

Abstract
This Review summarizes mechanistic investigations in faecal microbiota transplantation (FMT), which has increasingly been adapted into clinical practice as treatment for Clostridium difficile infection (CDI) that cannot be eliminated with antibiotics alone. Administration of healthy donor faecal microbiota in this clinical situation results in its engraftment and restoration of normal gut microbial community structure and functionality. In this Review, we consider several main mechanisms for FMT effectiveness in treatment of CDI, including direct competition of C. difficile with commensal microbiota delivered by FMT, restoration of secondary bile acid metabolism in the colon and repair of the gut barrier by stimulation of the mucosal immune system. Some of these mechanistic insights suggest possibilities for developing novel, next-generation CDI therapeutics. FMT might also have potential applications for non-CDI indications. The gut can become a reservoir of other potential antibiotic-resistant pathogens under pressure of antibiotic treatments, and restoration of normal microbial community structure by FMT might be a promising approach to protect against infections with these pathogens as well. Finally, FMT could be considered for multiple chronic diseases that are associated with some form of dysbiosis. However, considerable research is needed to optimize the FMT protocols for such applications before their therapeutic promise can be evaluated.
The germ theory of disease paradigm, as it was formulated in the late 19th and early 20th centuries, was centred on killing infectious pathogens with little consideration for the bystander effects on the indigenous microbial communities (microbiota) that inhabit the human body. The side effects of antibiotics, at least in the short-term, seemed to be quite favourable and many recalcitrant lethal infectious diseases became suddenly treatable. The problem of emerging antibiotic resistance, which was recognized quite early, had been largely solved by introduction of successive generations of antibiotics, which commonly had broader spectra of activity against a greater range of bacterial taxa1. Nevertheless, the problems posed by multidrug-resistant microorganisms have become one of the most urgent and growing issues in health care2. The increasing challenges presented by increasing failures of antibiotic treatments in clinical practice have also led to a new focus on the protective roles of the indigenous microbiota, which normally contrib utes substantially to colonization resistance against a wide range of infectious pathogens.
Clostridium difficile is one of the ‘superbugs’ that has grown in incidence, morbidity and mortality over the past 20 years3,4. This infection is typically enabled by suppression of native gut microbiota by antibiotic treatments. Upon reaching the colons of vulnerable individuals, C. difficile spores germinate into vegetative cells, which produce enterotoxins that cause inflammation and result in debilitating diarrhoeal symptoms5. Paradoxically, antibiotics also constitute the standard treatments for C. difficile infection (CDI), but can also perpetuate its recurrence due to progressive suppression of native gut microbiota. Chances of spontaneous relapse of the infection increase, usually at a rate of 20–30%3,6, with each round of antibiotic treatment until some patients develop an indefinite cycle of recurrent C. difficile infections (R-CDI) that cannot be broken with any known antibiotic regimen5.
Faecal microbiota transplantation (FMT) (BOX 1) normalizes the composition and functionality of gut microbiota7–13 and has now become widely accepted as a highly successful rescue treatment for R-CDI14. In addition, FMT is emerging as the best option for patients with acute, severe and complicated forms of CDI that fail to respond to antibiotic treatments15,16; an important issue as R-CDI is associated with high mortality following current standard surgical options as treatment, which includes removal or bypass of the diseased colon17,18.
Box 1. Definition of FMT.
Faecal microbiota transplantation (FMT) is a treatment that involves administration of minimally manipulated microbial community from stool of a healthy donor into the patient’s intestinal tract. The notion of ‘minimally manipulated’ material distinguishes FMT from defined consortia of microorganisms and explicitly acknowledges the high degree of complexity and functionality of natural microbiota that might be difficult to reproduce at this stage of microbiome science. Clinically, FMT is performed with the intent of restoring normal function of the gut microbiota. Most regulatory agencies in the world that have considered this form of treatment have categorized it as a ‘drug’. However, gut microbiota can be also viewed as an organ or tissue composed of complex microbial communities that have co-evolved with their human hosts. Many investigators, therefore, consider FMT a form of tissue transplantation106. The gut microbiota has the potential to affect many physiological functions, including energy metabolism, immunity and even neurological development. Thus, in addition to infectious risks, there are potential long-term risks for the recipient that should be considered in clinical practice of FMT. Careful donor selection, which is currently done solely through clinical evaluation of the donor rather than via metrics of gut microbiota composition, can theoretically mitigate these risks19,107.
The urgent need for alternatives to antibiotics by thousands of patients with refractory forms of CDI has led to the development of FMT as an increasingly standardized therapeutic agent. Faecal microbiota can be separated from stool of carefully selected donors, quantified in terms of viable bacteria, cryopreserved and banked19. The transplant material can be delivered by various routes, including an encapsulated form as an oral medication20. However, mechanistic understanding of how FMT actually works is needed for development of next-generation FMT-based therapeutics that can be standardized for efficacy and can be fully embraced by regulatory agencies. In addition, understanding mechanisms of FMT can inform development of more standard therapeutics, whether those might be specific molecules or highly defined microbial drugs. In this Review, we discuss the potential mechanisms that are involved in suppression and clearance of CDI following restoration of normal microbiota composition following FMT.
FMT-associated microbiota changes
The idea that FMT leads to donor-like normalization of the faecal microbial community structure derives primarily from studies including patients treated for the refractory R-CDI syndrome. Importantly, these studies have largely focused on patients who suffered multiple CDI recurrences and were exposed over many months to multiple courses of antibiotics with broad activity against the majority of colonic bacteria. Loss of microbial diversity has been shown to be progressive in patients with CDI and proportional to the number of rounds of antibiotic treatments21. In our centre, for example, an average patient has experienced around five episodes of CDI over a period of 1 year before FMT19. Thus, it is not surprising that such patients are found to have profound dysbiosis commonly characterized by complete disappearance of Bacteroidetes, a marked reduction in Firmicutes and massive increases in the relative abundances of Proteobacteria13. Colonoscopic administration of FMT results in prompt normalization of the microbial community structure that closely resembles the composition in donors as early as 24 h after the procedure12. The speed of normalization, a marked increase in the overall microbial diversity and the restoration of the dominance of faecal bacterial composition by members of Bacteroidetes and Firmicutes phyla make it nearly certain that donor microbiota become established and engrafted into recipients of FMT with refractory R-CDI. In fact, limited investigations suggest that donorlike microbiota composition persists in these patients for months and even years8,22.
