

Abstract
Exposure to ambient particulate matter (PM) causes cardiopulmonary morbidity and mortality through mechanisms that involve oxidative stress. 1,2-naphthoquinone (1,2-NQ) is a ubiquitous component of PM and a potent redox-active electrophile. We previously reported that 1,2-NQ increases mitochondrial H2O2 production through an unidentified mechanism. We sought to characterize the effects of 1,2-NQ exposure on mitochondrial respiration as a source of H2O2 in human airway epithelial cells. We measured the effects of acute exposure to 1,2-NQ on oxygen consumption rate (OCR) in the human bronchial epithelial cell line BEAS-2B and mitochondrial preparations using extracellular flux analysis. Complex-specific assays and NADPH depletion by glucose deprivation distinguished between mitochondrial and non-mitochondrial oxygen utilization. 1,2-NQ exposure of BEAS cells caused a rapid, marked dose-dependent increase in OCR that was independent of mitochondrial respiration, exceeded the OCR observed after mitochondrial uncoupling, and remained sensitive to NADPH depletion, implicating extra-mitochondrial redox cycling processes. Similar effects were observed with the environmentally relevant redox-cycling quinones 1,4-naphthoquinone and 9,10-phenanthrenequinone, but not with quinones that do not redox cycle, such as 1,4-benzoquinone. In mitochondrial preparations, 1,2-NQ caused a decrease in Complex I-linked substrate oxidation, suggesting impairment of pyruvate utilization or transport, a novel mechanism of mitochondrial inhibition by an environmental exposure. This study also highlights the methodological utility and challenges in the use of extracellular flux analysis to elucidate the mechanisms of action of redox-active electrophiles present in ambient air.
Keywords: mitochondria, bioenergetics, air pollution, quinones, extracellular flux
Introduction
Ambient particulate matter (PM) is a global public health concern. Exposure to ambient PM is estimated to cause 3.3 million premature deaths worldwide annually and is linked to increased cardiovascular and respiratory morbidity and mortality (Pope and Dockery, 2006; Lelieveld et al., 2015). Despite intensive investigation, the mechanism(s) that initiate the adverse health effects of PM exposure are not known. However, oxidative stress has been specifically implicated in the responses to PM inhalation (Kelly, 2003; Li et al., 2008).
Mitochondrial dysfunction has been identified as a key event in PM-induced cytotoxicity (Hiura et al., 2000; Li et al., 2003; Xia et al., 2004). In addition to their crucial role in cellular bioenergetics, mitochondria are important regulators of oxidative stress. In resting cells, mitochondria generate reactive oxygen species (ROS), the levels of which are highly controlled by multiple antioxidant systems (Murphy, 2009; Brand, 2010). A variety of xenobiotics disrupt mitochondrial function by inhibiting the electron transport chain or uncoupling mitochondrial membrane potential, processes which increase ROS generation (Kovacic et al., 2005).
Typically found on particles surfaces, quinones are ubiquitous organic components of PM that have been implicated in PM-induced mitochondrial dysfunction (Flowers-Geary et al., 1996). Quinones commonly found in ambient air include 1,2-naphthoquinone (1,2-NQ), 1,4-naphthoquinone (1,4-NQ), 9,10-phenanthrenequinone (9,10-PQ), and 1,4-benzoquinone (BQ) (Cho et al., 2004; Delgado-Saborit et al., 2013). Polyaromatic hydrocarbons (PAHs) can also be oxidized by cytochrome p450 to form quinones in vivo (Penning et al., 1999). Quinones have been proposed to contribute to the health effects of PM inhalation (Valavanidis et al., 2005). Previous work on environmentally relevant quinones has largely focused on their electrophilic properties, specifically their adduction of cellular macromolecules through Michael addition (Iwamoto et al., 2007; Kumagai et al., 2012). Less studied is the role played by ROS generated through redox cycling of specific quinone species (Watanabe and Forman, 2003). Redox-cycling quinones can undergo one-electron reduction to form unstable semiquinones (Figure S1). This reduction may be catalyzed by several flavin-containing enzymes, such as NADH:ubiquinone oxidoreductase (Complex I) of the mitochondrial electron transport chain and the cytochrome P450 system of the endoplasmic reticulum (Henry and Wallace, 1996). Semiquinones can donate an electron to oxygen (O2) to form superoxide (O2·−), which has an extremely short half-life due to its rapid dismutation to the more stable species, hydrogen peroxide (H2O2), by abundant superoxide dismutases. Individual quinone species vary in their propensity to adduct and redox cycle, dependent on their electrophilicity and redox potential, respectively (Song and Buettner, 2010). Thus, 1,2-NQ and 1,4-NQ are capable of both mechanisms, while BQ is exclusively an electrophile and 9,10-PQ only redox cycles in the cell (Kumagai et al., 2012). Prior studies have demonstrated that both redox-cycling and electrophilic quinones can depolarize mitochondrial membrane potential and deplete ATP production (reviewed in Henry and Wallace 1996).
Previous work implicated mitochondria as a source of H2O2 production in human airway epithelial cells exposed to environmentally relevant concentrations of 1,2-NQ (Cheng et al., 2012), but the mechanism remains to be elucidated. We hypothesized that 1,2-NQ impairs mitochondrial function, leading to increased mitochondrial H2O2. In the present study, we investigated whether 1,2-NQ disrupts mitochondrial function, as measured through extracellular flux analyses. We show here that 1,2-NQ exposure of the human bronchial epithelial cell line BEAS-2B impairs complex I-linked mitochondrial substrate oxidation, a novel mechanism of mitochondrial impairment by 1,2-NQ, and markedly increases oxygen consumption through a redox cycling mechanism, both potential sources of H2O2.
Methods
Materials
Tissue culture media and supplements were purchased from Lonza (Walkersville, MD, USA). The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO): 1,2-naphthoquinone (1,2-NQ), 1,4-naphthoquinone (1,4-NQ), 9,10-phenanthrenequinone (9,10-PQ), 1,4-benzoquinone (BQ), DMSO, oligomycin, Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), rotenone, Antimycin A, glucose, sodium pyruvate, fatty acid-free bovine serum albumin (BSA), mannitol, sucrose, potassium phosphate monobasic (KH2PO4), magnesium chloride (MgCl2), EGTA, H–2O2, and HEPES. XF Base Medium, XF Flux Paks, and XF PMP were purchased from Seahorse Bioscience (Billerica, MA, now Agilent Technologies). L-glutamine (Gibco) was purchased from ThermoFisher Scientific (Waltham, MA). Basic laboratory supplies were purchased from Fisher Scientific (Raleigh, NC, USA).
Cell Culture
SV40 large T antigen-transformed human airway epithelial cells [BEAS-2B cells, subclone S6 (Reddel et al., 1988)] were cultured in serum-free keratinocyte growth medium (KGM). BEAS-2B have been used extensively for in vitro testing of inhaled toxicants as a model of airway epithelium (Veljkovic et al., 2011; Persoz et al., 2012). BEAS-2B cell cultures were continually renewed from frozen stocks every 2–3 months for the duration of the study. Cells were deprived of growth factors overnight prior to experiment by changing the medium to keratinocyte basal medium (KBM), to induce cellular quiescence.
Quinone Preparation
100 mM stocks of 1,2-NQ and 1,4-NQ and 15 mM stocks of 9,10-PQ were prepared in DMSO. Working solutions were diluted in cell media with final DMSO concentrations less than 0.1%. 50 mM stocks of BQ were prepared directly in cell media or mitochondrial assay solution. All stocks and working solutions were made fresh on the same day as exposure.
