

Abstract
In this study, we investigated the applicability of using in vivo mouse micronucleus (MN) data to derive cancer potency information. We also present a new statistical methodology for correlating estimated potencies between in vivo MN tests and cancer studies, which could similarly be used for other systems (e.g. in vitro vs. in vivo genotoxicity tests). The dose–response modelling program PROAST was used to calculate benchmark doses (BMDs) for estimating the genotoxic and carcinogenic potency for 48 compounds in mice; most of the data were retrieved from the National Toxicology Program (NTP) database, while some additional data were retrieved from the Carcinogenic Potency Database and published studies. BMD05s (doses with 5% increase in MN frequency) were derived from MN data, and BMD10s (doses with 10% extra cancer risk) were derived from carcinogenicity data, along with their respective lower (BMDL) and upper (BMDU) confidence bounds. A clear correlation between the in vivo MN BMD05s and the cancer BMD10s was observed when the lowest BMD05 from the in vivo MN was plotted against the lowest BMD10 from the carcinogenicity data for each individual compound. By making a further selection of BMDs related to more or less equally severe cancer lesions, the correlation was considerably improved. Getting a general scientific consensus on how we can quantitatively compare different tumour lesion types and investigating the impact of MN study duration are needed to refine this correlation analysis. Nevertheless, our results suggest that a BMD derived from genotoxicity data might provide a prediction of the tumour potency (BMD10) with an uncertainty range spanning roughly a factor of 100.
Introduction
The 2-year rodent cancer bioassay plays an important role in cancer risk assessment for the evaluation of the carcinogenic potential of a chemical and the derivation of a point of departure (PoD) (1). Shortcomings of the 2-year cancer bioassay include the large number of animals utilised, the long time required to obtain the results and the high cost (~1 to several million euros depending on route of exposure) (2). Although worldwide efforts are being made to reduce the number of animals used in toxicity testing without compromising regulatory needs, few efforts are being made to investigate means of estimating the carcinogenic potential of substances in the absence of a full carcinogenicity study. One possible approach is the use of in vivo genotoxicity tests, given that the accumulation of DNA lesions plays a vital role in carcinogenesis. Our approach is novel in that we have analyzed genotoxicity data in a quantitative dose–response manner, in comparison to the traditional qualitative (yes/no) manner. Efforts by the International Workgroup on Genetic Toxicology (IWGT) quantitative workgroup (QWG) and the International Life Science Institute Health (ILSI) and Environmental Science Institute (HESI) Genetic Toxicology Technical Committee (GTTC) have evaluated the derivation of PoD estimates from genotoxicity studies using dose–response modelling and their potential application in cancer risk assessment (3–6). Here, we provide a proof-of-principle exercise for the potential of using PoDs from genotoxicity test data (in vivo micronucleus (MN) data) to derive estimates of carcinogenic potency.
To the best of our knowledge, two studies have explored the relationship between dose–response data from genotoxicity tests and carcinogenicity studies (7,8). One study compared the lowest effective dose (LED) for in vivo genotoxicity (MN, sister chromatid exchange, DNA adducts, chromosomal aberrations and comet assay) to the T25 (a dose measure assumed to relate to a 25% increase in tumour incidence) in mice and rats. Positive correlations were found both in mice and rats, and both for oral and inhalation exposure (7). A drawback of this study was that the LED and T25 are both highly imprecise estimates of equipotent doses. The LED is the lowest effective dose and the T25 is an estimate of the dose with 25% additional cancer risk obtained by linear inter- or extrapolation based on the observed responses at dose zero and the lowest significant dose; both potency estimates are generally not very accurate, and highly sensitive to experimental design differences, while they do not provide for a measure of uncertainty (9). Hernández et al. (8) performed a similar study but estimated equipotent doses using the benchmark dose (BMD) approach, which not only results in more precise estimates of equipotent doses, but also allows for evaluating the uncertainty associated with each potency estimate (in the form of a BMD confidence interval). The above-mentioned study (8) showed a positive correlation between genotoxic potency and tissue-matched carcinogenic potency (N = 18 compounds). This study aims to validate our initial findings (8) by using a larger sample of chemicals. In this study we focused on dose–response data from the in vivo MN study as a basis for estimating the genotoxic potency because the MN is the most common genotoxicity test performed.
In vivo genotoxicity tests are relevant candidates for exploring if a correlation between genotoxic and carcinogenic potency can be established. As opposed to in vitro tests, they account for metabolic disposition of a xenobiotic, including absorption, tissue distribution, metabolism and excretion; all these factors play a role in determining the carcinogenic potency of chemicals in vivo. The in vivo erythrocyte MN test measures the ability of a test compound to induce chromosomal damage in progenitor red blood cells in bone marrow (10). Increased incidences of micronucleated polychromatic erythrocytes (reticulocytes) in bone marrow (10), and micronucleated reticulocytes and micronucleated mature erythrocytes in peripheral blood (11) of chemical-exposed animals are indicators of chromosomal damage in the form of chromosomal breakage or chromosome loss at anaphase.
The majority of the studies used to derive carcinogenic potencies were obtained from the National Toxicology Program (NTP) database, and a few were retrieved from the Carcinogenic Potency Database (Table 1). Therefore, not all carcinogenicity data used in this analysis came from 2-year bioassays conducted according to OECD guidelines. Nonetheless, we included these studies and considered any protocol deviations that might influence the outcome or interpretation of our data analysis.
Table 1.
List of compounds
| Source | Cas # | IARC | Compound | AB | Cancer | In vivo MN |
|---|---|---|---|---|---|---|
| CPD | 53-96-3 | – | 2-acetylamino-fluorene | aaf | + | +a |
| TR-042 | 320-67-2 | 2A | 5-Azacytidine | acd | + | + |
| TR-447 | 08/05/1975 | – | Acetonitrile | ace | e | + |
| TR-376 | 106-92-3 | – | Allyl glycidyl ether | age | e | + |
| TR-543 | 98-83-9 | – | alpha-Methylstyrene | ams | + | − |
| CPD | 446-86-6 | 1 | Azathioprine | aza | + | +b |
| TR-469 | 30516-87-1 | 2B | 3′-Azido-3′-deoxythymidine | azt | + | + |
| CPD | 50-32-8 | 1 | Benzo(a)pyrene | bap | + | +3c,d |
| TR-289 | 71-43-2 | 1 | Benzene | ben | + | + |
| TR-288 | 106-99-0 | 1 | 1,3-Butadiene | but | + | + |
| CPD | 305-03-3 | 1 | Chlorambucil | cbc | + | ee |
| TR-502 | 302-17-0 | 3 | Chloral hydrate | chl | e | + |
| TR-479 | 68603-42-9 | 2B | Coconut oil acid diethanolamine condensate | coc | + | + |
| TR-063 | 95-83-0 | 2B | 4-Chloro-o-phenylene-diamine | cop | + | + |
| CPD | 50-18-0 | 1 | Cyclophosphamide | cpa | + | +f,g |
| TR-501 | 09/07/1980 | – | p,p′-Dichloro-diphenyl sulfone | cps | − | + |
| TR-028 | 08/12/1996 | 2B | 1,2-Dibromo-3-chloropropane | dbcp | + | ee |
| TR-206 | 08/12/1996 | 1,2-Dibromo-3-chloropropane | dbcp | + | ee | |
| TR-086 | 106-93-4 | 2A | 1,2-Dibromoethane | dbe | + | + |
| TR-513 | 91-17-8 | – | Decalin | dcn | + | + |
| TR-287 | 868-85-9 | 3 | Dimethyl hydrogen phosphite | dhp | + | + |
| CPD | 306-37-6 | 2A | 1,2-Dimethyl-hydrazine | dmh | + | eh |
| TR-316 | 513-37-1 | 2B | Dimethylvinyl chloride | dmvc | + | + |
| TR-493 | 518-82-1 | – | Emodin | emo | e | − |
| TR-237 | 630-20-6 | 3 | 1,1,1,2-Tetrachloro-ethane | eth | + | + |
| TR-000 (67-66-3) | 67-66-3 | 2B | Chloroform | for | + | + |
| TR-374 | 556-52-5 | 2A | Glycidol | gly | + | + |
| TR-330 | 136-77-6 | – | 4-Hexylresorcinol | hrc | e | + |
| TR-366 | 123-31-9 | 3 | Hydroquinone | hyd | e | + |
| TR-551 | 97-54-1 | – | Isoeugenol | ieg | + | − |
| TR-448 | 542-56-3 | – | Isobutyl nitrite | isn | + | + |
| TR-486 | 78-79-5 | 2B | Isoprene | iso | e | + |
| TR-247 | 50-81-7 | – | l-Ascorbic acid | las | − | + |
| TR-527 | 129-73-7 | – | Leucomalachite green | leu | e | + |
| TR-385 | 74-83-9 | 3 | Methyl bromide | mbr | − | + |
| CPD | 148-82-3 | 1 | Melphalan | mel | + | ee |
| CPD | 684-93-5 | 2A | N-Nitroso-N-methylurea | mnu | + | + |
