

Molecular dynamics simulations, equilibrium binding measurements, and fluorescence imaging reveal the influence of a key salt bridge in the mechanical activation of α-catenin at intercellular adhesions. Simulations reveal possible α-catenin conformational changes underlying experimental fluorescence and equilibrium binding data.
Abstract
Cadherin complexes transduce force fluctuations at junctions to activate signals that reinforce stressed intercellular contacts. α-Catenin is an identified force transducer within cadherin complexes that is autoinhibited under low tension. Increased force triggers a conformational change that exposes a cryptic site for the actin-binding protein vinculin. This study tested predictions that salt bridges within the force-sensing core modulate α-catenin activation. Studies with a fluorescence resonance energy transfer (FRET)-based α-catenin conformation sensor demonstrated that each of the salt-bridge mutations R551A and D503N enhances α-catenin activation in live cells, but R551A has a greater impact. Under dynamic force loading at reannealing cell–cell junctions, the R551A mutant bound more vinculin than wild-type α-catenin. In vitro binding measurements quantified the impact of the R551A mutation on the free-energy difference between the active and autoinhibited α-catenin conformers. A 2-μs constant-force, steered molecular dynamics simulation of the core force-sensing region suggested how the salt-bridge mutants alter the α-catenin conformation, and identified a novel load-bearing salt bridge. These results reveal key structural features that determine the force-transduction mechanism and the force sensitivity of this crucial nanomachine.
INTRODUCTION
Cadherin-mediated cell–cell adhesion is essential for tissue morphogenesis and for regulating the barrier functions of tissues such as the vascular endothelium (Gumbiner, 1996; Lecuit et al., 2011). Cell-surface cadherins bind the extracellular domains of cadherins on adjacent cells. The cadherin intracellular domain binds β-catenin, which binds the actin-binding protein α-catenin (Rimm et al., 1995; Pappas and Rimm, 2006; Buckley et al., 2014). At the plasma membrane, the N-terminal domain of α-catenin associates with cadherin-associated β-catenin, and the C-terminal domain binds F-actin (Rimm et al., 1995; Imamura et al., 1999). α-Catenin thus mechanically links the cadherin–catenin complex to the actin cytoskeleton (Imamura et al., 1999; Desai et al., 2013; Buckley et al., 2014). In solution, α-catenin dimerizes through its N-terminal domain, but the α-catenin dimer cannot bind β-catenin and F-actin simultaneously (Koslov et al., 1997; Drees et al., 2005).
Cadherin complexes both mechanically couple adjacent cells and transduce mechanical cues to activate signals that regulate cell functions (Ladoux et al., 2010; le Duc et al., 2010; Liu et al., 2010; Yonemura et al., 2010). Moreover, mechanically perturbed cadherin adhesions activate the accumulation of the actin-binding protein vinculin, a signature of cadherin-based force transduction (le Duc et al., 2010; Twiss et al., 2012; Desai et al., 2013; Thomas et al., 2013; Barry et al., 2014). Cadherin-dependent force transduction requires actin and α-catenin (le Duc et al., 2010; Twiss et al., 2012). In the current model of force transduction, α-catenin adopts an autoinhibited conformation at low tension (Yonemura et al., 2010). Increased force on cadherin adhesions triggers a conformational change, referred to as “unfurling,” which exposes the vinculin-binding site (VBS) of α-catenin (Yonemura et al., 2010; Rangarajan and Izard, 2012; Yao et al., 2014). α-Catenin unfurling and subsequent vinculin recruitment in turn promotes local actin polymerization thought to reinforce the stressed cell–cell junctions (Leerberg et al., 2014).
α-Catenin is homologous to vinculin, which adopts an autoinhibited conformation that is stabilized by a high-affinity association between its head and tail domains. The head–tail interaction regulates vinculin binding to F-actin and ligands, including α-catenin (Johnson and Craig, 1994, 1995; Watabe-Uchida et al., 1998). Despite their structural homology, the mechanisms of vinculin and α-catenin autoinhibition and force-induced activation differ. Figure 1A shows an autoinhibited structure of full-length α-catenin. Unlike vinculin, the N-terminal and C-terminal domains do not interact (Ishiyama et al., 2013; Rangarajan and Izard, 2013). The core, force-sensing modulatory (M) domain comprises three helical bundles, MI–MIII. The MI domain harbors the cryptic VBS, which is masked by intradomain interactions with other helices in MI. Several interdomain salt bridges within the α-catenin M domain (Figure 1B) were postulated to stabilize the autoinhibited conformation (Ishiyama et al., 2013). Intriguingly, each of the cancer-linked missense mutations R551Q and D503N disrupts an R551-D503 salt bridge, suggesting that the salt-bridge network may play a crucial role in α-catenin function (Cerami et al., 2012; Gao et al., 2013; Li et al., 2015).
FIGURE 1:
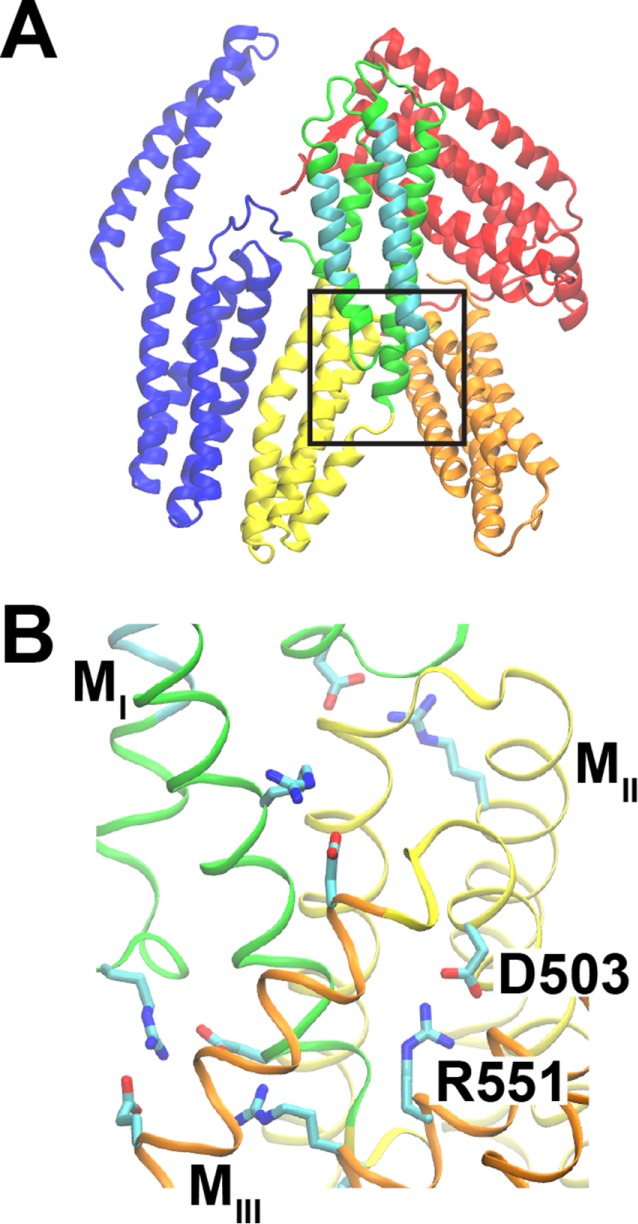
α-Catenin structure. Structure from PDB ID 4IGG (Rangarajan and Izard, 2013). (A) α-Catenin consists of three major domains: the N-terminal (N, blue), modulatory (M), and C-terminal (C, red) domains. The M domain contains three four-helix bundles: MI (green/cyan), MII (yellow), and MIII (orange). MI contains the vinculin-binding site (cyan). The box indicates the region shown in B. (B) The salt bridge between D503 (MII domain) and R551 (MIII) is part of a salt-bridge network that stabilizes the structure of the M domain.
Previously reported molecular dynamics (MD) simulations of the α-catenin M domain suggested that salt bridges form a cooperative network (Li et al., 2015); namely, in silico point mutations that disrupted single salt bridges destabilized the entire salt-bridge network and increased the conformational flexibility of the M domain (Li et al., 2015). None of the salt-bridge mutations investigated resulted in VBS exposure in either equilibrium MD simulations or in 325-ns constant-force (100 pN) steered molecular dynamics (SMD) simulations. However, the R551A mutation facilitated the rotation of MIII relative to MI–MII. We previously postulated that the resulting structure might represent an intermediate along the unfolding trajectory and that interdomain salt bridges between MI and MIII might tune the force sensitivity of α-catenin (Li et al., 2015). Consistent with this hypothesis, an in vitro vinculin pull-down assay showed that R551A bound more vinculin than wild-type (WT) α-catenin (Ishiyama et al., 2013). The latter assay was done in the absence of tension.
