

Abstract
B-lymphocyte differentiation is an exquisitely regulated homeostatic process resulting in continuous production of appropriately selected B-cells. Relatively small changes in gene expression can result in deregulation of this process, leading to acute lymphocytic leukemia, immune deficiency or autoimmunity. Translocation of Mll1 (Kmt2a) often results in a pro-B cell acute lymphocytic leukemia (B-ALL), but little is known about its role in normal B-cell differentiation. Using a Rag1-cre mouse knock-in to selectively delete Mll1 in developing lymphocytes, we show that B-cell, but not T-cell homeostasis depends on MLL1. Mll1-/- B-progenitors fail to differentiate efficiently through the pro- to pre-B cell transition, resulting in a persistent reduction in B-cell populations. Cells inefficiently transit the pre-B cell receptor (pre-BCR) checkpoint, despite normal to higher levels of pre-BCR components and rearranged IgH expression fails to rescue this differentiation block. Instead of IgH rearrangement defects, we find that Mll1−/− pre-B cells exhibit attenuated RAS/MAPK signaling downstream of the pre-BCR, resulting in reduced survival in physiologic levels of IL-7. Genome-wide expression data illustrate that MLL1 is connected to B-cell differentiation and IL-7-dependent survival through a complex transcriptional network. Overall, our data demonstrate that wild type MLL1 is a regulator of pre-BCR signaling and B-cell differentiation and further suggest that targeting its function in B-ALL may be more broadly effective than previously anticipated.
Keywords: H3K4 methyltransferase, KMT2A, IL-7 signaling, leukemia, differentiation
INTRODUCTION
The Mixed Lineage Leukemia (MLL, MLL1, KMT2A) gene is disrupted by chromosomal translocations that occur with >70% frequency in infant acute leukemia predominantly presenting with a pro-B/pre-B Acute Lymphocytic Leukemia (ALL) phenotype (1). Genome-wide sequencing efforts have illustrated that this disease, even among childhood leukemia subtypes, exhibits remarkably low mutational burden (2), suggesting that the majority of the leukemogenic process is driven by the fusion oncoprotein. In adults, MLL1 translocations or rearrangements are more commonly associated with the myeloid lineage and tend to harbor other mutations, most commonly NRAS and FLT3 (3). Gain-of-function RAS mutations are also the most common mutation in MLL-rearranged pediatric B-ALL and within this poor prognosis group predict even worse outcome (4, 5).
MLL1 encodes a histone methyltransferase that is the ortholog of the Drosophila Trithorax (Trx) protein. MLL1 and Trx function as positive epigenetic regulators of selective downstream target genes such as the well-characterized clustered homeodomain (Hox) or Hom-C genes. This specific gene regulatory role has been difficult to rationalize given the broadly acting histone modifying activity and overlapping expression patterns of expression of related enzymes. In mammals, six histone H3, lysine 4 (H3K4) methyltransferases are responsible for mono-(me), di-(me2), and tri-(me3) methyltation of H3K4. Whereas H3K4me3 enrichment at the transcriptional start site (TSS) of genes is associated with transcriptionally active or poised genes, H3K4me1 enrichment is predominantly associated with enhancers (6, 7). H3K4me2 enrichment has a more nuanced relationship with regulatory elements, but is closely linked to cell identity (8). Understanding which H3K4 methyltransferase performs which specific function is a major challenge since all 6 enzymes are frequently co-expressed in tissues (9). Furthermore, how each enzyme is specifically targeted to tissue-specific gene networks is poorly understood. One of the better characterized paradigms is represented by the recruitment of MLL3 and MLL4 by the sequence-specific PAX Transcription Activation Domain Interacting Protein (PTIP), which brings these complexes to IgH switch regions to control transcription and class-switching (10).
Inducible deletion of Mll1 in different hematopoietic populations demonstrated that this methyltransferase is non-redundant and uniquely required for hematopoietic stem cell (HSC) maintenance in late embryogenesis and adult animals (11–13). Pan-hematopoietic Vav-cre Mll1 deletion resulted in anemia, bone marrow failure and animal death approximately 3 weeks after birth (12). In these young Vav-cre;Mll1-deficient animals, the B-cell lineage was more severely reduced than the T-cell lineage; however, the gross defects in overall bone marrow cellularity confounded the assessment of MLL1 function in B-cells specifically. In contrast, previous studies using the late-induced CD19-cre knock-in to delete Mll1 showed no impact on B-cell numbers in adult animals (11). Therefore, to directly assess the normal role of MLL1 during early B-cell specification and differentiation, we crossed a Rag1-cre knock-in (14) to Mll1 floxed (f) allele animals and analyzed B-cell differentiation from late gestation to adult animals. This early lymphocyte lineage-specific deletion strategy circumvented gross perturbations of the bone marrow environment and illuminated a B-cell intrinsic requirement for MLL1 for efficient B-cell production in the bone marrow. This role was characterized by impaired survival, specifically at the pre-B-cell receptor (pre-BCR) checkpoint due to downstream signaling deficits in the RAS/MAPK pathway. These data suggest that sufficient MLL1 is necessary to maintain effective pre-BCR signaling, and that loss of MLL1 results in pressure on pre-B-cells to enhance RAS signaling. The connection between wild-type MLL1 and RAS signaling is particularly intriguing given that RAS pathway mutations are the most common genetic alteration to occur in MLL-rearranged pro/pre-B ALL (4).
MATERIALS AND METHODS
Animals
Mice were maintained in compliance with the Dartmouth Center for Comparative Medicine and Research and the University of Colorado, Denver IACUC policies. The B1–8i knock-in mice (#012642, obtained from The Jackson Laboratory) were crossed with Rag1-cre;Mll1f mice. Rag1-cre animals were obtained from Dr. Rabbitts, University of Oxford (14). Rag1 knockout mice were generated by intercrossing Rag1-cre animals since the knock-in disrupts the Rag1 gene (14). Rag1-cre;Mll1f mice were back-crossed to B6.SJL animals as described (15). Female C57Bl/6 animals between 6–12 weeks of age (#000664, obtained from The Jackson Laboratory) were used as recipients of transplanted cells and were either sublethally (450 Rads) or lethally (950 Rads) irradiated using a Cs137 source then maintained for 3 weeks on 0.1 mg/mL Baytril (Bayer) in their drinking water.
Cell culture
Sorted pro-B or fraction B-cells from the bone marrow were cultured in pro-B medium (Opti-MEM [Invitrogen] supplemented with 10% fetal bovine serum [FBS], 50 μM β-mercaptoethanol, 2 mM L-Glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and indicated concentrations of recombinant murine (rm) IL-7 (Peprotech). Lineage-negative bone marrow cells were prepared with a lineage-depletion kit (Miltenyi) and cultured in Iscove’s Modified Dulbecco Medium (IMDM) with 10% FBS, 50 μM β-mercaptoethanol, 10 mM HEPES, 10 μM non-essential amino acids, 2 mM L-Glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin (all from Mediatech), as well as 50 ng/mL rm SCF (R&D System), 40 ng/mL rm Flt3L, and 5 ng/mL rm IL-7 (all from Peprotech).
Flow cytometry and cell sorting
Bone marrow cells were prepared and lineage staining was performed as previously described (15), with the exception that the F’ab2 anti-rat conjugate was PE-Cy5.5 labeled (Invitrogen). Peritoneal cells were collected by injecting 10 mL FACS buffer (2% FBS in HBSS) into the peritoneal cavity and flushing back out into a collection tube. Flow cytometry was performed with a FACSCalibur or FACSAria (BD Biosciences), and data were analyzed using FlowJo Software (TreeStar, Inc.). Analyses used live cell gates that included most hematopoietic cells, but for sorting purified populations, non-B-cell lineages were depleted from the bone marrow cells prior to surface marker staining. For intracellular staining, cells were fixed for 20–30 minutes at room temperature with Cytofix/Cytoperm buffer (BD Biosciences) and were washed and stained in Perm/wash buffer (BD Biosciences). Bone marrow B-cells were defined as follows: fraction A (pre/pro-B; CD43+CD24-BP-1-), fraction B (early pro-B; CD43+CD24+BP-1-), fraction C (late pro-B; CD43+CD24+BP-1+), fraction C’ (large pre-B; CD43+CD24hiBP-1+), fraction D (small pre-B; CD43-IgM-IgD-), fraction E (B220+IgM+IgD-), and fraction F (B220+IgM+IgD+).