| TR-266 | 150-68-5 | 3 | Monuron | mon | + | + |
| CPD | 62-75-9 | 2A | N-Nitrosodimethyl-amine | nda | + | +i |
| TR-205 | 101-80-4 | 2B | 4,4′-Oxydianiline | oxy | + | + |
| TR-351 | 20265-96-7 | – | p-Chloraniline hydrochloride | pch | + | + |
| TR-515 | 57018-52-7 | 3 | Propylene glycol mono-t-butyl ether | pge | + | − |
| TR-203 | 108-95-2 | 3 | Phenol | phe | − | + |
| TR-465 | 08/09/1977 | 2B | Phenolphthalein | php | + | + |
| TR-403 | 108-46-3 | 3 | Resorcinol | rsc | − | + |
| TR-546 | 7789-12-0 | – | Sodium dichromate dihydrate (VI) | scd | + | + |
| TR-194 | 7446-34-6 | – | Selenium sulfide | sel | + | − |
| TR-200 | 15481-70-6 | – | 2,6-Toluene-diamine dihydrochloride | tac | − | + |
| TR-002 | 06/01/1979 | 2A | Trichloroethylene | tce | + | − |
| TR-473 | 58-55-9 | 3 | Theophylline | teo | − | + |
| TR-027 | 79-34-5 | 3 | 1,1,2,2-Tetrachloro-ethane | tet | + | + |
| TR-097 | 13463-67-7 | 3 | Titanium dioxide | tio | − | + |
| TR-510 | 51-79-6 | 2A | Urethane | ure | + | + |
AB, abbreviation; CPD, carcinogenic potency database (http://potency.berkeley.edu/); F, female; IARC, International Agency for Research on Cancer; M, male; MN, hematopoietic micronucleus test; NT, not tested; TR, National Toxicology Program technical report; +, positive; −, negative; e, equivocal.
a(12); b(13); c(14); d(15); e(16); f(17); g(18); h(19); i(20).
The main purpose of this study was to determine if the positive correlation between genotoxic and carcinogenic potency obtained in our previous analysis (8) could be confirmed using a larger database. The quantitative analysis of genotoxicity tests is an emerging field, and so far few groups have explored the quantitative relationship between shorter term genotoxicity tests and 2-year carcinogenicity studies. Therefore, the statistical methodology for performing this type of analysis continues to evolve. Indeed, the analyses presented here are not final, given the many challenges relating to the quantitative analysis of carcinogenicity studies. Therefore, refinement of the current methodology is in progress with the collaboration of international government agencies. Here, we present the results of our analyses using the current methodology.
Materials and methods
Data selection
A total of 222 technical reports published by the National Toxicology Program were surveyed to identify carcinogens that had both MN and carcinogenicity dose–response data; 44 compounds met this criterion (http://ntp.niehs.nih.gov/results/pubs/longterm/index.html). For another 10 compounds, dose–response carcinogenicity data were obtained from the Carcinogenic Potency Database (CPD, http://potency.berkeley.edu/) and missing in vivo MN data was extracted from published reports in the scientific literature in order to expand our database (12–15,19,21–23). Overall, 44 compounds were from NTP data and 10 compounds had a combination of CPD and MN data from published reports in the scientific literature (Table 1).
In vivo micronucleus test
In vivo MN data related to blood and/or bone marrow, examined in studies that employed a variety of protocols. Dose group sizes ranged from 5 to 15, the number of doses ranged from 3 to 8 (including controls), the exposure regimens varied from a single dose to repeated dosing up to182 days (26 weeks). Both sexes were used in some of the studies, but most studies only used males. While for some chemicals only a single dose–response dataset was available, other chemicals were tested more extensively by varying factors like sex, strain or exposure regimen, resulting in multiple dose–response datasets for the same compound. Thus, individual datasets were defined by the following factors: compound, sex, strain, route, tissue observed, exposure regimen, exposure duration and sampling time (24 or 48h) after the last dose treatment. BMD05s were estimated for each of these individual datasets, resulting in multiple BMD05s per compound.
Carcinogenicity tests
The carcinogenicity dose–response data varied considerably in group size (ranging between as few as 7 and as many as 999 (one dose group in the mega-mouse study with AAF) animals per group. The number of dose groups ranged from three to eight (including controls). Another important difference among studies is the type of lesions observed, varying from mild lesions to multiple malignant tumours in various tissues in an individual animal. Individual datasets were defined by the following factors: compound, sex, strain, route, exposure duration, tissue with lesion, type of lesion and lesion category (see below). Similar to the analysis of the in vivo MN, BMD10s were estimated for each individual dataset, resulting in multiple BMD10s per compound.
One of the complications of the dose–incidence data reported by carcinogenicity studies is that they may be based on different types of responses reflecting different stages of the carcinogenicity process, and hence with varying degrees of severity, such as non-neoplastic lesions, adenomas, carcinomas or tumour-bearing animals (tba). Clearly, a dose that causes a 10% increase in the fraction of animals with a pre-neoplastic lesion such as hyperplasia is not equipotent to a dose that causes a 10% increase in tbas or malignant carcinomas. To make a distinction between these differences, we used a pragmatic approach to assign ‘lesion category’ scores, roughly indicating the type of lesion in a given dose–response dataset. The lesions were designated as follows: (A) non-cancer effects (cytotoxicity) and compounds with no evidence for carcinogenicity; (B) non-malignant lesions (benign tumours like adenoma, papilloma, etc.); (C) malignant tumour at a single site (carcinoma, sarcoma, malignant lymphoma, blastoma); (D) mixed tumours (various tumour types in the same tissue); (E) Combination of tumours (various tumour types in different tissues); (F) Tumour-bearing animals. It should be noted that this definition of lesion categories is a pragmatic approach, which we intend to refine in the near future after consulting with relevant rodent pathology experts.
Dose–response analysis
Many of the dose–response datasets available did not contain much information on the shape of the dose–response (due to few doses with different responses) and, if analysed alone, would result in highly imprecise estimates of equipotent doses with large uncertainty in the BMDs. However, a recent re-analysis of a large number of toxicological datasets showed that the dose–responses for a given (continuous) endpoint from different chemicals tend to have similar shapes (24). A similar re-analysis of historical quantal data showed that cancer dose–response are often similar in shape even when different endpoints are analysed (Slob and Setzer, unpublished results). Note that in quantal endpoints, the maximum response (being the shape parameter that differed among continuous endpoints) is usually assumed to be 100%, leaving one single shape parameter reflecting the steepness of the curve (which did not differ greatly among continuous endpoints as well). In each analysis of a combined dataset, we visually inspected the fitted curves to the individual datasets, as a check of the assumption that the dose–response shapes were similar.
We used this result by fitting the model to combined clusters of datasets, where the common value of the shape parameter(s) is informed by all datasets in the cluster. This approach results in a considerable improvement of the precision of individual BMDs, i.e. smaller confidence intervals (CIs). One issue to be further examined is to what extent the estimated shape parameters might depend on the specific cluster. We intend to do a more comprehensive analysis of that in the future, in particular for the cancer data. Some results for MN data have already been reported (24). Roughly speaking, most clusters result in similar estimates of the shape parameters, with mild differences when comparing shorter and longer study durations in cancer data or when comparing lower cancer lesion categories with higher ones.
The continuous dose–response data from the MN tests were analysed by fitting the exponential model, which is one of the recommended models for continuous data (9), and known to be generally applicable to toxicity data:
where y is the response (proportion of cells with MN) and x the dose. In fitting the model to the combined cluster of datasets, separate values for parameters a (reflecting the response at dose 0) and b (reflecting the potency of the chemical) are estimated for each individual dose–response dataset, while parameters c and d are kept constant over all datasets within the cluster analyzed. The within-group variance was estimated separately for each individual dataset as well. See (25) for a more detailed discussion of this method.