The situation is much less clear with respect to attempts to assess engraftment of gut microbiota in conditions other than refractory R-CDI, in which the dysbiosis before the procedure is generally much less pronounced. For example, a broad pattern of dysbiosis in IBD has been noted, characterized by reduction in Bacteroidetes, reduced microbial diversity within the Firmicutes phylum and an increased proportion of Proteobacteria23. These observations suggested that FMT could be used therapeutically to correct these abnormalities and decrease gut inflammation. Thus far, however, these efforts have yielded largely disappointing results24–26, although some studies have been more encouraging and suggest further need for protocol development27. Notably, IBD-associated dysbiosis, which is substantially milder than that observed in refractory R-CDI, might not be sufficient to enable substantial engraftment of donor microbiota using simplistic protocols modelled in CDI experience. In addition, measurements of engraftment of donor microbiota following FMT for IBD indications alone are considerably less straightforward. Although it is possible to measure overall microbial diversity and relative proportions of bacterial taxa, attribution of these changes to donor gut microbiota engraftment versus microbial composition changes due to altered level of bowel inflammation is challenging. Improvements in dysbiosis could be seen in a fraction of patients with IBD who experienced clinical improvement following FMT24–26, but similar changes in microbiota composition can also be seen with TNF-targeted therapies and might simply be markers of reduced disease activity28,29.
Measurements of donor gut microbiota engraftment could be enhanced with deeper metagenomic sequencing, which might enable a greater level of taxonomic discrimination than possible with the currently more routine, small-size, variable segment 16S ribosomal RNA gene amplicon-based sequencing30. In addition, all engraftment computations also need to take into account the highly dynamic behaviour of the gut microbiota, which requires resource-intensive analysis of multiple samples from both donors and recipients to get a more accurate comparison12. Indeed, the method ology to measure donor microbiota engraftment remains an area of active development. Ultimately, standardized methods are needed to assess engraftment for optimization of various FMT protocols, especially those used outside of R-CDI treatment. In fact, it might arguably be inaccurate to apply the term ‘FMT’ to procedures that merely involve administration of donor gut microbiota in a variety of conditions, but fail to demonstrate consistent and substantial engraftment of donor gut microbiota coupled with failure to achieve clinical benefit.
The culture-independent methodologies used to study gut bacteria in the context of FMT have highlighted the importance of understanding the organizational structure and complexity of microbiota. In many respects, gut microbiota can be viewed as a microbial organ within the human body31, which itself is made of communities of highly specialized microorganisms representing all three known domains of life — the Archaea, Bacteria and Eukarya, as well as viruses. These diverse microorganisms form intricate interactive and communicating networks organized by complex ecosystem and metabolic drivers. Thus far, however, studies have focused their attention mainly on bacteria32, although initial investigations also demonstrated transmission of bacteriophages29,33.
Continued research is needed to gain detailed understanding of the metabolic interdependence between the different microbial constituents, including members of non-bacterial domains of life and the host. Our current understanding of these relationships is still rudimentary. However, it is clear that the simplistic notion held by many physicians and patients that a few individual species of ‘good microorganisms’ can repair antibiotic-induced severe dysbiosis is both naive and incorrect. Despite numerous attempts, it is therefore not surprising that simple probiotics (either single strains or a few microorganisms) have largely failed in clinical trials to contribute substantive benefit in treatment of CDI34. Holding C. difficile in check and bridging full recovery of microbial community structure using defined consortia of bacteria isolated from the human gut as basic scaffolding might be one possible approach35. However, rational design and consistent manufacturing of such simplified microbial assemblies requires a mechanistic understanding of the different parts played by individual microorganisms present in the preparation with respect to a desired therapeutic activity or contribution to the community stability and resilience in the human gut. Ultimately, it might be also important to demonstrate that such preparations do not result in potentially stable but dysbiotic ecosystem configurations that could lead to undesired adverse effects for the long-term health of the host. This potential problem could be of lesser concern in FMT with non-selected faecal microbiota, which uses minimally manipulated microbial communities from faeces that are already optimized by co-evolution with their human hosts and tested in individual healthy donors over decades.
Underlying mechanisms for FMT
The resurgence of FMT in the past few years in clinical practice was prompted by the epidemic rise of antibiotic-refractory CDI36,37, which remains the main indication for this treatment. This disease is also the only indication for FMT for which therapeutic efficacy is clear and undeniable, and why mechanistic studies in humans have also centred on this indication. Nevertheless, the general principles might be instructive for future research beyond treatment of C. difficile. Two broad, not mutually exclusive, mechanistic categor ies exist for the effectiveness of FMT that can be considered: the direct interaction of donor gut microbiota with C. difficile bacteria and micro biotamediated effects on host physiology and immune defences that are detrimental to C. difficile (FIG. 1). Gut microbiota can compete with C. difficile for nutritional and coloniza tion resources, interfere with its virulence factors and directly kill C. difficile bacteria. The gut micro biota can also activate multiple host immune defences and constrain C. difficile via secondary bile acids, which can be inhibitory of C. difficile germination and vegetative growth. Detailed understanding of these different mechanisms is critical for development of next-generation therapeutics against CDI.
Figure 1. Loss of indigenous intestinal microbiota leads to vulnerability to C. difficile infection.