Cell stimulation and Western blotting
B220+ or CD19+ bone marrow cells enriched by Miltenyi microbeads were cultured with 5–10 ng/mL IL-7 for 5–6 days to generate bone marrow-derived pro/pre-B-cells. Cultured cells were starved for IL-7 removal for 4–6 hours, and then pretreated with 30 μg/mL of biotin-conjugated Igβ mAb HM79 (Southern Biotech) for 20 minutes on ice. Stained cells were washed with cold Opti-MEM medium (Invitrogen) followed by addition of 30 μg/mL streptavidin (Sigma). Cells were then incubated at 37°C for 0.5–10 minutes, centrifuged at 4°C, and total cellular proteins were extracted with E1a lysis buffer (ELB, 50 mM HEPES pH 7.4, 250 mM NaCl, 5 mM EDTA, 0.1% NP-40) containing cOmplete™ mini EDTA-free protease inhibitor cocktail (Roche), plus Phosphatase Inhibitor Cocktails A and B (Santa Cruz Biotechnology). Cellular lysates were resolved using 12% SDS-PAGE gels and transferred to nitrocellulose. Blots were blocked in 5% non-fat milk in PBS containing 0.1% Tween-20 with the indicated antibodies. For detection of pERK and pAKT, the following antibodies were used: rabbit total anti-ERK1/ERK2 (9102); mouse anti-phospho-ERK (T202/Y204, E10); rabbit anti-AKT (C67E7), and rabbit anti-phosphor-AKT (S473) (all from Cell Signaling Technology). U0126 was also obtained from Cell Signaling Technology.
In vitro colony assays and retrovirus transduction
Retroviral supernatants were produced by co-transfection of an MSCV plasmid and a Psi-minus Eco packaging plasmid (Dr. Witte, UCLA) in 293T cells using FuGENE 6 (Promega). The Socs2 cDNA was amplified from C57Bl/6 bone marrow cDNA using the following primers: 5′-GGGACGTGTTGACTCATCTCCCAT-3′, and 5′-CGAAAAAGAGAGAGAAATACTTA-3′. The cDNA was TA cloned and transferred into a MSCV-IRES-hCD4 vector (16). Supernatants were collected 48 hours after transfection, filtered at 0.45 microns, and frozen at −80°C until use as described (16). Lineage-negative bone marrow cells were spin-infected on retronectin-coated plates (Takara), whereas fraction B-cells were spin-infected for 1.5 hours in pro-B medium containing 5 μg/mL polybrene (Sigma-Aldrich) then washed and transferred to new plates with fresh growth medium. To enumerate pre-B colonies, cells were first counted using polystyrene beads (Polysciences, Inc.) as a reference (15), and 3,000 cells were plated onto pre-B (M3630) methylcellulose according to the manufacturer’s recommendations (Stemcell Technologies). Colonies were counted at day 7–8 and characterized by flow cytometry. The MSCV-Igμ plasmid encoding the 383 cDNA was obtained from Drs. Malay Mandal and Marcus Clark (University of Chicago).
BCR-ABL transformation
Fraction B pro-B-cells were first sorted from Rag1-cre;Mll1F/F or Rag1-cre;Mll1F/+ mouse bone marrow and infected with MSCV-BCR-ABL p210 viral supernatant (gift from Dr. Shaoguang Li and Glen Raffel, University of Massachusetts Medical School) in pro-B-cell medium supplemented with 5–10 ng/mL IL-7. Spin-infection was performed as described above. Eight thousand transduced cells were mixed with 400,000 unfractionated C57Bl/6 bone marrow cells, and the mixture was injected periorbitally into lethally irradiated female C57Bl/6 recipients. Mice were closely monitored for any sign of disease and were sacrificed when moribund. A donor, B-cell phenotype (GFP+/CD45.1+/CD19+/HSA+/Mac-1neg), was confirmed for all moribund animals by flow cytometry.
Quantitative real-time PCR (qRT-PCR)
RNA was prepared using Trizol (Invitrogen) followed by RNeasy columns (Qiagen). cDNA was prepared using Superscript III (Invitrogen), and qRT-PCR was performed with Taqman PCR Master Mix or SYBR Green Mix (BioRad) and analyzed on an ABI Prism 7500 (Applied Biosystems). Relative transcript abundance was calculated using the ΔΔCt method after normalization using a rodent Gapdh Taqman assay (Applied Biosystems), Tbp, or Hprt1 SYBR assays. Primers used for qRT-PCR were: CD179a, 5′-CGTCTGTCCTGCTCATGCT-3′ and 5′-ACGGCACAGTAATACACAGCC-3′; CD179b, 5′-TGTGAAGTTCTCCTCCTGCTG-3′ and 5′-ACCACCAAAGTACCTGGGTAG-3′; Igμ, 5′-AAGGATGGGAAGCTCGTGGAATCT-3′ and 5′-TCAGGGTTTCATAGGTTGCCAGGT-3′; CD79a, 5′-CATCTTGCTGTTCTGTGCAGTG-3′ and 5′-TTCTCATTTTGCCACCGTTTC-3′; CD79b, 5′-GCTGTTGTTCCTGCTGCTGC-3′ and 5′-CTTCACCATGGAGCTCCGCTTT-3′.
PCR detection of IgH germline alleles
Genomic DNA from sorted pro-B-cells were purified with Wizard® SV Genomic DNA Purification System (Promega) and used for SYBR-based PCR to detect unrearranged DH-JH products as described previously (17). The Hprt genomic locus was used as control for normalization using 5′-GCTGGTGAAAAGGACCTCT-3′ and 5′-CACAGGACTAGAACACCTGC-3′ primers.
Cell cycle analysis and cell proliferation assays
DNA content was determined by staining sorted B-cell fractions with Krishan’s reagent (0.05 mg/mL propidium iodide; 0.1% sodium citrate; 0.02 mg/mL RNase A; 0.3% NP-40, pH 8.3). DNA content was analyzed by flow cytometry. For BrdU flux analysis, mice were injected with 0.6 mg BrdU (BD Biosciences) in 200 μL PBS intraperitoneally (IP) every 12 hours and were sacrificed at 24, 48, or 72 hours. Bone marrow was harvested and stained for surface phenotype and BrdU incorporation as described (18). The percentage of BrdU-labeled cells in each subset was determined and plotted as a function of time, and least squares regression analysis was performed to obtain the turnover and production rates as previously described (19).
Microarray experiments
One cohort of age- and sex-matched Rag1-cre;Mll1F/F and Mll1F/F mice (n=4 each genotype) were used to sort fraction B-cells. RNA was purified as above and amplified using MessageAmp™ II aRNA Amplification Kits (Ambion), labeled using BioArray HighYield RNA Transcript Labeling Kits (T7, Enzo Life Sciences) and fragmented and hybridized to Mouse 430 2.0 Arrays (Affymetrix) at the Dartmouth Genomics and Microarray Laboratory. Analysis was performed within Partek Genomic Suite (St. Louis, MO), where intensities were determined and normalized using GCRMA, and differentially-expressed probe sets were determined by class comparison using log2 expression values (Table S1). An additional table was generated using a p-value cut-off of 0.005, and probe sets collapsed to genes by retaining the probeset with the greatest fold change difference (Table S1). Heat maps and numbers of genes discussed in the text were determined using this p-value filtered data with a fold-change cut-off of 1.2.
MicroRNA expression assay
Total RNA was purified from sorted fraction B-cells with miRNeasy micro kit (Qiagen). A 2100 Bioanalyzer (Agilent) was used to assess the concentration and integrity of the RNA. One hundred nanograms of amplified DNA was the input for microRNA profiling using the nCounter Mouse microRNA expression assay (NanoString Technologies). Sample preparation and hybridization were performed following manufacturer’s recommendation at the Dartmouth Molecular Biology Core facility. Data analysis was performed using the nSolver Analysis Software 1.1. Expression counts were normalized to the sum of the counts of the 100 highest-expressed microRNA in each sample (complete data in Table S2).