For the quantal dose–response data from the carcinogenicity studies the log-logistic model was fitted.
where y is the response (fraction of affected animals) and x the dose. Again, parameters a (reflecting the response at dose zero) and b (reflecting the potency of the chemical) are estimated for each individual dose–response dataset, while (shape) parameter c is kept constant over all datasets within the analysed cluster.
As a more recent insight, model uncertainty should be taken into account in a BMD analysis by fitting various models to the data. The overall CI for the BMD is then obtained by integrating the results from the various models (for which various methods may be used (26)). In our approach of analysing clusters of combined datasets this is not needed, as the shape of the dose–response curve is estimated relatively precisely, by being based on all the dose–response data in the cluster of datasets (24). Therefore, we used only one model in this analysis.
Dose–response modelling was performed with the software package PROAST (www.proast.nl). This package allows for dose–response analysis with covariates. This makes it possible to perform combined analyses of clustered sets of similar datasets related to different chemicals by assuming that the potencies of the chemicals differ, but not the shape parameters of the dose–response model (see above). Visual inspection of the results indicated that this assumption was not violated in any clusters of datasets considered.
Deriving BMD confidence intervals
Equipotent doses are defined by BMDs at a given constant benchmark response (BMR) related to the same endpoint. For the carcinogenicity studies we used a BMR of 10% extra risk, which is the most commonly used value of the BMR in dose–response characterisation of quantal endpoints (26). For the continuous dose–response data from the in vivo MN test we used a 5% increase in the mean count in the controls as the BMR, being the recommended BMR for continuous response data by the European Food Safety Authority (26). In retrospect, a 5% increase in MN appears to be a small change relative to the maximum change that can be achieved in this parameter. The choice of the BMR is, however, not crucial in our analyses, as all BMDs for a higher BMR will be a constant factor higher, while the BMD CIs will only be slightly smaller (27). The reason of this relates to the fact that we applied dose–response modelling to combined datasets assuming the shape parameters to be constant among the constituent datasets, resulting in fairly precise estimates of the shape parameters. This is further substantiated in (27).
Rather than deriving BMDs as single values, we derived the (two-sided) 90% confidence intervals (CIs) to reflect the potency estimates. In this way, the potency of chemicals can be quantified even if they do not show a significant trend in the dose–response. For our purposes this is particularly important because this makes it possible to include chemicals not showing a significant dose–response trend. In those cases, the BMD CIs will have an infinite upper bound (BMDU), but a finite lower bound (BMDL). The information provided by a BMD CI, even in the case of an infinite upper bound, is very useful. To illustrate this, Table 2 shows some examples. In the first scenario, where the cancer BMDL10 is relatively high, but the MN BMDU05 is relatively low (Table 2, row A) it can be concluded that the genotoxic potency does not correlate with the cancer potency. Another scenario would be if both BMDLs are relatively high, while both BMDUs are infinite (Table 2, row B). Then, both potencies are low, and do correlate with each other. When the MN BMDU05 is finite, but the cancer BMDU10 infinite (Table 2, row C), the MN test would be considered as ‘positive’, but the cancer study as ‘negative’, and in the current approach this compound would be considered a false positive with respect to the MN outcome. However, in reality this is only a statistical artefact given that the CIs for MN and cancer both indicate a low potency, i.e. in the biological sense they are in agreement to each other. The example in row A (Table 2), in contrast, does provide evidence of a false positive in the biological sense, as the data show that the MN potency grossly overestimates the cancer potency.
Table 2.
Three hypothetical examples of BMD confidence intervals (mg/kg/day) for MN and Cancer, illustrating the usefulness of confidence intervals with infinite upper bounds
| Example | MN BMDL05 | MN BMDU05 | Cancer BMDL10 | Cancer BMDU10 | Correlation? Outcome? |
|---|---|---|---|---|---|
| A) | 0.01 | 0.05 | 500 | Infinite | No; |
| MN: +ve; Cancer: −ve | |||||
| MN: Biological false positive | |||||
| B) | 200 | Infinite | 500 | Infinite | Yes; |
| MN: −ve; Cancer: −ve | |||||
| C) | 200 | 800 | 500 | Infinite | Yes; |
| MN: +ve; Cancer: −ve | |||||
| MN: Statistical false positive |
An important factor to be considered in the context of classification of chemicals is that experimental data are never able to distinguish between zero potency and low potency. For that reason, the observation that a study is negative (no evidence for a genotoxic or carcinogenic response) is a highly imprecise observation because it implies that the chemical’s potency is somewhere between zero and low, without further quantification of ‘low’; where ‘low’ depends on the coincidental sensitivity of that particular study (and might not be that low). Therefore, the resulting ‘false positives’ or ‘false negatives’ can be merely the result of statistical imprecision (i.e., a statistical artefact). The approach of BMD CIs can filter those out, so that only the genuine (biological) false positives/negatives remain.
Correlation plots
Correlation plots of MN BMD05s against cancer BMD10s were created by plotting their CIs (in both the x- and y-direction) related to matching chemicals, including the ones that resulted in (one-sided) infinite CIs. Inside the correlation plots we plotted a dashed box, indicating the largest BMDU (and lowest BMDL) of all intervals assessed, so that it is directly visible which CIs have infinite bounds (i.e., when they cross outside the dashed box). We do not report correlation coefficients for characterizing the strength of the correlation. One reason is that we correlate CIs, some of which have infinite upper bounds, while correlation coefficients are defined for single values. Another reason is that a more useful way of characterizing the correlation is by a measure of the predictivity of the value on the x-axis (here: the MN BMD05’) for the value on the y-axis (here: the cancer BMD10). Therefore, we drew lines with unity slopes in the double-log plot that encompass most of the CIs (in both directions). A straight line with unity slope in a double log plot translates into a proportional relationship on the original scale. Given the fact that the present results are a first step (proof of principle), we purposely provide for this rough, visual evaluation, intended to illustrate the potential use of these correlations.
Steps of analysis
The overall analysis is summarised here. A large datasheet was created for the MN data with columns for dose (mg/kg/day), mean response, standard error of the mean (SEM), group size and a number of other relevant factors (chemical tested, sex, strain, route of exposure, tissue (blood or bone marrow), exposure duration, sampling time (24 or 48h) or vehicle used in controls). Another large datasheet was created for the tumour data with columns for dose (mg/kg/day), number of animals with response, group size and a number of relevant factors (chemical tested, sex, strain, route of exposure, exposure duration, duration of study, tissue, type of lesion, combination of lesion and tissue, severity of the response). In both datasheets, a column with dataset number was added, such that dose–response data differing in any of the associated factors were labelled with a unique number. As the total number of individual datasets was too large to be analysed as a single combined dataset, we composed clusters with a manageable number of clusters, with around 60 datasets as the maximum. Each cluster combined similar datasets like same route of administration, and/or same sex, etc. A dose–response model was fitted to each cluster of datasets, with individual dataset as a covariate for parameter a (estimated response in controls) and for parameter b (potency of the dose–response), and, in the case of MN data, for the within-group variance. In this way, the shape of the curves is estimated from all dose–response data in the cluster, since it is assumed to be constant among the chemicals. The confidence intervals (CIs) were calculated for the BMD related to each individual dataset. The sets of CIs resulting from each MN cluster were combined in one table. The same was done for the tumour clusters. The MN and tumour CIs were next combined in one single table, such that for matching chemicals each micronucleus BMD05 interval matched each tumour BMD10 interval, and vice versa. The factor levels (such as study duration, strain) related to both the MN and the tumour BMD intervals were maintained in additional columns. The CIs were finally plotted against each other, where the factor levels could be used for creating sub-selections of the data, or for labelling the data in the correlation plots (e.g. by compound).
Results
From the 52 compounds selected, 4 were omitted: TIO, EMO and DMH as they resulted in BMDU’s which were infinite both MN and cancer, and aza because it was tested in a genetically modified strain. Thus, 48 chemicals were left for further analysis.
All MN and carcinogenic BMDs related to the same chemicals were plotted against each other in Figure 1. Even though the scatter is large, it can be concluded that a positive correlation between MN potencies of compounds and their carcinogenic potencies exists. The variability in BMDs within the same compound (Figure 1) is particularly large in the Y-direction (carcinogenicity). The latter may be expected given the diversity in study conditions (e.g. exposure and study duration) and in type of lesions associated with the dose–response datasets for a given chemical.