Protein Assay
Where indicated, protein concentration was determined by a modified Bradford Assay (Bio-Rad Laboratories, Inc.).
Extracellular Flux Analysis
Oxygen consumption rate (OCR) was measured at 37°C using the Seahorse XFe96 Analyzer (Agilent Technologies). BEAS-2B were seeded at 16,000 cells per well two days prior to assay in XF96 cell plates with KGM. Four background wells without cells were included in all assays. XFe96 sensor cartridges were hydrated overnight with XF Calibrant at 37°C. Media was replaced the night before assay to KBM without growth factors. For intact cell experiments, XF Cell Mito Assay Media (XF Base Media with 10 mM glucose, 1 mM sodium pyruvate, 2 mM glutamine) was prepared fresh on the day of assay and adjusted to pH 7.4 with 100 mM NaOH. Cell media was changed 1 hour prior to assay and cells were placed in a 37°C incubator without CO2. Stock concentrations of oligomycin, FCCP, rotenone, and antimycin A were prepared in DMSO and stored at -20°C. All compounds used for in-assay injections were prepared in XF Cell Mito Assay Media with final DMSO concentrations less than 0.1%. The concentration of FCCP that effects maximal OCR was optimized in preliminary experiments. For Cell Mito Stress Tests, the injections were ordered as follows unless otherwise noted: A) quinone; B) 1 uM oligomycin; C) 0.25 uM FCCP; D) 1 uM rotenone, 1 uM antimycin A. Mix-wait-measure times were 3min-0min-3min.
Bioenergetic parameters were calculated through the standard Cell Mito Stress Test Assay (Agilent Technologies). Briefly, change in basal OCR after quinone addition was calculated as the measurement immediately after quinone addition minus the basal measurement directly before addition. ATP production was measured as the decrease in OCR after oligomycin addition. Non-mitochondrial respiration was measured as the minimum OCR after rotenone/antimycin A injection. Reserve capacity was measured as the increase in OCR after FCCP was added. Spare respiratory capacity was calculated as the maximal respiration after quinone treatment minus the basal respiration.
For glucose starvation experiments, cell media was changed to either XF Cell Mito Assay Media or XF Glycolysis media (XF Base Media supplemented with 2 mM glutamine) two hours prior to start of assay.
Cell Viability
Cell viability was measured by monitoring retention of pre-loaded calcein-AM (Molecular Probes, Eugene, OR, USA). BEAS-2B were plated in black-walled, clear-bottom 96-well plates (Costar, Corning Inc., Corning, NY) and grown until confluency. Cells were deprived of growth factors overnight before the experiment. Two hours before the assay, cell media was changed to XF Cell Mito Assay Media or XF Glycolysis media, and cells were placed in a non-CO2 incubator at 37°C. Cells were washed twice with warm PBS and incubated with 2.5 uM calcein AM for 20 min at 37°C. After incubation, excess calcein AM was removed through two washes of warm PBS. Cells were treated in triplicate with the following: a media control; DMSO; 3-50 uM 1,2-NQ; 3-50 uM 1,4-NQ; 50 uM 9,10-PQ; or 0.1% saponin (positive control). Cells were immediately measured for calcein fluorescence at excitation 490 nm/emission 520 nm using a Clariostar plate reader (BMG LABTECH, Cary, NC) at 37°C. Measurements were taken at 60 s intervals for 20 min. Cellular viability was expressed as percentage of media control.
Permeabilized Cell Preparation
Permeabilized cell experiments were conducted following a procedure similar to that described previously (Salabei et al., 2014). BEAS-2B were plated and prepared as described above in Extracellular Flux Analysis. Mitochondrial Assay Solution (MAS, 220 mM mannitol, 210 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, pH 7.2) was prepared. All substrates were diluted in MAS and adjusted to pH 7.2. Immediately prior to extracellular flux assay, media was changed to MAS with freshly added 0.6% BSA and 1 nM XF PMP. No equilibration step was included on the XFe96 Analyzer, and mix-wait-measure times were 0.5min-0min-2min. For Complex I substrate oxidation tests, injections were sequenced as follows: A) quinone; B) 10 mM pyruvate/2 mM malate or 10 mM glutamate/10 mM malate, and 4 mM ADP; C) 3.16 uM oligomycin; D) 1 uM rotenone. Complex II substrate oxidation injections were sequenced as follows: A) quinone; B) 10 mM succinate, 4 mM ADP, 2 uM rotenone; C) 3.16 uM oligomycin; D) 4 uM antimycin A, 100 uM TMPD, 10 mM ascorbate.
To track mitochondria during permeabilization procedure, BEAS cells expressing the redox potential sensor roGFP targeted to the mitochondria were imaged using a Nikon Eclipse C1si spectral confocal imaging system (Nikon Instruments Corporation, Melville, NY) and the Andor Zyla 4.2 camera (Andor Technology Ltd, Belfast, UK). In a live cell imaging experiment at 37°C and 5% CO2, cells were imaged prior to permeabilization and at 150 s intervals immediately after permeabilization using excitation of 404 nm and 488 nm and emission at 515 nm. Injections of 10 uM 1,2-NQ and subsequent mitochondrial inhibitors were performed at the same intervals and concentrations as extracellular flux analysis experiments.
Isolated Mitochondrial Preparation
Mouse hearts were the generous gift of Dr. Kathleen Caron. C57/BL6 mice were treated humanely under practices approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Mitochondria were isolated from hearts of middle-aged male C57/BL6 mice. Briefly, ventricles were minced in cold PBS and homogenized in 1 mL mitochondrial isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, 0.5% fatty-acid free BSA, pH 7.2) using a tissue processor (Tissue Tearer, Bio Spec Products Inc., Bartlesville, OK) on low speed for 2-3 strokes on ice, similar to as described in (Palmer et al., 1977). Homogenates were centrifuged at 800 × g for 10 min at 4°C. The fat/lipid layer was removed by aspiration and supernatant was centrifuged at 8000 × g for 10 min at 4°C. The resulting pellet was washed twice in ice-cold isolation buffer and resuspended in MAS and protein concentration was determined. Mitochondrial enrichment was verified by blotting for cytochrome c as a marker of the mitochondrial fractions (Figure S6), as previously described (Wages et al., 2014).
Extracellular flux analyses were conducted using a Seahorse XFe24 Analyzer (Seahorse Bioscience). Five ug of mitochondrial protein in 50 uL were added per well of an XF24 cell plate, with 4 background wells containing no mitochondria. The mitochondria were immobilized on the plate by centrifugation at 2000 × g for 20 min at 4°C using centrifuge equipped with a swinging bucket (Eppendorf, Hauppauge, NY). Pre-warmed MAS + 0.6% BSA was added to each well and the assay began immediately. For Complex I substrate oxidation tests, injections were sequenced as follows: A) quinone; B) 10 mM pyruvate, 2 mM malate, 4 mM ADP; C) 3.16 uM oligomycin; D) 1 uM rotenone. Mix-wait-measure times of 0.5 min-0 min-2 min were used and no equilibration step was included. After assay, cellular protein was extracted and subjected to Western Blotting analysis as previously described (Wages et al., 2015). A polyclonal antibody with affinity for 1,2-NQ protein adducts was developed by a custom antibody production service using a standard 70-day 2-rabbit immunization protocol (ThermoFisher Scientific, Waltham, MA). Unpurified rabbit serum was used at a concentration of 1:10,000 for immunoblotting.
