

Abstract
Objectives
Traumatic brain injury (TBI) has complex effects on the gastrointestinal tract that are associated with TBI-related morbidity and mortality. We examined changes in mucosal barrier properties and enteric glial cell response in the gut after experimental TBI in mice, as well as effects of the enteric pathogen Citrobacter rodentium (Cr) on both gut and brain after injury.
Methods
Moderate-level TBI was induced in C57BL/6 mice by controlled cortical impact (CCI). Mucosal barrier function was assessed by transepithelial resistance, fluorescent-labelled dextran flux, and quantification of tight junction proteins. Enteric glial cell number and activation were measured by Sox10 expression and GFAP reactivity, respectively. Separate groups of mice were challenged with Cr infection during the chronic phase of TBI, and host immune response, barrier integrity, enteric glial cell reactivity, and progression of brain injury and inflammation were assessed.
Results
Chronic CCI induced changes in colon morphology, including increased mucosal depth and smooth muscle thickening. At day 28 post-CCI, increased paracellular permeability and decreased claudin-1 mRNA and protein expression were observed in the absence of inflammation in the colon. Colonic glial cell GFAP and Sox10 expression were significantly increased 28 days after brain injury. Clearance of Cr and upregulation of Th1/Th17 cytokines in the colon were unaffected by CCI; however, colonic paracellular flux and enteric glial cell GFAP expression were significantly increased. Importantly, Cr infection in chronically-injured mice worsened the brain lesion injury and increased astrocyte- and microglial-mediated inflammation.
Conclusion
These experimental studies demonstrate chronic and bidirectional brain-gut interactions after TBI, which may negatively impact late outcomes after brain injury.
Keywords: traumatic brain injury, mucosal barrier function, enteric glial cells, brain-gut axis, neuroinflammation, neurodegeneration, Citrobacter rodentium
1. Introduction
Gastrointestinal (GI) consequences of traumatic brain injury (TBI) include symptoms of mucosal injury, barrier disruption and dysmotility along the intestinal tract, and impact posttraumatic morbidity and mortality1–3. Previous studies have reported intestinal injury, inflammation, and barrier dysfunction associated with endotoxemia up to 72 hours after TBI4–6. In the brain, secondary injury mechanisms initiated by trauma can continue for months to years, and include sustained neuroinflammatory processes that contribute to progressive neurodegeneration and neurological dysfunction7,8. The delayed systemic consequences of TBI, such as systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS), play a role in the increased morbidity and long-term mortality after TBI9,10. Cause-of-death analyses of TBI patients who have survived beyond one year after injury demonstrate that these individuals are 12 times more likely to die from septicemia and 2.5 times more likely to die of digestive system conditions than matched cohorts of the general population11. Mechanisms underlying these systemic consequences remain unclear, and the long-term impact of TBI on the intestinal tract is unknown.
Intestinal barrier dysfunction is implicated in the pathogenesis of inflammatory bowel diseases (IBD), metabolic syndrome, non-alcoholic fatty liver disease, diabetes, as well as SIRS and MODS12–14. Notably, indices of intestinal barrier disruption, such as increased intestinal permeability and endotoxemia, positively correlate with severity of brain injury and are associated with TBI-related morbidities15. As the intestinal mucosal barrier serves as the interface between vastly diverse external and internal environments, the maintenance of homeostasis is critical to its ability to respond to noxious or pathogenic stimuli. Intestinal epithelial cells, immune cells of the lamina propria, and enteric glial cells (EGCs) regulate mucosal barrier homeostasis though complex and dynamic interactions16,17.
Infections are a frequent co-morbidity in TBI patients18. Increased susceptibility to infections, such as Escherichia coli, is attributed to peripheral immune suppression as a direct consequence of CNS injury, not only during hospitalizations, but also long term19–21. Citrobacter rodentium (Cr) infection is a murine model for human enterohemorrhoragic E. coli and enteropathogenic E. coli infection, and has a well-characterized immune response with defined pathological hallmarks22.
In the present study, we use a murine model of moderate-level controlled cortical injury (CCI) that produces secondary injury progression through 28 days, as well as neurological impairment and chronic neurodegeneration that are evident even at 1 year post-injury23. Experimentally, persistent microglial activation and astrocyte reactivity after a single moderate-to-severe TBI or repeated mild TBI induces inflammation that contributes to long-term neuropathological and neurobehavioral changes24–26. The goal of this study was to investigate the hypotheses that TBI induces chronic changes in the intestinal tract, which impact responses to subsequent enteric challenge by a clinically relevant bacterial pathogen. Our results show a disruption of mucosal barrier function and EGC reactivity in the colon that persists through the chronic phase of TBI. These changes in the colon after brain trauma are maintained during the host protective immune response to Cr and are associated with exacerbation of chronic brain injury.
2. Material and methods
2.1 Animals
Animals used in these studies were 8- to 10-week-old male C57BL/6 mice weighing 20-25 grams (Taconic Biosciences, Hudson, NY). All experiments and procedures were conducted in accordance with the ethical standards and performed as approved by the Institutional Animal Care and Use Committee (IACUC). Mice were housed under a 12-hour light-dark cycle with ad libitum access to food and water at University of Maryland School of Medicine animal facilities in Baltimore, MD. Animals involved in Cr infection studies were transported and housed according to IACUC-approved procedures at the United States Department of Agriculture (USDA) in Beltsville, MD.
2.2 Controlled cortical impact (CCI) injury
Anesthesia with inhaled isoflurane (induction: 3%, maintenance: 1.5%), surgical site preparation, and post-surgical procedures were carried out as previously described28. Using a micro-drill with a trephine tip diameter of 5mm, a craniotomy was performed on the left parietal bone, centrally situated between bregma and lambda. A microprocessor-controlled device with a 3.5-mm diameter impactor tip driven by dry compressed air was used to induce moderate-level CCI injury at an impactor velocity of 6 m/s and a deformation depth of 2mm. Sham animals underwent the same procedure as CCI mice except for the impact.
2.3 Study Design
Injury groups were completely randomized in all experiments. In the first study, cohorts of sham- and CCI-injured mice were euthanized at 24 hours (n=5-8/group) and 28 days (n=6-8/group) after injury. In the second study, separate groups of sham- and CCI-injured mice were challenged with Cr infection (sham+Cr, n=10; CCI+Cr, n=10) or vehicle (sham, n=5; CCI, n=5) at 28 days post-injury (Fig 4A). Cohorts of mice (n=5/group) were euthanized on post-infection day (PID)-12. Additional cohorts of these mice (n=5/group) were euthanized on PID-25 following evidence of complete clearance of infection in both groups.
Figure 4. Host response and mucosal homeostasis after enteric Cr infection during the chronic phase of TBI.
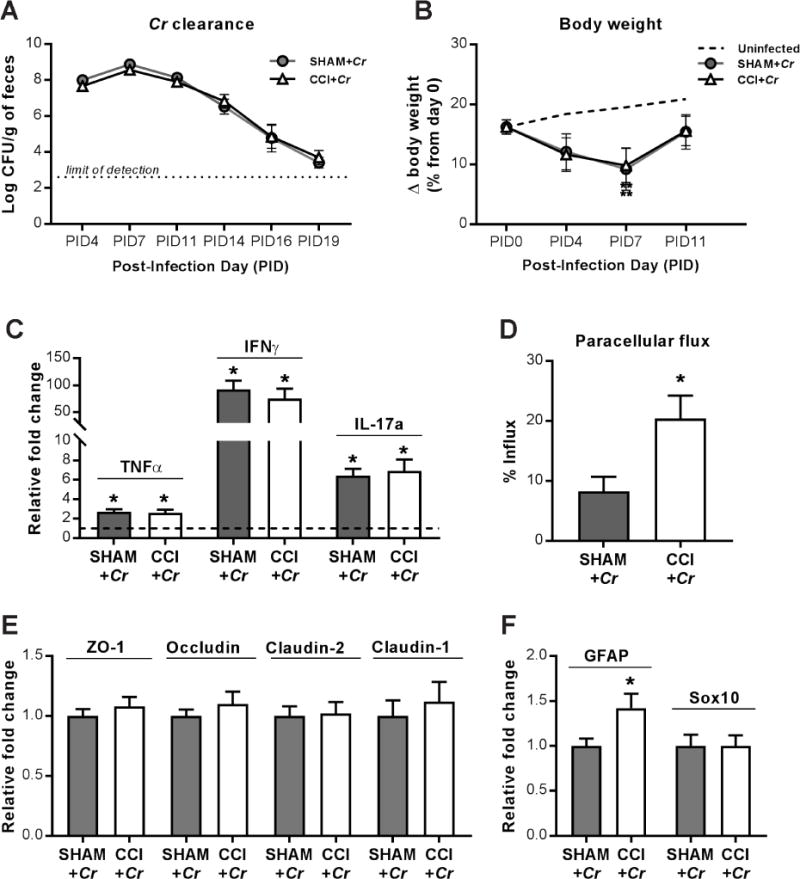
(A) The kinetics of fecal Cr excretion were monitored over time until samples reached the limit of detection (dotted line). (B) Body weight over time, on day 28 after CCI (PID0), and post-infection days (PID) 4, PID7, and PID11. Percent change in body weight relative to day 0 (day of CCI or sham injury). Changes in body weight between uninfected sham and uninfected CCI groups between day 0 and PID12 were identical and combined as a point of reference (dashed line).**P<0.01 relative to uninfected animals. (C) Th1/Th17 cytokine upregulation in colons on PID12, relative to uninfected counterparts (dashed line at y=1). Mean relative fold change is 1.0 for uninfected sham and uninfected CCI. Sham SEM for TNFα: 0.29, IL-1β: 0.09, IFNγ: 0.16, IL-17a: 0.24; CCI SEM for TNFα: 0.25, IL-1β: 0.21, IFNγ: 0.27, IL-17a: 0.24; *P<0.05, relative to respective uninfected control; n=8-10. (D) Mucosal paracellular flux of 3K dextran in Cr-infected colons on PID12. (E) ZO-1, occludin, claudin-2, and claudin-1 mRNA expression in Cr-infected colons on PID12. (F) GFAP and Sox10 mRNA expression in Cr-infected colons on PID12. Values (D–F) are means ± SEM; *P<0.05, relative to Sham+Cr; n=10.