Figure 1.

All MN BMD05s plotted against all carcinogenic BMD10s (n = 1452 pairs of BMDs, related to 48 compounds). Note the large variation in BMDs within chemicals, both for the MN and for the carcinogenic BMDs. The lower and left dashed lines of the inner block indicate the smallest lower confidence limit over all datasets; the right and upper dashed lines indicate the largest finite upper confidence limit over all datasets. Points outside these lines have BMD confidence intervals that are unbounded in that direction.
Normally, the lowest carcinogenic BMDL is selected as a PoD in risk assessment. Therefore, we selected for each compound the dataset resulting in the lowest BMD, both for the cancer and MN datasets, and plotted the associated BMD confidence intervals against each other, as shown in Figure 2A. Selecting the dataset with the lowest BMDL rather than the lowest BMD resulted in a very similar plot (data not shown). Some of the CIs in the right upper corner of Figure 2A have infinite upper bounds, but they are not observed to deviate from the overall correlation. Most BMDs in Figure 2A (Table 3) relate to lesion category E (combinations of tumours in different tissues; n = 21), followed by category C (single malignant lesions; n = 9).
Figure 2.
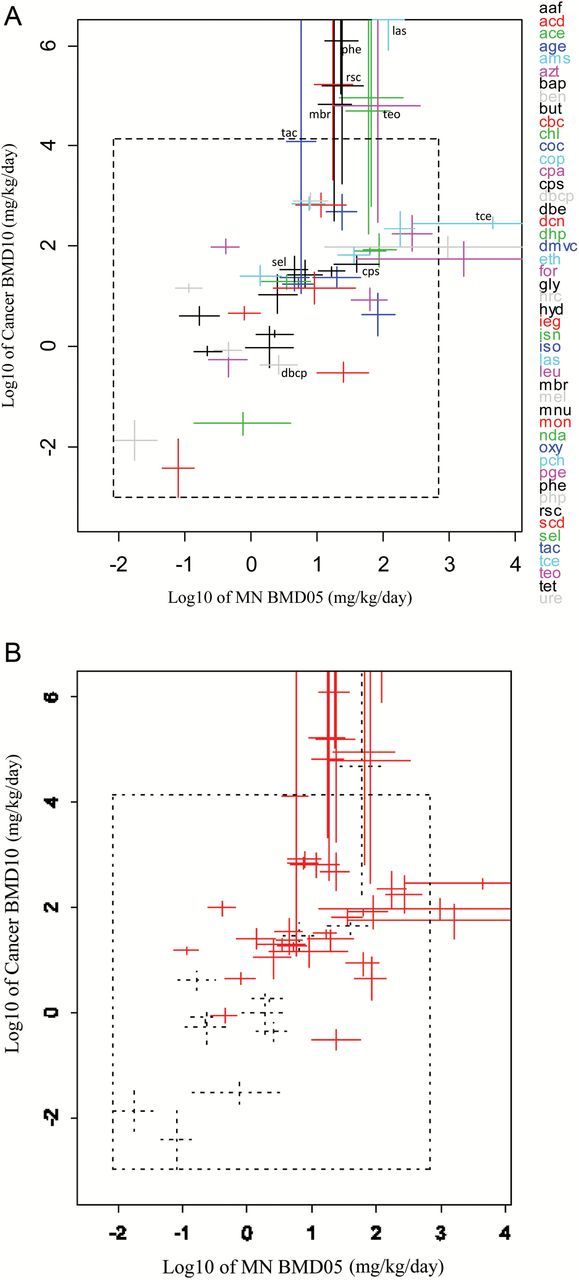
(A) The 90% CIs for the lowest carcinogenic BMD10s were plotted against the CIs for the lowest MN BMD05s for the 48 chemicals considered. The CIs that cross the inner dashed frame have infinite upper bounds in that direction. According to Table 1, CPS, LAS, MBR, PHE, RSC, TAC and TEO had positive results for MN and negative for carcinogenicity while DBCP, SEL and TCE were negative in the MN test and positive for carcinogenicity. With our methodology, all compounds do not deviate from the general correlation patterns. (B) Red BMD intervals relate to NTP studies and black (dashed) BMD intervals to studies obtained from the literature (CPD or PubMed). See Table 3 for the underlying data.
Table 3.
Summary of in vivo MN and carcinogenicity studies in mice presented Figure 2 (tba, tumour-bearing animals)
| MN (genotoxicity endpoint) | Carcinogenicity (tumour endpoint) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Strain | Sex | Route | Tissue | Duration exposure (days) | Vehicle control | Sampling time | Source | BMDL05 (mg/kg/day) | BMDU05 (mg/kg/day) | Source | Strain | Sex | Route | Exposure time (weeks) | Duration experiment (weeks) | Severity | Tissue | Tissue lesion | BMDL10 (mg/kg/day) | BMDU10 (mg/kg/day) |
| aaf | bdf1 | m | ip | Blood | 1 | na | 48 | Asano [12] | 1.2 | 4.4 | CPD | bcn | m | Feed | 96 | 104 | D | Bladder | Mixed | 1.6 | 2.0 |
| acd | B6 | m | ip | Blood | 1 | Corn oil | 48 | ntp A95392 | 10.1 | 60.5 | TR-042 | b6c | f | ip | 51 | 81 | A | Lymphoid | Lymphocytic and granulocytic neoplasm | 0.20 | 0.48 |
| ace | B6 | f | inh | Blood | 91 | Air | 24 | ntp A41659 | 21.7 | 201.0 | TR-447 | b6c | m | inh | 104 | 104 | A | NA | NA | 633.7 | NA |
| age | B6 | m | ip | bm | 3 | PBS | 24 | ntp 446759 | 2.8 | 7.6 | TR-376 | b6c | m | inh | 103 | 103 | B | Nasal cavity | Adenoma | 12.7 | 43.0 |
| ams | B6 | f | inh | Blood | 91 | Air | 24 | ntp A36497 | 104.3 | 302.6 | TR-543 | b6c | f | inh | 105 | 105 | E | Liver | Adenocarcinoma | 98.8 | 504.9 |
| azt | B6 | m | gav | Blood | 90 | na | 24 | ntp A88072 | 0.3 | 0.7 | TR-469 | b6c | f | gav | 105 | 105 | E | Vagina | Squamous carcinoma papilloma | 69.1 | 136.5 |
| bap | bdf1 | m | gav | Blood | 1 | na | 48 | Shimada [15] | 0.1 | 0.4 | CPD | b6c | f | Feed | 96 | 104 | D | Forestomach | Mixed | 0.6 | 1.0 |
| ben | P16 | m | gav | Blood | 32 | Corn oil | 24 | ntp A65367 | 0.1 | 0.2 | TR-289 | b6c | m | gav | 103 | 103 | C | Preputial gland | Carcinoma | 12.4 | 18.1 |
| but | B6 | m | inh | bm | 14 | Air | 24 | ntp A74454 | 3.6 | 11.8 | TR-288 | b6c | m | inh | 60 | 60 | E | Lung | Adenocarcinoma | 14.7 | 52.3 |
| cbc | bdf1 | m | ip | bm | 1 | na | 48 | Morita [16] | 0.05 | 0.14 | CDP | swiss | f | ip | 26 | 78 | F | tba | tba mix | 0.001 | 0.015 |
| chl | B6 | m | ip | bm | 3 | PBS | 24 | ntp 518954 | 38.0 | 107.2 | TR-502 | b6c | f | gav | 105 | 105 | B | Pituitary gland | adenoma | 70.0 | 96.3 |
| coc | B6 | f | dermal | Blood | 91 | Ethanol | 24 | ntp A44271 | 47.3 | 150.8 | TR-479 | b6c | f | Dermal | 104 | 104 | E | Liver | Adenocarcinoma hepatoblastoma | 1.7 | 11.6 |
| cop | cd1 | m | ip | bm | 2 | na | 48 | Morita [16] | 4.2 | 13.2 | TR-063 | b6c | m | Feed | 96 | 96 | C | Liver | Adenocarcinoma | 522.1 | 960.0 |
| cpa | bigblue | m | ip | blood | 1 | na | 48 | Gorelick [17] | 0.1 | 0.5 | CPD | swiss | f | ip | 26 | 79 | F | tba | tba mix | 0.25 | 1.19 |
| cps | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A64289 | 17.8 | 85.4 | TR-501 | b6c | f | Feed | 104 | 104 | C | Skin | Skin sarcoma | 30.4 | 66.0 |
| dbcp | cd1 | m | ip | bm | 1 | na | 48 | Morita [16] | 1.4 | 5.0 | TR-028 | b6c | f | inh | 91 | 104 | D | Mix | Mix | 0.28 | 0.68 |
| dbe | B6 | m | inh | Blood | 175 | Air | 24 | ntp A71010 | 0.8 | 4.4 | TR-086 | b6c | f | inh | 96 | 104 | F | tba | tba mix | 0.39 | 2.50 |
| dcn | B6 | m | inh | Blood | 91 | Air | 24 | ntp A40482 | 4.9 | 27.7 | TR-513 | b6c | f | inh | 105 | 105 | C | Uterus | Stromal polyp scarcoma | 380.7 | 1147.6 |
| dhp | B6 | m | ip | bm | 3 | PBS | 24 | ntp 224367 | 28.0 | 125.2 | TR-287 | b6c | m | gav | 103 | 103 | A | NA | NA | 178.1 | NA |
| dmvc | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A50288 | 3.0 | 8.9 | TR-316 | b6c | f | gav | 103 | 103 | C | Forestomach | Carcinoma | 14.9 | 22.1 |
| eth | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp 299785 | 21.0 | 62.9 | TR-237 | b6c | f | gav | 103 | 103 | E | Liver | Adenocarcinoma | 45.9 | 94.7 |
| for | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp 142011 | 33.5 | 113.1 | TR-000 (67-66-3) | b6c | f | gav | 93 | 93 | E | Liver | Carcinogenic myelosarcoma | 5.1 | 14.8 |