To permit comparisons between isolated mitochondria and BEAS-2B experiments, we empirically determined the mitochondrial protein content in BEAS-2B. We first determined the ratio of mitochondrial protein to total cellular protein as follows. BEAS-2B were grown on 8 × 150 cm2 flasks until confluent. Cells were disrupted using nitrogen cavitation (Gottlieb and Adachi, 1999). Mitochondrial fractions were obtained using differential centrifugation, at 800 × g and 8000 × g and the protein concentrations of cytosolic and mitochondrial fractions were determined. We determined the ratio to be 0.0253 ± 0.0064 ug of mitochondrial protein per 1 ug total cellular protein (mean ± SD, n=3 independent experiments). In separate experiments, we determined that approximately 64,000 cells were present per well in BEAS-2B experiments at the time of assay, therefore corresponding to 0.133 ± 0.060 ug of mitochondrial protein/well. OCR values from all experiments were thusly expressed normalized per ug of mitochondrial protein.
H2O2 Production Assay
Extracellular H2O2 was measured using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific, Waltham, MA) following the manufacturer’s instructions. Briefly, BEAS-2B were plated at 25,000 cells/well in a clear-bottom black-walled 96-well plate two days prior to assay. Eighteen hours prior to assay, the culture media was replaced with KBM. Cells were treated with DMSO, 3-25 uM 1,2-NQ, 3-25 uM 1,4-NQ, 3-25 uM 9,10-PQ, or 25 uM BQ for 18 min. at 37°C in phenol red-free KBM. 100-250 uM H2O2 was used as a positive control. The media was incubated with Amplex Red and HRP for 30 min and the fluorescence of the resorufin produced by the reaction with H2O2 was measured at excitation λ= 545 nm, emission λ= 590 nm on the Clariostar (BMG LABTECH, Inc., Cary, NC).
Complex I Activity Test
The rate of NADH oxidation to NAD+ was tested using the Complex I Enzyme Activity Microplate Assay Kit (Abcam, Cambridge, MA) per manufacturer’s instructions. Briefly, BEAS-2B were treated with KBM, DMSO, 10 uM or 50 uM 1,2-NQ for 15 min. Media was aspirated and cells were washed and scraped in ice-cold PBS. Following cell lysis and determination of protein content, 121.5 ug of total protein was added to each assay well and incubated for 3 hrs at room temperature. Background wells containing no sample and background control wells without assay solution were included. The NADH-dependent reduction of the dye was measured at 450 nm at 1 min intervals for 30 min with shaking between readings. Complex I activity was calculated as the rate of absorbance increase over time (mOD/min). Data was normalized to background wells.
Statistical Analyses
Data are expressed as means ± SEM from at least three separate experiments. Statistical significance (p < 0.05) was determined with Prism version 7.00 (Graphpad Software, La Jolla, CA), using one-way ANOVA with Dunnett’s post-test for comparisons to controls, Tukey’s post-test for comparisons within groups, and Sidak’s test for intergroup comparisons. Pairwise comparisons were conducted using Student’s t-tests.
Results
1,2-NQ Induces Dose-Dependent Increase in Oxygen Consumption Rate (OCR) of BEAS Cells
We previously reported that 1,2-NQ-induced H2O2 production could be blocked with inhibitors of mitochondrial respiration (Cheng et al., 2012). We therefore used extracellular flux analysis to characterize the effects of 1,2-NQ on indices of mitochondrial respiration. As shown in Figure 1, addition of 1,2-NQ (3-50 uM) resulted in a striking, dose-dependent increase in the oxygen consumption rate (OCR) by BEAS cells relative to baseline within 6 min. of exposure, with 25 uM 1,2-NQ inducing an approximately 80 pmol/min elevation in OCR (an increase of approximately 100%). Blank wells without cells showed no change in OCR, ruling out reactivity between 1,2-NQ and media components or the oxygen-sensing fluorophore as a possible signal artifact (data not shown). An increase in OCR is typically associated with mitochondrial membrane uncoupling, yet 1,2-NQ-induced OCR was higher than that produced by exposure to the model uncoupler FCCP (2.4-fold for 50 uM 1,2-NQ vs 1.2-fold for 0.25 uM FCCP). Addition of complex-specific inhibitors in the sequence prescribed by the standard Cell Mito Stress Test (Agilent Technologies; Figure S2A) showed decreased maximal respiration, unaffected ATP production, as well as a marked increase in spare respiratory capacity and non-mitochondrial respiration, in cells treated with 1,2-NQ (Figure S2).
Figure 1. 1,2-NQ increases basal Oxygen Consumption Rate (OCR, pmol/min).
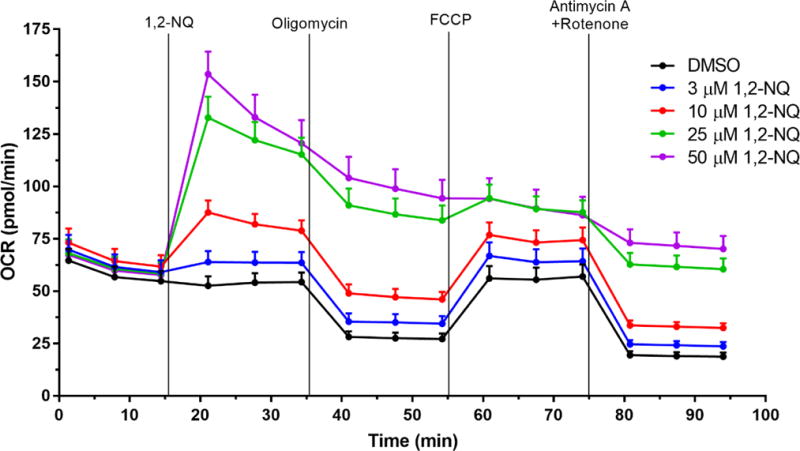
Bioenergetic effects of 1,2-NQ on BEAS cells were measured using the Cell Mito Stress Test on a XFe96 analyzer. An acute injection of 1,2-NQ was followed by additions of 1 uM oligomycin, 0.25 uM FCCP, and 1 uM antimycin A and rotenone at the indicated times. Data displayed as mean +/− SEM, n ≥ 10. One-sided error bars are used for clarity.
1,2-NQ-Induced OCR is Not Dependent on Mitochondrial Respiration
Endogenous quinones (i.e., ubiquinone) play an essential physiological role in the mitochondrial electron transport chain by shuttling electrons in the inner membrane between Complexes I, II, and III. As a redox-active quinone, 1,2-NQ could possibly increase the basal OCR by enhancing the rate of electron transport in the inner membrane. To test this hypothesis, we pretreated BEAS cells with rotenone (Complex I inhibitor) and antimycin A (Complex III inhibitor) for 13 min. prior to the addition of 1,2-NQ. As shown in Figure 2, the 1,2-NQ (10-50 uM)-induced dose-dependent rise in OCR was unaffected by pretreatment with these complex inhibitors. The efficacy of the rotenone and antimycin A pretreatment was demonstrated by the ablation of the OCR induced by the model uncoupler FCCP in cells not treated with 1,2-NQ. These findings suggested that a non-respirational process contributes significantly to the OCR induced by exposure of BEAS cells to 1,2-NQ.
Figure 2. 1,2-NQ-induced OCR increases are independent of mitochondrial electron transport.
