced immune response- DC maturation, migration and T cell activation in an integrated fashion. Given the growing interest in developing DC based vaccines geared towards cancer and infectious diseases, this can be a useful tool for testing and identifying DC maturation strategies for their efficacy in developing viable DC vaccines.
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available: Velocity distribution of mDCs exposed to control medium on both sides of the chemotaxis channels (no chemokine gradient).
References
- 1.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 2.Heath WR, Carbone FR. Nat Immunol. 2009;10:1237–1244. doi: 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Kupper TS. N Engl J Med. 1999;341:1817–1828. doi: 10.1056/NEJM199912093412407. [DOI] [PubMed] [Google Scholar]
- 4.Faley S, Seale K, Hughey J, Schaffer DK, VanCompernolle S, McKinney B, Baudenbacher F, Unutmaz D, Wikswo JP. Lab Chip. 2008;8:1700–1712. doi: 10.1039/b719799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirschbaum M, Jaeger MS, Duschl C. Lab Chip. 2009;9:3517–3525. doi: 10.1039/b911865a. [DOI] [PubMed] [Google Scholar]
- 6.Li Jeon N, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Nat Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 7.Abhyankar VV, Lokuta MA, Huttenlocher A, Beebe DJ. Lab Chip. 2006;6:389–393. doi: 10.1039/b514133h. [DOI] [PubMed] [Google Scholar]
- 8.Irimia D, Balazsi G, Agrawal N, Toner M. Biophys J. 2009;96:3897–3916. doi: 10.1016/j.bpj.2008.12.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irimia D, Liu SY, Tharp WG, Samadani A, Toner M, Poznansky MC. Lab Chip. 2006;6:191–198. doi: 10.1039/b511877h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keenan TM, Frevert CW, Wu A, Wong V, Folch A. Lab Chip. 2010;10:116–122. doi: 10.1039/b913494h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin F, Nguyen CM, Wang SJ, Saadi W, Gross SP, Jeon NL. Ann Biomed Eng. 2005;33:475–482. doi: 10.1007/s10439-005-2503-6. [DOI] [PubMed] [Google Scholar]
- 12.Breckenridge MT, Egelhoff TT, Baskaran H. Biomed Microdevices. 2010;12:543–553. doi: 10.1007/s10544-010-9411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamloo A, Ma N, Poo MM, Sohn LL, Heilshorn SC. Lab Chip. 2008;8:1292–1299. doi: 10.1039/b719788h. [DOI] [PubMed] [Google Scholar]
- 14.Haessler U, Kalinin Y, Swartz MA, Wu M. Biomed Microdevices. 2009;11:827–835. doi: 10.1007/s10544-009-9299-3. [DOI] [PubMed] [Google Scholar]
- 15.Irimia D, Charras G, Agrawal N, Mitchison T, Toner M. Lab Chip. 2007;7:1783–1790. doi: 10.1039/b710524j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faure-Andre G, Vargas P, Yuseff MI, Heuze M, Diaz J, Lankar D, Steri V, Manry J, Hugues S, Vascotto F, Boulanger J, Raposo G, Bono MR, Rosemblatt M, Piel M, Lennon-Dumenil AM. Science. 2008;322:1705–1710. doi: 10.1126/science.1159894. [DOI] [PubMed] [Google Scholar]
- 17.Quentmeier H, Duschl A, Hu ZB, Schnarr B, Zaborski M, Drexler HG. Immunology. 1996;89:606–612. doi: 10.1046/j.1365-2567.1996.d01-780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouwehand K, Santegoets SJ, Bruynzeel DP, Scheper RJ, de Gruijl TD, Gibbs S. Eur J Immunol. 2008;38:3050–3059. doi: 10.1002/eji.200838384. [DOI] [PubMed] [Google Scholar]
- 19.Santegoets SJ, Masterson AJ, van der Sluis PC, Lougheed SM, Fluitsma DM, van den Eertwegh AJ, Pinedo HM, Scheper RJ, de Gruijl TD. J Leukocyte Biol. 2006;80:1337–1344. doi: 10.1189/jlb.0206111. [DOI] [PubMed] [Google Scholar]
- 20.Hulme SE, Shevkoplyas SS, Whitesides GM. Lab Chip. 2009;9:79–86. doi: 10.1039/b809673b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santegoets SJ, Bontkes HJ, Stam AG, Bhoelan F, Ruizendaal JJ, van den Eertwegh AJ, Hooijberg E, Scheper RJ, de Gruijl TD. J Immunol. 2008;180:4540–4549. doi: 10.4049/jimmunol.180.7.4540. [DOI] [PubMed] [Google Scholar]
- 22.Scandella E, Men Y, Legler DF, Gillessen S, Prikler L, Ludewig B, Groettrup M. Blood. 2004;103:1595–1601. doi: 10.1182/blood-2003-05-1643. [DOI] [PubMed] [Google Scholar]
- 23.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimov IK, Kijanka G, Park Y, Ducree J, Kang T, Lee LP. Lab Chip. 2011;11:2701–2710. doi: 10.1039/c1lc20105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randolph GJ, Angeli V, Swartz MA. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 26.Yanagihara S, Komura E, Nagafune J, Watarai H, Yamaguchi Y. J Immunol. 1998;161:3096–3102. [PubMed] [Google Scholar]
- 27.Friedl P, den Boer AT, Gunzer M. Nat Rev Immunol. 2005;5:532–545. doi: 10.1038/nri1647. [DOI] [PubMed] [Google Scholar]
- 28.Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Cell. 2001;106:271–274. doi: 10.1016/s0092-8674(01)00448-2. [DOI] [PubMed] [Google Scholar]
- 29.Sharma NS, Jindal R, Mitra B, Lee S, Li L, Maguire TJ, Schloss R, Yarmush ML. Cell Mol Bioeng. 2012;5:52–72. doi: 10.1007/s12195-011-0189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair S, Archer GE, Tedder TF. Curr Protoc Immunol. 2012;Chapter 7:Unit7–32. doi: 10.1002/0471142735.im0732s99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feske S. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.