| gly | P16 | m | gav | Blood | 280 | Water | 24 | ntp A92101 | 1.3 | 5.1 | TR-374 | b6c | m | gav | 104 | 104 | E | Liver | Adenocarcinoma | 4.5 | 26.4 |
| hrc | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A27137 | 13.2 | NA | TR-330 | b6c | m | gav | 104 | 104 | E | Harderian gland | Adenocarcinoma | 57.7 | 156.5 |
| hyd | B6 | m | ip | bm | 3 | PBS | 24 | ntp A06686 | 2.8 | 7.4 | TR-366 | b6c | f | gav | 104 | 104 | E | Liver | Adenocarcinoma | 19.4 | 63.1 |
| ieg | B6 | f | gav | Blood | 91 | Corn oil | 24 | ntp A67394 | 2.2 | 38.6 | TR-551 | b6c | m | gav | 105 | 105 | E | Liver | Adenocarcinoma | 7.1 | 30.1 |
| isn | B6 | f | inh | Blood | 91 | Air | 24 | ntp A87798 | 50.6 | 153.9 | TR-448 | b6c | f | inh | 103 | 103 | E | Lung | Adenocarcinoma | 39.1 | 178.5 |
| iso | B6 | m | inh | bm | 91 | Air | 24 | ntp A63977 | 13.6 | 40.5 | TR-486 | b6c | m | inh | 72 | 72 | E | Liver | Hepatocellular adenocarcinoma | 211.2 | 1113.5 |
| las | B6 | m | ip | bm | 3 | PBS | 24 | ntp 521899 | 69.7 | 204.7 | TR-247 | b6c | m | Feed | 103 | 103 | A | NA | NA | 815520 | NA |
| leu | B6 | f | feed | Blood | 28 | Feed | 24 | ntp A31156 | 38.8 | NA | TR-527 | b6c | f | Feed | 104 | 104 | E | Liver | Adenocarcinoma | 25.6 | 121.0 |
| mbr | B6 | m | inh | Blood | 14 | Air | 24 | ntp A51339 | 10.3 | 33.2 | TR-385 | b6c | m | inh | 104 | 104 | A | NA | NA | 318.3 | NA |
| mel | cd1 | m | ip | bm | 1 | na | 24 | Morita [16] | 0.008 | 0.038 | CPD | swiss | f | ip | 26 | 79 | F | tba | tba mix | 0.0 | 0.0 |
| mnu | balb | m | ip | Blood | 1 | NaCl | 48 | Vrzoc [14] | 0.1 | 0.3 | CPD | c3h | m | Water | 30 | 54 | E | Stomach | Glandular adenocarcinoma | 2.6 | 6.3 |
| mon | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp 279574 | 8.9 | 34.9 | TR-266 | b6c | m | Feed | 104 | 104 | E | Liver | Adenocarcinoma | 2161 | NA |
| nda | 0 | m | ip | Blood | 1 | na | 48 | Suzuki [20] | 0.1 | 4.0 | CPD | cbl | f | gav | 50 | 72 | F | tba | tba mix | 0.018 | 0.048 |
| oxy | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp 427822 | 8.8 | 45.9 | TR-205 | b6c | f | Feed | 104 | 104 | E | Liver | Adenocarcinoma | 15.7 | 38.3 |
| pch | B6 | m | gav | bm | 3 | PBS | 24 | ntp A79778 | 0.7 | 2.9 | TR-351 | b6c | m | gav | 103 | 103 | C | Liver | Haemangiosarcoma | 16.3 | 41.3 |
| pge | B6 | f | inh | Blood | 91 | Air | 24 | ntp A04540 | 139.5 | 538.3 | TR-515 | b6c | m | inh | 104 | 104 | E | liver | Adenocarcinoma | 79.2 | 407.5 |
| phe | B6 | m | ip | bm | 3 | PBS | 24 | ntp 874983 | 13.3 | 40.8 | TR-203 | b6c | m | Water | 103 | 103 | A | NA | NA | 108690 | NA |
| php | P16 | f | feed | Blood | 42 | Feed | 24 | ntp A93723 | 4.3 | 14.4 | TR-465 | b6c | f | Feed | 104 | 104 | C | Haematopoetic system | Haematopoetic lymphosarcoma | 567.4 | 1187.2 |
| rsc | B6 | m | ip | bm | 3 | PBS | 24 | ntp A90027 | 12.1 | 48.8 | TR-403 | b6c | m | gav | 104 | 104 | A | NA | NA | 1774.6 | NA |
| scd | AM3 | m | feed | Blood | 65 | Water | 24 | ntp A73280 | 0.5 | 1.4 | TR-546 | b6c | f | Water | 104 | 104 | E | Small intestine | Adenocarcinoma | 3.4 | 6.1 |
| sel | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A25043 | 1.5 | 8.1 | TR-194 | b6c | f | gav | 103 | 103 | E | Liver | Adenocarcinoma | 14.0 | 27.6 |
| tac | B6 | m | ip | bm | 3 | PBS | 24 | ntp 256727 | 3.5 | 9.4 | TR-200 | b6c | f | Feed | 104 | 104 | B | Pituitary gland | Adenoma | 11.8 | NA |
| tce | B6 | m | gav | bm | 3 | Corn oil | 24 | ntp 679194 | 290.7 | NA | TR-002 | b6c | m | gav | 104 | 104 | C | Liver | Carcinoma | 230.5 | 377.8 |
| teo | B6 | m | gav | Blood | 91 | Corn oil | 24 | ntp A24621 | 18.5 | 355.4 | TR-473 | b6c | m | gav | 104 | 104 | E | Liver | Hepatocellular adenocarcinoma | 296.6 | NA |
| tet | B6 | f | feed | Blood | 91 | Feed | 24 | ntp A72450 | 10.6 | 25.7 | TR-027 | b6c | f | gav | 90 | 90 | C | Liver | Carcinoma | 25.9 | 39.0 |
| ure | B6 | f | water | Blood | 91 | Water | 24 | ntp A86637 | 0.28 | 0.73 | TR-510 | b6c | m | Water | 104 | 104 | E | Harderian gland | Adenocarcinoma | 0.62 | 1.2 |
As already noted, a large part of the scatter in the correlation might be due to the fact that BMDs may relate to different ‘lesion categories’, representing different degrees of severity. For instance, a BMD10 for a non-neoplastic lesion and a BMD10 for malignant tumours in different organs are not comparable and do not represent equipotent doses. As the BMDs in Figure 2A relate to different types of lesions in different lesions categories (4), the cancer BMD10s are not really equipotent doses. By selecting one lesion category, the scatter is indeed found to be reduced. Figure 3 (Table 4) shows the results for lesion category C (tissue-specific tumour). Figure 3 includes fewer compounds than Figure 2A, as some compounds did not show this lesion type. The two-sloped dashed lines were drawn by eye, such that they included most BMD intervals, but with the restriction that the slope of the line is unity. The reason is that a proportional relationship between the MN and cancer BMDs (on the original scale) translates in a straight line with unity slope in a double log plot. Therefore, it may be concluded from this correlation plot that the MN BMD05 and cancer BMD10 correlate proportionally to each other. The lower dashed line in Figure 3 represents the identity line (tumour BMD10 equals the MN BMD05), while the upper dashed line represents the situation where the tumour BMD10 is 100-fold higher than the MN BMD05. Therefore, given this particular correlation plot, the tumour BMD10 would be predicted to lie between the MN BMDL05 and 100-times the MN BMDU05. This illustrates how this type of correlation plots could be used for predicting the tumour BMD10 (with the indicated uncertainty range) in instances where no carcinogenicity studies are available.