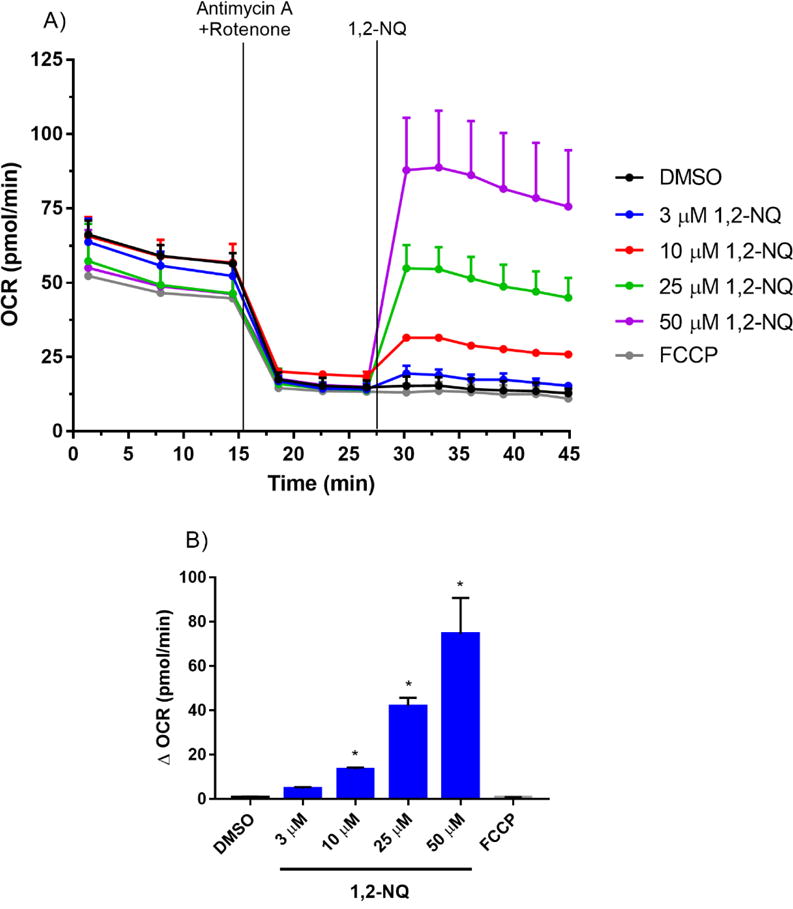
A. BEAS cells were treated with 1 uM antimycin A and 1 uM rotenone for 13 min prior to addition of DMSO (vehicle), the indicated concentration of 1,2-NQ, or 0.25 uM FCCP. B. The change in OCR after acute injection was calculated from rotenone/antimycin A-pretreated baseline, mean +/− SEM, n = 3, * indicates significant difference from the pretreated baseline (p < 0.05).
Environmentally Relevant Redox Cycling Quinones Increase OCR in BEAS Cells
To examine the role of redox cycling in 1,2-NQ-induced increase in OCR, we next compared the effect of 1,2-NQ to that induced by three structurally distinct quinones that are also commonly found as contaminants of ambient air (Cho et al., 2004). Like 1,2-NQ, 1,4-NQ is known to both redox cycle and adduct macromolecules within the cell. 9,10-PQ was examined as a quinone that is redox active but is not sufficiently electrophilic to participate in Michael addition. In contrast, BQ is potently electrophilic but incapable of intracellular redox cycling (Kumagai et al., 2012). As seen with 1,2-NQ, 9,10-PQ caused a dose-dependent increase in basal OCR, that exceeded that induced by FCCP by up to 15-fold (Figure 3). Similarly, 1,4-NQ also caused an increase in OCR that peaked at 25 uM but decreased at higher doses. Following treatment with either 1,4-NQ or 9,10-PQ, cells were no longer responsive to mitochondrial inhibitors, suggesting upstream inhibition of mitochondrial function (Figure S3). In marked contrast, treatment with BQ induced a comparatively small increase in OCR that was not dose-dependent and did not reach the level induced by FCCP (Figure 3). Unlike the cells treated with the redox active quinones, BEAS cells exposed to BQ remained responsive to the mitochondrial respiration inhibitors (Figure S3).
Figure 3. Exposure to environmentally relevant redox-cycling quinones increases OCR.
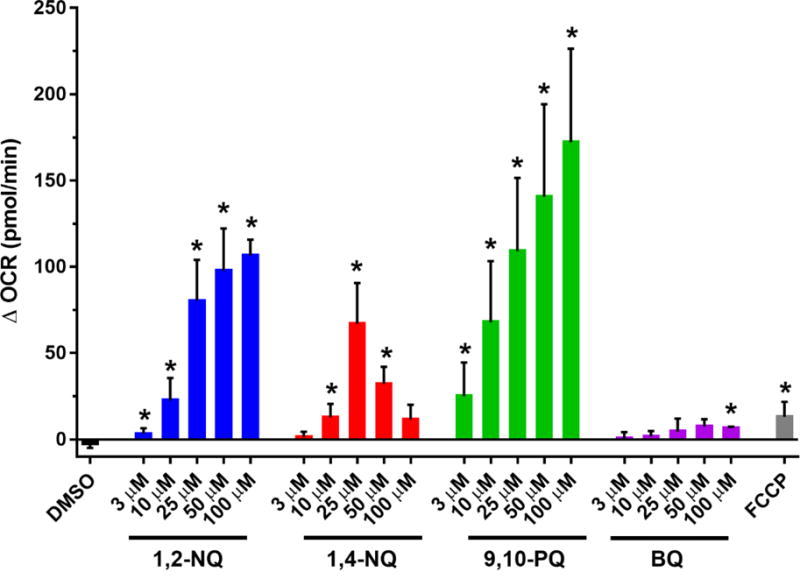
BEAS cells were treated with DMSO (vehicle), the indicated concentrations of 1,2-NQ, 1,4-NQ, 9,10-PQ, BQ, or 0.25 uM FCCP following a baseline OCR collection period of 18 min on the extracellular flux analyzer. Shown is the change in OCR at 6 min of exposure relative to basal value. * indicates the compound-induced OCR is significantly different from baseline OCR (p<0.05). For 1,2-NQ (3-50 uM), n ≥ 11. For 1,4-NQ, n ≥ 7. For 9,10-PQ (3-50 uM), n ≥ 6. For 100 uM of all quinones, n=3. For BQ, n =3. For FCCP, n = 4. Data displayed as mean +/− SEM.
1,2-NQ-induced Increase in OCR is NADPH-dependent
The rate-limiting step in quinone redox cycling is the flavoenzyme-mediated one-electron reduction of the quinone to the semiquinone, a process that is dependent on NADPH as a source of electrons (Henry and Wallace, 1996; Figure S1). Thus, we determined whether 1,2-NQ-induced increases in OCR are influenced by intracellular NADPH levels. BEAS cells were deprived of glucose for two hours prior to assay, a treatment which we have previously shown depletes NADPH levels by approximately 65% (Gibbs-Flournoy et al., 2013). Compared to control cells, the increase in OCR measured at 6 min following the addition of 25 uM or 50 uM 1,2-NQ was significantly blunted in glucose-deprived cells (Figure 4). For example, 25 uM 1,2-NQ increased OCR by approximately 30 pmol/min (~50%) after a two-hour glucose starvation, compared to 80 pmol/min (100% increase) in presence of normal glucose. Similarly, glucose deprivation also diminished the increase in OCR in BEAS cells treated with 1,4-NQ or 9,10-PQ (Figure 4). In contrast, the OCR increase induced by FCCP was not affected by glucose deprivation of the cells (Figure 4). To ensure that the blunted OCR after glucose starvation was not the result of cytotoxicity, cell viability was assessed. While exposure to the higher concentrations of 1,2-NQ and 1,4-NQ induced moderate loss of cell viability, we observed no significant differences in cell viability between glucose-deprived and glucose-sufficient BEAS cells (Figure S4).
Figure 4. Glucose starvation blunts quinone-induced increase in OCR.