ChIP-sequencing (ChIP-seq)
ChIP-seq libraries were prepared using sorted pro-B cells pooled from 3 mice per genotype following the manufacturer’s recommendations (Ovation Ultralow Library Systems, NuGEN). Multiplexed sequencing was performed on a HiSeq2000 instrument (Illumina). The resulting barcoded multiplexed sequences were debarcoded using Unix shell script and converted into Fastq format using qseq2fastq.pl perl script. Reads were mapped to the mouse genome (mm9) using Bowtie v0.12.7 and only reads that aligned to a unique position in the genome with no more than two mismatches were retained for further analysis. Bowtie outputted SAM files containing aligned reads were used to create bedgraph files using Homer with default parameters converted to bigwig format using bed Graph to Bigwig script and visualized on the Integrative Genomics Viewer (Broad Institute, MIT).
Statistical analysis
Unless otherwise indicated, unpaired Student’s t-tests were used to calculate p-values. All error bars represent 95% confidence intervals unless otherwise indicated in the Figure Legends. Graphs and statistical tests were performed using Prism (Graphpad) or Excel (Microsoft Corporation).
RESULTS
Mll1 deletion during lymphopoiesis reduces B- but not T-cell numbers
Our previous studies suggested a function for Mll1 in B-cells, however the severe effects of Mll1 loss on multiple bone marrow populations made it difficult to rule out cell-extrinsic effects on B-cell generation (12). To specifically examine the role of MLL1 in lymphocyte differentiation, we utilized a Rag1-cre knock-in strain to delete Mll1 exclusively in developing lymphocytes (14, 15). Bone marrow B-cell stages of differentiation were identified using the Hardy scheme (fraction A through fraction F (20)). Cre expression from the Rag1 locus resulted in nearly 100% deletion of a Rosa-YFP reporter by fraction B, or pro-B-cells (Figure 1A) and in the T-cell lineage by the double negative 1 population (not shown). Deletion of the floxed Mll1 gene paralleled what was observed using the YFP reporter (not shown). Thus, phenotypic analyses focused on only those populations in which Mll1 deletion was 90–100%.
Figure 1.
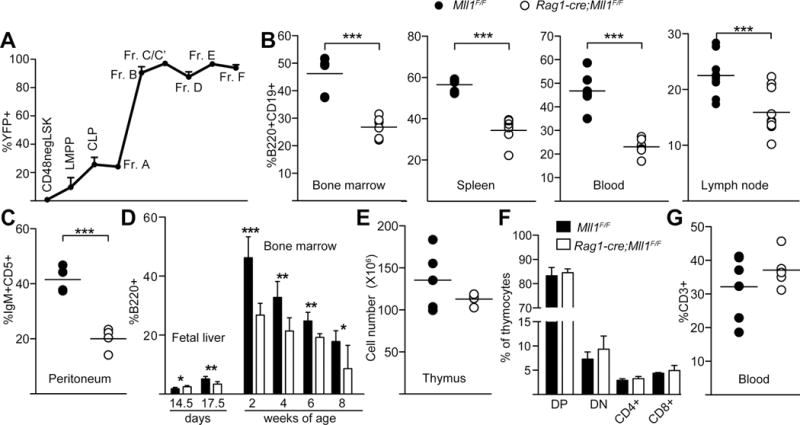
Lymphocyte specific deletion of Mll1 reduces B cell but not T cell numbers. A) Analysis of yellow fluorescent protein (YFP) expression as a read-out for cre activity during B-lymphopoiesis. The YFP+ percentage within progenitor or B cell populations from three Rag1-cre;RosaYFP reporter animals was determined by flow cytometry and average % YFP is shown. Populations are defined in the Methods. Error bars reflect 95% confidence intervals (CI), n=4–10 animals per genotype. B–C) Percent of B-cells in bone marrow and peripheral organs. Black circles represent control littermates and open circles are Rag1-cre;Mll1F/F animals, which were 2–3 weeks of age for most populations and 3–4 weeks of age for lymph node and peritoneum. Student’s t-tests were applied to determine significance; ***p< 0.001. D) Age dependence of B-cell reduction as measured by the fraction of B220+ cells in the indicated embryo or adult organs; n=3–6 animals per age/genotype, *p<0.05, **p<0.01, ***p< 0.001. Embryonic days or post-natal weeks are indicated under the graph. E–G) Total thymocyte numbers, subpopulations of thymocytes, and peripheral blood T-cells, n=4–10 animals per genotype. Double positive (DP), double negative (DN) and single positive CD4+ and CD8+ are shown, as well as the T-cell percentage (CD3+) in the peripheral blood. Black bars or circles represent control Mll1F/F littermates and open bars or circles are Rag1-cre;Mll1F/F animals.
Rag1-cre;Mll1F/F animals exhibited normal bone marrow cellularity, but B-cell populations were reduced approximately two-fold in the bone marrow and were similarly reduced in peripheral organs (Figure 1B–C). The B-cell reduction was most profound in 2–3 week-old animals but persisted as the animals aged (Figure 1D). Peripheral T-cell numbers and thymocyte subsets were not reduced, despite efficient Mll1 deletion (Figure 1D).
To determine more specifically the stage of B-cell differentiation affected in the bone marrow, we analyzed B-cell populations using the strategy shown in Figure 2A. Mll1-deficient B-lymphocyte progenitors appeared to accumulate slightly in fraction C, but all stages from fraction C’ onward were consistently reduced (Figure 2B). Since signals from the pre-B-cell receptor (BCR) are critical for transitioning to large pre-B-cells (C to C’ fraction), we considered whether reduced levels of the pre-BCR could explain this partial block in differentiation in Rag-cre;Mll1F/F animals. Levels of the pre-BCR measured by the SL156 antibody were normal to high relative to controls (Figure 2C–D), as was expression of most pre-BCR components, including Igμ, VpreB, λ5 and Igα/Igβ (Figure 2E). Therefore, failure to express pre-BCR components does not account for the reduction in developing B-cells. These data also demonstrate that MLL1 plays an important role in B-lymphopoiesis at the C-C’ transition, in contrast to its minimal role in T-cell differentiation.
Figure 2.
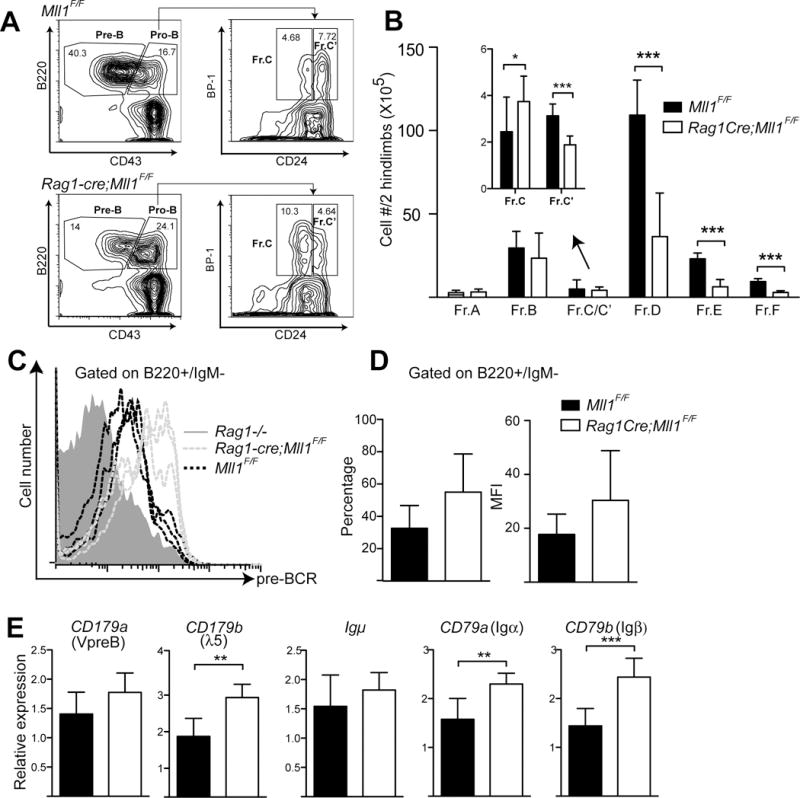
MLL1 is required for B-cell differentiation at the C–C’ transition of pro-B-cells. A) Gating strategy and representative FACS plot used to quantify Hardy fractions A–C’. Single cell suspensions from bone marrow were stained with B220, CD43, CD24, and BP-1 antibodies and analyzed as shown. B) Quantification of total B-cell numbers within fractions A–F. Three mice per genotype were analyzed and animals were 2–3 weeks of age. *p< 0.1; ***p< 0.001. Inset graph (arrow) shows C and C’ population quantification. C) Staining with the SL156 antibody to detect cell surface pre-BCR using cells sorted pro-B-cells cultured for 5 days in 5 ng/mL IL-7. The histogram shows representative staining example from three mice per genotype versus one Rag1 knockout as a control. D) Average and percentage of SL156+ cells and average mean fluorescence intensity (MFI) within the B220+IgMneg gated population, n=3–7 animals per genotype. E) Pre-BCR component transcript levels in sorted pro-B cells (n=5–6 animals of each genotype, IgM-/CD43+/CD19+). Student’s t-tests were applied to determine significance; *p<0.05, **p<0.01, ***p< 0.001.