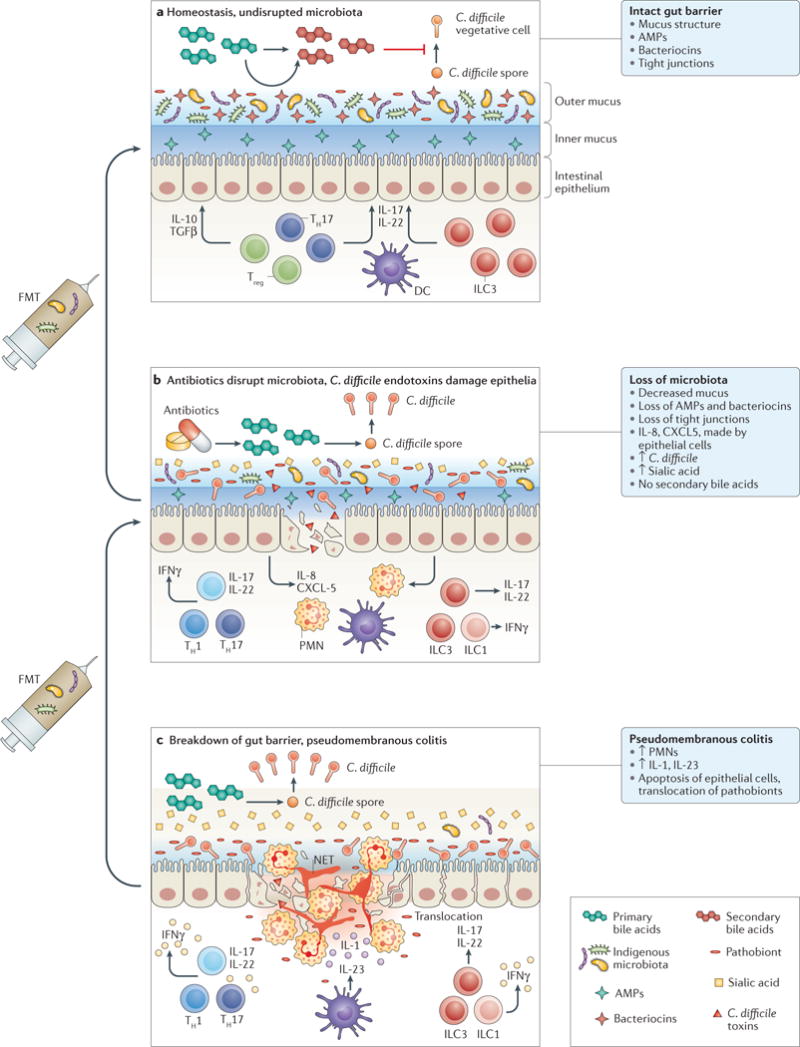
a At homeostasis, the indigenous gut microbiota is compartmentalized within the intestinal lumen. The gut barrier is maintained by organized mucus, antimicrobial peptides (AMPs) produced by the host and the microbiota, and tight junctions between the epithelial cells. Gut microbiota provides tonic stimulation to the mucosal immune system, leading to production of different cytokines, such as transforming growth factor (TGF)β and IL-22, which help to fortify the gut barrier defences. Competition for nutrients is intense. Gut microbiota also carry out transformation of primary bile acids, which lead to a secondary bile acid composition that is inhibitory to C. difficile spore germination and growth. b Antibiotic treatment suppresses most indigenous gut microbiota, although many drug-resistant microorganisms, which are also more likely to assume pathogenic roles themselves, remain intact. More nutrients, such as free sialic acid, become available to C. difficile. Primary bile acids are no longer converted to secondary bile acids, and the total bile acid composition becomes more favourable to C. difficile spore germination and growth. Production of mucus and AMPs is decreased. C. difficile enterotoxins lead to weakening of tight junctions and epithelial cell death. Neutrophils are recruited by chemokines, such as IL-8 and CXCL5. c Severe pseudomembranous colitis results from breakdown of gut barrier and translocation of residual microbiota. This step leads to activation of the inflammasome and production of IL-1. Massive recruitment of neutrophils occur, which form the pseudomembranes. Neutrophils release their DNA to form neutrophil extracellular traps (NETs), which form temporary patches in the broken gut barrier. T cells and innate lymphocytes produce more pro-inflammatory cytokines in response to loss of microbiota compartmentalization, which might further damage the gut epithelium and gut barriers. Faecal microbiota transplantation (FMT) restores the normal composition of gut microbiota. Severe pseudomembranous colitis typically requires multiple sequential applications of FMT. DC, dendritic cell; ILC, innate lymphoid cell; PMN, polymorphonuclear leukocyte; TH, T helper cell; Treg, regulatory T cell.
Competition with indigenous gut microbiota
The human colon provides a nutritionally rich environment for its microbiota: it is densely inhabited, being home to some 40–100 trillion bacteria31,38. The different resident microorganisms intensely compete with each other for nutrients and space, but also synergistically cooperate in digesting complex poly saccharides and other polymeric molecules. For example, Bacteroides thetaiotaomicron, a prominent, abundant commensal gut microorganism, liberates free sialic acid from mucin glycoproteins in the intestine, but cannot break it down further39. The free sialic acid can in turn be a nutrient for C. difficile because the organism possesses a sialic acid catabolic operon39. In fact, a mutant strain of C. difficile deficient in sialic acid consumption is compromised in its ability to expand in antibiotic-treated animals compared with wild-type C. difficile39. Intestinal microorganisms also liberate monomeric glucose and N-acetylglucosamine, which might become more accessible to C. difficile when indigenous microbiota are suppressed by antibiotics40. This syntrophic cross-feeding is common among gut microbiota. Notably, C. difficile is one of the first recognized autotrophic pathogens and can grow on merely carbon dioxide and hydrogen gases as sole carbon and energy sources, respectively41. This adaptation reduces competition with other microorganisms within narrowly differentiated nutritional niches and enables its survival in very unfavourable conditions41. Nevertheless, competitive niche exclusion is one plausible mech anism of FMT in treatment of CDI, and it has been the underlying principle hypothesis for the development of non-toxigenic C. difficile strains as therapeutics, which show promise in reducing recurrence of CDI42. However, the approach might be less successful with highly virulent, pathogenic, r-strategist (r-selection favours high fecundity rates and ability to disperse widely in unstable, unpredictable environments) strains of C. difficile characterized by greater efficiency of sporulation and growth43.
Competition between microorganisms is not limited to access for nutritional resources. Production of bacteriocins, which are antimicrobial peptides with bactericidal or bacteriostatic activity, is a more active strategy against competitors44. An example of a narrow-spectrum bacteriocin that is highly effective against C. difficile is thuricin CD, a compound produced by Bacillus thuringiensis isolated from the human intestine45. Another example is nisin, which is a lantibiotic, a class of bacteriocins produced by many Gram-positive bacteria with strong antimicrobial activity against other Gram-positive bacteria, including C. difficile46,47. Nisinproducing strains of Lactococcus lactis have been isolated from human faeces48. Generally, lantibiotics are active in very low concentrations and contribute to extending the shelf-life of a variety of foods, including dairy products, by fermentation49. Interestingly, lacticin 3147 is broad-spectrum lantibiotic that can be produced by L. lactis strains in kefir, which might benefit some patients with R-CDI50–52.
The role of secondary bile acid metabolism
C. difficile was first isolated from the stool of a healthy infant in 1935 and named for the difficulty of its isolation and culture53. Wilson reported in the early 1980s that addition of taurocholate greatly improved C. difficile spore germination and this primary bile acid has since been routinely used in laboratory C. difficile growth media54. Now, attention has once again been focused on the effects of bile acids on the life cycle of C. difficile. The spores of C. difficile germinate when they sense the appropriate host environment and favourable nutrient conditions55,56. A favourable host physicochemical environ ment is communicated to the spores by specific bile acids, and glycine can indicate nutrient availability. At least one germin ant receptor, CspC, has been identified in C. difficile spores that is stimulated by cholic acid class bile acids57 (BOX 2). Activation of CspC triggers a protease-dependent cascade that activates a lytic enzyme, SleC, which is stored in the spore as a zymogen. This enzyme degrades the spore cortex, allowing rehydration of the desiccated core. Mutations in SleC and CspC inhibited C. difficile spore germination and decreased pathogenicity in an animal model57,58.