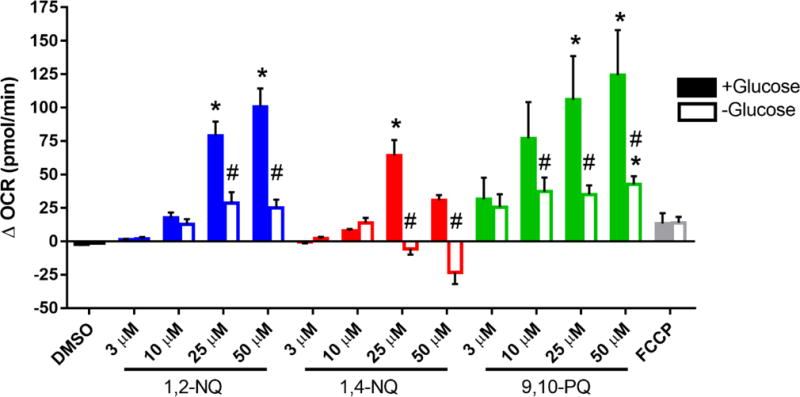
BEAS cells were starved of glucose (open bars) or provided with 10 mM glucose (closed bars) 2 hours prior to start of assay. BEAS cells were treated with DMSO (vehicle), the indicated concentrations of 1,2-NQ, 1,4-NQ, 9,10-PQ, BQ, or 0.25 uM FCCP following a baseline OCR collection period of 18 min on the extracellular flux analyzer. Shown is the change in OCR relative to basal value, * indicates the compound-induced OCR was significantly (p<0.05) different from baseline OCR. # indicates a significant (p<0.05) difference between glucose starved and supplemented groups. Data displayed as mean +/− SEM. For DMSO, 1,2-NQ and 1,4-NQ, n ≥ 4. For 9,10-PQ and FCCP, n = 3.
Redox-Cycling Quinones Increase H2O2 in BEAS cells
We hypothesized that the OCR increases induced in BEAS cells exposed to redox-cycling quinones is attributable to the conversion of dissolved oxygen in the media to reduced oxygen species that would not be detectable by the flux analyzer, leading to it being reported by the instrument as an increase in the rate of oxygen consumption. Superoxide produced during redox cycling is rapidly degraded to H2O2, making its detection challenging during an acute exposure. We therefore measured H2O2 levels in the medium of BEAS cells using Amplex Red after exposure to 3-25 uM 1,2-NQ, 1,4-NQ, 9,10-PAQ or BQ. As we described previously for 1,2-NQ (Cheng et al., 2012; Wages et al., 2015), exposure to each of the three redox-active quinones resulted in dose-dependent increases in H2O2 production. In contrast, 25 uM BQ induced a comparatively modest increase in H2O2 levels (Figure 5).
Figure 5. Exposure to redox cycling quinones induces H2O2 production.
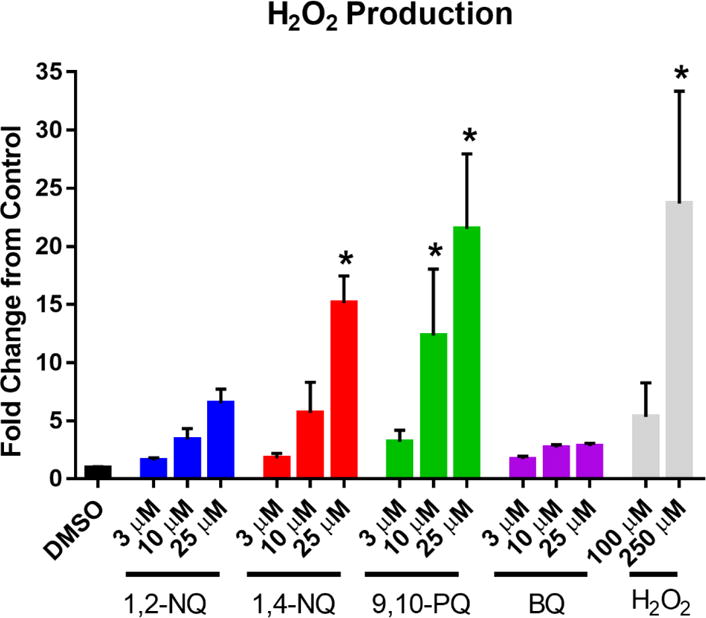
BEAS cells were exposed to DMSO (vehicle) or the indicated concentrations of 1,2-NQ, 1,4-NQ, 9,10-PQ, BQ, or H2O2 for 18 min. H2O2 was measured in apical media as fluorescence at excitation λ= 545 nm, emission λ= 590 nm following a 30 min incubation in the presence of HRP and Amplex Red. n = 3, mean +/− SEM.
1,2-NQ-Induced OCR is Dependent on an Extra-Mitochondrial Process
To further identify the intracellular site of oxygen consumption, we tested the effect of 1,2-NQ on OCR in BEAS cells pretreated with the cholesterol-specific permeabilizing reagent perfrinoglysin (XF PMP) to permeabilize the cells and allow the intracellular compartment to equilibrate with the extracellular medium (Divakaruni et al., 2013). Imaging analysis of BEAS cells expressing a mitochondrially-targeted fluorophore (roGFP) showed that the permeabilization procedure did not impact mitochondria number or integrity in the cells (Figure S5).
Relative to intact cells, permeabilized BEAS cells exposed to 10 uM or 50 uM 1,2-NQ showed an increase in OCR that was significantly diminished in magnitude (Figure 6), yet a significant dose-dependent increase in OCR in permeabilized BEAS cells remained, suggesting that reductive activity at another organelle, such as the mitochondria or the endoplasmic reticulum contributes to the redox cycling.
Figure 6. Mitochondrial preparations show a diminished 1,2-NQ-induced OCR increase.
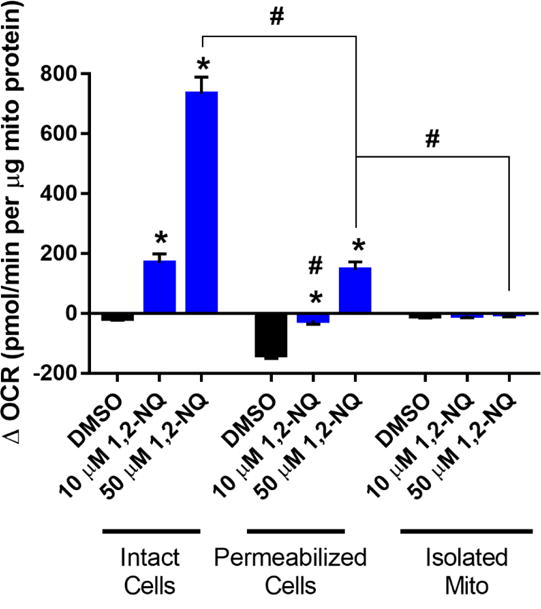
All values shown as a change from basal OCR observed immediately after addition of DMSO or the indicated concentration of 1,2-NQ. All values were normalized to the mass of mitochondrial protein in each preparation. * indicates the change in OCR was significantly (p<0.05) different from DMSO treatment within each preparation. # indicates the change in OCR was statistically different (p<0.05) between mitochondrial preparations. For intact cells, n ≥ 11; for permeabilized cells, n ≥ 13; for isolated mitochondria, n = 5. Data shown as mean +/− SEM.
To specifically examine the contribution of mitochondria further, we tested the effect of 1,2-NQ on OCR in mitochondria isolated by differential centrifugation of homogenates prepared from murine cardiac myocytes. Treatment of isolated mitochondria with 10 or 50 uM 1,2-NQ did not significantly increase OCR, suggesting that mitochondria are not a major site of quinone metabolism contributing to redox cycling in BEAS cells (Figure 6).