MLL1 is dispensable for heavy chain recombination and pre-BCR expression
Despite normal to high levels of pre-BCR components, we found that the Mll1-deficient pro-B-cells had more unrearranged IgH D-J alleles than rearranged alleles (Figure 3A) as well as higher levels of IgH germline transcripts (Figure 3B). Since MLL1 is a histone H3, lysine 4 (H3K4) methyltransferase and RAG-2 binding depends on H3K4me3 modification of its target chromatin (21), we tested whether H3K4me3 levels were reduced at the IgH locus in Mll1−/− pro-B cells. Chromatin immunoprecipitation-sequencing (ChIP-seq) experiments illustrated that there was no reduction in H3K4me3 along the entire IgH locus in Rag1-cre;Mll1F/F pro-B-cells, with the characteristic peak at the JH region appearing identical between control and Mll1-deficient cells (Figure S1A, arrow). To functionally test whether rearrangement of the IgH locus was limiting for B-cell differentiation, we used two strategies to introduce pre-rearranged IgH loci to the Rag1-cre;Mll1F/F cells or animals. First, we introduced a rearranged IgH cDNA by retroviral transduction to Mll1-deficient progenitors and enumerated pre-B colonies (22). Green fluorescent protein (GFP) was also expressed from the bicistronic virus providing a surrogate marker for retroviral IgH-expression. Mll1−/− cells transduced with the control virus exhibited reduced colonies in pre-B methylcellulose cultures, resulting in a lower percentage of B220+/GFP+ cells (Figure 3C). This reduced B220+/GFP+ cell number was not rescued by expression of the IgH cDNA (Figure 3C), although control experiments using Rag1−/− cells demonstrated Rag1 cDNA rescue (Figure S1B).
Figure 3.
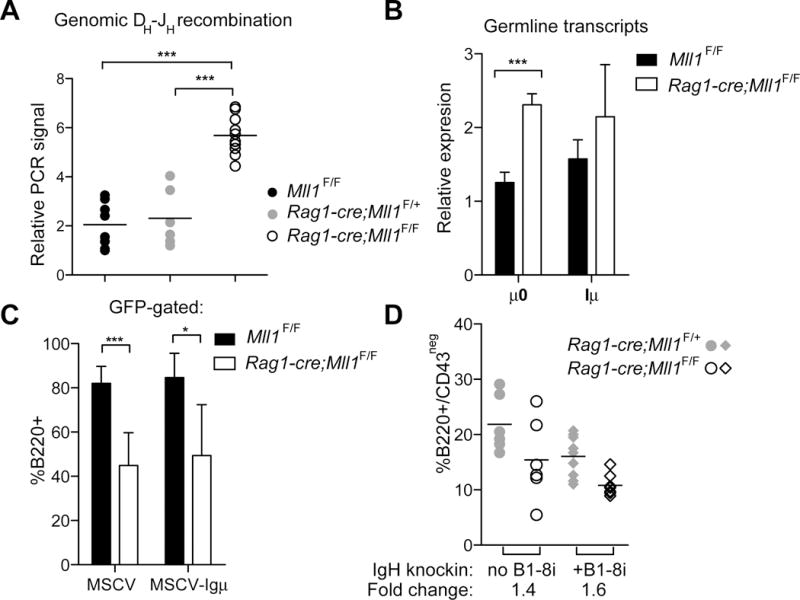
Reduced immunoglobulin heavy chain (IgH) rearrangement that cannot be rescued by expressing pre-rearranged IgH in Mll1-deficient pro-B-cells. A) Genomic DNA was purified from sorted CD19+CD43+IgM- pro-B-cells and used to perform quantitative SYBR-green based PCR assays. Hprt was used as an internal control to calculate the relative germline allele abundance. Six to eight animals at two weeks of age were used per genotype; to avoid confounding effects of cre expression and Rag1 haploinsufficiency both Mll1F/F (black circles) and Rag1-cre;Mll1F/+ controls were compared to Rag1-cre;Mll1F/F cells. B) Relative expression of germline transcripts from DH and JH gene segments. cDNA from pro-B cells isolated as above was used for qRT-PCR assays; n=3–4 animals per genotype. Hprt was used as an internal control for normalization. C) Expression of pre-rearranged immunoglobulin heavy chain was performed using an MSCV-IgH μ cDNA (μH383) viral supernatant. Lineage-depleted bone marrow cells were transduced then plated in pre-B methylcellulose after 2 days. After 7 days culture cells were harvested and stained with B220 and Mac1 antibodies; average percentage of GFP-gated cells that were B220+ is shown; n=3–6 individual donors per genotype. D) The B1-i8 knock-in IgH transgene was crossed into Rag1-cre;Mll1F/F or Rag1-cre;Mll1F/+ animals and pre-B cells (B220+/CD43neg) in the bone marrow of the indicated genotypes were analyzed; n=6–9 animals per genotype at 3 weeks of age. The fold reduction of the non-transgenic comparison is 1.4, p=0.109 whereas the fold reduction of the B1–8i-expressing transgenic pair is 1.6, p=0.0014. For other panels, *p<0.01, **p<0.01, ***p< 0.001.
The second strategy utilized a pre-rearranged IgH knock-in (B1-i8, (23)) which was crossed to Rag1-cre;Mll1F/F and Rag1-cre;Mll1F/+ animals to ensure that sufficient IgH was provided in a developmentally accurate time and expression level. We found that introduction of the B1-i8 knock-in reduced pre-B-cells regardless of Mll1 status (Figure 3D, grey symbols), which may reflect accelerated differentiation (23). Similar to the observations using the MSCV-IgH rescue strategy, B1-i8+;Rag1-cre;Mll1F/F still exhibited significantly fewer CD43neg/B220+ pre-B-cells than B1-i8+;Rag1-cre;Mll1F/+ animals (Figure 3D), although this strategy clearly rescued Rag1-knockout pre-B-cells (Figure S1C). Collectively, these results rule out inefficiencies in IgH recombination as a major explanation for the reduced B-cell numbers in animals lacking MLL1 in the B-cell lineage.
Reduced survival, not proliferation, accounts for reduced B-cell generation in the bone marrow
The amplification of B-cells after successful IgH rearrangement occurs downstream of pre-BCR signaling and involves both survival and proliferation and ultimately differentiation of pro-B-cells to pre-B-cells. Therefore, we assessed whether the lack of expansion after pre-BCR signaling in Rag1-cre;Mll1F/F was due to proliferation or cell survival defects. No differences in the number of cycling cells (S/G2/M) in any of the pro-pre-B-cell fractions were observed (Figure 4A). Similarly, a more dynamic analysis using bromo-deoxyuridine (BrdU) pulse-chase experiments revealed that flux through the pro-, pre-, and immature-B-cell populations was indistinguishable between control and Rag1-cre;Mll1F/F animals (Figure 4B). Together, these data suggest that altered proliferation does not account for the reduced output of B-cells after the pro-B-cell stage.