2.4 Histopathological Analysis
Longitudinal 4μm colon sections were prepared and stained with hematoxylin and eosin (H&E) for histopathological scoring and morphometric analyses. Sections were scored by two independent researchers who were unaware of the experimental groups. Histopathology was scored according to previously defined critera and included five different categories: inflammation, epithelial damage, edema, goblet cell loss, and mucosal hyperplasia29,30. The score of each category was based on severity and extent of the injury using a scale of 0 to 4 in 0.5 increments. The scores indicated degree of injury as follows: 0, absent; 1, mild; 2, moderate; 3, marked; 4, severe for each of the 5 categories, which were analyzed by category as well as combined for a total score of a maximum value of 20). In addition, morphometric measurements were conducted as described previously31. In brief, mucosal depth and smooth muscle thickness in the colon were measured in micrometers in digital brightfield images in ZEN Pro software (Carl Zeiss Microscopy, Thornwood, NY). Morphometric parameters were averaged in a minimum of 10 random, well-oriented fields per section and were performed by investigators who were unaware of the treatment groups.
2.5 Permeability Assays
In order to isolate the barrier properties of the mucosal epithelium, segments of jejunum and colon were carefully stripped of smooth muscle, opened longitudinally along the mesenteric border, and mounted into microsnapwells, as described previously32. Resistance was measured with an EVOM Voltohmmeter in 30min intervals over 3h for transepithelial electrical resistance (TEER), and TEER values for each mounted section were averaged across these six time points. Changes in TEER reflect permeability to the net flux of ions across the mucosa. To determine specifically the component of TEER that was attributed to paracellular flux, cascade blue-labeled dextran to the apical aspect of the mucosae of the same sections of jejunum (10,000 mol wt, 200mg/ml, Invitrogen, Carlsbad, CA) and colon (3,000 mol wt, 200mg/ml, Invitrogen). After 3h of incubation, media was collected from basolateral and apical sides and plated in duplicates in black 96-well microplates. Fluorescence (Fl) was measured at excitation and emission wavelengths of 355 and 425 nm.
2.6 Quantitative Real-Time PCR (qPCR)
Total RNA was isolated and purified from whole tissue homogenates of colon in Trizol using RNeasy Mini Kit (Qiagen, Valencia, CA). Equal amounts of purified RNA were reverse transcribed using the Verso cDNA Synthesis Kit (Thermo Scientific). Quantitative real-time PCR was performed using TaqMan gene expression assays for mouse (TNFα, Mm00443258_m1; IFNγ, Mm01168134_m1; IL-6, Mm00446190_m1; IL-1β, Mm01336189_m1; GFAP, Mm01253033_m1; Sox10, Mm00569909_m1; and GAPDH, Mm99999915_g1; Applied Biosystems), and amplification reactions were run using TaqMan Universal Master Mix II (Applied Biosystems). For tight junction analyses, qPCR was performed using custom designed primers sets for mouse ZO-1, occludin, claudin-1, and claudin-2, as described previously33, and amplification reactions were run using SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA), according to the manufacturer’s protocol. Samples were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or 18S RNA, and relative gene expression levels were calculated.
2.7 Immunofluorescence
Transverse frozen tissue sections (20μm) were rinsed, blocked, and incubated with polyclonal rabbit anti-GFAP (1:500, Dako, Glostrup, Denmark), or monoclonal rabbit anti-Claudin-1 (1:50, Cell Signaling Technology, Danvers, MA) overnight in 4°C. Sections were washed, incubated with AlexaFluor-conjugated goat anti-rabbit IgG (1:1000, Life Technologies, Carlsbad, CA) and counter-stained with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (1:50,000, Sigma-Aldrich, St. Louis, MO). Images were acquired with 20× and 63× objectives by confocal microscopy (Leica Microsystems, Wetzlar, Germany) with acquisition parameters kept constant. Analyses were performed on images captured with the 20× objective using ImageJ software (https://imagej.nih.gov/ij/). Mucosal regions indicated by apical epithelial cells and the submucosal border were outlined using the freehand tool. Parameters for image adjustments were set based on threshold signals and kept constant for all images. Mean gray value and area of outlined regions were measured, and integrated density of immunofluorescent signal was calculated.
2.8 Citrobacter rodentium infection, clearance, and sample collection
Mice were inoculated with a naladixic acid-resistant Citrobacter rodentium (Cr) strain derived from DBS100 (American Type Culture Collection 51459; Manassas, VA), as described previously34. Briefly, the bacteria were cultured, concentration was calculated from an optical density standard curve, and resuspended for a final concentration of 5 × 1010 colony forming units (CFU) per milliliter of Luria-Bertani (LB) broth. Animals were administered 0.2 mL containing 1010 CFU of resuspended bacteria or LB broth alone by oral gavage at 28 days after CCI or sham injury. For chase studies, fecal samples were collected on post-injury day (PID) 4, 7, 11, 15, 19, 23. To monitor the kinetics of the infection, fecal samples were homogenized and serially diluted in PBS, then plated in duplicate onto LB/agar plates supplemented with 50 μg/mL naladixic acid. Cr load in stool was quantified as CFU/gram of feces. Based on the course of infection determined by chase studies, groups of mice were euthanized on PID12 and colons, brains, and spleens were collected for gross, molecular, and/or functional examinations. To assess bacterial translocation to the systemic compartment, spleens were collected under sterile conditions, homogenized, and plated onto LB/agar plates to detect both translocated Cr and non-Cr bacterial strains.
2.9 BrdU immunostaining
Epithelial proliferation after Cr infection was assessed by BrdU incorporation in colonic epithelial cells. Two hours prior to euthanasia, mice were injected intraperitoneally with 10mg/kg of 5-bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich) diluted in warm PBS. Samples were collected, fixed, and processed, as described previously35. BrdU-positive cells were counted in 8-10 well-oriented crypts per section in images acquired with a 10× objective. BrdU incorporation was expressed as mean number of BrdU-positive cells per crypt.
2.10 Brain collection and sectioning
Brains were fixed in 4% paraformaldehyde for 24h in 4°C, then transferred to 20-30% sucrose for cryoprotection prior to embedding into O.C.T. compound (VWR International, Radnor, PA). Coronal sections beginning at +1.78 mm from bregma were serially collected (three × 60 μm, three × 20 μm), mounted on charged glass slides (Globe Scientific, Paramus, NJ), and stored in −80°C until use.
2.11 Stereological assessment of brain injury
One series of 60 μm coronal brain sections were incubated in double strength cresyl violet (FD Neurotechnologies, Columbia, MD) for 1-3 min to visualize neural structures and cell bodies. Stereoinvestigator software (MBF Bioscience, Williston, VT) was used for stereological assessment of lesion volume and neuronal loss analysis, as previously described36,37. On digital virtual images of whole brain sections, lesion contours were traced, and lesion volumes calculated based on a section evaluation interval of 4 across 24 sections per sample using the cavalieri estimator probe with a grid spacing of 100μm. In the same sections, hippocampal neuronal loss was assessed by the optical fractionator method. Hippocampal subregions cornu ammonis 1 (CA1) and dentate gyrus (DG) were demarcated by contour tracings, and neuronal cell bodies were counted within sampled regions. The volume of hippocampal subregions were calculated using the cavalieri estimator probe with a grid spacing of 50μm. The estimated number of neurons within a subregion was divided by the volume of the region, and neuronal cell densities were expressed in counts/mm3.
2.12 Immunohistochemical assessment of brain microglia/macrophages
To assess microglial/macrophage activation, a separate series of 60μm coronal brain sections were probed with rat monoclonal anti-mouse CD68 (1:200; Bio-Rad), mouse adsorbed biotinylated goat anti-rat IgG antibody (10μg/ml, Vector Labs), followed by ABC Vectastain Reagent and a DAB peroxidase substrate (Vector Labs), according to the manufacturer’s instructions. Digitalized brightfield images captured with a 20× objective (Leica) were processed and analyzed using the Fiji package of ImageJ (http://fiji.sc), as described previously38. All images were processed with the Colour Deconvolution plug-in using the H-DAB vector to remove background and to separate the DAB-only image panel for analysis. CD68+ cells were counted within ipsilateral cortex areas on 3 sections spanning the injury site per brain, and averages were expressed as cells/mm2.