The present study tested the prediction that disrupting the M domain salt-bridge network lowers the barrier to α-catenin unfurling in cells. We achieved this by introducing point mutations that disrupt the R551-D503 salt bridge in a fluorescence resonance energy transfer (FRET)-based α-catenin conformation sensor (Kim et al., 2015). Enhanced cyan fluorescent protein (ECFP; donor) and yellow fluorescent protein YPet (acceptor) were inserted at the N and C termini, respectively, of the M domain within full-length α-catenin. In this construct, α-catenin unfurling (activation) separates the fluorophores and decreases the FRET between them (see Figure 4A later in this article). A prior study demonstrated that the FRET/ECFP ratio responds to differences in tension on cadherin adhesions (Kim et al., 2015). The present study quantified differences in tension-induced conformational changes in the R551A, D503N, and WT forms of the α-catenin sensor. We further validated the increased activation of R551A α-catenin relative to WT with a conformation-specific antibody. BioLayer Interferometry (BLI) and immunofluorescence confocal imaging investigated vinculin binding to full-length R551A and WT α-catenin at equilibrium in vitro and under tension in vivo, respectively. Equilibrium MD simulations of the core M domain assessed the effect of the R551A and D503N mutations on the autoinhibited conformation of α-catenin. Finally, a 2-μs, all-atom, constant-force (100-pN) SMD simulation of the α-catenin M domain revealed the force-dependent unfurling trajectory and identified potential roles of R551-D503 and other salt bridges in force-induced α-catenin activation. These findings reveal novel insights into the structural basis of α-catenin stability and its activation under force.
FIGURE 4:

Relative populations of unfurled α-catenin in cells expressing the WT, R551A, or D503N α-catenin conformation sensor. (A) Cartoon of cells expressing the α-catenin conformation sensor. Under tension, α-catenin undergoes a conformational change that separates the ECFP and YPet and reduces the FRET/ECFP ratio. (B) DIC (left) and FRET/ECFP (right) images for untreated or CytoD-treated MDCK cells transfected with WT, R551A, or D503N α-catenin sensor and cultured on collagen-coated glass. White boxes indicate ROIs at cell-cell junctions. Color scale indicates the absolute FRET/ECFP ratio. (C, D) FRET/ECFP ratios measured in cells expressing the WT, R551A, or D503N α-catenin conformation sensor. All ratios are normalized to the FRET/ECFP signal for untreated MDCK cells expressing the WT α-catenin conformation sensor. FRET/ECFP ratios at junctions and in the cytosol are normalized separately. For the +EGTA condition, FRET/ECFP ratios were measured after a 2-min treatment with 2 mM EGTA to disrupt cadherin-mediated junctions. For the +CytoD condition, cells were treated with 1 μM CytoD for 10 min before imaging. FRET/ECFP ratios represent the averages of N = 4 independent experiments for untreated cells and N = 2 independent experiments for EGTA- or CytoD-treated cells. Error bars represent SEM. * indicates p < 0.001. The numbers of analyzed cells and junctions for each condition are given in Supplemental Table S1. (C) FRET/ECFP in ROIs at junctions. (D) FRET/ECFP in cytosolic ROIs in MDCK cells. (E) Intensity ratios of α18 (activated) to anti–α-catenin (total) immunostained α-catenin at junctions between R2/7 cells cultured overnight on collagen-coated glass substrates. Intensity ratios are normalized to the α18/α-catenin ratio at junctions between cells expressing the WT conformation sensor. Intensity ratios represent the average of N = 2 independent experiments. Error bars represent SEM. * indicates p < 0.001.
RESULTS
The R551A mutation enhances equilibrium binding between the constitutively active vinculin head domain and α-catenin in vitro
To demonstrate that the increased conformational flexibility of R551A α-catenin suggested by simulations (Li et al., 2015) increases the apparent affinity for vinculin, we measured dissociation constants between full-length WT or R551A α-catenin and constitutively active vinculin (vinculin head domain, VHD), using BLI (Figure 2). The measured dissociation constant 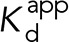 of 1.8 ± 0.2 μM for binding between VHD and WT α-catenin agrees with the previously reported value of 1.8 ± 0.2 μM for binding between monomeric α-catenin and the constitutively active vinculin D1 domain, determined with isothermal titration calorimetry (Choi et al., 2012). The dissociation constant between the VHD and R551A α-catenin that we measured is ∼13-fold lower at 0.14 ± 0.01 μM.
of 1.8 ± 0.2 μM for binding between VHD and WT α-catenin agrees with the previously reported value of 1.8 ± 0.2 μM for binding between monomeric α-catenin and the constitutively active vinculin D1 domain, determined with isothermal titration calorimetry (Choi et al., 2012). The dissociation constant between the VHD and R551A α-catenin that we measured is ∼13-fold lower at 0.14 ± 0.01 μM.
FIGURE 2:

Apparent affinities between the vinculin head domain and WT or R551A α-catenin. The apparent dissociation constants between constitutively active vinculin (VHD, residues 1–252) and WT or R551A α-catenin measured using BLI. Traces on the left are the binding signal over time for different concentrations of VHD binding full-length α-catenin immobilized on a sensor by an N-terminal His6 tag. Graphs on the right show the steady-state analysis fits.
These dissociation constants were used to estimate the equilibrium constant for the α-catenin conformational change–-that is, for the equilibrium between the autoinhibited and unfurled states–-for both WT and R551A. The dissociation constant Kd between the VHD (V) and the α-catenin VBS (A) is the following:
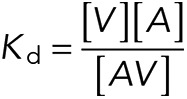 |
If the unfurled conformation is U and the autoinhibited conformation is I, then the unfurling equilibrium constant KU is the following:
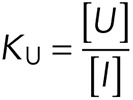 |
It can be shown that the apparent dissociation constant 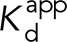 between the full-length α-catenin and the VHD–that is, the dissociation constant observed for vinculin binding to α-catenin under conformational equilibrium–-is the following:
between the full-length α-catenin and the VHD–that is, the dissociation constant observed for vinculin binding to α-catenin under conformational equilibrium–-is the following:
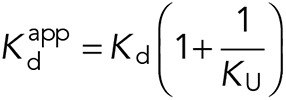 |
We assumed that the unfurled conformation of full-length α-catenin (U) has the same affinity for VHD (V) as the isolated VBS (A) and thus used the reported value of 5.2 ± 0.3 nM for Kd (Choi et al., 2012). Using our measured apparent affinities, the calculated values for the unfurling equilibrium constants KU of WT and R551A α-catenin are 0.0029 ± 0.0004 and 0.039 ± 0.004, respectively. From the relationship for the free energy, 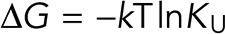 , the calculated decrease in the R551A unfurling free energy (I→U) relative to WT is ΔΔG = −2.6 kT ± 0.2 kT (−1.6 ± 0.1 kcal/mol at 37°C), consistent with the destabilization of the autoinhibited conformation by this salt-bridge mutant.
, the calculated decrease in the R551A unfurling free energy (I→U) relative to WT is ΔΔG = −2.6 kT ± 0.2 kT (−1.6 ± 0.1 kcal/mol at 37°C), consistent with the destabilization of the autoinhibited conformation by this salt-bridge mutant.
Salt-bridge disruption alters dynamic vinculin recruitment to reannealing junctions
We tested whether the lower (relative to WT) unfurling free energy calculated for R551A α-catenin alters vinculin recruitment to cell–cell junctions, by quantifying the intensity ratio of immunostained vinculin to α-catenin at reannealing cell–cell junctions, following a calcium switch (Figure 3). We did not measure FRET/ECFP because initial studies showed that fixation alters the FRET/ECFP ratios of the sensor (data not shown). The vinculin/α-catenin intensity ratio was also measured for control cells that were cultured in remove minus sign (DMEM) (2 mM Ca) overnight but were not treated with ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) (Figure 3, “control”).
FIGURE 3:
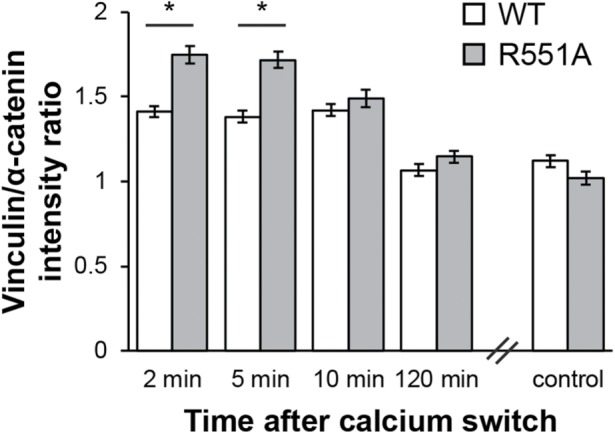
Vinculin recruitment at junctions between R2/7 cells expressing the WT or R551A α-catenin conformation sensor. Intensity ratios of immunostained vinculin and α-catenin at reannealing junctions between R2/7 cells cultured on collagen-coated glass substrates at intervals following a calcium switch. For the control condition, cells were not calcium switched but instead cultured on collagen-coated glass dishes overnight before fixation and immunostaining for vinculin and α-catenin. Intensity ratios represent the average of N = 3 independent experiments. Error bars represent SEM. * indicates p < 0.05.
Within the first 5 min after activating cell–cell junction formation by adding calcium to EGTA-treated cells, the ratio of vinculin to α-catenin at R2/7 cell–cell junctions was significantly higher (p < 0.001) for cells that expressed the R551A sensor versus the WT sensor (Figure 3). The vinculin-to-α-catenin ratio was statistically similar for WT and R551A at time points longer than 5 min and reached the steady-state level within 120 min.