Figure 4.
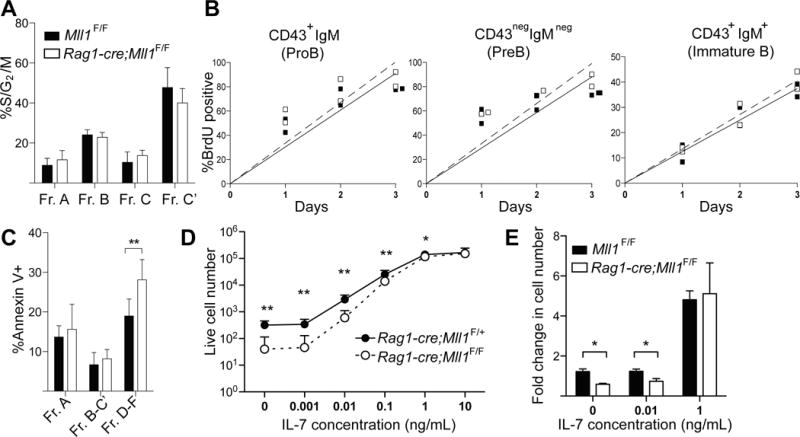
Mll1-deficiency affects pro-/pre-B-cell survival, not proliferation. A) B-cell progenitors were isolated from the bone marrow of control Mll1F/F and Rag1-cre;Mll1F/F animals using non-B lineage markers plus B220, CD43, BP-1 and CD24. Cells were then fixed, permeabilized and incubated with Krishan’s reagent to assess DNA content. Four animals per genotype were used for the analysis and average percentage of cells with greater than 2N DNA content (S/G2/M) are shown in the bar graph. B) Production rates of B cell progenitor pools. Control (black squares, Mll1F/F) or Rag1-cre;Mll1F/F (open squares) mice were injected with BrdU, and bone marrow was harvested at the indicated time and stained for surface phenotype and BrdU incorporation. Each point represents an individual mouse, n=2 per genotype. Experiments were repeated three times with similar results. Solid and dashed lines represent linear regression lines for control and mutant, respectively. C) Fraction A (B220+/CD43+/CD19 neg), fraction B–C’ (B220+/CD43+/CD19+) and fraction D–F (B220+/CD43neg) cells were gated and apoptotic cells identified by Annexin V-FITC staining. Five animals per genotype were used. D) Survival in low IL-7 is impaired. Pro-B-cells (fraction B) were sorted from the bone marrow of 2–3 week old mice and cultured with a range of IL-7 concentrations for 7 days. Live cells were enumerated by flow cytometry. Each point represents average viable cells (n=4 per genotype) with error bars representing standard error of the mean (SEM). E) Fraction B cells were cultured and enumerated as above (except on day 4 of culture) using the IL-7 concentrations indicated. Fold increases were normalized to the input cell number, n=4 per genotype. Bars represent average fold increase with error bars representing SEM. For all panels, *p<0.05; **p<0.01.
To determine whether cell survival was affected in Rag1-cre;Mll1F/F developing B-cells, we sorted pooled populations from the bone marrow and observed a slight increase in the annexin V+ cell percentage in Mll1-deficient B220+CD43neg pre-B-cells (fractions D–F, Figure 4C). Given the difficulty detecting annexin V-binding cells in vivo due to their rapid clearance, as well as the imperfect correlation with cells committed to apoptosis (24, 25), we also isolated fraction B pro-B-cells (Lin neg/CD19+/CD43+/BP-1 neg/CD24+) and assessed their survival in vitro using a range of IL-7 concentrations (26). IL-7 is critical for the survival of pro-B-cells in vivo and in vitro and synergizes with signals from the pre-BCR to promote expansion of cells upon successful IgH rearrangement (27). Assessment of cell numbers after 7 days in culture revealed that Rag1-cre;Mll1F/F cells exhibited an IL-7 concentration-dependent reduction in cell accumulation (Figure 4D). Interestingly, the reduction in cell accumulation could be overcome by supra-physiologic levels of IL-7 (Figure 4D–E), similar to Rag2−/− pro-B-cells (28). Furthermore, low (0–1 ng/mL) IL-7 cultures consistently contained more propidium iodide (PI)-permeable cells, yet induced CD2 and CD22 similarly to wild-type cells (not shown). Collectively, these data suggest that the reduced B-cell output in Rag1-cre;Mll1F/F animals is the result of reduced B-cell survival particularly downstream of IL-7 and not reduced proliferation or differentiation.
MLL1 contributes to pre-BCR-mediated signaling
Given the defects in survival at or near the pre-BCR checkpoint (pro-pre-B transition), we explored further potential defects in pre-BCR signaling. The pre-BCR triggers a complex signaling network that both promotes survival, proliferation, and differentiation of pre-B-cells in synergy with IL-7 signaling. These signals converge at the level of ERK activation (28). We found that IL-7 receptor levels were not reduced on Mll1-deficient pro-B-cell populations, and also determined that STAT5 phosphorylation in response to either 0.5 or 50 ng/mL IL-7 was not affected by Mll1 deficiency (Figure S2A–E). Furthermore, AKT basal phosphorylation or response to pre-BCR stimulation did not differ between control Rag1-cre;Mll1F/+ and Rag1-cre;Mll1F/F pro-B-cells expanded in vitro (Figure S2F). In contrast, investigation of the RAS-MEK-ERK pathway revealed deficiencies in signaling upon pre-BCR stimulation. Although the levels of total ERK1/2 were similar to controls in cultured Rag1-cre;Mll1F/F animals, phospho-ERK1/2 (detected with anti-pT202/Y204) was consistently attenuated (Figure 5A). H-, K-, and N-RAS are all expressed in pro-B-cells, however, K-RAS plays a dominant role downstream of the pre-BCR (29). The level of K-RAS, phosphorylation status of MEK1/2, and B-RAF did not account for the attenuated phospho-ERK1/2 (Figure S2G–H). Furthermore, Mll1-deficient pro-B-cells exhibited enhanced sensitivity to the MAP kinase inhibitor U0126, demonstrating that the impaired survival can be further inhibited (Figure 5B). These data suggest that the defect resulting in poor survival in physiologic IL-7 and reduced B-cell output in Rag1-cre;Mll1F/F animals originates from a signaling inefficiency, but not block, downstream of the pre-BCR culminating in insufficient phosphorylation of ERK1/2.
Figure 5.
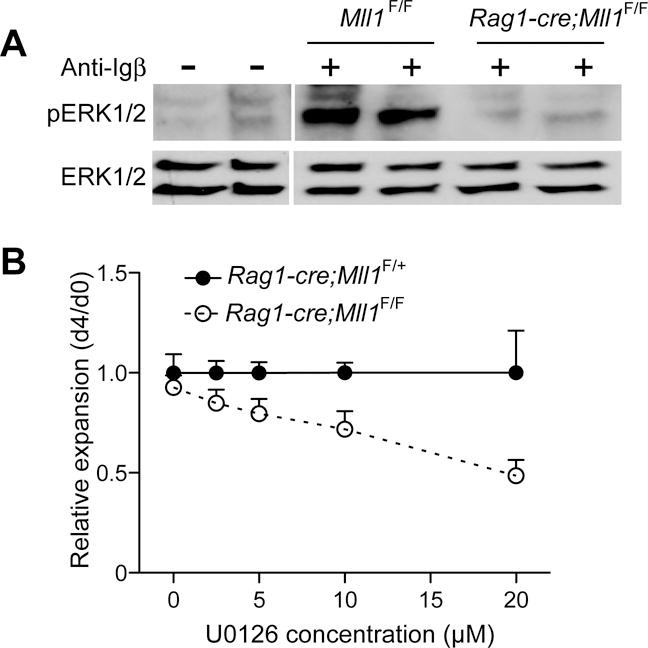
ERK phosphorylation downstream of the pre-BCR is attenuated in Mll1−/− cells. A) CD19+ B-cells were magnetically enriched from Rag1-cre;Mll1F/+ and Rag1-cre;Mll1F/F animals and expanded for 5 days with 10 ng/ml IL-7. After withdrawal of IL-7 for 4–6 hours, cells were stimulated with anti-Igβ and the level of total ERK1 and 2 (bottom panel) and phospho-ERK 1 and 2 (T202/Y204) determined by Western blotting. Data from two individual animals of the indicated genotypes are representative of four independent experiments. B) Fraction B pro-B-cells were sorted and cultured with 10 ng/mL of IL-7 and concentrations of the MEK kinase inhibitor U0126 as indicated. Relative expansion was measured at day 4 using bead normalization as indicated in the Methods. Six 2–3 week old animals per genotype were used for the analysis. Error bars represent SEM.