2.13 Immunofluorescent assessment of brain astrocytes
For immunofluorescent staining of astrocytes, 20μm coronal brain sections were blocked and incubated in polyclonal rabbit anti-GFAP antibody (1:500, Dako, Glostrup, Denmark) overnight in 4°C, followed by AlexaFluor 488-conjugated goat anti-rabbit IgG (1:1000, Life Technologies), and counterstaining with DAPI. Images were acquired using a fluorescent Nikon Ti-E inverted microscope (Nikon Instruments, Inc., Melville, NY) with a 20× objective, under constant acquisition parameters. All images were quantified using NIS-Elements Advanced Research imaging software (Nikon) under constant parameters, and intensity of GFAP was normalized to the total area of the ipsilateral cortex.
2.14 Statistical Analysis
Data are expressed as mean ± SEM. Differences between sham and CCI groups were analyzed by unpaired Student t-test or Mann-Whitney test. Immunofluorescence data were analyzed across time by one-way analysis of variance (ANOVA), followed by post hoc adjustments using a Bonferroni correction. Data in Cr infection studies were analyzed by two-way ANOVA for injury, infection, and interaction. Statistical analyses were performed using Prism 7.01 for Windows (GraphPad Software, San Diego, CA); P<0.05 was considered significant.
3. Results
3.1 Chronic TBI induces morphopathological changes in the colon
To assess TBI-induced morphological changes along the intestinal tract, H&E-stained sections of jejuna and colons were evaluated at acute (24 hours post-injury) and chronic phases (28 days post-injury). Jejunal morphology evaluated for villus length and crypt depth was unchanged at 24 hours or 28 days after moderate CCI (data not shown). In colons, total histopathology scores were not significantly different between sham and CCI groups at 24 hours or 28 days after injury (Supplemental Table 1). When individual categories were assessed, no differences were seen in inflammatory infiltrate, epithelial damage, goblet cell loss, or edema in colons at either 24 hours or 28 days after CCI (Supplemental Table 1). Pathology scores for mucosal hyperplasia, visualized by increased mucosal depth, were unchanged at 24 hours after CCI, but were significantly increased in colons of CCI-injured mice at 28 days after injury (Figure 1A–D). Morphometric analyses demonstrated quantitative evidence of hyperplasia by significantly increased mucosal depth in colons of CCI-injured mice at 28 days post-injury (Figure 1E). In addition, colonic smooth muscle thickness was increased significantly at 28 days after CCI compared to sham (Figure 1F). These data indicate that moderate TBI induces morphological abnormalities within the colon, but not in the jejunum, at 28 days after injury.
Figure 1. Histopathological evaluation in the colon at 24 hours and 28 days after TBI.
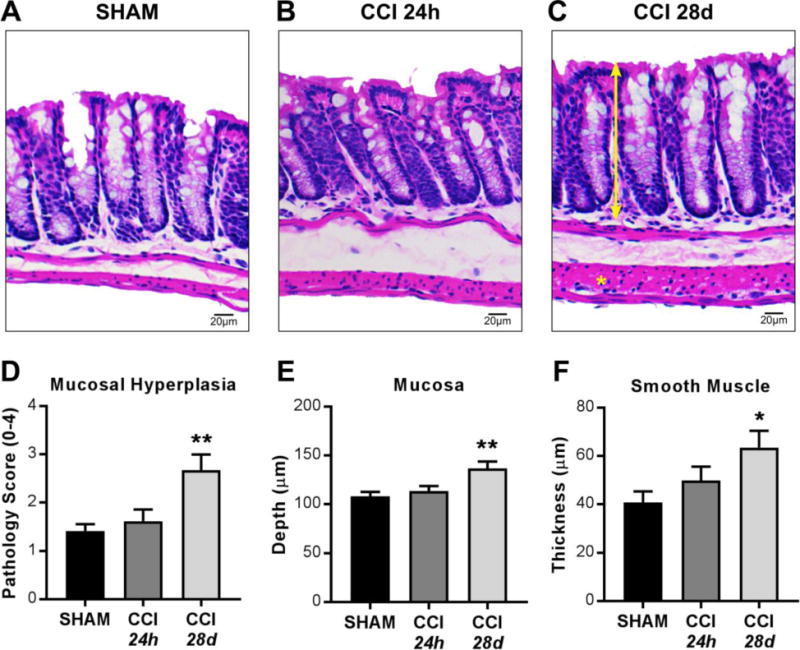
(A–C) Representative H&E staining and microscopy of colon sections from sham, 24 hours post-CCI, and 28 days post-CCI mice. (D) Pathology index for mucosal hyperplasia scored for severity and extent from 0 to 4. Morphometric analyses reveal (E) increased mucosal depth (double-headed arrrow) and (F) increased smooth muscle thickness (asterisk) in colons 28 days after CCI. Values (D–F) are means ± SEM; **P<0.01,*P<0.05, compared to shams; n=6-8.
3.2 TBI increases colonic paracellular permeability at 28 days post-injury
Increased incidence of GI-related disorders among TBI patients implicates changes in GI function as a consequence of TBI15,39,40; therefore, we measured indices of mucosal barrier permeability in sham and CCI mice. Transepithelial electrical resistance (TEER) in the jejunum was significantly decreased at 24 hours after CCI when compared to shams; however, by post-injury day 28, jejunal TEER was not different from shams (Supplemental Figure 1A). In the colon, no differences in TEER were detected at either 24 hours or 28 days post-injury (Figure 2A). Paracellular flux was unchanged in jejuna (Supplemental Figure 1B), but was significantly enhanced in colons at 28 days post-CCI when compared to shams (Figure 2B). These data are consistent with previous observations of increased permeability in the jejunum at 24 hours after brain injury4,41. Importantly, these results also demonstrate that moderate TBI results in specific alterations in colonic mucosal barrier permeability at 28 days post injury.
Figure 2. Mucosal barrier properties and tight junction proteins in colons after TBI.
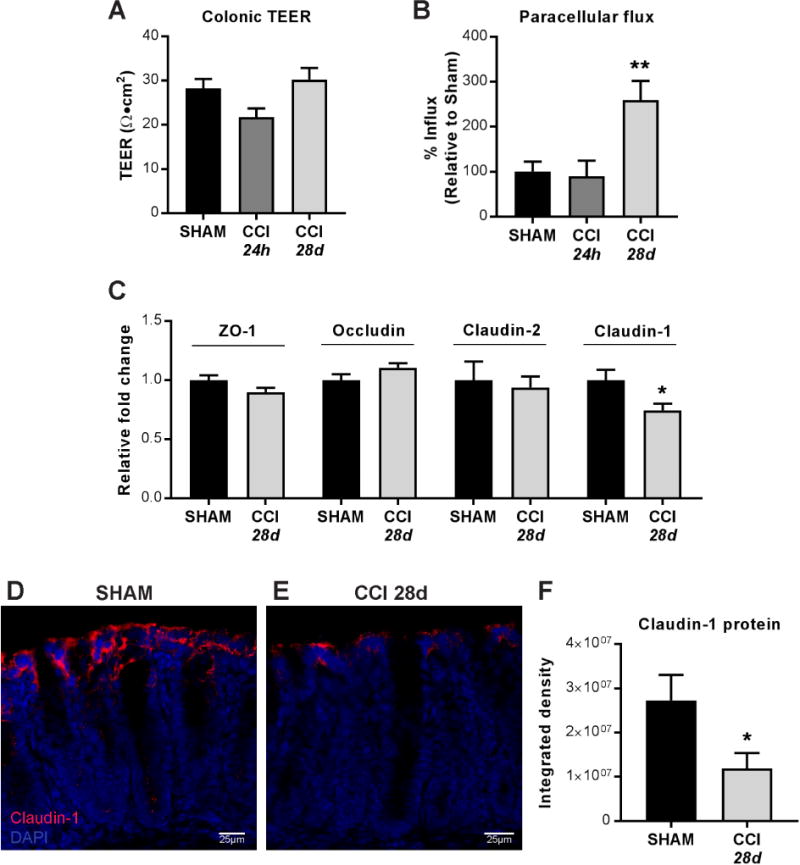
(A) Transepithelial electric resistance (TEER) measurements at either 24 hours or 28 days after CCI in muscle-stripped colons mounted in duplicates in microsnapwells. Individual TEER values were averaged across time for measurements taken every 30 minutes for 3 hours. (B) Paracellular flux of 3K dextran molecules across mounted colonic mucosa. (C) ZO-1, occludin, claudin-2, and claudin-1 mRNA expression and (D–F) Immunofluorescent claudin-1 staining in colons 28 days after CCI compared to sham. Quantification of mucosal claudin-1 protein expressed as integrated density of fluorescent signal. Values (A–C, F) are means ± SEM; (B) are normalized to control (sham) values, set to 100%; **P<0.01, compared to sham; n=6–10; (C, F) are relative to sham expression; *P<0.05; n=6–8.
3.3 Mucosal abnormalities in the colon during chronic TBI are not associated with colonic inflammation
To investigate whether functional and histopathological changes in the colon during chronic TBI were associated with inflammation, we determined expression levels of the cytokines IL-1β, TNF-α, IFN-γ, IL-6, and IL-10. There was a small, but significant, increase in IL-1β gene expression in the colon at 24 hours post-injury, but this increase was not observed in the chronic phase of CCI at 28 days post-injury (Table 1). There were no significant changes in the gene expression of the pro-inflammatory cytokines, TNFα, IFNγ, and IL-6, or the anti-inflammatory cytokine IL-10 in the colon either at 24 hours or 28 days post-injury (Table 1). Thus, TBI is associated with chronic changes in colonic barrier function and morphology in the absence of an overt inflammatory response.