The R551A and D503N mutations increase the population of unfurled α-catenin in live cells
To test the importance of the M domain salt-bridge network in α-catenin autoinhibition, we compared FRET/ECFP ratios at junctions between live cells transfected with either the WT, R551A, or D503N form of the full-length α-catenin conformation sensor (Figure 4). A decrease in the FRET/ECFP ratio reflects an increase in the population of unfurled α-catenin (Figure 4A). In experiments with Madin-Darby canine kidney (MDCK) cells, analyzed junctions typically involved only one transfected cell. When the sensor was stably expressed in R2/7 cells, which lack endogenous α-catenin, we analyzed junctions between cells that both expressed the sensor. Figure 4B shows the DIC and fluorescence images of MDCK cells that express the different sensor variants. In Figure 4, C and D, each of the quantified FRET/ECFP ratios in regions of interest at junctions or in the cytosol are normalized to the FRET/ECFP ratios obtained with untreated MDCK cells that express the WT α-catenin conformation sensor. The absolute FRET/ECFP values are given in Supplemental Table S1.
For MDCK cells cultured on collagen-coated glass, the average FRET/ECFP ratio in regions of interest (ROIs) at cell–cell junctions was 15 ± 1% lower (mean ± SEM; p < 0.001) for the R551A sensor than the WT sensor (Figure 4C). Similar results were obtained with R2/7 cells: the FRET/ECFP for R551A was 16 ± 2% lower (p < 0.001) than the WT sensor (Supplemental Figure S2). Thus, endogenous α-catenin in MDCK cells expressing the α-catenin sensor does not significantly affect the extent of α-catenin unfurling at junctions. More R551A than WT α-catenin was unfurled at junctions between cells in both cell types.
To verify that the FRET/ECFP ratios reflect α-catenin conformation switching, we used a conformation-specific, anti–α-catenin antibody (α18) that recognizes an epitope in the unfurled protein (Nagafuchi et al., 1994; Yonemura et al., 2010). Measurements compared the intensity ratio of α18 to anti–α-catenin antibody in R2/7 cells that expressed the WT or R551A α-catenin sensor. Cells were cultured in DMEM overnight, as described above for the control condition used in the vinculin recruitment measurements. Consistent with the FRET data, the ratio of α18 (activated α-catenin) to anti–α-catenin (total α-catenin) at junctions was 10 ± 2% (p < 0.001) higher for the R551A sensor compared with WT (Figure 4E). Thus, the R551A mutation promotes the activated conformation of α-catenin and enhances initial vinculin binding at reannealing junctions.
To support the interpretation that the R551A mutation promotes unfurling by disrupting the R551–D503 salt bridge, we also measured FRET/ECFP ratios for the D503N mutant of the α-catenin conformation sensor. At junctions, the average FRET/ECFP ratio was 9 ± 1% lower (p < 0.001) than that of the WT sensor. Thus mutating each residue in the R551–D503 salt bridge increases the population of unfurled α-catenin at cell–cell junctions. The greater extent of α-catenin unfurling observed with the R551A mutant compared with D503N (p < 0.001) is consistent with the ability of R551 to also interact with several negatively charged residues near the R551–D503 salt bridge, as observed in molecular dynamics simulations (see below).
To verify the response of the α-catenin conformation sensor to differences in tension, cadherin adhesions were disrupted with EGTA, or the actin cytoskeleton was disrupted with cytochalasin D (CytoD). Both treatments were previously demonstrated to result in the formation of the more compact, low-tension α-catenin conformation at cell–cell contacts (Kim et al., 2015). EGTA reduces tension across cell–cell junctions by chelating calcium, which is essential for cadherin adhesion. In a previous report, the increase in the FRET/ECFP ratio at junctions was complete within 2 min of EGTA treatment (Kim et al., 2015). We therefore compared FRET/ECFP measurements after 2 min of treatment with 2 mM EGTA. Cells were treated with 1 μM CytoD for 10 min in order to disrupt actin and lower tension across the junctions. Although intercellular gaps sometimes formed within the timeframe of the measurements, we limited our analysis to intact junctions. As expected, EGTA treatment increased FRET/ECFP ratios at junctions between MDCK cells, relative to untreated controls maintained in 2 mM calcium (Figure 4C). Actin disruption by CytoD similarly increased FRET/ECFP ratios at junctions (Figure 4C). Neither treatment triggered statistically significant FRET changes in cytosolic ROIs (p > 0.05 in each case; Figure 4D).
Under each condition studied, the FRET/ECFP ratios were lower in cells that expressed the α-catenin sensor salt-bridge mutants compared with WT (p < 0.001 for each condition; Figure 4, C and D). Importantly, FRET/ECFP ratios for R551A and D503N were lower both in the cytosol and at cell–cell junctions, compared with the WT sensor. Thus R551A and D503N are more likely to adopt the unfurled conformation than WT α-catenin in cells, even in the absence of tension. To assess whether the salt-bridge disrupting mutations facilitate a tension-dependent increase in unfurling, we compared the difference in FRET/ECFP at junctions under high (untreated) and low (EGTA- or CytoD-treated) tension. The increase in FRET/ECFP due to EGTA treatment was 9 ± 1% for WT, 12 ± 1% for R551A, and 7 ± 1% for D503N (p < 0.001 in each case). Similar increases were observed for CytoD treatment (9 ± 1% for WT, 12 ± 1% for R551A, 7 ± 1% for D503N; p < 0.001 in each case). For each sensor, the magnitude of the observed increase was ∼9% following either EGTA or CytoD treatment. Thus the extent of force-induced unfurling was similar for WT α-catenin and the salt-bridge mutants, within experimental error. Nevertheless, the data clearly show that the R551A and D503N mutants unfurl more readily than WT α-catenin under the same mechanical conditions, and establish that disrupting the salt-bridge network in the M domain promotes this conformational change.
Atomic-level details of force-induced α-catenin activation
Salt-bridge contributions to the stability of autoinhibited α-catenin.
Comparisons of the equilibrium simulations of the WT, R551A, and D503N α-catenin M domains (Table 1) evaluated the effect of the R551A and D503N mutations on the α-catenin structure. Data from the longer simulations in this study reinforce a previous postulate that the R551A mutation increases the conformational flexibility of the M domain by disrupting the salt-bridge network (Li et al., 2015).
TABLE 1:
The R551A and D503N mutations destabilize the α-catenin MII–MIII interface.
| Internal energy (kcal/mol) | |||
|---|---|---|---|
| M | |||
| WT | −4900 ± 200 | ||
| R551A | −4500 ± 200 | ||
| D503N | −4700 ± 300 |
| Interaction energy (kcal/mol) | |||
|---|---|---|---|
| MI–MII | MI–MIII | MII–MIII | |
| WT | −300 ± 80 | −270 ± 70 | −240 ± 80 |
| R551A | −300 ± 60 | −270 ± 90 | −80 ± 80 |
| D503N | −320 ± 70 | −220 ± 50 | −130 ± 70 |
| Angle (°) | |||
|---|---|---|---|
| MI–MII | MI–MIII | MII–MIII | |
| WT | 26 ± 6 | 47 ± 9 | 41 ± 7 |
| R551A | 18 ± 6 | 55 ± 9 | 50 ± 8 |
| D503N | 23 ± 5 | 50 ± 10 | 50 ± 10 |
Internal energy of the autoinhibited α-catenin M domain, and interdomain angles and interaction energies between the MI–MIII domains, during five (WT) or four (R551A and D503N) pooled 195-ns (WT and R551A) or 95-ns (D503N) equilibrium simulation trajectories. Values reported as average ± SD.
In equilibrium simulations of the α-catenin M domain, the total internal energies of the R551A and D503N mutants were higher than that of WT α-catenin (Table 1). The calculated internal energy determines simple pairwise interactions within the autoinhibited configuration and differs from the Gibbs free energy, which is the free-energy difference between the autoinhibited and unfurled states. The largest contribution to the M domain destabilization was the MII–MIII interaction energy.
Importantly, electrostatic contributions to the interaction energies, rather than altered van der Waals interactions, dominated the difference between WT and the salt-bridge mutants (Table 2). Although the electrostatic contribution is overestimated in the simulations (see the Discussion), the computed van der Waals energies are actually slightly lower in the D503N and R551A mutants relative to WT. These results therefore suggest that the computed destabilization arises exclusively from changes in the electrostatic interaction energies. This result is expected if disrupting salt bridges between MII and MIII destabilizes the M domain. The destabilization of the MII-MIII interface also correlated with the increased rotation of MIII, evident as an increase in the MII-MIII angle (Table 1).
TABLE 2:
Contributions to the MII–MIII interaction energy of WT, R551A, and D503N α-catenin.
| α-Catenin | Total interaction energy (kcal/mol) | Electrostatic IE (kcal/mol) | van der Waals IE (kcal/mol) |
|---|---|---|---|
| WT | −240 ± 80 | −220 ± 80 | −21 ± 6 |
| R551A | −80 ± 80 | −60 ± 80 | −25 ± 6 |
| D503N | −130 ± 70 | −100 ± 70 | −26 ± 6 |
Interaction energies (IE) calculated by NAMD from five (WT) or four (R551A and D503N) pooled 195-ns (WT and R551A) or 95-ns (D503N) equilibrium simulation trajectories of the autoinhibited α-catenin M domain. Values reported as average ± SD.
These results suggest that the R551A and D503N mutations facilitate the formation of the intermediate by disrupting the salt-bridge network at the MII–MIII interface, thereby destabilizing the autoinhibited conformation. The intermediate and unfurled states were not sampled during equilibrium MD simulations, so we cannot quantify the effect of either the R551A or the D503N mutation on the free energies of these two conformations. Nevertheless, the higher experimentally determined unfurling equilibrium constant KU for R551A relative to WT α-catenin confirms that the R551A mutation destabilizes the autoinhibited conformation.