Mll1-deficiency limits B-ALL induced by BCR-ABL
MLL1 translocations produce proteins with neomorphic functions but fusion oncoproteins also lack normal MLL1 functions (e.g. the histone methyltransferase activity at the C-terminus). Thus it is possible that both gain- and loss-of-function activity of total MLL1 in the developing B-cell occurs upon MLL1 translocation. The selective disruption in B-cell differentiation observed in the loss-of-function model described here prompted us to examine whether the differentiation block could contribute to leukemogenesis, similar to the mechanisms by which reduction in PAX5, EBF, or IKAROS can contribute to leukemogenesis in human B-ALL (30). On the other hand, if the signaling deficiencies cannot be overcome by a particular oncogenic signal, MLL1 loss may limit leukemogenesis. To determine the impact of MLL1 loss in a well-characterized B-ALL model system, we transduced Rag1-cre;Mll1F/F and Rag1-cre;Mll1F/+ pro-B-cells with BCR-ABL (31) and transplanted cells into irradiated syngeneic recipients (Figure 6A) or expanded these cells in IL-7 cultures (Figure 6B). As shown in Figure 6A, recipients of BCR-ABL/Mll1−/− cells exhibited extended survival relative to their Mll1+/− controls (p=0.0152), demonstrating that MLL1 facilitates B-ALL driven by BCR-ABL.
Figure 6.
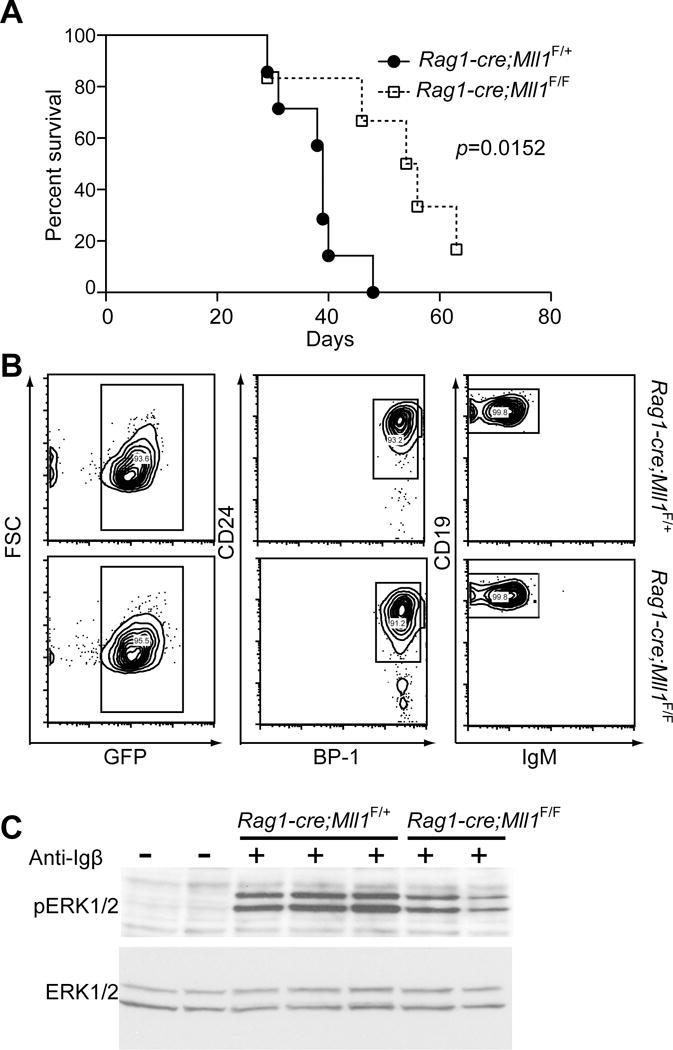
BCR-ABL-driven B-ALL is limited by Mll1 deficiency. A) Fraction B pro-B-cells from the indicated genotypes were sorted and transduced with p210 BCR-ABL and injected into sublethally irradiated C57Bl/6 recipients. Animals were sacrificed when moribund; B cell identity was confirmed by flow cytometry; n=5–6 animals per genotype and the p-value was calculated using the Mantel-Cox test. B) Mll1-deficient B-ALL is phenotypically similar to control B-ALL. Representative FACS plots are shown illustrating that control Rag1-cre;Mll1F/+ or Rag1-cre;Mll1F/F B-ALL from the bone marrow of moribund recipients in (A) exhibits a CD19+/CD24+/BP-1+/IgM- immunophenotype. Cells were previously gated as CD45.1+ (donor origin) and PI-negative. C) B-ALL cells from moribund mice were expanded in vitro, serum and IL-7 starved for 4 hours then stimulated with anti-Igβ as described in the Methods for 3 minutes. Phospho-ERK 1 and 2 (T202/Y204) and total ERK were detected by Western blotting; n=2–3 individual recipient B-ALL populations.
To examine the status of pre-BCR signaling in the BCR-ABL transformed cells, we utilized in vitro expanded cultures of transformed cells. Surface marker expression demonstrated that both Mll1-deficient and control Mll1+/− cells were immortalized at the fraction C/C’ stage, and no phenotypic difference was observed between the two genotypes (Figure 6B and data not shown). Upon anti-Igβ crosslinking, the level of pERK1/2 in the BCR-ABL transformed cells was significantly more detectable than that observed in primary pro-B cells. However, we still observed attenuated phosphorylation of ERK1/2 in Mll1-deficient BCR-ABL cells relative to controls (Figure 6C), suggesting that the delayed latency of B-ALL obtained with Mll1-deficient B-ALL cells is due to a signaling defect at the level of ERK phosphorylation that cannot be overcome by BCR-ABL.
Perturbations in gene expression in Mll1-deficient pro-B-cells
To gain insight into the phenotypic defects in Mll1−/− pro-B-cells, we performed genome-wide transcriptome and microRNA (miR) surveys using sorted fraction B pro-B-cells. These analyses revealed 224 genes to be down-regulated and 129 genes up-regulated in Mll1−/− pro-B-cells using a fold-change threshold of 1.2 and p-value cutoff of 0.005 (Figure 7A–B, Table S1). Several well-described MLL1 direct target genes were among the most strongly down-regulated genes (Hoxa9, Eya1, Meis1, Pbx1, red in Figure 7A), but surprisingly, many well-studied B-cell regulators, including Ebf1, Pax5, Ikzf1, Spi1, Blk1, and Irf4 were unchanged (Table S1 and data not shown). Several hypotheses were tested based on these findings.
Figure 7.