Table 1.
Relative gene expression of cytokines in colons after moderate TBI.
| Colon | Sham 24h | CCI 24h | Sham 28d | CCI 28d |
|---|---|---|---|---|
| TNF-α | 1.00 ± 0.28 | 0.96 ± 0.11 | 1.00 ± 0.12 | 1.12 ± 0.17 |
| IFN-γ | 1.00 ± 0.18 | 0.57 ± 0.17 | 1.06 ± 0.33 | 1.02 ± 0.15 |
| IL-6 | 1.00 ± 0.07 | 0.85 ± 0.12 | 1.00 ± 0.08 | 1.13 ± 0.23 |
| IL-1β | 1.00 ± 0.08 | 1.33 ± 0.12* | 1.00 ± 0.08 | 0.92 ± 0.09 |
| IL-10 | 1.00 ± 0.12 | 1.10 ± 0.10 | 1.00 ± 0.14 | 0.86 ± 0.10 |
P<0.05 relative to Sham 24h
3.4 TBI-induced increase in colonic permeability is associated with decreased expression of specific tight junction proteins
To investigate whether disrupted tight junction proteins (TJP) were associated with long-term colonic barrier dysfunction after TBI, we assessed expression of key TJPs in colon tissue. There were no changes in colonic mRNA expression of ZO-1, occludin, or the pore-forming claudin-2; however, expression of the barrier-forming claudin-1 was significantly decreased in the colon at 28 days post-injury (Figure 2C). Consistent with this observation, immunofluorescence staining showed significantly reduced claudin-1 protein expression at the apical mucosal barrier of the colon 28 days post-injury (Figure 2D–F). These data indicate that chronic TBI is associated with dysregulation of claudin-1 synthesis and expression in the colon.
3.5 Enteric glial cells demonstrate altered GFAP reactivity after TBI
Parallels drawn from known astrocytic responses in the brain have been used to investigate the role of EGCs in the maintenance of epithelial barrier integrity42. Astrocytes and EGCs both upregulate the expression of glial fibrillary acidic protein (GFAP) upon activation. Peripherally, varying isoforms of GFAP are expressed among glial cells; within the intestinal mucosa, GFAP is a specific marker for EGCs43. Immunofluorescent quantification of GFAP-positive cells in mucosa of colon sections revealed a significant decrease in mucosal GFAP expression at 24 hours post-injury relative to shams (Figure 3A,B,D). In contrast, at 28 days post-injury, mucosal GFAP expression levels were significantly increased compared to sham levels (Figure 3A,C,D). Colonic GFAP mRNA expression was unchanged at 24 hours post-injury, but was significantly increased at 28 days post-injury when compared to shams (Figure 3E). Moreover, colonic Sox10 mRNA expression was significantly increased 28 days post-injury (Figure 3E), indicating that GFAP upregulation was associated with an increase in the number of activated EGC in the colon. Thus, TBI produces robust acute and chronic changes in EGC reactivity in the colon.
Figure 3. Enteric glial cells in the colon are significantly altered after TBI.
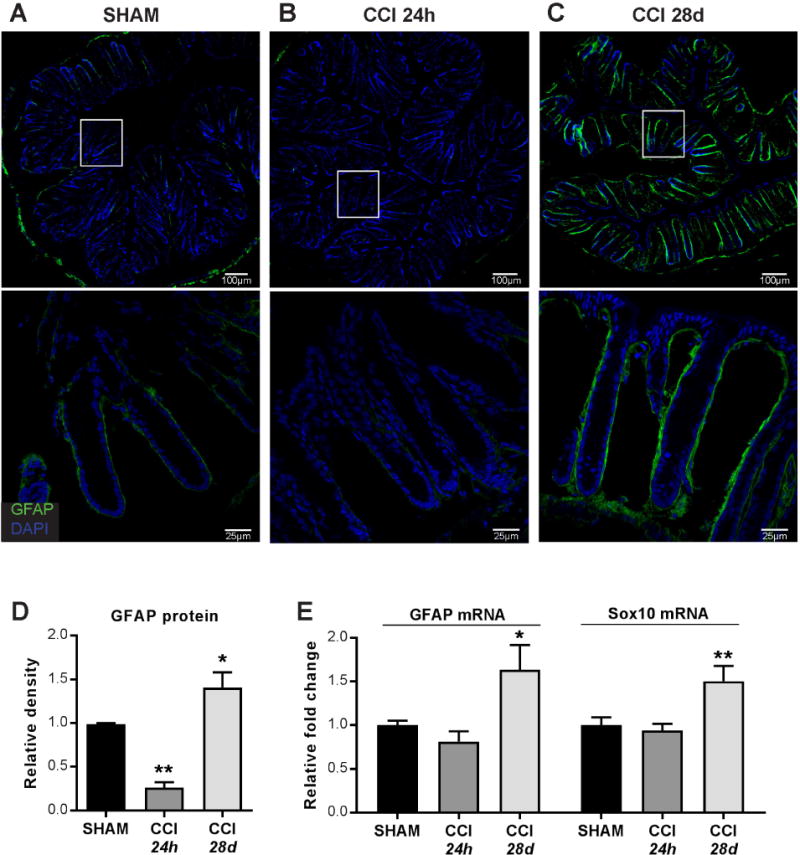
(A–C) Representative immunofluorescent staining and microscopy of glial cells in colons. (D) Mucosal GFAP+ quantification by integrated density. (E) GFAP and Sox10 mRNA expression in colon whole tissue isolates. Images (A–C, top) acquired with a 20× objective. Images (A–C, bottom) acquired with a 63× objective. Values (D–E) are means ± SEM, relative to shams; **P<0.01, compared to sham; *P<0.05, compared to sham; n=5–8.
3.6 TBI does not alter host immune responses to enteric bacterial infection by Citrobacter rodentium
To investigate whether chronic TBI led to peripheral changes and altered immune responses in response to an enteric microbial infection, sham and CCI-injured mice were infected by pathogenic Citrobacter rodentium (Cr) at 28 days post-injury, and the kinetics of the infection were monitored by fecal Cr excretion over time. No difference in Cr load was observed between sham and CCI groups at any point post-infection (Figure 4A). Thus, neither peak colonization (PID7) nor clearance (PID11-PID19) was affected by TBI. Consistent with this conclusion, the characteristic cytokine responses to Cr infection, which include upregulation of TNFα, IFNγ, and IL-17α, were comparable in the colons of both sham- and CCI-injured mice (Figure 4C). Changes in body weight were monitored over time, and no differences were seen up to 28 days after CCI (Figure 4B). Given that dietary content remained constant, this data suggests that longitudinal growth and associated food consumption were comparable between sham and CCI-injured animals prior to enteric infection by Cr. In response to Cr infection, changes in body weight followed the predicted time course and were significantly lower in Cr-infected groups on the day of peak infection (PID7), with no difference between sham+Cr and CCI+Cr groups (Figure 4B). By the late peak phase of infection, four days later (PID11), overall body weights were comparable among all experimental groups. Increases in colon/body weight ratios, a feature of Cr infection44, were comparable in both sham+Cr and CCI+Cr mice, and colon lengths were similar among all uninfected and infected groups (Supplemental Table 2). Spleen/body weight ratios were significantly increased by Cr infection in sham and CCI groups; however, spleen/body weight ratios of CCI+Cr mice were significantly lower than sham+Cr (Supplemental Table 2).
3.7 Citrobacter rodentium infection leads to sustained barrier dysfunction and histopathological changes in the colon during chronic TBI
To investigate the impact of Cr infection on TBI-induced changes in the intestinal tract, we assessed mucosal barrier function and morphopathology in the colon at PID12. Measures of mucosal paracellular permeability in response to Cr infection showed significantly enhanced labelled-dextran flux across colonic mucosa in CCI+Cr mice when compared to sham+Cr (Figure 4D). Spleen homogenate cultures, however, were negative for bacterial growth in all groups (Supplemental Table 2), indicating that moderate-level CCI and ensuing pathologies in the colon were not sufficient to cause systemic dissemination of Cr or commensal bacteria. In support of this, expression of TJPs ZO-1, occludin, claudin-2, and claudin-1 were unchanged in colons of CCI+Cr mice (Figure 4E). Notably, GFAP mRNA levels were significantly increased in colons of CCI+Cr compared to sham+Cr, whereas Sox10 mRNA expression was unchanged (Figure 4F). Therefore, Cr infection during chronic TBI was associated with greater activation of existing EGCs.
On PID12, total histopathology scores were significantly increased in Cr-infected colons of sham and CCI-injured animals, which were attributed to significantly increased lesion severity of inflammation, epithelial damage, goblet cell loss, mucosal hyperplasia, and edema (Supplemental Table 3). Compared to Cr-infected shams, chronic CCI did not further worsen colon histopathology during enteric infection by any of these criteria. Considering the sustained paracellular barrier dysfunction evident in CCI-injured colons during Cr infection, additional indices of mucosal pathology were assessed. BrdU incorporation into proliferating epithelial cells was increased similarly in colons of both sham+Cr and CCI+Cr groups (Figure 5A–C, E). Morphometric analyses demonstrated that Cr infection significantly increased mucosal depth in colons of both sham+Cr and CCI+Cr groups (Figure 5F), and there was no difference between the two infected groups. While Cr infection alone did not alter colonic smooth muscle thickness in sham mice, the hypertrophy was augmented further in CCI+Cr animals compared to shams or CCI alone (Figure 5G). The increases in both mucosal permeability and smooth muscle hypertrophy following Cr show that TBI chronically impacts the maintenance of colonic homeostasis in response to a subsequent potentially harmful stimulus.