Although both mutations destabilized the MII–MIII interface relative to WT, the effect was larger for the R551A α-catenin M domain compared with the D503N mutant (Table 2). As mentioned above in the context of the FRET measurements, this is consistent with the interaction of R551 with several negatively charged residues near the R551–D503 salt bridge. During equilibrium simulations of the WT M domain, the only salt bridge in MII-MIII that involved D503 was D503–R551. In contrast, R551 formed salt bridges with four different acidic amino acids. These additional salt bridges could partially compensate for the loss of the D503–R551 interaction in the D503N mutant.
Salt-bridge contributions to α-catenin activation under force.
An all-atom, constant-force (100 pN) SMD simulation of the α-catenin M domain revealed atomic-level details of force-induced α-catenin activation. As we previously described, the N- and C-terminal domains of full-length α-catenin do not contact the M domain under tension. Therefore, these simulations also focused on the isolated M domain (Li et al., 2015). The reorientation of the MIII helix bundle during constant-force simulations was reported previously and postulated to be an intermediate step that precedes the exposure of the VBS in MI (Li et al., 2015). Distinct from the earlier report, the much longer 2-μs simulation reported here revealed the force-induced exposure of the VBS (α-catenin activation). This uniquely long time scale allowed us to simulate α-catenin unfurling at relatively low force. The trajectory showing the domain reorientations and activation is shown in Supplemental Movie S1.
Movie S1.
Related to Fig. 6. Trajectory of the reported 2-μs, constant-force (100 pN) steered molecular dynamics (SMD) simulation.
Figure 5 shows the number of intact salt bridges between the MIII domain and the MI or MII domain during the SMD simulation. During the first 200 ns of the simulation, the MIII domain rotated relative to the MI–MII bundles (see Figure 6A). This rotation resulted in the formation of an intermediate conformation with drastically altered MI–MIII and MII–MIII interfaces. In this conformation, MI interacted with the opposite end of the MIII bundle, so that specific salt bridges between MI and MIII in the autoinhibited and intermediate conformations were mutually exclusive. However, the number of intact MI–MIII salt bridges was similar in each conformation (Figure 5). In contrast, the rotation of MIII brought MII and MIII into an approximately parallel configuration, in which there were 21 potential salt bridges between MII and MIII, compared with 4 that existed before the rotation of MIII. The average number of intact salt bridges between MII and MIII also increased following this rotation (Figure 5). These observations suggest that the MII-MIII interface is more robust in the intermediate conformation than in autoinhibited α-catenin.
FIGURE 5:
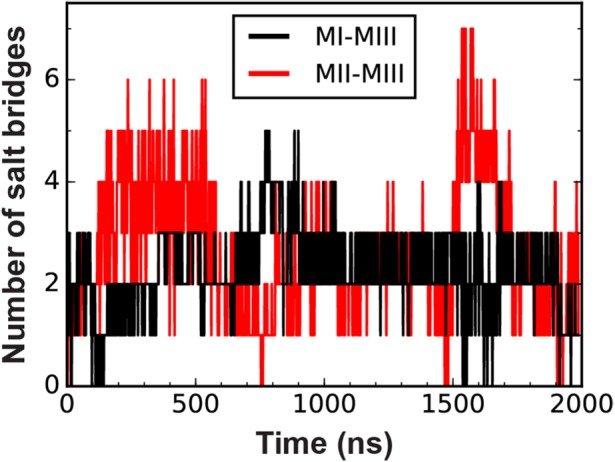
Salt bridges at interdomain interfaces in the α-catenin M domain. Traces indicate the number of salt bridges between MIII and either MI or MII during the 2-μs constant-force SMD simulation of the α-catenin M domain.
FIGURE 6:

Simulated unfolding of WT α-catenin under constant force. (A) Snapshots of α-catenin conformations at selected time points during a 2-μs SMD simulation of WT α-catenin M domain subject to a constant force of 100 pN. (See Supplemental Movie S1 for the entire trajectory.) Unraveled N-terminal helices are not shown for clarity. The light or dark gray box in the snapshot at 920 or 400 ns indicates a typical inset for the MI-MIII or MII-MIII interface, shown in B or C, respectively. (B, C) Insets showing salt bridges at interdomain interfaces at the simulation time points corresponding to the snapshots in A. (B) Salt bridges involving R326 at the MI-MIII interface of α-catenin. (C) Salt bridges involving R551 at the MII–MIII interface of α-catenin. (D, E) Evolution of distances between selected pairs of residues as a function of simulation time. Arrows indicate time points of snapshots in A–C. (D) D503-R551 is an interdomain salt bridge between MII and MIII; R326–D536 is a force-bearing contact between MI and MIII. (E) A316 and P635 are located near the ECFP and YPet of the α-catenin conformation sensor and are represented in A as blue and yellow spheres, respectively. G273 and P635 are the N- and C-terminal residues, respectively, of the M domain.
The snapshots of α-catenin configurations and plots of interresidue distances in Figure 6 highlight specific amino acids that played key roles in α-catenin unfurling. During the SMD simulation, R326 (located in the VBS, helix 3 of MI) participated in salt bridges with either MIII or helix 4 of MI. R551 (located in MIII) formed salt bridges with five different residues in MII, most frequently D503 or E492. This plasticity of the MII-MIII interface was also observed in shorter, 325-ns SMD simulations (Li et al., 2015). Several different salt-bridge configurations at the MI–MIII and MII–MIII interfaces are shown in Figure 6, B and C, respectively. Figure 6D highlights two particularly important salt bridges. R326–D536 (MI-MIII) appears to be a primary load-bearing interaction that ruptured immediately prior to α-catenin activation at 1909 ns, when the VBS separated from helix 4 in MI. D503–R551 is the only core MII–MIII salt bridge that forms both before and after the rotation of MIII.
The R326–E521 salt bridge (MI–MIII; Figure 6B, snapshot at 1 ns) ruptured during the rotation of MIII, and E521 rotated to the outside of the MIII domain such that R326–E521 could not reform. At the same time, R326 formed new salt bridges within MI; namely, with D392 (Figure 6B, 98 ns) or D388. The R326–D536 salt bridge (MI–MIII) first formed at 690 ns (see Figure 6D). By 920 ns, R326–D536 was the only remaining salt bridge between the VBS and MIII (Figure 6B). Immediately after R326–D536 broke at 1909 ns, α-catenin unfurled (Figure 6, A and D). Salt bridges between MIII and helix 4 of MI persisted during α-catenin activation (see Figure 5 and 6B), and might stabilize the remaining MI–MIII interface.
The D503–R551 salt bridge (MII–MIII) must break to allow for the full MIII rotation (see snapshots at 98 ns in Figure 6, A and C). However, D503–R551 reformed quickly after the rotation was complete (Figure 6D). At the new MII–MIII interface created by this rotation, R551 was located near several acidic amino acids in the inner helix of MII, including E492 (Figure 6C, 400 and 920 ns). During the simulation, R551 fluctuated between several salt bridges with five different amino acids, whereas D503 was only involved in salt bridges with either R551 or R548. Although D503–R551 formed only intermittently (Figure 6D), R551 or D503 was involved in salt bridges with at least one residue in MII or MIII for ∼53% or ∼26%, respectively, of the ∼1.5-μs lifetime of the intermediate state. These observations suggest that R551 plays a particularly important role in stabilizing both the intermediate and the autoinhibited conformation. Unexpectedly, the D503–R551 salt bridge forms again on VBS exposure, which might indicate that R551 also contributes to the stability of the unfurled conformation. Nonetheless, the higher apparent affinity between the VHD and R551A versus WT α-catenin (Figure 2) confirms that the loss of the D530–R551 salt bridge destabilizes the autoinhibited conformation.
Comparisons with FRET measurements.
To relate the simulated α-catenin conformational changes to the FRET results, we monitored the A316–P635 distance during the SMD simulation. Comparison of the A316–P635 (“FRET”) and G273–P635 (N- to C-terminal, “N-C”) distances over time suggests the structural basis of force-dependent changes in FRET signals (Figure 6E).
The computed FRET and N–C distances were both approximately constant from 400 ns to 1.9 μs, consistent with the formation of a relatively stable intermediate in the activation pathway. This intermediate is slightly extended relative to the autoinhibited conformation, with a 3-nm increase in the N–C extension. This extension can be seen in Figure 6A: helix 4 of MI (green helix) was wedged between MII and MIII in the autoinhibited conformation shown at 1 ns but not at later time points. In the intermediate conformation, the computed FRET distance is 2.6 nm longer than in the autoinhibited conformation, largely due to the separation of P635 and A316 on rotation of MIII. Using the reported value of R0 = 4.9 nm for the ECFP–YPet FRET pair (Patterson et al., 2000), we estimated that formation of this extended conformation would decrease by up to 80% the calculated FRET efficiency 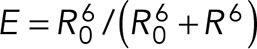 , where R is the interchromophore distance. Exposure of the VBS during α-catenin unfurling at 1909 ns results in a separation between A316 and P635 of >15 nm, a distance that would completely abrogate FRET. Although the simulated A316–P635 distance merely estimates the interchromophore distance in the α-catenin sensor, the above analyses suggest that the decreased FRET observed at high junction tension could reflect contributions from both the intermediate and the unfurled conformations.
, where R is the interchromophore distance. Exposure of the VBS during α-catenin unfurling at 1909 ns results in a separation between A316 and P635 of >15 nm, a distance that would completely abrogate FRET. Although the simulated A316–P635 distance merely estimates the interchromophore distance in the α-catenin sensor, the above analyses suggest that the decreased FRET observed at high junction tension could reflect contributions from both the intermediate and the unfurled conformations.