Box 2. Bile acid production and role.
Bile acids and their conjugates are amphipathic end products of cholesterol metabolism. In addition to their role in lipid digestion and absorption, bile salts are signalling molecules that have multiple physiological effects in energy metabolism and immune responses. Primary bile acids produced in the liver are modified in the colon by indigenous gut microbiota to produce secondary bile acids. Bile acids can modulate composition of indigenous microbiota, and different bile acids can have major effects, both stimulatory and inhibitory, on the lifecycle of C. difficile61.
Bile acids in the cholic acid class generally promote C. difficile spore germination. The primary bile salt taurocholate is typically used in laboratory C. difficile growth media.
Lithocholic acid, a secondary bile acid, is an inhibitor of C. difficile spore germination.
Combinations of bile acids present at physiological concentration in healthy adult faeces (lithocholic acid and deoxycholate) are inhibitory to C. difficile61.
Combinations of bile acids found in faeces of patients with refractory R-CDI before faecal microbiota transplantation (taurocholate, cholate, chenodeoxycholic acid) are stimulatory to C. difficile61.
Although bile acids in the cholic acid class generally stimulate germination of C. difficile spores, those in the chenodeoxycholic acid class generally inhibit germination of C. difficile spores59. Thus, the growth and reproductive responses of C. difficile depend on the combined concentration of all the bile acids present in the intestinal lumen. Colonic bile acid composition is normally dominated by secondary bile acids60. These secondary bile acids are generated following deconjugation of taurine and glycine, and 7α-dehydroxylation by some bacteria, yielding deoxycholic and lithocholic acids from cholic and chenodeoxycholic acids, respectively. Antibiotic treatments used to treat CDI inhibit bacteria involved in secondary bile acid metabolism, and secondary bile acids are essentially absent in patients with refractory R-CDI syndrome13. FMT promptly restores secondary bile acid metabolism and the total composition of bile acids in the faeces becomes donor-like13. Furthermore, C. difficile spore germination and vegetative growth are stimulated by the combinations of bile acids present in pre-FMT patients with R-CDI and inhibited by the combinations of bile acids present in faeces of post-FMT patients with R-CDI and their donors61.
The relative concentrations of stimulatory and inhibitory bile acids present in the intestinal lumen is determined by their production rates in the liver, microbial metabolism and their differential uptake into the enterohepatic circulation60. The potential germinant deoxycholate is efficiently absorbed in the colon62, which contributes to a colonic environment that is normally inhibitory to C. difficile. Interestingly, the pool size of inhibitory chenodeoxycholic class bile acids, relative to the cholic class bile acids, is smaller in women than in men63. This finding might be one potential explanation for the disproportionate fraction of female patients with R-CDI in FMT cohorts: the overall mix of bile acids in women should be more stimulatory to C. difficile because of the lower content of inhibitory bile acids64–66.
The composition of bile acids might also be favourable to the growth of C. difficile in infants, since their ability to produce secondary bile acids develops only gradually during the first year of life67. In fact, approx imately one-third of infants carry C. difficile asympto matically up to 6 months of age, after which the carriage rate gradually decreases68. By 3 years of age, carriage rates reach 0–3%, similar to those seen in non-hospitalized adults68,69. Fortunately, clinical disease in children is rare before 12–24 months of age, perhaps due to the non-expression of the C. difficile toxin receptor in the infant colon70.
Bile-acid-based approaches
The seemingly prominent role of bile acid metabolism in CDI suggests development of novel therapeutics beyond FMT. Possible directions for development include: defined, live microorganisms as probiotic drugs71 that can mediate secondary bile acid metabolism; targeting host metabolism to diminish availability of germinant bile acids in the intestinal lumen; and development of bile acid analogues that are inhibitory to C. difficile.
Microorganisms involved in secondary bile acid metabolism are largely undefined. One of the key desired chemical transformations for inhibitory activity against C. difficile is the 7α-dehydroxylation step72. Clostridium scindens, one of several bacteria known to carry out this step, is able to inhibit CDI in a mouse model72. Additional microorganisms with similar capacity are likely to exist. Defined microbial-based therapeutics will potentially be developed to treat CDI in humans, although the final formulation might also require additional microorganisms to optimize therapeutic engraftment into the intestine.
Interestingly, the bile acid sequestrant cholestyramine has been, anecdotally, successfully used in the past in treatment of CDI73. An analogous resin colestipol was tested in combination with vancomycin in treatment of primary infection in a randomized trial74. That trial failed to show any benefit, and currently bile acid sequestrants are not recommended in treatment of active CDI because they can bind and neutralize the anti biotic75. However, it is possible that bile acid sequestrants might help to prevent recurrence of CDI rather than treat active disease. Notably, administration of bile acid sequestrants leads to a compensatory increase in the production of primary bile acids in the liver from cholesterol and negates the desired effects76,77. Concurrent administration of statin drugs can blunt the compensatory increase in hepatic bile acid production78. We have success fully treated several patients with R-CDI who had failed FMT with a combination of cholestyramine and lovastatin (A. Khoruts and M.J. Sadowsky, unpublished data). Emerging new drugs that can decrease hepatic bile acid production might have some potential utility in treatment of CDI. One example is a potent agonist of the farnesoid X receptor, obeticholic acid, which inhibits bile acid synthesis by stimulating the ileal hormone fibroblast growth factor 19 (REFS 79,80).
Finally, bile acid analogues could be developed with enhanced inhibitory activity and favourable pharmacokinetic properties to ensure adequate and prolonged concentration in the colon. We have successfully treated a patient with refractory, recurrent C. difficile pouchitis with ursodeoxycholic acid (UDCA) after demonstrating that this minor secondary bile acid was inhibitory to her C. difficile isolate81. Unfortunately, UDCA is efficiently absorbed into the enterohepatic circulation and might not be applicable to the majority of patients with CDI in the colon. However, certain modified bile analogues can be made resistant to intestinal uptake and achieve high intracolonic concentrations82. Thus, it is highly likely that novel drugs inhibitory to C. difficile can be developed based on bile acid chemistry.