1,2-NQ Impairs Complex I-Linked Respiration
The preceding experiments strongly implicated extra-mitochondrial redox cycling as a major contributor to the large OCR induced in BEAS cells exposed to 1,2-NQ. However, the reduction in mitochondrial maximum respiration shown in Figure S2 suggested that 1,2-NQ may also impair mitochondrial respiratory function. We therefore next examined the effect of 1,2-NQ exposure on mitochondrial respiration in permeabilized BEAS cells unimpeded by redox cycling. Pyruvate, malate, and ADP were added as substrates to support respiration through Complex I. Exposure of permeabilized BEAS cells to 1,2-NQ for 10 min prior to substrate addition, resulted in a dose-dependent reduction in pyruvate-dependent respiration (Figure 7A), with exposure to 25 uM 1,2-NQ resulting in an impairment of pyruvate-driven OCR by approximately 75 pmol/min (equivalent to a 60% decrease). While exposure to 1,4-NQ produced results similar to those induced by 1,2-NQ, pyruvate-driven respiration was not significantly impaired by exposure of permeabilized BEAS cells to either 9,10-PQ or BQ (Figure 7A). Compared to the permeabilized cells, virtually the same result was obtained in isolated mitochondria exposed to 1,2-NQ (Figure S7). Additionally, numerous protein adducts were detected by blotting with a 1,2-NQ-specific antibody, suggesting that 1,2-NQ binds to multiple mitochondrial proteins (Figure S7). Substitution of glutamate for pyruvate in the medium as a Complex I-linked substrate partially rescued the 1,2-NQ-induced inhibition of OCR, suggesting that 1,2-NQ preferentially inhibits pyruvate-linked respiration (Figure 7A).
Figure 7. 1,2-NQ impairs Complex I-linked respiration in permeabilized cells.

A. BEAS cells were permeabilized with XF PMP and treated as indicated with buffer, DMSO,1,2-NQ, 1,4-NQ, 9,10-PQ, BQ for 10 min, followed by an injection of 10 mM pyruvate/2 mM malate or 10 mM glutamate/10 mM malate, in a buffer containing 4 mM ADP, and mitochondrial respiration was measured as OCR. * indicates activity was significantly (p<0.05) different from buffer control. # indicates a significant (p<0.05) difference between glutamate and pyruvate supplementation. For 1,2-NQ, 1,4-NQ, Buffer, and DMSO, n ≥ 4. For 9,10-PQ and BQ, n ≥ 3. B. Complex II-linked mitochondrial respiration measured by OCR after addition of 10 mM succinate/4 mM ADP/2 uM rotenone. BEAS cells were permeabilized with XF PMP and treated as indicated with DMSO,1,2-NQ, 1,4-NQ, 9,10-PQ, BQ for 10 min, and substrate oxidation was measured as OCR. * indicates activity was significantly (p<0.05) different from buffer control. For DMSO and 1,2-NQ, n = 4. For 1,4-NQ and BQ, n = 3. C. BEAS cells were treated with 10 uM or 50 uM 1,2-NQ for 15 min and Complex I activity was assayed as the NADH-dependent reduction of a chromophore over 30 min. * indicates activity was significantly (p<0.05) different from media control, n = 3. Data shown as mean +/− SEM.
To further test whether 1,2-NQ inhibits downstream complexes in the electron transport chain, we used rotenone (Complex I inhibitor) and succinate (Complex II substrate) to bypass Complex I. As shown in Figure 7B, 1,2-NQ treatment did not affect Complex II-linked respiration using succinate as a substrate in permeabilized BEAS cells, suggesting no impairment of electron transport occurs downstream of Complex I and thus indicating that the functional loss occurs at or upstream of Complex I (Figure 7B).
1,2-NQ Does Not Impair Complex I Activity
Given the observed decrease in Complex I-linked substrate oxidation induced by 1,2-NQ, we next determined whether this was due to 1,2-NQ directly inhibiting Complex I activity. We assayed the effect of 1,2-NQ exposure on Complex I activity by measuring NADH oxidation by BEAS cell proteins immunocaptured with a mouse monoclonal antibody directed to bovine Complex I. As shown in Figure 7C, exposure of BEAS cells to 1,2-NQ did not inhibit Complex I activity, suggesting that 1,2-NQ-induced inhibition observed in permeabilized cells occurs upstream of Complex I.
Discussion
In the present study, we utilized extracellular flux analysis to characterize the effect of environmentally relevant doses of 1,2-NQ exposure on mitochondrial function in human airway epithelial cells. We report that 1,2-NQ induces a large, dose-dependent increase in BEAS cell OCR that masks a frank impairment in mitochondrial respiration.
While initial observations suggested that 1,2-NQ could be a mitochondrial uncoupler, further study revealed that the increase in OCR induced by 1,2-NQ is primarily attributable to a non-mitochondrial NADPH-dependent redox-cycling process. This is supported by multiple lines of evidence. First, the magnitude of the 1,2-NQ-induced increase in OCR exceeded that produced by the model mitochondrial uncoupler FCCP. Second, inhibition of the electron transport chain with mitochondrial respiration inhibitors rotenone and antimycin A did not prevent 1,2-NQ-induced OCR increases. Third, only exposure to quinones with redox cycling potential produced elevations in OCR and H2O2 production. Fourth, prior depletion of levels of NADPH, a cofactor required to support redox-cycling (Cohen and Doherty, 1987), significantly blunted 1,2-NQ-induced OCR increases. Given that the degradation of H2O2 produced by the dismutation of O2.− generates O2, the data in this study may underrepresent the true magnitude of the OCR increase induced by 1,2-NQ exposure.
Despite the masking effect of redox cycling, in this study we elucidate a novel mechanism of mitochondrial inhibition by 1,2-NQ. Our data show that that 1,2-NQ exposure impaired mitochondrial respiration through a mechanism that is dependent on oxidation of pyruvate and to a lesser extent, glutamate, two substrates that feed electrons to Complex I, in a dose-dependent manner. When succinate (Complex II substrate) was provided, mitochondrial respiration was not affected by 1,2-NQ exposure, suggesting that the electron transport chain was intact downstream of Complex I. That the enzymatic activity of Complex I itself was not inhibited by 1,2-NQ, leads us to conclude that 1,2-NQ inhibits upstream pyruvate transporters or Kreb’s cycle enzymes (Brand and Nicholls, 2011). We have detected 1,2-NQ adducts in mitochondrial proteins (Figure S6), indicating that 1,2-NQ adducts mitochondrial targets. However, since neither 9,10-PQ nor BQ caused inhibition in pyruvate-driven mitochondrial respiration, we are unable to infer the extent to which quinone-induced effects are mediated by ROS generation of by electrophilic adduction. An inherent limitation of comparing the effects of different quinones is that they may vary in solubility and not compartmentalize similarly. Nonetheless, given the electrophilicity of 1,2-NQ, it is possible to speculate that key substrate transporters or dehydrogenases are attacked by 1,2-NQ. Additionally, we cannot exclude the possibility that H2O2 produced extra-mitochondrially damages mitochondria, leading to the observed mitochondrial inhibition and further H2O2 production. However, the lack of an inhibition of substrate oxidation by exposure to 9,10-PQ, a more potent redox cycler than 1,2-NQ, mediated, suggests that this is not a likely to be a major contribution. An unavoidable limitation of our study is that the assays to measure activity of specific electron transport Complexes require a disrupted cell preparation. Further investigation using novel methodological approaches is needed to identify potential sites of substrate oxidation inhibition by 1,2-NQ and determine the likelihood of it occurring in intact cells.