Figure 5. Histopathological evaluation in colons after Cr infection.
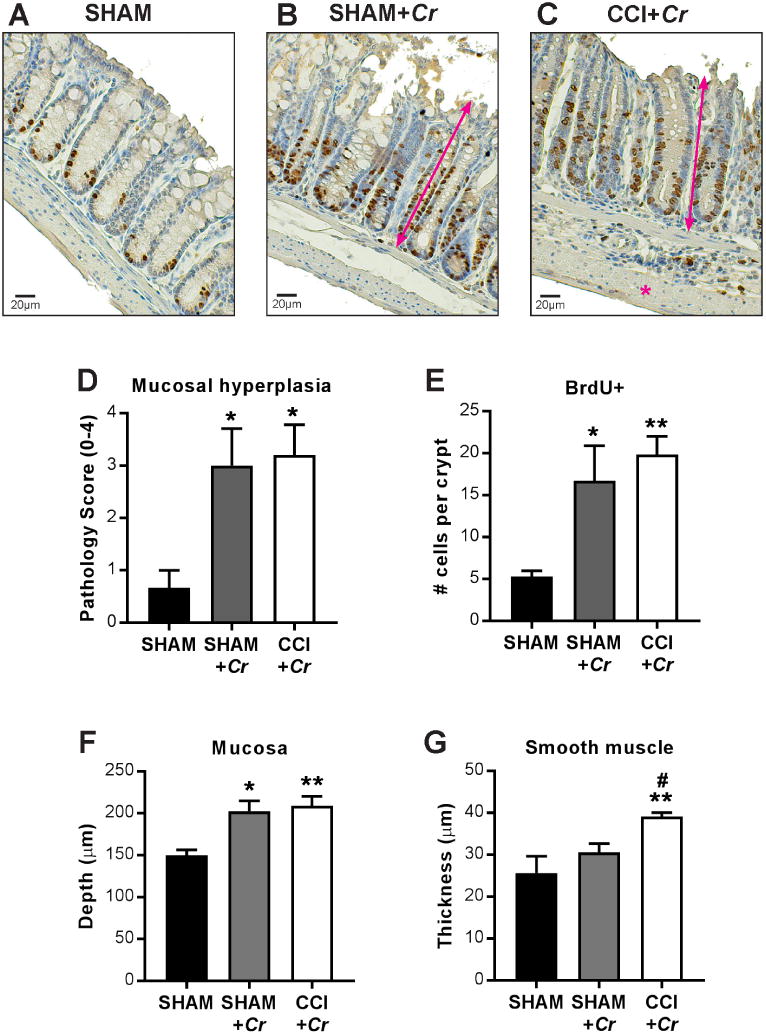
(A–C) Representative BrdU immunohistochemistry and hematoxylin counter-stained sections and microscopy of colons at PID12 after Cr infection. (D) Pathology index for mucosal hyperplasia scored for severity and extent from 0 to 4. (E) Epithelial crypt proliferation indicated by BrdU incorporation. Morphometric analyses reveal (F) increased mucosal depth (double-headed arrows) in response to Cr in both sham- and CCI-injured mice and (G) increased colonic smooth muscle response (asterisk) to Cr in CCI+Cr groups. Values are means ± SEM; (D–F) *P<0.05, **P<0.01, compared to uninfected shams; (G) **P<0.01, compared to uninfected controls; #P<0.05, compared to sham+Cr; n=6–8.
3.8 Enteric Citrobacter rodentium infection exacerbates chronic TBI neuropathology
To examine whether pathogenic bacterial infection in the gut during chronic TBI may impact the progression of brain injury, TBI neuropathology was evaluated in CCI and CCI+Cr mice at PID12. CCI-induced lesion volumes were significantly increased following enteric Cr infection when compared to CCI alone (Figure 6A–C). Hippocampal neuronal densities in the dentate gyrus and CA1 were comparable in CCI and CCI+Cr mice (Figure 6D–E). The larger brain lesions in CCI+Cr groups were associated with enhanced number of peri-lesional CD68+ immunoreactive cells, indicating that enteric challenge by Cr exacerbates the microglial/macrophage activation response during chronic stage of brain injury (Figure 7A–C). In addition, the peri-lesional GFAP+ response was significantly increased in CCI+Cr brains when compared to CCI alone, indicating that Cr infection also enhances astrocyte reactivity and glial scar formation after TBI (Figure 7D–F). Moderate-level TBI is associated with enhanced neuroinflammation and glial scar formation surrounding the lesion during the chronic phase of TBI, and the data in the present study demonstrate that enteric infection, secondary to brain injury, exacerbates cortical tissue loss and neuroinflammation.
Figure 6. Enteric Cr infection during chronic TBI leads to greater cortical loss.
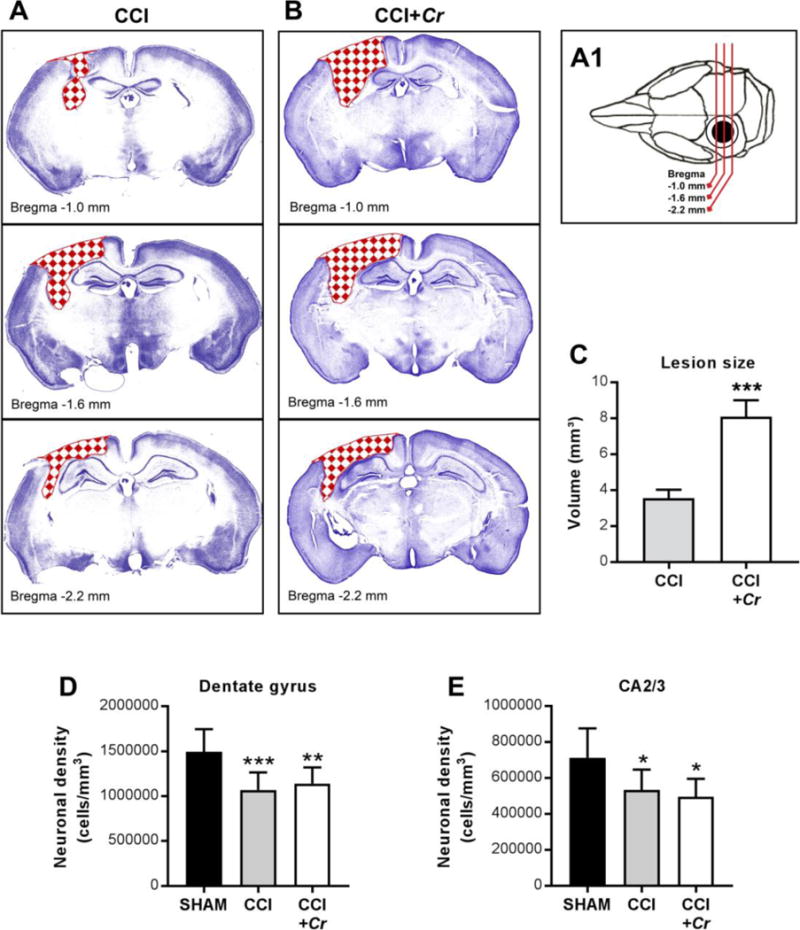
(A1–B) Schematic and representative cresyl violet-stained coronal sections showing CCI-induced lesions (red checkerboard fill) across injury site in brains of CCI and CCI+Cr mice. (C) Stereological analyses of lesion volume in whole brains on day 40 after CCI. Values are means ± SEM; ***P<0.001, compared to CCI; n=8–10. (D-E) Neuronal cell densities in the dentate gyrus and CA2/3 regions of the hippocampus. Values are means ± SEM; One-way ANOVA with Sidak correction for multiple comparisons *P<0.05, **P<0.01, ***P<0.001, compared to sham; n=8–10.
Figure 7. Exacerbation of TBI-induced microglia/macrophages and astrocyte activation in brains after enteric Cr infection.