Immune-mediated colonization resistance
Continuous signalling from indigenous microbiota is required to maintain the optimal tone of gut barrier function and the mucosal immune system83. Multiple mucosal defences maintain compartmentalization of gut micro biota within the intestinal lumen, including the specialized mucus layer, antimicrobial peptides, secreted immunoglobulins and a diverse array of mucosal lymphocytes83. Antibiotic treatments weaken virtually all elements of this barrier, causing collapse of the mucus layer that separates epithelial cells and microbiota and reduced expression of antimicrobial peptides such as RegIIIγ, which targets Gram-positive bacteria that probably include C. difficile84. Once in contact with the epithelia, C. difficile toxins TcdA and TcdB inactivate small Rho-family regulatory GTPases by glycosylation, leading to disruption of essential cellular signalling pathways85. This step leads to weakening of the tight junctions and apoptotic death of colonocytes, which results in opening of the epithelial barrier and interaction of toxins and C. difficile pathogen-associated molecular patterns (PAMPs) with resident mucosal immune cells.
The mucosal immune response is the last defence before the host succumbs to systemic infections resulting from translocation of commensal microbiota across the gut wall. However, the immune response can be both protective and detrimental, which is probably dependent on multiple factors associated with the speci fic C. difficile strain and attributes of the individual host. For instance, the intensity of cytokine production in patients can correlate with worse outcomes. Thus, higher levels of faecal IL-8 and CXCL5, cytokines important in neutrophil recruitment, correlate with CDI severity and worse outcomes, whereas the burden of C. difficile bacteria does not86.
A number of interesting insights into the immune mechanisms in CDI have emerged from studying the infection in mouse models, which highlighted the roles of C. difficile PAMPs, innate immune receptors, the inflammasome, as well as key cytokines and different immune cells. C. difficile toxins activate the inflammasome, which cleaves pro-IL-1β and pro-IL-18 to generate their mature and secreted forms87. The severity of toxin-induced inflammation in the colon can be reduced by blocking IL-1 or genetic deletion of different components of the inflammasome, including specific innate immune receptors in the nucleotide-binding domain leucine-rich repeat (NLR) family, the adaptor protein ASC or caspase-1 (REF. 88). However, outcomes of pure toxin-mediated colitis can differ from actual infection with C. difficile. In fact, CDI-associated mortality is increased in the absence of IL-1 or various inflammasome components due to increased translocation of commensals across the gut barrier89. Normally, the commensal microorganisms promote neutrophil recruitment by increasing IL-1 production via a positive feedback loop89. The massive neutrophil infiltration that is classically associated with CDI-induced pseudomembranes probably results in rapid deployment of neutrophil extracellular traps (NETs) following release of granule proteins and chromatin90. These NETs can patch the epithelial barrier defects and prevent microbial translocation91. IL-1 also enhances production of IL-23, another cytokine that protects survival of mice infected with C. difficile92,93. Importantly, IL-1 and IL-23 regulate secretion of IL-22, a cytokine that has a central role in virtually all elements of barrier maintenance, including epithelial proliferation, wound repair, as well as production of antimicrobial peptides and mucins94. CDI in mice lacking IL-22 results in increased systemic infections caused by commensal γ-Proteobacteria that breach the gut barrier95.
The need to repair the disrupted gut barrier and dampen the exuberant immune response that has been modelled in mice might be especially relevant in the context of acute pseudomembranous colitis in humans. FMT has the potential to restore the gut barrier by providing the necessary tonic signals for epithelial regeneration and production of mucins and anti microbial peptides. Indeed, it is emerging as a potentially preferable treatment approach compared with surgical options in many patients15,16. The need for sequential administration of multiple rounds of FMT in these situations is consistent with the time needed to repair the gut mucosa15,16. However, these patients can also demonstrate remarkably quick, although not necessarily sustained, favourable responsiveness to FMT in terms of haemodynamic parameters that enable prompt stabil ization of the patient15. This phenomenon cannot easily be explained by mechanisms of gut repair or bile-acid-mediated inhibition of C. difficile germination and growth. However, it is possible that certain elements of healthy, donor gut microbiota can induce rapid production of anti-inflammatory mediators that might counteract pro-inflammatory cytokines. Investigation of such possibilities are needed for patients with severe and complicated CDI being treated with FMT.
Future applications of FMT
Beyond C. difficile treatment
The increased recognition of gut microbiota as a true organ, contained within the intestine and integral to human physiology, has suggested that FMT can be applied to many diseases, including many problems pertaining to the digestive tract such as IBD and IBS96. This recognition has also generated speculation that FMT might be beneficial as a treatment for problems with metabolism, autoimmunity and nervous system development96,97. However, many challenges remain before we learn whether this approach can offer any benefit in these indications. In some respects, the issues often revolve around questions of cause versus effect. Even if dysbiotic gut microbiota are causally linked to pathophysiology of some of these problems, it is not known whether ‘normalizing’ the gut microbial community structure will result in clinical improvement once the clinical disorder is established. Possibly, certain immunological and metabolic consequences are set during a critical developmental time window in microbiota–host interaction, and cannot be reversed98–100. In addition, optimal protocols for gut microbiota engraftment have not yet been developed for many of these indications, and cannot be simply extrapolated from R-CDI experience with FMT. This aspect is in large part due to unique antibiotic conditioning intrinsic to current standard treatment of R-CDI75. Despite what might be assumed, replacing all of the gut microbiota of a recipient with that of a donor is ecologically not trivial, and requires displacement of the recipient’s autochthonous gut microbiota. Finally, the characteristics of optimal therapeutic microbiota, if such exist, might vary between different conditions. For example, gut micro biota that could be therapeutic in anorexia nervosa might be detrimental in obesity, and vice versa.
Antibiotic resistance and complications
The next major applications of FMT will probably continue to be related to complications of antibiotic treatments and emergence of increasingly more virulent, multidrug-resistant pathogens such as vancomycin-resistant enterococci (VRE), extended-spectrum β-lactamase-producing bacteria, carbepenem-resistant Enterobacteriaceae, and others. Many of these pathogens form reservoirs within the gastrointestinal tract under antibiotic pressure that is routinely applied in the context of intense medical care101. One example is routine antibiotic usage in patients undergoing allogeneic haematopoietic stem cell transplantation (HSCT), which leads to striking loss of microbial diversity in the gut and dominance by potential pathogens that include Enterococcus and γ-Proteobacteria102. Importantly, blooms of these specific bacteria within the intestinal tract correlate with blood-borne infections in patients undergoing HSCT, whom also undergo myeloablative conditioning with chemotherapy and radiation that are also disruptive to the gut barrier102. Autologous FMT using cryopreserved microbiota is already being tested in a clinical trial (NCT02269150) at the Memorial Sloan Kettering Cancer Center, USA, with the goal of testing reduction in incidence of CDI and blood-borne infections103. Similar issues exist within the solid organ transplant recipient population, and loss of VRE dominance has been documented following FMT performed in treatment of CDI in this setting104.