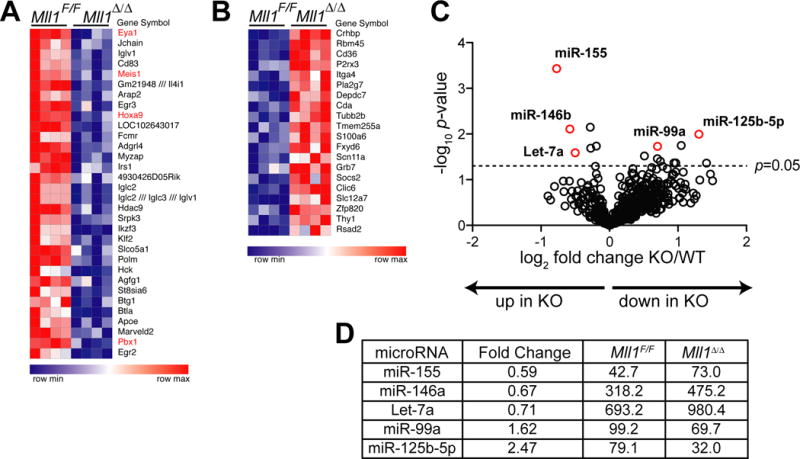
Perturbations in gene expression and miR levels in Mll1-deficient pro-B-cells. A) Heat map showing top 15% of down-regulated genes in Mll1-deficient (Mll1Δ/Δ) fraction B pro-B-cells. Differentially expressed genes were filtered using a p-value cutoff of 0.005. Genes shown in red are direct targets. B) The top 15% up-regulated genes in Mll1-deficient cells, analyzed as in panel A. Complete data are found in Table S1. C) Results of a mature miR screen using the Nanostring nCounter Mouse microRNA expression platform. Small RNAs were purified from sorted fraction B cells of control (Rag1-cre;Mll1F/+) and Rag1-cre;Mll1F/F animals (n=3 animals per genotype). Volcano plot of log2-transformed expression ratios (control/knockout) plotted against the p-value (-log10 transformed). Labeled circles highlight the miRs showing at least a 1.4-fold difference in expression with p<0.05. D) Summary list of significant up-regulated (miR-155, miR-146a, Let7a) and down-regulated (miR-99a, miR-125b-5p) microRNAs. Values reflect average normalized digital counts for average of 3 samples per genotype.
First, Hoxa9 germline knockout animals exhibit reduced pro-B populations and pre-B-cell colony frequency, particularly in conjunction with Meis1 deficiency (32, 33), whereas Eya1 was consistently and significantly down-regulated, but is not known to play a role at the pro/pre-B-cell transition. Thus we attempted to rescue pre-B-cell colony frequency by re-introducing Hoxa9 or Eya1 expression to Rag1-cre;Mll1F/F progenitors, but neither of these individual genes restored pre-B-cell colony numbers. In fact, Hoxa9 re-expression reduced pre-B colonies and tended to promote myeloid differentiation (Figure S3A and data not shown). Second, supressor of cytokine signaling 2 (Socs2) is significantly increased in Rag1-cre;Mll1F/F pro-B-cells (Figure 7B), and given its potential in negatively regulating IL-7 signaling (34), we tested whether overexpression of SOCS2 similar to the level we observe in Mll1−/− pro-B-cells could recapitulate the IL-7-dependent survival defect. We found that neither high nor low (comparable to Mll1−/− pro-B) levels of SOCS2 overexpression had any impact on IL-7 dependent survival (Figure S3B–D), suggesting that this single gene expression alteration is not sufficient to recapitulate the Mll1−/− phenotype (or to reduce B-cell viability) in low IL-7. Thus, it is likely that the high Socs2 expression occurred as a result of attenuated RAS-MEK-ERK signaling and was not causative.
Analysis of mature miRs revealed that two miRs were significantly decreased and three significantly increased in Mll1-deficient pro-B-cells relative to their wild-type counterparts, leading us to test two major hypotheses (Figure 7C–D, Table S2). First, the increase in Let7a and the fact that LET7 family members can reduce K-RAS levels and activity (35, 36) prompted us to determine K-RAS protein levels. We found that K-RAS total protein was unaffected in Mll1−/− pro-B-cells (Figure S2H). Second, both miR-155 and miR-146b are part of a feedback loop regulating NFκB activity (37, 38). Since NFκB activity can coordinate differentiation and survival downstream of the pre-BCR (39), we determined whether Mll1-deficient pro-B-cells exhibit altered NFκB activation downstream of the pre-BCR. As shown in Figure S3E–F, the turnover of IκBα and the nuclear translocation of p65/RelA (both are responses to NFκB activation) were indistinguishable between control and Mll1-deficient pro-B-cells. Thus, despite analysis of the earliest population to achieve 100% deletion of Mll1, it is likely that some of the gene expression changes observed were indirect or compensatory. Our data are consistent with the hypothesis that altered expression of multiple transcripts and miRs collectively impact the effectiveness of pre-BCR signaling. Overall, our data demonstrate that the role of wild-type MLL1 in B-cell differentiation is to appropriately tune pre-BCR signals by impacting the RAS-MEK-ERK axis and thereby survival of pro-B-cells in physiologic IL-7 during homeostasis.
Discussion
In this study, we show that MLL1 plays a significant role in B-cell differentiation, specifically at the pre-BCR checkpoint. Surprisingly, this role appears not to involve classic transcriptional regulators of B-cell differentiation that also act as tumor supressors in B-ALL but rather involves coordinating the response to pre-BCR signaling. The attenuated pre-BCR signaling observed in Mll1-deficient pro/pre-B-cells results in reduced cell survival and inefficient, but not completely blocked, B-cell differentiation that apparently cannot be compensated at later stages of differentiation to restore peripheral B-cell numbers. The exact molecular mechanism linking MLL1 to MEK/ERK signaling at this stage of differentiation appears to involve a complex network of mRNA and miR’s, as no single candidate deregulated gene could account for the selective phenotype observed upon MLL1 loss. Interestingly, high IL-7, but not BCR-ABL-mediated transformation, could overcome the survival and phospho-ERK defects imparted by Mll1-deficiency.
Both the IL-7 and pre-B-cell receptors are critical for B-cell differentiation and can be subverted in B-cell malignancy (40). The requirement for IL-7 during B-cell differentiation is highly dynamic, tuning survival and differentiation at the pro- to pre-B transition (41, 42). When a high level (ng/mL) of IL-7 is present in the environment, JAK-STAT5 and PI3K-AKT pathways activated solely by IL-7 are sufficient to sustain the expansion of pre-B-cells. Developing B-cells in a niche with limited IL-7 availability (pg/mL) exhibit attenuated STAT5 and AKT, thus requiring pre-BCR signaling to augment PI3K and RAS-ERK signaling to enable the survival of cells harboring functional pre-BCRs (27, 43). Using sorted fraction B pro-B-cells, we found that MLL1 is specifically required for cells to respond to low IL-7, similar to observations using pre-BCR-deficient pro-B-cells (43). Given the fact that Mll1-deficient cells still responded to high levels of IL-7, we speculate that these cells do not have an absolute defect associated with IL-7 signaling, rather exhibit inefficient signaling. Complete ERK1/2 double knockout in developing B-cells results in severe proliferation and survival defects (44), whereas Mll1−/− pre-B-cells exhibit survival, but not proliferation defects, suggesting that an effect on proliferation requires a more complete reduction in total phosphorylated ERK. In addition, one unique feature of the RAS-MEK-ERK pathway in B-cell differentiation is that effects on survival are wired differently depending on differentiation stage. For example, the survival of mature B-cells during mitogenic stimulation requires MEK/ERK-mediated phosphorylation of Bim (45), whereas inhibition of RAS activity significantly reduced the survival of pre-B-cells partially due to down-regulation of Bcl-xL (46). In the current study, neither of these transcripts were consistently and significantly reduced in Mll1−/− pro-/pre-B-cells (data not shown). Therefore it remains to be determined through which pathways cell death is deregulated. Nonetheless, the control of pre-BCR-dependent RAS-MAPK signaling by the proto-oncogene, MLL1, is intriguing given the B-cell propensity of infant ALL harboring MLL1 translocations as well as the pressure to acquire gain-of-function RAS mutations (4) in this disease.
Supplementary Material
Acknowledgments
We thank lab members, Michael Farrar, Jing Zhang, and Demin Wang for commenting on the manuscript. Drs. Malay Mandal, Marcus Clark, Shaoguang Li, and Glen Raffel provided critical reagents and advice. Reagents and mice donated by Professors Klaus Rajewsky and Terence Rabbitts were important for this work.