Figure 3.
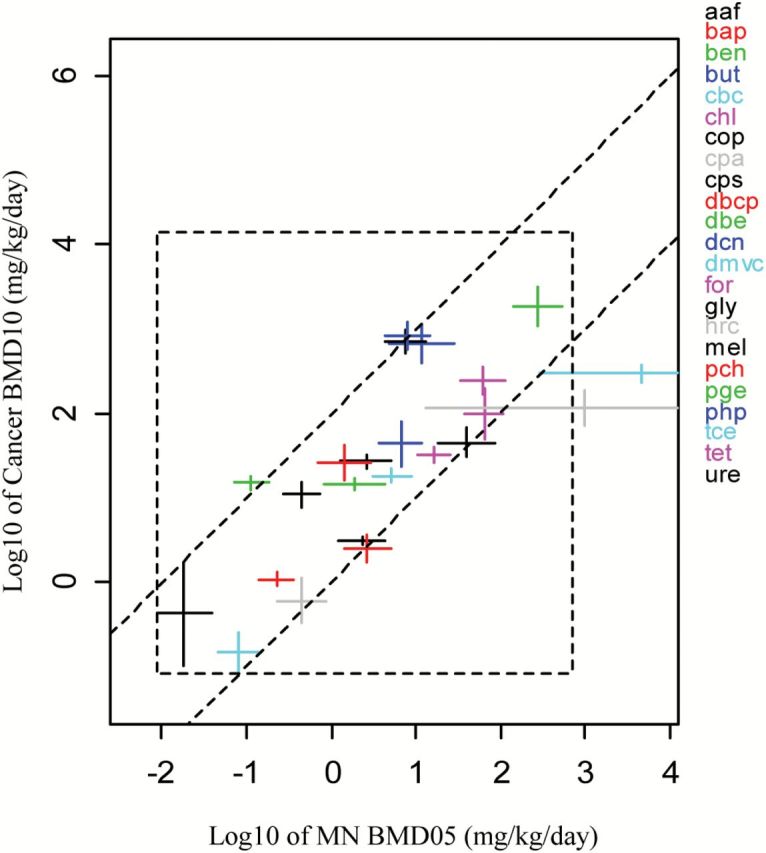
Lowest carcinogenic BMD10s for lesion category C against lowest MN BMD05s (n = 23 compounds). The 90% CIs are plotted in both directions. The lower dashed line represents the case where the carcinogenic BMD10 is equal to the MN BMD05. At the upper dashed line the carcinogenic BMD10s is 100 times higher than the MN BMD05. See Table 4 for the underlying data.
Table 4.
Summary of in vivo MN and carcinogenicity studies in Figure 3
| MN (genotoxicity endpoint) | Carcinogenicity (tumour endpoint) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Strain | Sex | Route | Tissue | Duration exposure (days) | Vehicle control | Sampling time | Source | BMDL05 (mg/kg) | BMDU05 (mg/kg/day) | Source | Strain | Sex | Route | Exposure time (weeks) | Duration experiment (weeks) | Severity | Tissue | Tissue lesion | BMDL10 (mg/kg/day) | BMDU10 (mg/kg/day) |
| aaf | bdf1 | m | ip | Blood | 1 | na | 48 | Asano [12] | 1.19 | 4.37 | CPD | bcn | m | Feed | 96 | 104 | C | Bladder | Carcinoma | 2.7 | 3.4 |
| bap | bdf1 | m | gav | Blood | 1 | na | 48 | Shimada [15] | 0.14 | 0.36 | CPD | b6c | f | Feed | 96 | 104 | C | Forestomach | Squamous carcinoma | 0.9 | 1.3 |
| ben | P16 | m | gav | Blood | 32 | Corn oil | 24 | ntp A65367 | 0.07 | 0.18 | TR-289 | b6c | m | gav | 103 | 103 | C | Preputial gland | Carcinoma | 12.4 | 18.1 |
| but | B6 | m | inh | bm | 14 | Air | 24 | ntp A74454 | 3.64 | 11.81 | TR-288 | b6c | f | inh | 36 | 36 | C | Forestomach | Papilloma | 23.3 | 80.5 |
| cbc | bdf1 | m | ip | bm | 1 | na | 48 | Morita [16] | 0.05 | 0.14 | CPD | swiss | m | ip | 26 | 78 | C | Lymphoid system | Lymphoma | 0.08 | 0.25 |
| chl | B6 | m | ip | bm | 3 | PBS | 24 | ntp 518954 | 38.05 | 107.22 | TR-502 | b6c | m | gav | 104 | 104 | C | Liver | Hepatocellular carcinoma | 49.7 | 198.3 |
| cop | cd1 | m | ip | bm | 2 | na | 48 | Morita [16] | 4.24 | 13.21 | TR-063 | b6c | m | Feed | 96 | 96 | C | Liver | Adenocarcinoma | 522.1 | 960.0 |
| cpa | cd1 | m | ip | Blood | 1 | na | 48 | Hatanaka [18] | 0.23 | 0.89 | CPD | swiss | f | ip | 26 | 79 | C | Lung | Malignant carcinoma | 0.3 | 1.1 |
| cps | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A64289 | 17.83 | 85.40 | TR-501 | b6c | f | Feed | 104 | 104 | C | Skin | Sarcoma | 30.4 | 66.0 |
| dbcp | cd1 | m | ip | bm | 1 | na | 48 | Morita [16] | 1.40 | 5.01 | TR-028 | b6c | f | inh | 91 | 104 | C | Nasal cavity | Carcinoma | 1.7 | 3.5 |
| dbe | B6 | m | inh | Blood | 175 | Air | 24 | ntp A71010 | 0.83 | 4.41 | TR-086 | b6c | f | gavage | 53 | 90 | C | Stomach | Squamous carcinoma | 12.3 | 16.7 |
| dcn | B6 | m | inh | Blood | 91 | Air | 24 | ntp A40482 | 4.87 | 27.71 | TR-513 | b6c | f | inh | 105 | 105 | C | Uterus | Stromal polyp sarcoma | 380.7 | 1147.6 |
| dmvc | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A50288 | 3.01 | 8.88 | TR-316 | b6c | f | gav | 103 | 103 | C | Forestomach | Carcinoma | 14.9 | 22.1 |
| for | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp 142011 | 33.49 | 113.1 | TR-000 (67-66-3) | b6c | m | gav | 93 | 93 | C | Haematopoetic system | Lymphoma | 168.9 | 347.3 |
| gly | P16 | m | gav | blood | 280 | Water | 24 | ntp A92101 | 1.29 | 5.08 | TR-374 | b6c | f | gav | 104 | 104 | C | Skin | Fibrosarcoma | 21.5 | 32.5 |
| hrc | B6 | m | ip | bm | 3 | Corn oil | 24 | ntp A27137 | 13.20 | NA | TR-330 | b6c | m | gav | 104 | 104 | C | Adrenal gland | Pheochromocytoma | 71.1 | 187.1 |
| mel | cd1 | m | ip | bm | 1 | na | 24 | Morita [16] | 0.01 | 0.04 | CPD | swiss | f | ip | 26 | 79 | C | Adrenal gland | Fibrosarcoma | 0.1 | 1.7 |
| pch | B6 | m | gav | bm | 3 | PBS | 24 | ntp A79778 | 0.69 | 2.87 | TR-351 | b6c | m | gav | 103 | 103 | C | Liver | Haemangiosarcoma | 16.3 | 41.3 |
| pge | B6 | f | inh | Blood | 91 | Air | 24 | ntp A04540 | 139.45 | 538.29 | TR-515 | b6c | m | inh | 104 | 104 | C | Liver | Hepatoblastoma | 1102.8 | 3166.6 |
| php | P16 | f | Feed | Blood | 42 | Feed | 24 | ntp A93723 | 4.34 | 14.36 | TR-465 | b6c | f | Feed | 104 | 104 | C | Haematopoetic system | Lymphoma | 567.4 | 1187.2 |
| tce | B6 | m | gav | bm | 3 | Corn oil | 24 | ntp 679194 | 290.7 | NA | TR-002 | b6c | m | gav | 104 | 104 | C | Liver | Carcinoma | 230.5 | 377.8 |
| tet | B6 | f | Feed | Blood | 91 | Feed | 24 | ntp A72450 | 10.56 | 25.69 | TR-027 | b6c | f | gav | 90 | 90 | C | Liver | Carcinoma | 25.9 | 39.0 |
| ure | B6 | f | Water | Blood | 91 | Water | 24 | ntp A86637 | 0.28 | 0.73 | TR-510 | b6c | f | water | 104 | 104 | C | Liver | Haemangiosarcoma | 7.5 | 15.2 |
Figure 2B distinguishes between BMDs that are based on NTP studies, and those that are not. This plot makes it clear that the compounds studied by NTP were overall less potent than the additional chemicals that we added from the published literature. The added studies appear to be in line with the correlation resulting from the NTP studies. This is somewhat surprising given that the cancer studies from the published literature used shorter exposure (and study) durations in the case of eight compounds (ACD:51 weeks, BUT:60 weeks, CBC:26 weeks, CPA:26 weeks, ISO:72 weeks, MEL:26 weeks, MNU:30 weeks, NDA: 50 weeks; see (4)). The two points in the lower left corner in Figure 2B were derived from studies that used a 26-week exposure duration (compounds: CBC and CPA; both in Swiss mice, see (4)).