The evolutionary forces that encourage antibiotic resistance and pathogenic potential among some commensal organisms are probably intrinsically linked. The basic symbiotic relationship based on mutual interests between the host and its gut microbiota is broken under antibiotic pressure, and commensal microorganisms seek existence outside the individual host. Many pathogens are fairly promiscuous and are able to easily move between different host species. Some are able to survive in harsh environments outside the host as spores101,105. Rapid evolution of antibiotic-resistant pathogens is compounded by common coexistence of both anti biotic resistance and virulence factors on mobile genetic elements that can be transferred between bacteria. FMT has the potential to restore the normal microbial gut ecology and might contribute to the range of innovative therapeutic approaches to cure infectious diseases that does not drive antibiotic resistance and might actually decrease it. We can anticipate emergence of next-generation FMT-based therapeutics that will increasingly focus on this growing challenge in medicine.
Conclusions
Rapid emergence of FMT in management of CDI has brought into focus the limitations of standard antibiotic therapies and the important roles of gut microbiota in human physiology. Mechanistic investigations of FMT have demonstrated its restorative potential for both composition and functionality of gut microbiota and suggested directions of novel therapeutic development. However, the field remains in its infancy, and interventional clinical trials coupled with mechanistic investigations will remain critical for continued progress that is needed for applications beyond C. difficile.
Key points.
Faecal microbiota transplantation (FMT) involves administration of the whole microbial community from healthy donor stool into the recipient’s intestinal tract to normalize or modify intestinal microbiota composition and function
Overall suppression of microbiota and disruption of its community structure in the colon, most commonly resulting from antibiotic therapies, is the fundamental problem underlying the pathogenesis of Clostridium difficile infection (CDI)
FMT results in normalization of microbial diversity and community structure in patients being treated for CDI, with high rates of clinical cure
The restored colon microbial community could inhibit C. difficile by multiple mechanisms: competition for nutrients; direct suppression by antimicrobial peptides; bile-acid-mediated inhibition of spore germination and vegetative growth; and activation of immune-mediated colonization resistance
Acknowledgments
The authors were supported in part by the NIH Grant 1R21-AI114722-01 and Minnesota’s Discovery, Research and Innovation Economy grant.
Competing interests statement
A.K. and M.J.S. have received research grant support from Crestovo, a company working to commercialize faecal microbiota transplantation. A.K. and M.J.S. have also filed a patent application that relates to the composition and methods for transplantation of colon microbiota.
Footnotes
Author contributions
Both authors made substantial discussions to content and reviewed/edited the manuscript before submission. A.K. researched data and wrote the article.
References
- 1.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laxminarayan R, et al. Antibiotic resistance — the need for global solutions. Lancet Infect Dis. 2013;13:1057–1098. doi: 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- 3.Kelly CP, LaMont JT. Clostridium difficile — more difficult than ever. N Engl J Med. 2008;359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 4.Lessa FC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. 2012;9:88–96. doi: 10.1038/nrgastro.2011.244. [DOI] [PubMed] [Google Scholar]
- 6.McFarland LV, Elmer GW, Surawicz CM. Breaking the cycle: treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am J Gastroenterol. 2002;97:1769–1775. doi: 10.1111/j.1572-0241.2002.05839.x. [DOI] [PubMed] [Google Scholar]
- 7.Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol. 2010;44:354–360. doi: 10.1097/MCG.0b013e3181c87e02. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes. 2013;4:125–135. doi: 10.4161/gmic.23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 10.Shankar V, et al. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome. 2014;2:13. doi: 10.1186/2049-2618-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seekatz AM, et al. Recovery of the gut microbiome following fecal microbiota transplantation. mBio. 2014;5:e00893–e00814. doi: 10.1128/mBio.00893-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weingarden A, et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3:10. doi: 10.1186/s40168-015-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weingarden AR, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol. 2014;306:G310–G319. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drekonja D, et al. Fecal microbiota transplantation for Clostridium difficile infection: a systematic review. Ann Intern Med. 2015;162:630–638. doi: 10.7326/M14-2693. [DOI] [PubMed] [Google Scholar]
- 15.Weingarden AR, Hamilton MJ, Sadowsky MJ, Khoruts A. Resolution of severe Clostridium difficile infection following sequential fecal microbiota transplantation. J Clin Gastroenterol. 2013;47:735–737. doi: 10.1097/MCG.0b013e31829004ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer M, et al. Faecal microbiota transplantation plus selected use of vancomycin for severecomplicated Clostridium difficile infection: description of a protocol with high success rate. Aliment Pharmacol Ther. 2015;42:470–476. doi: 10.1111/apt.13290. [DOI] [PubMed] [Google Scholar]
- 17.Markelov A, Livert D, Kohli H. Predictors of fatal outcome after colectomy for fulminant Clostridium difficile Colitis: a 10-year experience dr.markelov@gmail.com. Am Surg. 2011;77:977–980. doi: 10.1177/000313481107700813. [DOI] [PubMed] [Google Scholar]
- 18.Lamontagne F, et al. Impact of emergency colectomy on survival of patients with fulminant Clostridium difficile colitis during an epidemic caused by a hypervirulent strain. Ann Surg. 2007;245:267–272. doi: 10.1097/01.sla.0000236628.79550.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol. 2012;107:761–767. doi: 10.1038/ajg.2011.482. [DOI] [PubMed] [Google Scholar]
- 20.Youngster I, et al. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA. 2014;312:1772–1778. doi: 10.1001/jama.2014.13875. [DOI] [PubMed] [Google Scholar]
- 21.Chang JY, et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 22.Broecker F, et al. Long-term changes of bacterial and viral compositions in the intestine of a recovered Clostridium difficile patient after fecal microbiota transplantation. Cold Spring Harb Mol Case Stud. 2016;2:a000448. doi: 10.1101/mcs.a000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelberger S, et al. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol. 2013;108:1620–1630. doi: 10.1038/ajg.2013.257. [DOI] [PubMed] [Google Scholar]