The nearly 30-nm increase in the N–C distance during the constant-force simulation exceeds the 16-nm extension reported for the force-induced activation of α-catenin in vitro (Yao et al., 2014). This difference can be accounted for by the unraveling of the first two helices of the MI bundle. The second helix is partially unstructured in crystal structures of α-catenin in complex with the VHD (Choi et al., 2012; Rangarajan and Izard, 2012). Thus unfolding of the N-terminal helices during the simulation likely reflects their structural instability relative to the rest of the M domain, but complete unraveling of these helices is unlikely at physiological levels of force.
DISCUSSION
α-Catenin is essential for intercellular adhesion and force transduction. The results reported here demonstrate that disrupting the salt-bridge network postulated to stabilize the M domain facilitates α-catenin unfurling. The higher energy of the M domain of the R551A and D503N salt-bridge mutants compared with WT in equilibrium MD simulations (Table 1) suggested that the mutants destabilize the autoinhibited conformation of the M domain thermodynamically. Both the higher equilibrium constant KU determined in vitro for R551A and the increase in unfurled cytosolic R551A and D503N compared with WT α-catenin (Figure 4D) bear out this prediction.
Under the highest-tension conditions in our FRET measurements–-that is, at junctions between untreated cells–-E-cadherin experiences pN-scale forces (Borghi et al., 2012). Changes in FRET/ECFP ratios following the disruption of cadherin adhesion or changes in endogenous cell contractility, which affect junction tension, previously confirmed the sensitivity of the α-catenin conformation to physiological forces (Yonemura et al., 2010; Kim et al., 2015; see also Figure 4, B and C). Here, use of the conformation-specific α18 anti–α-catenin antibody (Nagafuchi et al., 1994; Yonemura et al., 2010) also verified that the FRET/ECFP ratios reflect the sensor conformation (Figure 4E). The insensitivity of cytosolic FRET/ECFP ratios to EGTA or CytoD treatment confirmed that the observed FRET changes at junctions reflect tension-dependent α-catenin conformation changes (Kim et al., 2015; see also Figure 4D). In addition, prior studies ruled out the contribution of intermolecular FRET to measured changes in FRET/ECFP, based on the absence of FRET/ECFP changes in measurements with sensor mutants that quenched either ECFP or YPet fluorescence (Kim et al., 2015). Thus the lower FRET/ECFP ratios at junctions between cells expressing the R551A or D503N sensor (Figure 4) reflect the increased population of unfurled α-catenin mutants relative to WT under physiological tension.
Given the estimated FRET decrease on rotation of MIII and the relatively long ∼1.5-μs in silico lifetime of the intermediate under a 100-pN force, the measured FRET/ECFP ratios likely reflect contributions from formation of the intermediate as well as complete activation. Nevertheless, correlations between decreased FRET and vinculin recruitment during acute mechanical perturbations previously demonstrated that the FRET sensor reports the real-time activation of α-catenin at cell–cell junctions (Kim et al., 2015). The constant-force SMD simulation rules out further decreases in FRET due to postactivation conformational changes, in contrast to a recent suggestion (Biswas et al., 2016).
One might initially expect a greater difference between the FRET/ECFP ratios of R551A or D503N and WT α-catenin at high-tension (untreated) versus low-tension (EGTA- or CytoD-treated) junctions. However, the percentage differences were statistically similar for WT and the salt-bridge mutants under each condition studied (see Figure 4, C and D). Predicting how a constant, steady-state force might affect the free energy ΔG of α-catenin unfurling requires knowledge of how force affects the free-energy landscape of the conformational change (Yew et al., 2008). We do not know how force or the salt-bridge mutations alter the free-energy landscape, but our results suggest that the tension-independent term ΔΔG0 dominates observed differences between R551A or D503N and WT at established junctions. This observation is consistent with the predicted key role of salt bridges in the force-induced activation of α-catenin because the R551A and D503N mutants mimic force-induced salt-bridge disruption in WT α-catenin.
Apparent differences between the extent of α-catenin unfurling in the cytosol relative to junctions under low tension (EGTA- or CytoD-treated; see Supplemental Table S1) are attributed to the restricted mobility of α-catenin tethered at junctions versus freely rotating in the cytosol. Restricted mobility would contribute to quantitative differences in FRET measured in these environments through its effect on the orientation factor κ2 (Lakowicz, 2006). However, the FRET/ECFP ratios might suggest that some cytosolic α-catenin adopts a force-independent, activated conformation. This possibility was suggested by immunostaining results (Biswas et al., 2016) with the conformation-specific α18 antibody. The existence of force-independent, unfurled α-catenin pools is not surprising. Cytosolic α-catenin dimerization (Koslov et al., 1997; Drees et al., 2005) and/or interactions with proteins such as α-actinin or formins (Nieset et al., 1997; Kobielak et al., 2004) could shift the conformational equilibrium. However, such tension-independent pools of activated α-catenin would not rule out real-time, force-activated conformational changes at junctions, as confirmed experimentally (Yonemura et al., 2010; Yao et al., 2014; Kim et al., 2015).
The computed increase in the MII–MIII interaction energy in the salt-bridge mutants R551A and D503N relative to WT (Table 2) is larger than the destabilization (ΔΔG) of R551A relative to WT α-catenin in solution binding measurements. The internal energies cannot be compared quantitatively with experimentally determined free-energy differences for several reasons. First, the computed interdomain energy is the interaction energy of the interface between the two domains, whereas the Gibbs free energy (obtained from solution binding data) reflects the free-energy difference between fully solvated, dissociated versus associated domains. The internal energy calculations thus do not account for charge screening by nearby ions or water molecules. In addition, the dielectric constant at the interface is not known, and the dielectric constant of unity used in the calculations would overestimate electrostatic contributions. Furthermore, the overestimation of the association constant KA for salt-bridge formation in molecular dynamics simulations was reported previously (Debiec et al., 2014). Despite these limitations, the computed interdomain interaction energies enable qualitatively meaningful comparisons of the impact of structural perturbations on interdomain interactions that can be used to interpret experimental data.
Under dynamic force loading at resealing junctions, during the initial 5 min after calcium addition, more vinculin bound R551A than WT α-catenin. The decrease in the vinculin/α-catenin ratio at t > 5 min after cadherin activation is consistent with the reported kinetics of vinculin phosphorylation at tyrosine 822 (pY822), which directs vinculin to cell–cell junctions (Bays et al., 2014). The ratio of pY822 vinculin to total vinculin reportedly peaks at 5 min and is significantly lower at stable junctions. We therefore speculate that vinculin phosphorylation dynamics contribute to the kinetics of vinculin accumulation shown in Figure 3. Clearly, the extent of α-catenin unfurling is one of many factors that affect vinculin recruitment to junctions. Nonetheless, our data demonstrate that salt-bridge disruption enhances vinculin binding under dynamic force loading in live cells, an important functional consequence of enhanced α-catenin unfurling.
The 2-μs, all-atom SMD simulation revealed a potential mechanism for tension-dependent α-catenin activation. The rotation of MIII triggers the formation of an intermediate conformation with altered MI–MIII and MII–MIII interfaces that appears to allow the redistribution of force across α-catenin. This intermediate, which is extended by ∼3 nm relative to the autoinhibited conformation, may correspond to the structure that was suggested by a 2.8-nm increase in the protein extension prior to the force-induced unfolding of α-catenin by atomic force microscopy (Maki et al., 2016). The MI–MIII interface in the intermediate structure includes a newly identified salt bridge between R326 and D536, which appears to be a key force-bearing interaction that ruptures immediately before α-catenin activation. The MII–MIII interface persists during α-catenin activation, which involves the unbundling of MI to expose the VBS. The higher number of salt bridges at the MII–MIII interface in the intermediate relative to the autoinhibited conformation (Figure 5) might contribute to the ability of MII–MIII to remain intact while MI unfurls. The D503–R551 salt bridge seems to play a particularly important role in stabilizing the α-catenin structure throughout activation, in addition to stabilizing the salt-bridge network in the autoinhibited conformation (Li et al., 2015). Although we did not observe the intermediate or unfurled states of the R551A or D503N α-catenin M domain in our simulations, the higher experimentally determined unfurling equilibrium constant KU for R551A relative to WT confirms that the R551A mutation destabilizes autoinhibited α-catenin relative to the unfurled conformation. Future studies will examine the potential role of the R326–D536 salt bridge in α-catenin activation.
The simulated activation trajectory is consistent with magnetic tweezers measurements, in which exposure of the VBS preceded the disruption of a relatively stable structure (Yao et al., 2014). We postulated that the latter structure was the rotated MII–MIII structure (Li et al., 2015), in which the constant-force simulation revealed a higher number of intact salt bridges (Figure 5). In the simulation, the N-to-C-terminal distance increases by ∼3 nm during formation of the intermediate and by an additional ∼10 nm during VBS exposure. This accounts for most of the 16-nm extension observed during the force-induced activation of α-catenin by magnetic tweezers (Yao et al., 2014). The remaining extension is likely due to unbundling and/or partial unraveling of helices 1 and 2 of MI, which completely unraveled under the simulated force of 100 pN, but are unlikely to do so at physiological forces.