Discussion
In this study, we extended our initial study (8), which was based on 18 compounds, by using a larger data base of 48 compounds with both in vivo MN and cancer data. Our previous study demonstrated a positive correlation between MN and cancer potency. Here, we found even stronger evidence of a positive correlation between in vivo MN and tumour potency, with the best correlation found when the lowest cancer BMD for single specific tumours (lesion category C) was selected for each compound (Figure 3). When selecting other lesion categories, or combinations of them (while again selecting the lowest BMD from the remaining datasets for each chemical), the correlations showed more scatter, mainly caused by a few chemicals that visually appeared to be outliers, by deviating more from the general pattern than the bulk of the chemicals. These relatively large deviations suggest that the chemicals involved have specific characteristics, such as other modes of action that make them different from the majority of chemicals considered. For example, a chemical might have carcinogenic pathways contributing to potency that are not reflected in the MN test.
Prior to seeking biological explanations for the deviations, it is however important to first determine if they are related to other issues. For example, one of the most difficult problems in the correlation exercises described here is the selection of methods for estimating equipotent cancer doses for chemicals that induced highly distinct lesions. As an additional problem, there may be different reporting procedures among laboratories. Apart from these qualitative differences in responses measured in cancer studies, there is also a hidden quantitative difficulty in estimating equipotent doses. The reason is that the background incidences related to both BMDs may differ. A BMD10 could relate to an increase from 0.1% to 10.1% in one study but from an increase from 20 to 30% in another (in this example, an additional rather than extra risk of 10% was used, for the sake of the argument). Intuitively, the former increase would require a more potent carcinogen than the latter. Indeed, epidemiologists would argue that an increase from 0.1 to 10% is much larger than from 20 to 30% as they measure a risk increment in terms of relative risk. However, other numerical examples can be composed showing that relative risk does not solve the issue of correcting for background such that the resulting risk increments may be regarded as equivalent (28).
Both the qualitative and quantitative problems inherent in quantifying results from carcinogenicity studies makes it difficult to measure equipotent doses from different carcinogens. We are currently trying to derive solutions to this problem. One of the refinements we are currently exploring with pathology experts is the characterisation of lesion categories. In interpreting the correlation plots presented here, it is important to keep in mind that equipotent doses for cancer cannot be precisely defined. For the MN test, where the proportion of MN is measured in either blood or bone marrow, equipotent doses are more precisely defined. In addition, differences in background are not a problem for defining an equipotent dose, as with carcinogenicity studies: a given percent increase in MN over background may be considered as an equivalent change, independent of the background value. The advancements in technology with flow cytometry techniques have made it possible to rapidly score up to 1 million cells, and thus more precise MN frequencies can be achieved (29). Obviously, differences in study conditions will also remain in MN tests, but these can often be addressed, or may be assumed to have a relatively small impact. Therefore, in general, a large part of the scatter in the correlation plots might be related to the large variability in reported lesions and in background responses in carcinogenicity studies. In other words, the error in predicting cancer potencies from MN potencies, as discussed with Figure 3, is at least partly due to errors in the estimated cancer potencies themselves. Theoretically, it could even be that a well-performed MN test (or some combination of various genotoxicity tests) is a better predictor of the carcinogenic potency of a chemical than a carcinogenicity study.
This analysis differed in approach to our previous study (8) in several ways. First, we analyzed 48 compounds, in comparison to 18 compounds in our previous study. Second, the dose–response analysis was applied to combined datasets as opposed to separate datasets in our previous study, resulting in more precise BMD estimates. Third, in this study we only used MN tests, while in the previous study we included various types of genotoxicity assays, including the transgenic rodent mutation assay and the comet assay. Fourth, in this study we made a first attempt to take into account the differences between severities in observed (reported) lesions in the cancer studies. As already discussed, the definition of lesion categories was a pragmatic attempt, and may need revision after consulting the relevant experts. In this regard, a general scientific consensus on how we can quantitatively compare different tumour lesion types is needed.
The theoretical examples provided in Table 2 illustrate how our methodology can shed some light on whether false positive or negative MN tests (as a predictor of carcinogenicity) are due to biological or statistical reasons. By considering the two quantitative estimates of the potencies related to both systems (in this study: MN test vs. carcinogenicity test), it can be made visible which of these two (biological or statistical) is the most likely explanation. A compound with a high MN potency but low cancer potency would be a false positive in the biological sense, or if vice versa, these compounds would be false negatives. Such compounds would show up in the lower right, or the upper left corner of the correlation plot, respectively. However, none of the compounds in the database we considered ended up in one of these two corners of any of the correlation plots. The compounds we considered included seven compounds (CPS, LAS, MBR, PHE, RSC, TAC, TEO) with positive results for MN and negative for carcinogenicity (indicated in Figure 2). All seven compounds comply with the overall correlation (Figure 2) and there is no basis for considering these compounds as false positives in the biological sense. Three compounds (DBCP, SEL and TCE) were negative in the MN test and positive for carcinogenicity (Figure 2). All three compounds do not deviate from the general correlation patterns. These results indicate that a considerable fraction of currently labelled false positives or negatives may be simply due to statistical artefacts.
In conclusion, our results suggest that tumour BMD10s can be predicted from in vivo MN BMD05s using the statistical correlations as observed in this study, be it with a considerable uncertain margin (around two orders of magnitude). Our approach might be a very useful tool in those cases where an in vivo MN test is available in the absence of carcinogenicity data. We intend to further examine the impact of various factors on the correlation, including the lesion categories observed in cancer studies, exposure duration, sex and strain. It is expected that the strength of this correlation would be improved if mutation and chromosomal aberration data were available from cancer target tissues in the same strain, species and sex of animals exposed by the same routes using similar protocols. Nevertheless sufficient data to obtain the quantitative metrics necessary to examine this assumption have not been found by the ILSI/HESI GTTC and a recommendation was made for the generation and analysis of such data (4,5). To fill this gap, we are currently collaborating with international government agencies (Health Canada, US FDA) and international organisations (ILSI/HESI GTTC) to expand our database with more chemicals and to refine the methodology.
Funding
Dr Slob and Dr Soeteman-Hernández (RIVM) have received contract funding from the Government of Canada (project number E/133510) for the development of advanced quantitative methods for the analysis of genetic toxicity dose–response data and from The Dutch Ministry of Welfare and Sport (VWS) (project 340700). The National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs, contract number NC/K500033/1) provided funding for Dr Johnson.