Funding sources: ACS RSG-10-242-LIB, NIH HL009036 and AI129426
Footnotes
Genomic data has been deposited in the Gene Expression Omnibus database under the following accession numbers: GSE108093 (https://www.ncbi.nlm.nih.gov/geo/)
References
- 1.Jansen MW, Corral L, van der Velden VH, Panzer-Grumayer R, Schrappe M, Schrauder A, Marschalek R, Meyer C, den Boer ML, Hop WJ, Valsecchi MG, Basso G, Biondi A, Pieters R, van Dongen JJ. Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia. 2007;21:633–641. doi: 10.1038/sj.leu.2404578. [DOI] [PubMed] [Google Scholar]
- 2.Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, Nakitandwe J, Holmfeldt L, Parker M, Easton J, Huether R, Kriwacki R, Rusch M, Wu G, Li Y, Mulder H, Raimondi S, Pounds S, Kang G, Shi L, Becksfort J, Gupta P, Payne-Turner D, Vadodaria B, Boggs K, Yergeau D, Manne J, Song G, Edmonson M, Nagahawatte P, Wei L, Cheng C, Pei D, Sutton R, Venn NC, Chetcuti A, Rush A, Catchpoole D, Heldrup J, Fioretos T, Lu C, Ding L, Pui CH, Shurtleff S, Mullighan CG, Mardis ER, Wilson RK, Gruber TA, Zhang J, Downing JR, P. St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330–337. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369:1473. doi: 10.1056/NEJMc1308782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Driessen EM, van Roon EH, Spijkers-Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013;98:937–944. doi: 10.3324/haematol.2012.067983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trentin L, Bresolin S, Giarin E, Bardini M, Serafin V, Accordi B, Fais F, Tenca C, De Lorenzo P, Valsecchi MG, Cazzaniga G, Kronnie GT, Basso G. Deciphering KRAS and NRAS mutated clone dynamics in MLL-AF4 paediatric leukaemia by ultra deep sequencing analysis. Sci Rep. 2016;6:34449. doi: 10.1038/srep34449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 8.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, Friedman N, Amit I. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, Ernst P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr Opin Hematol. 2017;24:322–328. doi: 10.1097/MOH.0000000000000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, Cho YW, Sun HW, Ge K, Peng W, Nussenzweig MC, Casellas R, Dressler GR, Zhao K, Nussenzweig A. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and Independent Roles for MLL in Adult Hematopoietic Stem Cells and Progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gan T, Jude CD, Zaffuto K, Ernst P. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24:1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Artinger EL, Ernst P. Cell context in the control of self-renewal and proliferation regulated by MLL1. Cell Cycle. 2013;12:2969–2972. doi: 10.4161/cc.26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCormack MP, Forster A, Drynan L, Pannell R, Rabbitts TH. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Mol Cell Biol. 2003;23:9003–9013. doi: 10.1128/MCB.23.24.9003-9013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122:2039–2046. doi: 10.1182/blood-2013-03-486647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Artinger EL, Mishra BP, Zaffuto KM, Li BE, Chung EK, Moore AW, Chen Y, Cheng C, Ernst P. An MLL-dependent network sustains hematopoiesis. Proc Natl Acad Sci U S A. 2013;110:12000–12005. doi: 10.1073/pnas.1301278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlissel MS, Corcoran LM, Baltimore D. Virus-transformed pre-B cells show ordered activation but not inactivation of immunoglobulin gene rearrangement and transcription. J Exp Med. 1991;173:711–720. doi: 10.1084/jem.173.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas MD, Kremer CS, Ravichandran KS, Rajewsky K, Bender TP. c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity. 2005;23:275–286. doi: 10.1016/j.immuni.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Opstelten D, Osmond DG. Pre-B cells in mouse bone marrow: immunofluorescence stathmokinetic studies of the proliferation of cytoplasmic mu-chain-bearing cells in normal mice. J Immunol. 1983;131:2635–2640. [PubMed] [Google Scholar]
- 20.Opstelten D, Osmond DG. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimazaki N, Lieber MR. Histone methylation and V(D)J recombination. Int J Hematol. 2014;100:230–237. doi: 10.1007/s12185-014-1637-4. [DOI] [PubMed] [Google Scholar]
- 22.Mandal M, Powers SE, Ochiai K, Georgopoulos K, Kee BL, Singh H, Clark MR. Ras orchestrates exit from the cell cycle and light-chain recombination during early B cell development. Nat Immunol. 2009;10:1110–1117. doi: 10.1038/ni.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pewzner-Jung Y, Friedmann D, Sonoda E, Jung S, Rajewsky K, Eilat D. B cell deletion, anergy, and receptor editing in "knock in" mice targeted with a germline-encoded or somatically mutated anti-DNA heavy chain. J Immunol. 1998;161:4634–4645. [PubMed] [Google Scholar]
- 24.Dillon SR, Constantinescu A, Schlissel MS. Annexin V binds to positively selected B cells. J Immunol. 2001;166:58–71. doi: 10.4049/jimmunol.166.1.58. [DOI] [PubMed] [Google Scholar]
- 25.Dillon SR, Mancini M, Rosen A, Schlissel MS. Annexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activation. J Immunol. 2000;164:1322–1332. doi: 10.4049/jimmunol.164.3.1322. [DOI] [PubMed] [Google Scholar]
- 26.Lu L, Osmond DG. Apoptosis and its modulation during B lymphopoiesis in mouse bone marrow. Immunol Rev. 2000;175:158–174. [PubMed] [Google Scholar]
- 27.Fleming HE, Paige CJ. Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity. 2001;15:521–531. doi: 10.1016/s1074-7613(01)00216-3. [DOI] [PubMed] [Google Scholar]
- 28.Milne CD, Paige CJ. IL-7: a key regulator of B lymphopoiesis. Semin Immunol. 2006;18:20–30. doi: 10.1016/j.smim.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Zheng Y, You X, Yu M, Fu G, Su X, Zhou F, Zhu W, Wu Z, Zhang J, Wen R, Wang D. Kras Is Critical for B Cell Lymphopoiesis. J Immunol. 2016;196:1678–1685. doi: 10.4049/jimmunol.1502112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med. 2015;373:1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 32.Hu YL, Fong S, Ferrell C, Largman C, Shen WF. HOXA9 modulates its oncogenic partner Meis1 to influence normal hematopoiesis. Mol Cell Biol. 2009;29:5181–5192. doi: 10.1128/MCB.00545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.So CW, Karsunky H, Wong P, Weissman IL, Cleary ML. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood. 2004;103:3192–3199. doi: 10.1182/blood-2003-10-3722. [DOI] [PubMed] [Google Scholar]
- 34.Li LX, Goetz CA, Katerndahl CD, Sakaguchi N, Farrar MA. A Flt3- and Ras-dependent pathway primes B cell development by inducing a state of IL-7 responsiveness. J Immunol. 2010;184:1728–1736. doi: 10.4049/jimmunol.0903023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 2011;3:159–166. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaileh M, Sen R. NF-kappaB function in B lymphocytes. Immunol Rev. 2012;246:254–271. doi: 10.1111/j.1600-065X.2012.01106.x. [DOI] [PubMed] [Google Scholar]
- 40.Buchner M, Swaminathan S, Chen Z, Muschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev. 2015;263:192–209. doi: 10.1111/imr.12235. [DOI] [PubMed] [Google Scholar]
- 41.Jacobsen K, Osmond DG. Microenvironmental organization and stromal cell associations of B lymphocyte precursor cells in mouse bone marrow. Eur J Immunol. 1990;20:2395–2404. doi: 10.1002/eji.1830201106. [DOI] [PubMed] [Google Scholar]
- 42.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marshall AJ, Fleming HE, Wu GE, Paige CJ. Modulation of the IL-7 dose-response threshold during pro-B cell differentiation is dependent on pre-B cell receptor expression. J Immunol. 1998;161:6038–6045. [PubMed] [Google Scholar]
- 44.Yasuda T, Sanjo H, Pages G, Kawano Y, Karasuyama H, Pouyssegur J, Ogata M, Kurosaki T. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 45.O’Reilly LA, Kruse EA, Puthalakath H, Kelly PN, Kaufmann T. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J Immunol. 2009;183:261–269. doi: 10.4049/jimmunol.0803853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagaoka H, Takahashi Y, Hayashi R, Nakamura T, Ishii K, Matsuda J, Ogura A, Shirakata Y, Karasuyama H, Sudo T, Nishikawa S, Tsubata T, Mizuochi T, Asano T, Sakano H, Takemori T. Ras mediates effector pathways responsible for pre-B cell survival, which is essential for the developmental progression to the late pre-B cell stage. J Exp Med. 2000;192:171–182. doi: 10.1084/jem.192.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.