In summary, these equilibrium binding studies, FRET measurements, and molecular dynamics simulations demonstrate how salt bridges within the core M domain gate α-catenin activation. The disrupted salt-bridge network in the R551A mutant increased the force-independent equilibrium constant for unfurling 13-fold. Use of a FRET-based α-catenin conformation sensor in cells demonstrated that the R551A and D503N mutants increased the population of unfurled α-catenin in different mechanical environments, relative to WT. Molecular dynamics simulations revealed possible atomic-level details that underlie the force-induced activation of α-catenin and the role of salt bridges in this process. The simulations agree remarkably well with these and other experimental findings and deepen our understanding of the mechanism of molecular force transduction at critical intercellular adhesions.
MATERIALS AND METHODS
Cell culture and transfection
All cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 1 U/ml penicillin, 100 μg/ml streptomycin, and 1 mM sodium pyruvate (“culture medium”) unless otherwise indicated.
MDCK cells were from the American Type Culture Collection (ATCC). MDCK cells were transiently transfected with the FRET-based α-catenin conformation sensor (Kim et al., 2015) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
The R2/7 line is an α-catenin-null subclone of the DLD-1 cell line derived from human colorectal adenocarcinoma (Vermeulen et al., 1995). R2/7 cells were a gift from Johan deRooij (University Medical Center Utrecht). The absence of α-catenin was verified by immunofluorescence imaging and by Western blot. R2/7 cells stably expressing either the WT or the R551A mutant of the α-catenin FRET sensor were maintained by culturing the cells in culture medium supplemented with 6 μg/ml puromycin. Expression of the sensor was verified by fluorescence imaging (Supplemental Figure S1).
Cloning and creation of stable cell lines
The α-catenin conformation sensor was subcloned from the pcDNA3.1 plasmid (Kim et al., 2015) into the pCDH-CMV-MCS-EF1-Puro lentiviral expression vector (System Biosciences) between the Nhe1 and Not1 restriction sites. The R551A mutation was generated by PCR amplification of the lentiviral α-catenin conformation sensor construct using Phusion high-fidelity DNA polymerase (New England Biolabs, NEB). Primer sequences are provided in the Supplemental Experimental Procedures. Following treatment with restriction enzyme Dpn1 (NEB), the PCR product was transformed into competent Escherichia coli DH5α cells. The R551A mutation was verified by sequencing of the isolated plasmid.
VSV-G pseudotyped virus particles were generated by transfecting human embryonic kidney 293T (HEK293T) cells with the α-catenin conformation sensor in the pCDH vector backbone, together with packaging plasmids psPAX2 and pMD2.G. HEK293T cells were a gift from Cara Gottardi (Northwestern). pMD2.G and psPAX2 were gifts from Didier Trono (École Polytechnique Fédérale de Lausanne; Addgene plasmid #12259 and #12260). HEK293T cells were transfected using a standard calcium phosphate protocol. Plasmid DNA (10 μg α-catenin pCDH expression construct, 7.5 μg psPAX2, 5 μg pMD2.G) was combined with 50 μl of 2 M CaCl2 and diluted to 500 μl with nuclease-free water. The plasmid-CaCl2 solution was added to 500 μl of HEPES-buffered saline (140 mM NaCl, 1.5 mM Na2HPO4, 50 mM HEPES) and incubated 30 min at room temperature before being added dropwise onto HEK293T cells cultured on a 100-mm cell culture dish. At 6 h posttransfection, the cell culture medium was removed by aspiration and replaced with 5 ml of virus harvesting medium (DMEM supplemented with 10% FBS, 6 mM glutamine, 1× MEM Non-Essential Amino Acids Solution [Life Technologies], 1.1 g/l bovine serum albumin [BSA], and 1 mM sodium pyruvate). Packaged viral particles collected from 12 100-mm dishes at 24 h posttransfection were concentrated by centrifugation at 20,000 rpm for 2 h in a Beckman L8-70 ultracentrifuge and then resuspended in 300 μl of virus harvesting medium.
R2/7 cells were grown to 60% confluence in T25 cell culture flasks and infected for 24 h with 50 μl of concentrated virus diluted in 5 ml culture medium supplemented with 10 μg/ml Polybrene (Santa Cruz Biotechnology). Infected cells were washed with phosphate-buffered saline (PBS) and cultured for an additional 48 h. At 72 h postinfection, cells were rinsed in PBS and detached by with TrypLE Express with phenol red (1×; ThermoFisher Scientific) and resuspended in PBS. The cells were then sorted according to the intensities of the ECFP and YPet fluorophores in a FACSAria cell sorter (BD Biosciences) at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana–Champaign (UIUC). Cells with highest fluorescence intensities for both the donor and acceptor fluorophores (top 5% of the total population) were selected and cultured in T25 flasks with culture medium supplemented with 8 μg/ml puromycin.
Live-cell FRET imaging
In the α-catenin conformation sensor, ECFP is inserted between residues 315 and 316 and YPet is inserted between residues 639 and 640 (Kim et al., 2015). Residues 315 and 316 are located N-terminal to the VBS within the MI helix bundle. Although the insertion of ECFP disrupts the second helix (h2) within the MI bundle, it is not expected to perturb the key interactions between h3 and h4 that encrypt the VBS, as observed in the crystal structure (Ishiyama et al., 2013) and in the constant-force SMD simulation (Figure 6A). This expectation is borne out in prior functional studies that verified that the sensor both recapitulates α-catenin function in α-catenin-depleted cells and responds to changes in tension at the junction (Kim et al., 2015). Residues 639 and 640 are part of the unstructured linker region between MIII and the C-terminal domain (Ishiyama et al., 2013; Rangarajan and Izard, 2013), such that the insertion of YPet should not significantly affect α-catenin unfurling. The function of the R551A mutant was verified by its localization at MDCK and R2/7 cell–cell junctions (Figure 4 and Supplemental Figure S1), as well as by the increased recruitment of vinculin to resealing junctions between R2/7 cells (Figure 3).
Each of MDCK and R2/7 cells expressing the α-catenin conformation sensor were detached with TrypLE Express (1×; ThermoFisher Scientific) and seeded on collagen-coated glass. Petri dishes with #1.5 cover glass (Cell E&G) were functionalized with collagen by incubation with 20 μg/ml collagen overnight at 4°C. Live-cell fluorescence images were obtained at 6 h after seeding, using a Zeiss Axiovert 200 inverted microscope and MetaFluor 6.2 software (Molecular Devices). For the +EGTA condition, cells were treated with 2 mM EGTA 2 min before imaging. For the +CytoD condition, cells were treated with 1 μM CytoD 10 min before imaging. The background-subtracted ratio of FRET to donor (ECFP) emission (FRET/ECFP) was determined in regions of interest (ROIs) at cell–cell junctions or in the cytosol with MetaFluor. ROIs used for background subtraction were defined near each transfected cell. Average FRET/ECFP ratios of triplicate images taken at each ROI and average FRET/ECFP ratios for each condition were calculated with Excel (Microsoft). The number of ROIs analyzed and the number of independent experiments performed for each condition are provided in Supplemental Table S1. The statistical significance of differences between mean values was assessed by one-way analysis of variance supported by OriginPro software (OriginLab). A p value < 0.001 defines a statistically significant difference at the 99.9% confidence level.
Immunofluorescence imaging
The ratio of vinculin to α-catenin was determined in ROIs at reannealing junctions, following a calcium switch. Subconfluent R2/7 cells expressing the α-catenin conformation sensor were treated with 2 mM EGTA for 2 min to disrupt cadherin junctions. Next, the cells were bathed with DMEM (∼2 mM Ca2+) to induce junction reannealing. At defined time points after calcium addition, cells were fixed and immunostained. In the control condition, cells were cultured in DMEM (∼2 mM Ca2+) but were not subjected to calcium removal before fixation. Cells were fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.1% (wt/vol) Triton X-100 in PBS for 5 min. After blocking with 1% (wt/vol) BSA in PBS for 20 min, cells were immunostained for vinculin (mouse anti-vinculin, Sigma cat. # V9131, 1:200 dilution; goat anti-mouse immunoglobulin G [IgG] Alexa 647, Invitrogen cat. # A21236, 1:200 dilution) and α-catenin (rabbit anti–α-catenin, Sigma cat. # C2081, 1:200 dilution; goat anti-rabbit IgG FITC, Sigma cat. # F9887, 1:200 dilution).
In measurements of the relative intensities of α18/anti–α-catenin (activated/total α-catenin), R2/7 cells stably expressing either WT or R551A forms of the α-catenin sensor were seeded on collagen-coated glass (∼80% confluence) and cultured overnight in DMEM. The cells were then fixed and stained for total α-catenin with rabbit anti–α-catenin (as above) and for activated α-catenin with the conformation-specific α18 antibody (rat anti–α-catenin, gift from Akira Nagafuchi, Nara Medical University, Japan, 1:400 dilution; goat anti-rat Alexa 647, Abcam, cat. no. ab150159, 1:400 dilution).
Images were acquired with a Zeiss LSM 700 laser-scanning confocal microscope equipped with a Plan Apochromat 63 × 1.4 NA oil immersion objective lens (Zeiss) and ZEN 2008 software (Zeiss). The fluorescence intensities of vinculin (Alexa 647) or α-catenin (FITC) at junctions were quantified using ImageJ software (version 1.44; National Institutes of Health). Average vinculin/α-catenin intensity ratios were calculated with Excel. At least 20 cells and at least 41 ROIs were analyzed for each condition. The statistical significance of differences between two mean values was assessed by a Welch’s t test supported by Excel. A p value < 0.05 defines a statistically significant difference at the 95% confidence level.