Inhibition of mitochondrial substrate oxidation could disrupt cellular ATP production and increase H2O2 production. Though Complexes I and III are the most well-known, there are at least ten different sites of superoxide/H2O2 generation in mitochondria, including upstream substrate oxidation processes (Quinlan et al., 2013). Increased H2O2 and resulting oxidative stress can induce downstream inflammatory responses, as we previously observed (Cheng et al., 2012).
The intracellular localization of redox cycling by quinones in human airway epithelial cells has not been identified. A number of different flavin-containing enzymes are capable of 1-electron quinone reduction, including cytochrome P450 reductases located in the endoplasmic reticulum and Complex I in the mitochondrion. It has previously been suggested that quinone redox cycling is preferentially compartmentalized within mitochondria, leading to permeabilization of the inner mitochondrial membrane and disruption of calcium homeostasis in hepatocytes (Henry and Wallace, 1996). Hydrophilic quinones have also been found to redox cycle at a rotenone-insensitive allosteric site in Complex I (King et al., 2009). Furthermore, we have previously reported that 1,2-NQ-induced H2O2 production was of mitochondrial origin (Cheng et al., 2012). In contrast, the data presented herein support an extra-mitochondrial site for 1,2-NQ redox cycling leading to H2O2 production. The ineffectiveness of electron transport chain inhibition in blunting the 1,2-NQ-induced OCR increase, combined with the diminished magnitude of the OCR in isolated mitochondria, further suggest that the mitochondrion plays a relatively minor role in the 1,2-NQ-induced OCR in BEAS cells. Reductases bound to the endoplasmic reticulum and possibly other cytosolic enzymes are likely key players in quinone redox cycling. Sepiapterin reductase is a cytosolic NADPH-dependent enzyme found to readily reduce 1,2-NQ in murine lung epithelial cells and could also be responsible for mediating 1,2-NQ redox cycling in human airway epithelial cells (Yang et al., 2013).
To our knowledge, this is the first application of extracellular flux technology to elucidate the bioenergetic effects of ambient air pollution in human airway epithelial cells. Technological innovations in cellular metabolism have increased accessibility and standardized approaches to measure mitochondrial function (Ferrick et al., 2008; Pelletier et al., 2014). Extracellular flux technology can advance our understanding of the role of bioenergetic processes as both targets of environmental agents and mediators of response, and has the potential to revolutionize screening for mitochondrial toxicity. While the technology is rapidly gaining in popularity, this report shows the need for caution when interpreting extracellular flux findings in toxicologic research. A decrease in OCR is not the only indicator of mitochondrial toxicity. On the other hand, as demonstrated in this study, an increase in OCR may not always indicate an uncoupling effect. It is important to consider alternative pathways that consume oxygen, including redox cycling and the activities of xanthine oxidase, NADPH oxidase and cytochrome p450 oxidase.
By consuming oxygen, redox-active compounds can induce OCR, confounding changes in respiration and bioenergetics when using extracellular flux analysis. Organic and inorganic redox cycling compounds of toxicological concern include paraquat, menadione, doxorubicin and transition metals. Use of the NADPH oxidoreductase inhibitor diphenyleneiodonium (DPI) has been recommended as an adequate control for redox cycling to measure bioenergetic function using extracellular flux analysis (Dranka et al., 2010). However, we found that DPI and other pharmacologic redox cycling interventions (e.g., dicoumarol) inhibit mitochondrial respiration at doses lower than those required to suppress 1,2-NQ-induced redox cycling (data not shown). As demonstrated in this study, the use of a permeabilized cell system allows examination of the contribution of mitochondrial and extra-mitochondrial processes to OCR changes induced by redox-active compounds. In spite of these limitations, extracellular flux technology can be used as a powerful tool to measure O2 consumption and bioenergetic effects in cells exposed to redox-active compounds. The burden is therefore on the investigator to be aware of these limitations and devise appropriate experimental strategies to address them in order to better interpret findings for toxicological research applications.
The environmental relevance of the concentrations of 1,2-NQ used in this study are based on our previously published calculations modelling a plausible in vivo exposure (Cheng et al., 2012). Briefly, a realistic 3 hr inhalational exposure to diesel exhaust (e.g., at a bus depot) could result in human airway epithelial cells experiencing concentrations of 1,2-NQ between 0.6 and 6 uM, comparable to the levels that showed clear effects in this study. Moreover, ambient air pollution contains a varied mixture of redox-active components that includes multiple quinone species and precursors to which in vivo airway epithelial cells are exposed simultaneously (Mastral and Callen, 2000; Alves et al., 2015). For example, naphthalene, the most prevalent and abundant polycyclic aromatic hydrocarbon in the air is rapidly converted in airway cells to 1,2-NQ through cytochrome p450 oxidation (Zheng et al., 1997; Lanza et al., 1999), and thus represents an additional source of exposure to 1,2-NQ and the accompanying oxidative burden on the cells. Additional dose modeling is needed to gain a more accurate concentration of the total quinone burden experienced by airway epithelial cells during PM exposure.
The data presented here suggest that 1,2-NQ induces H2O2 production in human airway epithelial cells through extra-mitochondrial redox cycling, though mitochondrial impairment also contributes to the oxidative stress induced by quinone exposure. This indicates that an organic PM component can have significant oxidative effects in human airway epithelial cells and is a potential initiating mechanism leading to downstream adverse effects. This study also highlights the strengths and limitations of extracellular flux technology for investigating the bioenergetics effects of environmental agents.
Supplementary Material
Acknowledgments
We express our gratitude to Danielle Suarez for Seahorse instrument training and support. We thank Claire Trincot and Dr. Kathleen Caron for providing the mouse hearts used in the isolated mitochondria experiments. We are also grateful to thank Dr. Haiyan Tong for her assistance with mitochondrial isolation.
Funding
This work was supported by the EPA-UNC Toxicology Training Agreement [CR-83591401-0] and NIEHS [NRSA T32 ES007126].
Footnotes
Disclaimer
The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. The contents of this article should not be construed to represent agency policy nor does mention of trade names or commercial products constitute endorse-ment or recommendation for use.
Conflict of Interest
The authors declare they have no actual or potential competing financial interests.
Supplementary Data
Supplemental figures are available as a resource to readers but are not essential to the main findings of the manuscript.