- 25.Kump PK, et al. Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis. 2013;19:2155–2165. doi: 10.1097/MIB.0b013e31829ea325. [DOI] [PubMed] [Google Scholar]
- 26.Rossen NG, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;149:110–118.e4. doi: 10.1053/j.gastro.2015.03.045. [DOI] [PubMed] [Google Scholar]
- 27.Moayyedi P, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–109.e6. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Kolho KL, et al. Fecal microbiota in pediatric inflammatory bowel disease and its relation to inflammation. Am J Gastroenterol. 2015;110:921–930. doi: 10.1038/ajg.2015.149. [DOI] [PubMed] [Google Scholar]
- 29.Busquets D, et al. Anti-tumour necrosis factor treatment with adalimumab induces changes in the microbiota of Crohn’s disease. J Crohns Colitis. 2015;9:899–906. doi: 10.1093/ecco-jcc/jjv119. [DOI] [PubMed] [Google Scholar]
- 30.Damman CJ, et al. Low level engraftment and improvement following a single colonoscopic administration of fecal microbiota to patients with ulcerative colitis. PLoS ONE. 2015;10:e0133925. doi: 10.1371/journal.pone.0133925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host–bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 32.Fuentes S, et al. Reset of a critically disturbed microbial ecosystem: faecal transplant in recurrent Clostridium difficile infection. ISME J. 2014;8:1621–1633. doi: 10.1038/ismej.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chehoud C, et al. Transfer of viral communities between human individuals during fecal microbiota transplantation. mBio. 2016;7:e00322–e00316. doi: 10.1128/mBio.00322-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller M. The fascination with probiotics for Clostridium difficile infection: lack of evidence for prophylactic or therapeutic efficacy. Anaerobe. 2009;15:281–284. doi: 10.1016/j.anaerobe.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Petrof EO, et al. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome. 2013;1:3. doi: 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology. 2009;136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 37.Lessa FC, Gould CV, McDonald LC. Current status of Clostridium difficile infection epidemiology. Clin Infect Dis. 2012;55:S65–S70. doi: 10.1093/cid/cis319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164:337–340. doi: 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Ng KM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson KH, Perini F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun. 1988;56:2610–2614. doi: 10.1128/iai.56.10.2610-2614.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopke M, Straub M, Durre P. Clostridium difficile is an autotrophic bacterial pathogen. PLoS ONE. 2013;8:e62157. doi: 10.1371/journal.pone.0062157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerding DN, et al. Administration of spores of nontoxigenic Clostridium difficile strain M3 for prevention of recurrent C. difficile infection: a randomized clinical trial. JAMA. 2015;313:1719–1727. doi: 10.1001/jama.2015.3725. [DOI] [PubMed] [Google Scholar]
- 43.Lawley TD, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lenski RE, Riley MA. Chemical warfare from an ecological perspective. Proc Natl Acad Sci USA. 2002;99:556–558. doi: 10.1073/pnas.022641999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rea MC, et al. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci USA. 2010;107:9352–9357. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breukink E, de Kruijff B. The lantibiotic nisin, a special case or not? Biochim Biophys Acta. 1999;1462:223–234. doi: 10.1016/s0005-2736(99)00208-4. [DOI] [PubMed] [Google Scholar]
- 47.Le Lay C, Dridi L, Bergeron MG, Ouellette M, Fliss I. Nisin is an effective inhibitor of Clostridium difficile vegetative cells and spore germination. J Med Microbiol. 2016;65:169–175. doi: 10.1099/jmm.0.000202. [DOI] [PubMed] [Google Scholar]
- 48.Lakshminarayanan B, et al. Prevalence and characterization of Clostridium perfringens from the faecal microbiota of elderly Irish subjects. J Med Microbiol. 2013;62:457–466. doi: 10.1099/jmm.0.052258-0. [DOI] [PubMed] [Google Scholar]
- 49.Ross RP, Morgan S, Hill C. Preservation and fermentation: past, present and future. Int J Food Microbiol. 2002;79:3–16. doi: 10.1016/s0168-1605(02)00174-5. [DOI] [PubMed] [Google Scholar]
- 50.Rea MC, et al. Antimicrobial activity of lacticin 3,147 against clinical Clostridium difficile strains. J Med Microbiol. 2007;56:940–946. doi: 10.1099/jmm.0.47085-0. [DOI] [PubMed] [Google Scholar]
- 51.McAuliffe O, et al. Lacticin 3147, a broad-spectrum bacteriocin which selectively dissipates the membrane potential. Appl Environ Microbiol. 1998;64:439–445. doi: 10.1128/aem.64.2.439-445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bakken JS. Staggered and tapered antibiotic withdrawal with administration of kefir for recurrent Clostridium difficile infection. Clin Infect Dis. 2014;59:858–861. doi: 10.1093/cid/ciu429. [DOI] [PubMed] [Google Scholar]
- 53.Heinlen L, Ballard JD. Clostridium difficile infection. Am J Med Sci. 2010;340:247–252. doi: 10.1097/MAJ.0b013e3181e939d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson KH. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J Clin Microbiol. 1983;18:1017–1019. doi: 10.1128/jcm.18.4.1017-1019.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhattacharjee D, et al. Reexamining the germination phenotypes of several Clostridium difficile strains suggests another role for the CspC germinant receptor. J Bacteriol. 2015;198:777–786. doi: 10.1128/JB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Browne HP, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533:543–546. doi: 10.1038/nature17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Francis MB, Allen CA, Shrestha R, Sorg JA. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog. 2013;9:e1003356. doi: 10.1371/journal.ppat.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burns DA, Heap JT, Minton NP. SleC is essential for germination of Clostridium difficile spores in nutrient-rich medium supplemented with the bile salt taurocholate. J Bacteriol. 2010;192:657–664. doi: 10.1128/JB.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weingarden AR, et al. Changes in colonic bile acid composition following fecal microbiota transplantation are sufficient to control Clostridium difficile germination and growth. PLoS ONE. 2016;11:e0147210. doi: 10.1371/journal.pone.0147210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mekhjian HS, Phillips SF, Hofmann AF. Colonic absorption of unconjugated bile acids: perfusion studies in man. Dig Dis Sci. 1979;24:545–550. doi: 10.1007/BF01489324. [DOI] [PubMed] [Google Scholar]
- 63.Bennion LJ, et al. Sex differences in the size of bile acid pools. Metabolism. 1978;27:961–969. doi: 10.1016/0026-0495(78)90140-3. [DOI] [PubMed] [Google Scholar]