When comparing the levels of activated α-catenin at static junctions between R2/7 cells, the expression levels of the α-catenin sensor was found to affect cell packing and junction organization, both of which would likely affect the intercellular tension. To minimize variability in α18 α-catenin measurements that could arise from such heterogeneity, we focused on cells with similar sensor expression levels, based on the intensity of FITC fluorescence. We further limited measurements (and comparisons) to cells within similar-sized clusters and away from edges of the clusters where cells could be more contractile. These measures significantly reduced variability in determined α18/α-catenin ratios that were obtained with cells expressing either the WT or R551A sensor. For comparisons of the α18/ α-catenin ratios, at least 220 junctions from two independent experiments were analyzed.
Protein expression and purification
Escherichia coli BL21(DE3) cells expressing either full-length mouse His6-αE-catenin (WT or R551A, gifts from Mitsu Ikura, University of Toronto) or human VHD (residues 1–252 with N-terminal GST tag, gift from Mitsu Ikura, University of Toronto) were grown in Lennox LB medium (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl). Expression of His6-α-catenin or VHD was induced with 0.1 or 1 mM isopropyl β-d-1-thiogalactopyranoside, respectively. The bacterial cultures were pelleted by centrifugation at 5000 rpm for 10 min in a Sorvall RC-5C Plus centrifuge equipped with a GS3 rotor. Pellets were frozen at –20°C at least overnight, then thawed and resuspended in lysis buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 5 mM MgCl2, 10 mm β-mercaptoethanol, 1 mM Tris(2-carboxyethyl)phosphine [TCEP], 1× Complete EDTA-free protease inhibitor cocktail [Roche], 0.67 mg/ml lysozyme) and incubated on ice for 30 min. The resuspended pellets were sonicated using a Qsonica sonicator at 70% intensity, using six 1-s pulses per min for 4 min total sonication time. The cell lysate was clarified by centrifugation at 17,000 rpm for 45 min in a Sorvall RC-5C Plus centrifuge equipped with an SS34 rotor. The clarified lysate was filtered through a 0.45-μm filter (Sarstedt) before loading onto an affinity column. The His6-α-catenin was bound to a Ni-NTA agarose (Qiagen), washed with 20 mM imidazole, and eluted with 100 mM imidazole. The constitutively active vinculin head domain was bound to glutathione resin (GenScript) and washed with wash buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 5 mM MgCl2, 1 mM TCEP). The GST tag was cleaved on-column by overnight incubation at 4°C with 200 U bovine thrombin (BioPharm Laboratories) in two column volumes wash buffer without TCEP. Both proteins were further purified by anion exchange chromatography on a Pharmacia HiTrap Q Sepharose HP column. Buffer was exchanged with storage buffer (15 mM Tris, pH 8.0, 300 mM NaCl, 5 mM MgCl2) using Amicon Ultra-15 10 kDa MWCO centrifugal filters (EMD Millipore).
BioLayer Interferometry
Solution binding measurements were performed with the hexahistidine-tagged, full-length αE-catenin. Purified WT α-catenin and the R551A mutant both retain vinculin-binding activity, as documented previously (Ishiyama et al., 2013). BLI measurements were carried out on a ForteBio Octet QK system. All protein solutions were diluted in assay buffer (15 mM Tris, pH 8.0, 300 mM NaCl, 5 mM MgCl2, 0.1 mg/ml BSA). BSA was included in the assay buffer to minimize nonspecific VHD binding to the sensor. Ni-NTA biosensors (ForteBio) were hydrated at least 20 min in assay buffer before beginning the assay. All assay steps were carried out at 30°C. After a 10-min delay with shaking to mix the 96-well plate containing the protein solutions, hydrated biosensors were equilibrated 1 min in assay buffer, loaded with 50–100 nM His6-α-catenin (WT or R551A) until binding reached ∼1 nm, and washed 2 h in assay buffer. A 15-min baseline was then acquired in assay buffer. Next, parallel α-catenin-loaded sensors were submerged in a range of concentrations of VHD (200 nM to 20 μM for WT, 50 nM to 3.2 μM for R551A). The highest concentration corresponds to ∼10× Kd in each case. Any nonspecific adsorption of BSA to the sensors was controlled for by subtracting from all association traces the trace for a loaded sensor immersed in assay buffer.
It should be noted that although the α-catenin in solution exists as a mix of monomer and homodimer, we expect the protein on the sensor to be primarily monomeric because the 50–100 nM concentrations used for loading are low relative to the 25 μM Kd for α-catenin dimerization (Pokutta et al., 2014). At these concentrations, the α-catenin in solution is 99% monomeric at equilibrium. However, because the oligomeric state of immobilized α-catenin was not verified explicitly, we cannot rule out the presence of some dimers.
Dissociation constants (Kd) were determined using Octet 8.0 Data Analysis software (ForteBio) to fit the steady-state response to the following formula, in which Rmax is the response maximum and A is the analyte concentration:
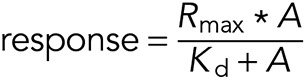 |
The dissociation constants determined from each independent experiment, as well as the R2 and χ2 goodness-of-fit measures for the steady-state fits to the data, are provided in Supplemental Table S2. The uncertainty in the average Kd value was determined by the propagation of error from the individual fits.
Molecular dynamics simulations
Molecular dynamics simulations were performed with the M domain (residues 273–635) of the human αE-catenin monomer (chain A), the structure of which was obtained from the crystal structure of the dimer (PDB ID 4IGG; Rangarajan and Izard, 2013). This study did not investigate the dimer because only the α-catenin monomer binds to β-catenin at cadherin adhesions. The core M domain (MI–MIII) was essentially identical in both protomers in the asymmetric dimer of human αE-catenin (Rangarajan and Izard, 2013), as well as in the mouse αE-catenin dimer structure (Ishiyama et al., 2013; Ishiyama, personal communication). We used the monomer structure extracted from the human α-catenin dimer because it contains a more complete M domain structure (residues 273–635, as opposed to 290–631) and was determined at higher resolution (3.7 Å, compared with 6.5 Å) compared with the mouse α-catenin structure. We used this same structure in our prior simulations (Li et al., 2015). Slight differences in the N-terminal domains of the full-length protein structures were not relevant to these simulations, because we previously showed that the M-domain unfolds independently of the N-terminal domain (Li et al., 2015).
All ionizable residues in the α-catenin M domain were assigned their default protonation states. The peptide bond between G274 and G275 was changed from the cis to trans configuration using the Cispeptide plug-in of VMD (Schreiner et al., 2011). The Solvate and Autoionize plug-ins of VMD were used to solvate the system in a water box with at least 15 Å between the protein and the boundary of the box and to add 150 mM NaCl and neutralize the net charge of the system, respectively (Humphrey et al., 1996).
All-atom MD simulations were performed using NAMD2 (Phillips et al., 2005), the CHARMM27 force field for proteins and ions (MacKerell et al., 1998, 2004; Foloppe and MacKerell, 2000), and the TIP3P model for explicit water (Jorgensen et al., 1983). NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana–Champaign. All simulations were performed using periodic boundary conditions and a time step of 2 fs. A constant temperature of 310 K was maintained using Langevin dynamics with a damping coefficient of 0.5 ps−1 (Martyna et al., 1994; Feller et al., 1995). Short-range, nonbonded interactions were calculated using a cutoff distance of 12 Å, and long-range electrostatic forces were described with the particle mesh Ewald (PME) method (Darden et al., 1993). Throughout the simulations, bond distances involving hydrogen atoms were fixed using the SHAKE algorithm (Ryckaert et al., 1977). After an initial 10,000 steps of energy minimization with all alpha carbon (Cα) atoms fixed, the system was equilibrated in a constant number, volume, and temperature (NVT) ensemble for 500 ps, during which all protein Cα atoms were fixed to allow relaxation of the side chains and water. Subsequent equilibrium simulations were performed in an NVT ensemble. In constant-force SMD simulations of forced unfolding of α-catenin, a 100-pN pulling force was applied to the Cα atom of N-terminal residue 273. The Cα atom of C-terminal P635 was fixed to prevent the overall translation of the system in response to the applied external force. The force was directed along the x-axis between the two anchoring points, and the C-to-N direction was defined as positive. The water box containing the equilibrated structure was extended by an extra 300 Å in the positive x direction to accommodate the unfolded structure.
Simulation outputs were analyzed using VMD (Humphrey et al., 1996) and plotted using the matplotlib plotting library (Hunter, 2007). The first 5 ns of each equilibrium simulation were excluded from analysis to avoid biasing the results toward the initial coordinates. Internal and interdomain energies were computed using the NAMD Energy plug-in built in to VMD (Humphrey et al., 1996; Phillips et al., 2005). We used the A316–P635 distance during the SMD simulation as an approximation of the interchromophore distance in the α-catenin conformation sensor. We used residue 635 as a proxy for the YPet position (between residues 639 and 640) because the amino acids 636–665 in the α-catenin crystal structure are disordered (Rangarajan and Izard, 2013). Snapshots of α-catenin conformations were created using the Tachyon ray tracing library built-in to VMD (Stone, 1998).
Supplementary Material
Acknowledgments
This work was supported, in whole or in part, by National Science Foundation Grant CHE 12-13755 (DEL), by National Institutes of Health Grants P41-GM104601 and 1 RO1 GM097443-01A1, and by a supercomputing allocation through XSEDE Grant MCA06N060 and the Illinois Campus Cluster Program. This research is part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana–Champaign and its National Center for Supercomputing Applications. We acknowledge Saiko Rosenburger for technical assistance, Mitsu Ikura and Noboru Ishiyama for useful discussions, and Steve Huber for use of the BLI instrument. We acknowledge the flow cytometry facility of the Roy J. Carver Biotechnology Center, UIUC, for assistance with cell sorting. We dedicate this article to the memory of Klaus Schulten.