References
- Alves CA, Barbosa C, Rocha S, Calvo A, Nunes T, Cerqueira M, Pio C, Karanasiou A, Querol X. Elements and polycyclic aromatic hydrocarbons in exhaust particles emitted by light-duty vehicles. Environmental Science and Pollution Research. 2015;22:11526–11542. doi: 10.1007/s11356-015-4394-x. [DOI] [PubMed] [Google Scholar]
- Brand MD. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochemical Journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WY, Currier J, Bromberg PA, Silbajoris R, Simmons SO, Samet JM. Linking oxidative events to inflammatory and adaptive gene expression induced by exposure to an organic particulate matter component. Environmental Health Perspectives. 2012;120:267. doi: 10.1289/ehp.1104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho AK, Di Stefano E, You Y, Rodriguez CE, Schmitz DA, Kumagai Y, Miguel AH, Eiguren-Fernandez A, Kobayashi T, Avol E. Determination of four quinones in diesel exhaust particles, SRM, 1649a, and atmospheric PM2. 5 special issue of aerosol science and technology on findings from the fine particulate matter supersites program. Aerosol Science and Technology. 2004;38:68–81. [Google Scholar]
- Cohen G, Doherty MDA. Free radical mediated cell toxicity by redox cycling chemicals. The British Journal of Cancer Supplement. 1987;8:46. [PMC free article] [PubMed] [Google Scholar]
- Delgado-Saborit JM, Alam MS, Pollitt KJG, Stark C, Harrison RM. Analysis of atmospheric concentrations of quinones and polycyclic aromatic hydrocarbons in vapour and particulate phases. Atmospheric Environment. 2013;77:974–982. [Google Scholar]
- Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proceedings of the National Academy of Sciences. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radical Biology and Medicine. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discovery Today. 2008;13:268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Flowers-Geary L, Bleczinski W, Harvey RG, Penning TM. Cytotoxicity and mutagenicity of polycyclic aromatic hydrocarbon o-quinones produced by dihydrodiol dehydrogenase. Chemico-Biological Interactions. 1996;99:55–72. doi: 10.1016/0009-2797(95)03660-1. [DOI] [PubMed] [Google Scholar]
- Gibbs-Flournoy EA, Simmons SO, Bromberg PA, Dick TP, Samet JM. Monitoring intracellular redox changes in ozone-exposed airway epithelial cells. Environmental Health Perspectives. 2013;121:312. doi: 10.1289/ehp.1206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Adachi S. Nitrogen cavitation for cell disruption to obtain mitochondria from cultured cells. Methods in Enzymology. 1999;322:213–221. doi: 10.1016/s0076-6879(00)22022-3. [DOI] [PubMed] [Google Scholar]
- Henry TR, Wallace KB. Differential mechanisms of cell killing by redox cycling and arylating quinones. Archives of Toxicology. 1996;70:482–489. doi: 10.1007/s002040050302. [DOI] [PubMed] [Google Scholar]
- Hiura TS, Li N, Kaplan R, Horwitz M, Seagrave JC, Nel AE. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. The Journal of Immunology. 2000;165:2703–2711. doi: 10.4049/jimmunol.165.5.2703. [DOI] [PubMed] [Google Scholar]
- Iwamoto N, Sumi D, Ishii T, Uchida K, Cho AK, Froines JR, Kumagai Y. Chemical knockdown of protein-tyrosine phosphatase 1B by 1, 2-naphthoquinone through covalent modification causes persistent transactivation of epidermal growth factor receptor. Journal of Biological Chemistry. 2007;282:33396–33404. doi: 10.1074/jbc.M705224200. [DOI] [PubMed] [Google Scholar]
- Kelly FJ. Oxidative stress: its role in air pollution and adverse health effects. Occupational and Environmental Medicine. 2003;60:612–616. doi: 10.1136/oem.60.8.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MS, Sharpley MS, Hirst J. Reduction of Hydrophilic Ubiquinones by the Flavin in Mitochondrial NADH: Ubiquinone Oxidoreductase (Complex I) and Production of Reactive Oxygen Species†. Biochemistry. 2009;48:2053–2062. doi: 10.1021/bi802282h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic P, Pozos RS, Somanathan R, Shangari N, O’Brien PJ. Mechanism of mitochondrial uncouplers, inhibitors, and toxins: focus on electron transfer, free radicals, and structure-activity relationships. Current Medicinal Chemistry. 2005;12:2601–2623. doi: 10.2174/092986705774370646. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Shinkai Y, Miura T, Cho AK. The chemical biology of naphthoquinones and its environmental implications. Annual Review of Pharmacology and Toxicology. 2012;52:221–247. doi: 10.1146/annurev-pharmtox-010611-134517. [DOI] [PubMed] [Google Scholar]
- Lanza DL, Code E, Crespi CL, Gonzalez FJ, Yost GS. Specific dehydrogenation of 3-methylindole and epoxidation of naphthalene by recombinant human CYP2F1 expressed in lymphoblastoid cells. Drug Metabolism and Disposition. 1999;27:798–803. [PubMed] [Google Scholar]
- Lelieveld J, Evans J, Fnais M, Giannadaki D, Pozzer A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature. 2015;525:367–371. doi: 10.1038/nature15371. [DOI] [PubMed] [Google Scholar]
- Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environmental Health Perspectives. 2003;111:455. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radical Biology and Medicine. 2008;44:1689–1699. doi: 10.1016/j.freeradbiomed.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastral AM, Callen MS. A review on polycyclic aromatic hydrocarbon (PAH) emissions from energy generation. Environmental Science & Technology. 2000;34:3051–3057. [Google Scholar]
- Murphy M. How mitochondria produce reactive oxygen species. Biochemistry. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. Journal of Biological Chemistry. 1977;252:8731–8739. [PubMed] [Google Scholar]
- Pelletier M, Billingham LK, Ramaswamy M, Siegel RM. Extracellular flux analysis to monitor glycolytic rates and mitochondrial oxygen consumption. Methods in Enzymology. 2014;542:125–149. doi: 10.1016/B978-0-12-416618-9.00007-8. [DOI] [PubMed] [Google Scholar]
- Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chemical Research in Toxicology. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- Persoz C, Achard S, Momas I, Seta N. Inflammatory response modulation of airway epithelial cells exposed to formaldehyde. Toxicology letters. 2012;211:159–163. doi: 10.1016/j.toxlet.2012.03.799. [DOI] [PubMed] [Google Scholar]
- Pope CA, Dockery DW. Health effects of fine particulate air pollution: lines that connect. Journal of the Air & Waste Management Association. 2006;56:709–742. doi: 10.1080/10473289.2006.10464485. [DOI] [PubMed] [Google Scholar]
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biology. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Research. 1988;48:1904–1909. [PubMed] [Google Scholar]
- Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nature Protocols. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Buettner GR. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radical Biology and Medicine. 2010;49:919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A, Fiotakis K, Bakeas E, Vlahogianni T. Electron paramagnetic resonance study of the generation of reactive oxygen species catalysed by transition metals and quinoid redox cycling by inhalable ambient particulate matter. Redox Report. 2005;10:37–51. doi: 10.1179/135100005X21606. [DOI] [PubMed] [Google Scholar]
- Veljkovic E, Jiricny J, Menigatti M, Rehrauer H, Han W. Chronic exposure to cigarette smoke condensate in vitro induces epithelial to mesenchymal transition-like changes in human bronchial epithelial cells, BEAS-2B. Toxicology in Vitro. 2011;25:446–453. doi: 10.1016/j.tiv.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Wages PA, Lavrich K, Zhang Z, Cheng WY, Corteselli E, Gold A, Bromberg P, Simmons SON, Samet J. Protein Sulfenylation: A Novel Readout of Environmental Oxidant Stress. Chemical Research in Toxicology. 2015 doi: 10.1021/acs.chemrestox.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wages PA, Silbajoris R, Speen A, Brighton L, Henriquez A, Tong H, Bromberg PA, Simmons SO, Samet JM. Role of H2O2 in the oxidative effects of zinc exposure in human airway epithelial cells. Redox Biology. 2014;3:47–55. doi: 10.1016/j.redox.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Forman HJ. Autoxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells. Archives of Biochemistry and Biophysics. 2003;411:145–157. doi: 10.1016/s0003-9861(02)00716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, Nel A. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environmental Health Perspectives. 2004:1347–1358. doi: 10.1289/ehp.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Jan YH, Gray JP, Mishin V, Heck DE, Laskin DL, Laskin JD. Sepiapterin reductase mediates chemical redox cycling in lung epithelial cells. Journal of Biological Chemistry. 2013;288:19221–19237. doi: 10.1074/jbc.M112.402164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Cho M, Jones AD, Hammock BD. Evidence of quinone metabolites of naphthalene covalently bound to sulfur nucleophiles of proteins of murine Clara cells after exposure to naphthalene. Chemical Research in Toxicology. 1997;10:1008–1014. doi: 10.1021/tx970061j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.