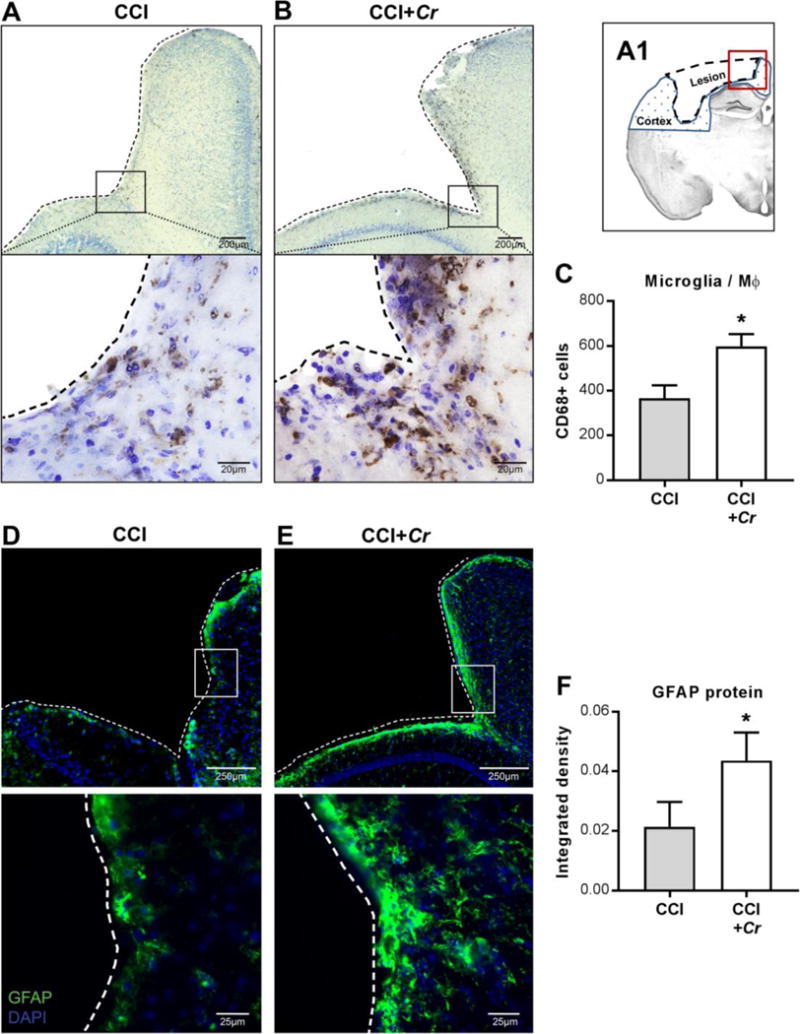
(A1) Schematic representation of a coronal section of the injured brain, illustrating the lesion site (dashed outline), the ipsilateral cortex (blue dotted fill), and the perilesional area depicted in the following images (red square). (A–B) Representative images of 60 μm brain sections stained for the microglia/macrophage activation marker CD68 taken with 20× (upper panels) and 63× (lower panels) objectives. Black squares in upper panels indicate the regions used for high-magnification images. Dashed tracings indicate lesion perimeter. (C) Perilesional CD68+ cells in ipsilateral cortex of injured brains after CCI+Cr compared to CCI alone. Values are means ± SEM; *P<0.05, compared to CCI; n=8–10. (D–E) Representative images of 20 μm brain sections stained for the activated astrocyte marker GFAP taken with 20× (top) and 63× (bottom) objectives. White squares in upper panels indicate the regions used for high-magnification images. Dashed tracings indicate lesion perimeter. (F) GFAP+ density in ipsilateral cortical hemispheres of injured brains after CCI+Cr compared to CCI alone. Values are means ± SEM; *P<0.05; n=8–10.
4. Discussion
In this study, we report for the first time that experimental TBI induces chronic changes in mucosal barrier function and histopathology in the colon, and impacts permeability during the response to pathogenic microbial infection in the gut. These studies also identify EGCs as potential contributors to the long term sequelae of TBI. Importantly, the sustained barrier dysfunction and gut inflammation during Cr infection are linked to the exacerbation of chronic TBI neuropathology, demonstrating a bidirectional brain-gut communication that may impact long-term recovery from TBI. Therefore, brain-gut effects may underlie the increased morbidity and mortality that occur late after brain trauma in humans, and may expand therapeutic targets for improving outcomes after TBI.
The most identifiable GI symptoms in brain-injured patients are consistent with mucosal abnormalities such as ulcerations, GI bleeding, intolerance to enteral feeding, and leaky gut1,3,45. Several studies showed previously that moderate-to-severe experimental TBI in rodents acutely increased mucosal damage and permeability in the small intestine up to 72 hours after brain injury4,41. Using a clinically relevant murine model of TBI (moderate-level CCI), we observed similar acute changes in jejunal barrier function. In contrast to earlier studies, however, increased permeability in the jejunum occurred in the absence of mucosal damage or inflammation, a difference that can be attributed to the known correlation between severity of brain injury and severity of intestinal symptoms15. To date, there is only a single study on the effects of TBI on the colon that reported no significant changes in permeability or histopathology in the colon 6 hours after brain injury46. In the present study, TBI also did not alter permeability or histopathology in the colon at 24 hours, but induced chronic changes in morphology and function at day 28 following brain injury.
Chronically reduced barrier dysfunction in the colon can be debilitating, leading to the inability to maintain an effective barrier against the microbiota concentrated in this region of the GI tract, thereby increasing susceptibility to systemic disease and risk of life-threatening complications13. Chronic pathologies that affect the GI tract, such as IBD, diabetes, and obesity, are characterized by reduced mucosal barrier function, tissue damage, and up-regulation of pro-inflammatory cytokines47. Features of colonic tissue injury in IBD patients and in experimental models of chronic colitis include increased thickness of mucosa and smooth muscle, as well as inflammatory infiltration during the active phase of disease48. The morphometric changes in the colons of mice at 28 days post-injury are consistent with these generalized mucosal hyperplasia and smooth muscle hypertrophy in response to insult or injury. There were, however, no indications of increased inflammatory infiltrate and no upregulation of pro-inflammtory cytokines TNFα, IFNγ, IL-β or IL-6 or downregulation of the anti-inflammatory cytokine IL-10 in the colon, suggesting that the pathological response to TBI develops over time and occurs in the absence of overt inflammation. Thus, colonic barrier dysfunction and mucosal hyperplasia represent a delayed pathophysiological response to TBI, coinciding with the delayed and chronic secondary injury responses in the brain7.
The stimulus for the changes in colon morphology at 28 days post-injury may be linked to altered mucosal barrier integrity. Movement across the intestinal mucosa consists of transepithelial exchanges through specific membrane transporters and paracellular passage through the space between adjacent epithelial cells, which is regulated primarily by TJPs49. Values of TEER provide an index of the net flow of ions through both trans- and paracellular pathways, whereas the mucosal flux of dextran molecules directly measures paracellular permeambility. Although TBI did not produce changes in colonic TEER, enhanced dextran passage across colonic mucosa indicates that TBI leads to disruption of the paracellular barrier. Increased paracellular flux in the colon at 28 days post-injury was associated with decreased claudin-1 expression with no changes in other TJPs ZO-1, occludin, or the pore-forming claudin-2. Although occludins and ZO have a targeted regulatory role in low-volume leakage of larger molecules, claudins are indispensable for maintaining structural and functional integrity of the tight junctional barrier 50. In reconstituting epithelial tight junctions, the ZO− and occludin-families of proteins undergo rapid and reversible modifications, whereas claudin proteins undergo alterations in synthesis and trafficking that impact tight junction integrity over longer periods of time49. Similarly, in the present study, downregulation of claudin-1 mRNA synthesis and expression at apical sites coincided with paracellular barrier dysfunction, both of which may have developed over time following TBI.
Given the interplay between the enteric nervous system (ENS) and colonic mucosa in the maintenance of homeostasis51, we investigated the effects of TBI on EGCs. EGC populations are divided into distinct subtypes based primarily on their location in enteric plexuses, smooth muscle, or mucosa, and play different roles in the regulation of GI function52. Changes in myenteric plexus- and intramuscular-associated EGC are implicated in disordered motility53, whereas mucosal EGC are strongly implicated in altered epithelial barrier integrity16,42,54. In the adult gut, EGCs can be specifically identified by Sox10, a marker useful for quantifying the number of differentiating glial cells and mature EGCs, and by GFAP, an index of functional activation55. In the present study, we found that the colonic EGC response to TBI is consistent with an increased number of activated cells. The increase in Sox10 expression in the colon suggests that TBI triggers adult gliogenesis, giving rise to newly differentiated EGCs. Rates of gliogenesis are increased after ENS injury or inflammation56,57; thus, it is possible that TBI induces underlying injury at the level of the ENS that manifests as mucosal barrier dysfunction and triggers the proliferation and activation of mucosal EGCs in the colon, which persist during the chronic phase of brain injury.
In the brain, TBI results in recruitment and activation of glial cells at the lesion site as a neuroprotective and reparative response to local injury and blood-brain-barrier disruption. Over time, however, chronic astrocyte reactivity results in the formation of a glial scar, which can impair resolution of brain injury23. In the gut, increased activation of EGCs may be either destructive or beneficial depending on the model system, the timing, and level of activity; however, these different roles of mucosal EGC responses to injury are not well defined. In this study, inflammation was not evident at the time of barrier leakage, suggesting that the over-activation of mucosal EGCs at 28 days post-injury may occur in response to permeability changes in the colon. Others have shown that the suppression of EGC responses following gut barrier dysfunction leads to the development of fulminant inflammation58,59. Therefore, it may be that increased mucosal EGC influx and activation during the chronic phase of TBI occurs as a compensatory mechanism, serving to promote mucosal homeostasis and mitigate overt tissue injury/inflammation in response to colonic barrier disruption.
The known long-term intestinal and systemic co-morbidities among TBI patients11 may be attributed to altered brain-gut interactions. As TBI patients exhbit and increased susceptibility to peripheral infection60, we investigated the response to Cr, a clinically relevant enteric infection, in the context of chronic brain injury. Moderate TBI did not alter the host immune reponse to Cr infection in the colon as rates of colonization, clearance, and upregulation of Th1/Th17 cytokines in response to Cr were similar in infected sham and TBI mice. There was, however, a notable reduction in paracellular flux in colons of TBI animals compared to shams during enteric Cr infection that could not be attributed to differences in mucosal damage scores, epithelial proliferation (BrdU), inflammation, or expression of TJPs. The observation that increased paracellular flux coincided with the increased GFAP in the colon further links reactive EGCs to permeability changes in the gut.