Abbreviations used:
- VBS
vinculin-binding site
- VHD
vinculin head domain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-03-0168) on November 15, 2017.
REFERENCES
- Barry AK, Tabdili H, Muhamed I, Wu J, Shashikanth N, Gomez GA, Yap AS, Gottardi CJ, de Rooij J, Wang N, Leckband DE. α-Catenin cytomechanics–role in cadherin-dependent adhesion and mechanotransduction. J Cell Sci. 2014:1779–1791. doi: 10.1242/jcs.139014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays JL, Peng X, Tolbert CE, Guilluy C, Angell AE, Pan Y, Superfine R, Burridge K, DeMali KA. Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol. 2014;205:251–263. doi: 10.1083/jcb.201309092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas KH, Hartman KL, Zaidel-Bar R, Groves JT. Sustained α-catenin activation at E-cadherin junctions in the absence of mechanical force. Biophys J. 2016;111:1044–1052. doi: 10.1016/j.bpj.2016.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, Nelson WJ, Dunn AR. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc Natl Acad Sci USA. 2012;109:12568–12573. doi: 10.1073/pnas.1204390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 2014;346:1254211. doi: 10.1126/science.1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Pokutta S, Cadwell GW, Bobkov AA, Bankston LA, Liddington RC, Weis WI. αE-catenin is an autoinhibited molecule that coactivates vinculin. Proc Natl Acad Sci USA. 2012;109:8576–8581. doi: 10.1073/pnas.1203906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Debiec KT, Gronenborn AM, Chong LT. Evaluating the strength of salt bridges: a comparison of current biomolecular force fields. J Phys Chem B. 2014;118:6561–6569. doi: 10.1021/jp500958r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Sarpal R, Ishiyama N, Pellikka M, Ikura M, Tepass U. Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat Cell Biol. 2013;15:261–273. doi: 10.1038/ncb2685. [DOI] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband DE, de Rooij J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189:1107–1115. doi: 10.1083/jcb.201001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103:4613. [Google Scholar]
- Foloppe N, MacKerell ADJ. All-atom empirical force field for nucleic acids. I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comput Chem. 2000;21:86–104. [Google Scholar]
- Gao J, Aksoy B, Dogrusoz U, Dresdner G. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science. 2013;6:1–20. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14:27-28-38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:99–104. [Google Scholar]
- Imamura Y, Itoh M, Maeno Y, Tsukita S, Nagafuchi A. Functional domains of α-catenin required for the strong state of cadherin-based adhesion. J Cell Biol. 1999;144:1311–1322. doi: 10.1083/jcb.144.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Tanaka N, Abe K, Yang YJ, Abbas YM, Umitsu M, Nagar B, Bueler SA, Rubinstein JL, Takeichi M, Ikura M. An autoinhibited structure of α-catenin and its implications for vinculin recruitment to adherens junctions. J Biol Chem. 2013;288:15913–15925. doi: 10.1074/jbc.M113.453928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, Craig SW. An intramolecular association between the head and tail domains of vinculin modulates talin binding. J Biol Chem. 1994;269:12611–12619. [PubMed] [Google Scholar]
- Johnson RP, Craig SW. F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature. 1995;373:261–264. doi: 10.1038/373261a0. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kim T, Zheng S, Sun J, Muhamed I, Wu J, Lei L, Kong X, Leckband DE, Wang Y. Dynamic visualization of α-Catenin reveals rapid, reversible conformation switching between tension states. Curr Biol. 2015;25:218–224. doi: 10.1016/j.cub.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak A, Pasolli HA, Fuchs E. Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol. 2004;6:21–30. doi: 10.1038/ncb1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslov ER, Maupin P, Morrow JS, Rimm DL, Pradhan D. α-Catenin can form asymmetric homodimeric complexes and/or heterodimeric complexes with β-catenin. J Biol Chem. 1997;272:27301–27306. doi: 10.1074/jbc.272.43.27301. [DOI] [PubMed] [Google Scholar]
- Ladoux B, Anon E, Lambert M, Rabodzey A, Hersen P, Buguin A, Silberzan P, Mège R-M. Strength dependence of cadherin-mediated adhesions. Biophys J. 2010;98:534–542. doi: 10.1016/j.bpj.2009.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy, 3rd Ed. New York: Plenum Press; 2006. [Google Scholar]
- Lecuit T, Lenne P-F, Munro E. Force generation, transmission, and integration during cell and tissue morphogenesis. Annu Rev Cell Dev Biol. 2011;27:157–184. doi: 10.1146/annurev-cellbio-100109-104027. [DOI] [PubMed] [Google Scholar]
- Leerberg JM, Gomez GA, Verma S, Moussa EJ, Wu SK, Priya R, Hoffman BD, Grashoff C, Schwartz MA, Yap AS. Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr Biol. 2014;24:1689–1699. doi: 10.1016/j.cub.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Newhall J, Ishiyama N, Gottardi C, Ikura M, Leckband DE, Tajkhorshid E. Structural determinants of the mechanical stability of α-catenin. J Biol Chem. 2015;290:18890–18903. doi: 10.1074/jbc.M115.647941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci USA. 2010;107:9944–9949. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell AD Jr, Feig M, Brooks CL 3rd. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- MacKerell ADJ, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Maki K, Han S-W, Hirano Y, Yonemura S, Hakoshima T, Adachi T. Mechano-adaptive sensory mechanism of α-catenin under tension. Sci Rep. 2016;6:24878. doi: 10.1038/srep24878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- Nagafuchi A, Ishihara S, Tsukita S. The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin-α catenin fusion molecules. J Cell Biol. 1994;127:235–245. doi: 10.1083/jcb.127.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieset JE, Redfield AR, Jin F, Knudsen KA, Johnson KR, Wheelock MJ. Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin. J Cell Sci. 1997;110:1013–1022. doi: 10.1242/jcs.110.8.1013. [DOI] [PubMed] [Google Scholar]
- Pappas DJ, Rimm DL. Direct interaction of the C-terminal domain of alpha-catenin and F-actin is necessary for stabilized cell-cell adhesion. Cell Commun Adhes. 2006;13:151–170. doi: 10.1080/15419060600726142. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Piston DW, Barisas GB. Förster distances between green fluorescent protein pairs. Anal Biochem. 2000;284:438–440. doi: 10.1006/abio.2000.4708. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Choi H-J, Ahlsen G, Hansen SD, Weis WI. Structural and thermodynamic characterization of cadherin-β-catenin-α-catenin complex formation. J Biol Chem. 2014;289:13589–13601. doi: 10.1074/jbc.M114.554709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan ES, Izard T. The cytoskeletal protein α-catenin unfurls upon binding to vinculin. J Biol Chem. 2012;287:18492–18499. doi: 10.1074/jbc.M112.351023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan ES, Izard T. Dimer asymmetry defines α-catenin interactions. Nat Struct Mol Biol. 2013;20:188–193. doi: 10.1038/nsmb.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. α1(E)-catenin is an actin-binding and actin-bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with contraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- Schreiner E, Trabuco LG, Freddolino PL, Schulten K. Stereochemical errors and their implications for molecular dynamics simulations. BMC Bioinform. 2011;12:190. doi: 10.1186/1471-2105-12-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE. An Efficient Library for Parallel Ray Tracing and Animation. MS Thesis. 1998. University of Missouri–Rolla.
- Thomas WA, Boscher C, Chu YS, Cuvelier D, Martinez-Rico C, Seddiki R, Heysch J, Ladoux B, Thiery JP, Mege RM, Dufour S. α-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J Biol Chem. 2013;288:4957–4969. doi: 10.1074/jbc.M112.403774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss F, le Duc Q, Van Der Horst S, Tabdili H, Van Der Krogt G, Wang N, Rehmann H, Huveneers S, Leckband DE, de Rooij J. Vinculin-dependent Cadherin mechanosensing regulates efficient epithelial barrier formation. Biol Open. 2012;1:1128–1140. doi: 10.1242/bio.20122428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen SJ, Bruyneel EA, Bracke ME, De Bruyne GK, Vennekens KM, Vleminckx KL, Berx GJ, van Roy FM, Mareel MM. Transition from the noninvasive to the invasive phenotype and loss of alpha-catenin in human colon cancer cells. Cancer Res. 1995;55:4722–4728. [PubMed] [Google Scholar]
- Watabe-Uchida M, Uchida N, Imamura Y, Nagafuchi A, Fujimoto K, Uemura T, Vermeulen S, Van Roy F, Adamson ED, Takeichi M. α-Catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J Cell Biol. 1998;142:847–857. doi: 10.1083/jcb.142.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Qiu W, Liu R, Efremov AK, Cong P, Seddiki R, Payre M, Lim CT, Ladoux B, Mege RM, Yan J. Force-dependent conformational switch of α-catenin controls vinculin binding. Nat Commun. 2014;5 doi: 10.1038/ncomms5525. [DOI] [PubMed] [Google Scholar]
- Yew ZT, Krivov S, Paci E. Free-energy landscapes of proteins in the presence and absence of force. J Phys Chem B. 2008;112:16902–16907. doi: 10.1021/jp807316e. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. α-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12:533–542. doi: 10.1038/ncb2055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.