- 64.Lee CH, et al. The outcome and long-term follow-up of 94 patients with recurrent and refractory Clostridium difficile infection using single to multiple fecal microbiota transplantation via retention enema. Eur J Clin Microbiol Infect Dis. 2014;33:1425–1428. doi: 10.1007/s10096-014-2088-9. [DOI] [PubMed] [Google Scholar]
- 65.Agrawal M, et al. The long-term efficacy and safety of fecal microbiota transplant for recurrent, severe, and complicated Clostridium difficile infection in 146 elderly individuals. J Clin Gastroenterol. 2015;146:S-42–S-43. doi: 10.1097/MCG.0000000000000410. [DOI] [PubMed] [Google Scholar]
- 66.Khoruts A, et al. Inflammatory bowel disease affects the outcome of fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin Gastroenterol Hepatol. 2016 doi: 10.1016/j.cgh.2016.02.018. http://dx.doi.org/10.1016/j.cgh.2016.02.018. [DOI] [PMC free article] [PubMed]
- 67.Hammons JL, Jordan WE, Stewart RL, Taulbee JD, Berg RW. Age and diet effects on fecal bile acids in infants. J Pediatr Gastroenterol Nutr. 1988;7:30–38. doi: 10.1097/00005176-198801000-00008. [DOI] [PubMed] [Google Scholar]
- 68.Jangi S, Lamont JT. Asymptomatic colonization by Clostridium difficile in infants: implications for disease in later life. J Pediatr Gastroenterol Nutr. 2010;51:2–7. doi: 10.1097/MPG.0b013e3181d29767. [DOI] [PubMed] [Google Scholar]
- 69.Hafiz S, Oakley CL. Clostridium difficile: isolation and characteristics. J Med Microbiol. 1976;9:129–136. doi: 10.1099/00222615-9-2-129. [DOI] [PubMed] [Google Scholar]
- 70.Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280:G178–G183. doi: 10.1152/ajpgi.2001.280.2.G178. [DOI] [PubMed] [Google Scholar]
- 71.Hill C, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506–514. doi: 10.1038/nrgastro.2014.66. [DOI] [PubMed] [Google Scholar]
- 72.Buffie CG, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gerding DN, Muto CA, Owens RC. Jr Treatment of Clostridium difficile infection. Clin Infect Dis. 2008;46:S32–S42. doi: 10.1086/521860. [DOI] [PubMed] [Google Scholar]
- 74.Mogg GA, et al. Randomized controlled trial of colestipol in antibiotic-associated colitis. Br J Surg. 1982;69:137–139. doi: 10.1002/bjs.1800690306. [DOI] [PubMed] [Google Scholar]
- 75.Cohen SH, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31:431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 76.Bertolotti M, et al. Regulation of bile acid synthesis in humans: effect of treatment with bile acids, cholestyramine or simvastatin on cholesterol 7α-hydroxylation rates in vivo. Hepatology. 1991;14:830–837. doi: 10.1002/hep.1840140515. [DOI] [PubMed] [Google Scholar]
- 77.Reihner E, et al. Regulation of hepatic cholesterol metabolism in humans: stimulatory effects of cholestyramine on HMG-CoA reductase activity and low density lipoprotein receptor expression in gallstone patients. J Lipid Res. 1990;31:2219–2226. [PubMed] [Google Scholar]
- 78.Bertolotti M, et al. Influence of newly synthesized cholesterol on bile acid synthesis during chronic inhibition of bile acid absorption. Hepatology. 2003;38:939–946. doi: 10.1053/jhep.2003.50377. [DOI] [PubMed] [Google Scholar]
- 79.Walters JR, et al. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther. 2015;41:54–64. doi: 10.1111/apt.12999. [DOI] [PubMed] [Google Scholar]
- 80.Zhang JH, et al. Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G940–G948. doi: 10.1152/ajpgi.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weingarden AR, et al. Ursodeoxycholic acid inhibits Clostridium difficile spore germination and vegetative growth, and prevents the recurrence of ileal pouchitis associated with the infection. J Clin Gastroenterol. 2015 doi: 10.1097/MCG.0000000000000427. http://dx.doi.org/10.1097/MCG.0000000000000427. [DOI] [PMC free article] [PubMed]
- 82.Rodrigues CM, Kren BT, Steer CJ, Setchell KD. The site-specific delivery of ursodeoxycholic acid to the rat colon by sulfate conjugation. Gastroenterology. 1995;109:1835–1844. doi: 10.1016/0016-5085(95)90750-5. [DOI] [PubMed] [Google Scholar]
- 83.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vaishnava S, et al. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.El Feghaly RE, et al. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis. 2013;56:1713–1721. doi: 10.1093/cid/cit147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu H, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 88.Ng J, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–552.e3. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 89.Hasegawa M, et al. Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J Immunol. 2012;189:3085–3091. doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 91.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 92.Buonomo EL, et al. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis. 2013;208:917–920. doi: 10.1093/infdis/jit277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cowardin CA, et al. Inflammasome activation contributes to interleukin-23 production in response to Clostridium difficile. mBio. 2015;6:e02386–e02314. doi: 10.1128/mBio.02386-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hasegawa M, et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity. 2014;41:620–632. doi: 10.1016/j.immuni.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aroniadis OC, Brandt LJ. Fecal microbiota transplantation: past, present and future. Curr Opin Gastroenterol. 2013;29:79–84. doi: 10.1097/MOG.0b013e32835a4b3e. [DOI] [PubMed] [Google Scholar]
- 97.Smits LP, Bouter KE, de Vos WM, Borody TJ, Nieuwdorp M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology. 2013;145:946–953. doi: 10.1053/j.gastro.2013.08.058. [DOI] [PubMed] [Google Scholar]
- 98.Backhed F, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:852. doi: 10.1016/j.chom.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 99.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khoruts A. First microbial encounters. Nat Med. 2016;22:231–232. doi: 10.1038/nm.4042. [DOI] [PubMed] [Google Scholar]
- 101.Modi SR, Collins JJ, Relman DA. Antibiotics and the gut microbiota. J Clin Invest. 2014;124:4212–4218. doi: 10.1172/JCI72333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taur Y, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55:905–914. doi: 10.1093/cid/cis580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02269150.
- 104.Stripling J, et al. Loss of vancomycin-resistant enterococcus fecal dominance in an organ transplant patient with Clostridium difficile colitis after fecal microbiota transplant. Open Forum Infect Dis. 2015;2:ofv078. doi: 10.1093/ofid/ofv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human–microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Smith MB, Kelly C, Alm EJ. Policy: how to regulate faecal transplants. Nature. 2014;506:290–291. doi: 10.1038/506290a. [DOI] [PubMed] [Google Scholar]
- 107.Bakken JS, et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol. 2011;9:1044–1049. doi: 10.1016/j.cgh.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]