During the chronic phase of TBI, Cr infection exacerbated neuroinflammation and increased lesion volume, emphasizing the importance of gut-brain communication on long-term outcomes after TBI. In the injured brain, recruitment of CD68+ microglia/macrophages and sustained GFAP reactivity at the site of injury are known mechanisms of persistent, cytotoxic neuroinflammation that contributes to long-term secondary injury and related neurodegeneration61. In experimental models of chronic TBI, these neurodegenerative and neuroinflammatory processes are linked to alterations in cognitive and behavioral functions62,63. In the present study, neuronal densities in the hippocampus were not reduced further in the TBI+Cr group, indicating that the exacerbation of brain injury was confined to the cortical lesion. It should be noted that increased EGC reactivity and mucosal barrier dysfunction are also features of chronic neurodegenerative disorders, such as Parkinson’s disease64. In parallel, altered EGC reactivity was evident in TBI+Cr colons in conjunction with increased paracellular permeability during active inflammation in the gut and chronic neuroinflammation in the brain. Interestingly, experimental induction of a systemic immune response by LPS administration not only increases enteric glial reactivity in the gut, but also to activates microglial-driven inflammation in the brain65,66. TBI-induced colon dysfunction was not severe enough to allow bacterial translocation during Cr infection; however, the overt mucosal inflammation and injury after Cr in conjunction with barrier leakage after TBI points to a likely role for the release of gut-derived endotoxins or signaling molecules in the perpetuation of chronic disease. Thus, the present studies establish a bidirectional link between cortical neurodegeneration, microglial/macrophage and astrocyte activation in the brain, and EGC reactivity and mucosal barrier permeability in the gut.
There are a number of factors that may play a role in the bidirectional brain-gut effects reported here. Further studies are needed to investigate TBI-induced changes in morphology and function at the level of the ENS, as well as in the time course of immune/inflammatory responses in the gut and in the brain. Immune responses are known to differ between males and females27. Therefore, future studies investigating sex-dependent development and progression of immune response to TBI, both in the brain and in the gut, are warranted.
The region-specific findings reported here as well as the worsening of brain injury and neuroinflammation in response to Cr infection support a role for the gut microbiome in mediating bidirectional brain-gut effects of TBI. The colon harbors the vast majority of the total bacteria in the human body; in fact, relative to the high bacterial density of the colon, the bacterial content within small intestinal regions is considered negligible67. Interestingly, the migratory influx of mature mucosal EGCs during adult gliogenesis in response to injury has been shown to rely on signals from the gut microbiota68. It has recently been postulated that a better understanding of the contributions and responses by the brain-gut-microbiota axis to chronic inflammation and brain injury would expand opportunities for therapeutics interventions for the long-term sequelae of TBI69. Acute and chronic changes in the gut microbiome following CNS injuries such as spinal cord injury and ischemic brain injury are associated with intestinal barrier dysfunction, immune dysregulation, and functional neurologic deficits70,71. Moreover, induction of gut dysbiosis has been demonstrated to have deleterious effects on the outcome of acute ischemic brain injury, including increased lesion volume and neuroinflammation72–74. Remarkably, these gut-to-brain effects are paralleled in our study which show that gut dysbiosis induced by Cr infection during chronic TBI exacerbated injury outcomes in the brain.
5. Conclusions
In summary, chronic TBI induces changes in colon morphology and mucosal barrier function that develop over time and are associated with reduced expression of claudin-1 and increased activation of sub-epithelial EGCs. These delayed alterations could not be ascribed to inflammatory disease-states in the gut, but do parallel known increases in neuroinflammation and neurodegeneration in the injured cortex. Similar changes in both the brain and gut are also observed in response to enteric infection by Cr, suggesting that TBI-induced changes in the colon represent an adaptation to a chronic brain-derived stimulus. A subsequent challenge by inflammation induced by pathogenic Cr in the gut worsened ongoing injury brain tissue injury and neuroinflammation and inflammatory processes, demonstrating the importance of the brain-gut axis in chronic TBI.
Supplementary Material
Highlights.
Experimental TBI induces chronic structural and functional changes in the colon.
Barrier dysfunction after TBI is linked to decreased colonic claudin-1 expression.
TBI alters the number of activated enteric glial cells in the colon mucosa.
Enteric C.rodentium infection weeks after TBI exacerbates brain neuropathology.
Acknowledgments
This work was supported by the National Institutes of Health [4R01NS052568 (A.I.F.); 5R01NS037313-16 (A.I.F.); 5R01DK083418-05 (T.S-D.); R01NS082308 (D.J.L.); T32DK067872(E.L.M.)]; and the USDA CRIS project [8040-51000-058 (A.S.)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential Conflicts of Interest: None.
References
- 1.Kao CH, ChangLai SP, Chieng PU, Yen TC. Gastric emptying in head-injured patients. Am J Gastroenterol. 1998;93(7):1108–12. doi: 10.1111/j.1572-0241.1998.00338.x. [DOI] [PubMed] [Google Scholar]
- 2.Tan M, Zhu J-C, Yin H-H. Enteral nutrition in patients with severe traumatic brain injury: reasons for intolerance and medical management. British journal of neurosurgery. 2011;25(1):2. doi: 10.3109/02688697.2010.522745. [DOI] [PubMed] [Google Scholar]
- 3.Olsen AB, Hetz RA, Xue H, et al. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol Motil. 2013;25(7):593–e463. doi: 10.1111/nmo.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin W, Wang H, Ji Y, et al. Increased intestinal inflammatory response and gut barrier dysfunction in Nrf2-deficient mice after traumatic brain injury. Cytokine. 2008;44(1):135. doi: 10.1016/j.cyto.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Katzenberger R, Chtarbanova S, Rimkus S, et al. Death following traumatic brain injury in Drosophila is associated with intestinal barrier dysfunction. eLife. 2015:4. doi: 10.7554/eLife.04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bansal V, Costantini T, Kroll L, et al. Traumatic Brain Injury and Intestinal Dysfunction: Uncovering the Neuro-Enteric Axis. J Neurotrauma. 2009;26(8):1353. doi: 10.1089/neu.2008.0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics. 2015;12(1):143–50. doi: 10.1007/s13311-014-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masel B, DeWitt D. Traumatic Brain Injury: A Disease Process, Not an Event. Journal of Neurotrauma. 2010;27(8):1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 9.Lim HB, Smith M. Systemic complications after head injury: a clinical review. Anaesthesia. 2007;62(5):474. doi: 10.1111/j.1365-2044.2007.04998.x. [DOI] [PubMed] [Google Scholar]
- 10.Anthony D, Couch Y. The systemic response to CNS injury. Experimental Neurology. 2014;258:105–111. doi: 10.1016/j.expneurol.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Harrison-Felix C, Whiteneck G, Jha A, et al. Mortality Over Four Decades After Traumatic Brain Injury Rehabilitation: A Retrospective Cohort Study. Archives of Physical Medicine and Rehabilitation. 2009;90(9):1506–1513. doi: 10.1016/j.apmr.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 12.César Machado MC, da Silva FP. Intestinal Barrier Dysfunction in Human Pathology and Aging. Curr Pharm Des. 2016;22(30):4645–4650. doi: 10.2174/1381612822666160510125331. [DOI] [PubMed] [Google Scholar]
- 13.Turner J. Intestinal mucosal barrier function in health and disease. Nature Reviews Immunology. 2009;9(11):799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 14.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nature clinical practice Gastroenterology & hepatology. 2005;2(9):416–22. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 15.Faries PL, Simon RJ, Martella AT, et al. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44(6):1031–5. doi: 10.1097/00005373-199806000-00016. discussion 1035–6. [DOI] [PubMed] [Google Scholar]
- 16.Sharkey KA. Emerging roles for enteric glia in gastrointestinal disorders. J Clin Invest. 2015;125(3):918–25. doi: 10.1172/JCI76303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y-B, Li Y-Q. Enteric glial cells and their role in the intestinal epithelial barrier. World journal of gastroenterology. 2014;20(32):11273–80. doi: 10.3748/wjg.v20.i32.11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffin G. The injured brain: TBI, mTBI, the immune system, and infection: connecting the dots. Military Medicine. 2011;176(4):364. doi: 10.7205/milmed-d-10-00021. [DOI] [PubMed] [Google Scholar]
- 19.Hazeldine J, Lord J, Belli A. Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus. Frontiers in neurology. 2015;6:235. doi: 10.3389/fneur.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwab J, Zhang Y, Kopp M, et al. The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Experimental Neurology. 2014;258:121–129. doi: 10.1016/j.expneurol.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao Y, Liu P, Guo F, et al. Oxidative Burst of Circulating Neutrophils Following Traumatic Brain Injury in Human. PLoS ONE. 2013;8(7):e68963. doi: 10.1371/journal.pone.0068963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith AD, Cheung L, Botero S. Long-term selenium deficiency increases the pathogenicity of a Citrobacter rodentium infection in mice. Biol Trace Elem Res. 2011;144(1–3):965–82. doi: 10.1007/s12011-011-9071-4. [DOI] [PubMed] [Google Scholar]