Acknowledgements
We wish to thank Dr Dan Levy from the U.S. Food and Drug Administration and Dr Kristine L. Witt from the U.S. National Institute of Environmental Health Sciences for their expert advise and critical comments with regards to this manuscript. We also wish to thank Mrs Lea Patrice McDaniel from the United States Food and Drug Administration for retrieving the data from the NTP database.
Conflict of interest statement: None declared.
References
- 1. OECD (2008) Draft OECD Guideline for the Testing of Chemicals, Test Guideline 451: Carcinogenicity Studies http://www.oecd.org/dataoecd/30/46/41753121.pdf (accessed January 15, 2014).
- 2. Jacobson-Kram D. Sistare F. D. and Jacobs A. C (2004) Use of transgenic mice in carcinogenicity hazard assessment. Toxicol. Pathol., 32 (Suppl 1), 49–52. [DOI] [PubMed] [Google Scholar]
- 3. Johnson G.E., Soeteman-Hernández L.G., et al. (2013) Derivation of points of departure (PoD) estimates in genetic toxicology studies and their potential application in risk assessment. Environ. Mol. Mutag., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacGregor, J. T., Frötschl, R., White, P. A., et al (2014) IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure-response relationships and points of departure (PoDs). Mutat. Res. Genet. Toxicol., 783, 55–65. [DOI] [PubMed]
- 5.MacGregor, J. T., Frötschl, R., White, P. A., et al. (2014) IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat. Res. Genet. Toxicol., 783, 66–78. [DOI] [PubMed] [Google Scholar]
- 6. Gollapudi B. B., Johnson G. E., Hernandez L. G., et al. (2013) Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ. Mol. Mutagen., 54, 8–18. [DOI] [PubMed] [Google Scholar]
- 7. Sanner T. and Dybing E (2005) Comparison of carcinogenic and in vivo genotoxic potency estimates. Basic Clin. Pharmacol. Toxicol., 96, 131–139. [DOI] [PubMed] [Google Scholar]
- 8. Hernández L. G. Slob W. van Steeg H. and van Benthem J (2011) Can carcinogenic potency be predicted from in vivo genotoxicity data?: a meta-analysis of historical data. Environ. Mol. Mutagen., 52, 518–528. [DOI] [PubMed] [Google Scholar]
- 9. EFSA (2009) Risk assessment methodologies and approaches for genotoxic and carcinogenic substances http://ec.europa.eu/health/ph_risk/committees/04_scher/docs/scher_o_113.pdf (accessed July 10, 2014).
- 10. Heddle J. A. Hite M. Kirkhart B. Mavournin K. MacGregor J. T. Newell G. W. and Salamone M. F (1983) The induction of micronuclei as a measure of genotoxicity. A report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutat. Res., 123, 61–118. [DOI] [PubMed] [Google Scholar]
- 11. Hamada S., Sutou S., Morita T., et al. (2001) Evaluation of the rodent micronucleus assay by a 28-day treatment protocol: Summary of the 13th Collaborative Study by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Environmental Mutagen Society of Japan (JEMS)-Mammalian Mutagenicity Study Group (MMS). Environ. Mol. Mutagen., 37, 93–110. [DOI] [PubMed] [Google Scholar]
- 12. Asano N. and Hagiwara T (1992) The mouse peripheral blood micronucleus test with 2-acetylaminofluorene using the acridine orange supravital staining method. Mutat. Res., 278, 153–157. [PubMed] [Google Scholar]
- 13. Smith C. C. Archer G. E. Forster E. J. Lambert T. R. Rees R. W. and Lynch A. M (1999) Analysis of gene mutations and clastogenicity following short-term treatment with azathioprine in MutaMouse. Environ. Mol. Mutagen., 34, 131–139. [PubMed] [Google Scholar]
- 14. Vrzoc M. and Petras M. L (1997) Comparison of alkaline single cell gel (Comet) and peripheral blood micronucleus assays in detecting DNA damage caused by direct and indirect acting mutagens. Mutat. Res., 381, 31–40. [DOI] [PubMed] [Google Scholar]
- 15. Shimada H. Suzuki H. Itoh S. Hattori C. Matsuura Y. Tada S. and Watanabe C (1992) The micronucleus test of benzo[a]pyrene with mouse and rat peripheral blood reticulocytes. Mutat. Res., 278, 165–168. [PubMed] [Google Scholar]
- 16. Morita T., Asano N., Awogi T., et al. (1997) Evaluation of the rodent micronucleus assay in the screening of IARC carcinogens (groups 1, 2A and 2B) the summary report of the 6th collaborative study by CSGMT/JEMS MMS. Collaborative Study of the Micronucleus Group Test. Mammalian Mutagenicity Study Group. Mutat. Res., 389, 3–122. [DOI] [PubMed] [Google Scholar]
- 17. Gorelick N. J. Andrews J. L. deBoer J. G. Young R. Gibson D. P. and Walker V. E (1999) Tissue-specific mutant frequencies and mutational spectra in cyclophosphamide-treated lacI transgenic mice. Environ. Mol. Mutagen., 34, 154–166. [PubMed] [Google Scholar]
- 18. Hatanaka Y. Kitagawa Y. Toyoda Y. Kawata T. Ando N. Kawabata Y. Iwai M. and Arimura H (1992) Micronucleus test with cyclophosphamide using mouse peripheral blood reticulocytes. Mutat. Res., 278, 99–101. [DOI] [PubMed] [Google Scholar]
- 19. Meli C. and Seeberg A. H (1990) Activity of 1,2-dimethylhydrazine in the mouse bone marrow micronucleus assay using a triple- and a single-dosing protocol. Mutat. Res., 234, 155–159. [DOI] [PubMed] [Google Scholar]
- 20. Suzuki T. Itoh T. Hayashi M. Nishikawa Y. Ikezaki S. Furukawa F. Takahashi M. and Sofuni T (1996) Organ variation in the mutagenicity of dimethylnitrosamine in Big Blue mice. Environ. Mol. Mutagen., 28, 348–353. [DOI] [PubMed] [Google Scholar]
- 21. Durling L. J. and Abramsson-Zetterberg L (2005) A comparison of genotoxicity between three common heterocyclic amines and acrylamide. Mutat. Res., 580, 103–110. [DOI] [PubMed] [Google Scholar]
- 22. Suzuki T. Hayashi M. Ochiai M. Wakabayashi K. Ushijima T. Sugimura T. Nagao M. and Sofuni T (1996) Organ variation in the mutagenicity of MeIQ in Big Blue lacI transgenic mice. Mutat. Res., 369, 45–49. [DOI] [PubMed] [Google Scholar]
- 23. Levy D. D., McDaniel L. P., Witt K. L. (2010) Analysis of micronucleus data in the NTP database http://www.regulations.gov/#!documentDetail;D=FDA-2009-N-0519-0008 (accessed 1, 2013).
- 24. Slob W. and Setzer R. W (2014) Shape and steepness of toxicological dose-response relationships of continuous endpoints. Crit. Rev. Toxicol., 44, 270–297. [DOI] [PubMed] [Google Scholar]
- 25. Slob W. (2002) Dose-response modeling of continuous endpoints. Toxicol. Sci., 66, 298–312. [DOI] [PubMed] [Google Scholar]
- 26. EFSA (2009) European Food Safety Authority. Guidance of the scientific committee on use of the benchmark dose approach in risk assessment. The EFSA J., 1150, 1–72. [Google Scholar]
- 27. Bemis J. C., Willis J. W., Bryce S. M., Torous D. K., Dertinger S. D., Slob W. (2015) Comparison of in vitro and in vivo clastogenic potency based on benchmark dose analysis of flow cytometric micronucleus data. Mutagenesis, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.IPCS. (2014) World Health Organization (WHO) International Programme on Chemical Safety (IPCS): Guidance document on evaluating and expressing uncertainty in hazard characterization. Harmonization Project Document 11. http://www.who.int/ipcs/methods/harmonization/areas/hazard_assessment/en/.
- 29. Johnson G.E., Rees B., Gollapudi B., et al. (2015) New Approaches to Advance the use of Genetic Toxicology Analyses for Human Health Risk Assessment and Regulatory Decision-Making. Toxicol. Res., 4, 667–676. [Google Scholar]