- 23.Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173(4):681–91. doi: 10.1111/bph.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aungst SL, Kabadi SV, Thompson SM, et al. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014;34(7):1223–32. doi: 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loane DJ, Kumar A, Stoica BA, et al. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73(1):14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mouzon BC, Bachmeier C, Ferro A, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75(2):241–54. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- 27.Klein S, Flanagan K. Sex differences in immune responses. Nature Reviews Immunology. 2016;16(10):626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 28.Loane DJ, Pocivavsek A, Moussa CE, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med. 2009;15(4):377–9. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg D, Davidson N, Kühn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. 1996;98(4):1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackos A, Eubank T, Parry N, Bailey M. Probiotic Lactobacillus reuteri Attenuates the Stressor-Enhanced Severity of Citrobacter rodentium Infection. Infection and Immunity. 2013;81(9):3253–3263. doi: 10.1128/IAI.00278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLean L, Smith A, Cheung L, et al. Type 3 muscarinic receptors contribute to intestinal mucosal homeostasis and clearance of Nippostrongylus brasiliensis through induction of TH2 cytokines. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2016;311(1):G130–G141. doi: 10.1152/ajpgi.00461.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asmar R, Panigrahi P, Bamford P, et al. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology. 2002;123(5):1607–15. doi: 10.1053/gast.2002.36578. [DOI] [PubMed] [Google Scholar]
- 33.Buzza M, Netzel-Arnett S, Shea-Donohue T, et al. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(9):4200–5. doi: 10.1073/pnas.0903923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith A, Botero S, Shea-Donohue T, Urban J. The pathogenicity of an enteric Citrobacter rodentium Infection is enhanced by deficiencies in the antioxidants selenium and vitamin E. Infection and immunity. 2011;79(4):1471–8. doi: 10.1128/IAI.01017-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLean LP, Smith A, Cheung L, et al. Type 3 Muscarinic Receptors Contribute to Clearance of Citrobacter rodentium. Inflamm Bowel Dis. 2015;21(8):1860–71. doi: 10.1097/MIB.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrnes KR, Loane DJ, Stoica BA, et al. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Stoica BA, Sabirzhanov B, et al. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34(5):1397–411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuhrich DG, Lessey BA, Savaris RF. Comparison of HSCORE assessment of endometrial beta3 integrin subunit expression with digital HSCORE using computerized image analysis (ImageJ) Anal Quant Cytopathol Histpathol. 2013;35(4):210–6. [PMC free article] [PubMed] [Google Scholar]
- 39.DOIG C, SUTHERLAND L, SANDHAM D, et al. Increased Intestinal Permeability Is Associated with the Development of Multiple Organ Dysfunction Syndrome in Critically Ill ICU Patients. American Journal of Respiratory and Critical Care Medicine. 1998;158(2):444–451. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 40.Kharrazian D. Traumatic Brain Injury and the Effect on the Brain-Gut Axis. Alternative therapies in health and medicine. 2015;21(Suppl 3):28–32. [PubMed] [Google Scholar]
- 41.Hang C-H, Shi J-X, Li J-S, et al. Alterations of intestinal mucosa structure and barrier function following traumatic brain injury in rats. World journal of gastroenterology. 2003;9(12):2776–81. doi: 10.3748/wjg.v9.i12.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savidge T, Sofroniew M, Neunlist M. Starring roles for astroglia in barrier pathologies of gut and brain. Laboratory investigation; a journal of technical methods and pathology. 2007;87(8):731. doi: 10.1038/labinvest.3700600. [DOI] [PubMed] [Google Scholar]
- 43.Uesaka T, Nagashimada M, Enomoto H. Neuronal Differentiation in Schwann Cell Lineage Underlies Postnatal Neurogenesis in the Enteric Nervous System. The Journal of Neuroscience. 2015;35(27):9879–9888. doi: 10.1523/JNEUROSCI.1239-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vallance B, Deng W, Jacobson K, Finlay Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infection and immunity. 2003;71(6):3443–53. doi: 10.1128/IAI.71.6.3443-3453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krakau K, Omne-Pontén M, Karlsson T, Borg J. Metabolism and nutrition in patients with moderate and severe traumatic brain injury: A systematic review. Brain injury: [BI] 2006;20(4):345. doi: 10.1080/02699050500487571. [DOI] [PubMed] [Google Scholar]
- 46.Feighery L, Smyth A, Keely S, et al. Increased Intestinal Permeability in Rats Subjected to Traumatic Frontal Lobe Percussion Brain Injury. Journal of Trauma and Acute Care Surgery. 2008;64(1):131. doi: 10.1097/TA.0b013e3181568d9f. [DOI] [PubMed] [Google Scholar]
- 47.Lopetuso, Scaldaferri, Bruno, et al. The therapeutic management of gut barrier leaking: the emerging role for mucosal barrier protectors. European review for medical and pharmacological sciences. 2015;19(6):1068–76. [PubMed] [Google Scholar]
- 48.Kiesler P, Fuss I, Strober W. Experimental Models of Inflammatory Bowel Diseases. Cellular and molecular gastroenterology and hepatology. 2015;1(2):154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen L, Weber C, Raleigh D, et al. Tight junction pore and leak pathways: a dynamic duo. Annual review of physiology. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bischoff S, Barbara G, Buurman W, et al. Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterology. 2014;14(1):189. doi: 10.1186/s12876-014-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snoek S, Verstege M, Boeckxstaens G, et al. The enteric nervous system as a regulator of intestinal epithelial barrier function in health and disease. Expert Rev Gastroenterology Hepatology. 2014;4(5):637–651. doi: 10.1586/egh.10.51. [DOI] [PubMed] [Google Scholar]
- 52.Gulbransen B, Sharkey K. Novel functional roles for enteric glia in the gastrointestinal tract. Nature Reviews Gastroenterology and Hepatology. 2012;9(11):625. doi: 10.1038/nrgastro.2012.138. [DOI] [PubMed] [Google Scholar]
- 53.McClain J, Grubiai V, Fried D, et al. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology. 2014;146(2):497. doi: 10.1053/j.gastro.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neunlist M, Aubert P, Bonnaud S, et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. American journal of physiology Gastrointestinal and liver physiology. 2007;292(1):G231. doi: 10.1152/ajpgi.00276.2005. [DOI] [PubMed] [Google Scholar]
- 55.Gulbransen B. Enteric glia. Morgan & Claypool Life Sciences. 2014;1(2):1–70. [Google Scholar]
- 56.Joseph NM, He S, Quintana E, et al. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121(9):3398–411. doi: 10.1172/JCI58186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laranjeira C, Sandgren K, Kessaris N, et al. Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. The Journal of clinical investigation. 2011;121(9):3412–24. doi: 10.1172/JCI58200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bush TG, Savidge TC, Freeman TC, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93(2):189. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 59.Savidge T, Newman P, Pothoulakis C, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132(4):1344. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 60.Gaddam SS, Buell T, Robertson CS. Systemic manifestations of traumatic brain injury. Handb Clin Neurol. 2015;127:205–18. doi: 10.1016/B978-0-444-52892-6.00014-3. [DOI] [PubMed] [Google Scholar]
- 61.Loane DJ, Kumar A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol. 2016;275(Pt 3):316–27. doi: 10.1016/j.expneurol.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Z, Faden AI, Loane DJ, et al. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2013;33(12):1897–908. doi: 10.1038/jcbfm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loane DJ, Stoica BA, Tchantchou F, et al. Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics. 2014;11(4):857–69. doi: 10.1007/s13311-014-0298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clairembault T, Kamphuis W, Leclair-Visonneau L, et al. Enteric GFAP expression and phosphorylation in Parkinson’s disease. Journal of neurochemistry. 2014;130(6):805–15. doi: 10.1111/jnc.12742. [DOI] [PubMed] [Google Scholar]
- 65.Da Cunha Franceschi R, Nardin P, Machado CV, et al. Enteric glial reactivity to systemic LPS administration: Changes in GFAP and S100B protein. Neurosci Res. 2017 doi: 10.1016/j.neures.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 66.Sandiego C, Gallezot J-D, Pittman B, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(40):12468–73. doi: 10.1073/pnas.1511003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sender R, Fuchs S, Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell. 2016;164(3):337–40. doi: 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 68.Kabouridis P, Lasrado R, McCallum S, et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron. 2015;85(2):289–95. doi: 10.1016/j.neuron.2014.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sundman M, Chen N, Subbian V, Chou Y. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain, Behavior, and Immunity. 2017 doi: 10.1016/j.bbi.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 70.Kigerl KA, Hall JC, Wang L, et al. Gut dysbiosis impairs recovery after spinal cord injury. J Exp Med. 2016;213(12):2603–2620. doi: 10.1084/jem.20151345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houlden A, Goldrick M, Brough D, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benakis C, Brea D, Caballero S, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nature medicine. 2016;22(5):516–23. doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winek K, Meisel A, Dirnagl U. Gut microbiota impact on stroke outcome: Fad or fact? Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2016;36(5):891–8. doi: 10.1177/0271678X16636890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh V, Roth S, Llovera G, et al. Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke. J Neurosci. 2016;36(28):7428–40. doi: 10.1523/JNEUROSCI.1114-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.