

The tricellular junction (TCJ) forms at the convergence of three neighboring epithelia. The targeting of Gliotactin, an essential TCJ protein, to the TCJ is controlled by phosphorylation and endocytosis. C-terminal Src kinase controls endocytosis of Gliotactin in an Src-independent manner.
Abstract
Tricellular junctions (TCJs) are uniquely placed permeability barriers formed at the corners of polarized epithelia where tight junctions in vertebrates or septate junctions (SJ) in invertebrates from three cells converge. Gliotactin is a Drosophila TCJ protein, and loss of Gliotactin results in SJ and TCJ breakdown and permeability barrier loss. When overexpressed, Gliotactin spreads away from the TCJs, resulting in disrupted epithelial architecture, including overproliferation, cell delamination, and migration. Gliotactin levels are tightly controlled at the mRNA level and at the protein level through endocytosis and degradation triggered by tyrosine phosphorylation. We identified C-terminal Src kinase (Csk) as a tyrosine kinase responsible for regulating Gliotactin endocytosis. Increased Csk suppresses the Gliotactin overexpression phenotypes by increasing endocytosis. Loss of Csk causes Gliotactin to spread away from the TCJ. Although Csk is known as a negative regulator of Src kinases, the effects of Csk on Gliotactin are independent of Src and likely occur through an adherens junction associated complex. Overall, we identified a new Src-independent role for Csk in the control of Gliotactin, a key tricellular junction protein.
INTRODUCTION
Permeability barriers are critical to protect the body from pathogens and to generate body compartmentalization to provide specific fluid environments for each organ (Tyler, 2003; Furuse and Tsukita, 2006). Barrier function is achieved by the septate junctions in invertebrate epithelia and tight junctions in vertebrate epithelia (Auld et al., 1995; Genova and Fehon, 2003; Paul et al., 2003; Tyler, 2003). Furthermore, these junctions are required to maintain cell polarity in epithelial tissues by preventing movement of proteins/molecules along the apico-basal axis or by anchoring polarity domains within cells (Tepass et al., 2001; Nakajima et al., 2013). A subdomain of the septate or tight junction necessary for the formation of epithelia barriers is the tricellular junction, which forms at the convergence point of three epithelial cells. At this point, the bicellular junction strands perform a turn at the corners and run parallel to the apico-basal axis of the cells, where they connect to a series of plugs in insects (Fristrom, 1982; Noirot-Timothee et al., 1982). Many core proteins and associated proteins of septate junctions have been identified and include Neurexin IV (NrxIV) (Baumgartner et al., 1996), Coracle (Cora) (Fehon et al., 1994), Na/K ATPase (both alpha [ATPα] and beta [Nrv2] subunits) (Genova and Fehon, 2003; Paul et al., 2003), Neuroglian (Nrg) (Paul et al., 2003), Contactin (Cont) (Faivre-Sarrailh et al., 2004), and Mcr (Bätz et al., 2014; Hall et al., 2014).
In Drosophila epithelia, two proteins are uniquely concentrated at the tricellular junctions (TCJ). Gliotactin (Gli) is a single-pass transmembrane protein from the Neuroligin family (Schulte et al., 2003) and Bark-beetle (Bark)/Anakonda is a transmembrane protein with a large extracellular domain harboring a tripartite structural motif that is proposed to form the plugs sealing the TCJ (Byri et al., 2015; Hildebrandt et al., 2015). In vertebrates, the MARVELD family member Tricellulin, and the Angulin family member lipolysis-stimulated lipoprotein receptor (LSR) are concentrated at the tricellular tight junctions (Ikenouchi et al., 2005; Masuda et al., 2011). Loss of TCJ proteins disrupts the barrier function of epithelia in both invertebrates and vertebrates (Schulte et al., 2003; Ikenouchi et al., 2005; Nayak et al., 2013). In Gli null mutant animals, core septate junction proteins spread basally, fewer septa are formed, and the septa are not tightly packed, resulting in compromised barrier function and death by late embryogenesis (Schulte et al., 2003). Conversely, when overexpressed using apterous-GAL4 in the wing imaginal disk, Gliotactin spreads around the cell and along the basolateral membrane, triggering deleterious effects such as apoptosis paired with delamination, migration, or extrusion along the basal epithelium, as well as increased cell proliferation (Padash-Barmchi et al., 2010). Gliotactin-positive cells become concentrated beneath the columnar epithelium and move into the ventral (nonapterous) compartment. Prior work established that these displaced cells were undergoing apoptotic death and that the set of Gliotactin-induced phenotypes can be blocked by reducing C-Jun Kinase (JNK) (Padash-Barmchi et al., 2010). Overall, loss-of-function and overexpression experiments show that the level and the localization of Gliotactin to the TCJ must be tightly controlled to ensure proper barrier function and normal epithelial development. One mechanism of regulation is through the Gliotactin mRNA, which is degraded by miR-184, in response to a feedback loop controlled by bone morphogenetic protein (BMP) signaling (Sharifkhodaei et al., 2016). Another mechanism is employed at the Gliotactin protein level, where the phosphorylation of two highly conserved tyrosine residues leads to endocytosis and lysosome-mediated degradation (Padash-Barmchi et al., 2010). Paradoxically, the tyrosine phosphorylation of Gliotactin is also responsible for the deleterious phenotypes observed when Gliotactin is overexpressed (Padash-Barmchi et al., 2010). While Gliotactin can be phosphorylated by Src in vitro (Padash-Barmchi et al. 2010), the kinase(s) that regulates Gliotactin in vivo is not known. Another known point of kinase control is the interaction of Gliotactin and the MAGUK protein Discs large (Dlg). The disruption of the epithelium generated by Gliotactin overexpression is mediated through an interaction with Dlg, and this interaction is dependent on phosphorylation of Dlg at Ser797 by an unidentified Ser/Thr kinase (Padash-Barmchi et al., 2013).
To identify the kinase(s) responsible for controlling Gliotactin endocytosis, location, and epithelial disruption, we carried out an RNA interference (RNAi) screen of the majority of Drosophila kinases. We identified C-terminal Src kinase (Csk) as a tyrosine kinase that can modulate the Gliotactin-induced phenotypes and Gliotactin location. Csk and Src are closely related kinases, and Csk is well known as a negative regulator of Src family kinases (Nada et al., 1991; Okada et al., 1991; Okada, 2012; Imamoto and Soriano, 1993). We found that loss of Csk enhanced the Gli overexpression phenotypes, while increased Csk expression completely suppressed the Gli phenotypes. In contrast to our expectations, the two isoforms of Drosophila Src (Src42A and Src64B) did not suppress the Gliotactin overexpression phenotypes, showing that Csk functions independently of Src in Gliotactin protein regulation. The degree of tyrosine phosphorylation associated with Gliotactin and Gliotactin endocytosis was increased in parallel with increasing Csk expression. When Csk was down-regulated in an otherwise wild-type (WT) background, Gliotactin spread away from the TCJ, showing that Csk regulates not only overexpressed Gliotactin but also endogenous Gliotactin. Overall, our study highlights a Src-independent role of Csk in the regulation and localization of the tricellular junctional protein Gliotactin.
RESULTS
Gliotactin overexpression phenotypes are changed with changes to Csk level
In a wild-type background (w1118 in our experiments), Gliotactin is restricted to the tricellular corners of the columnar epithelial cell layer of the Drosophila wing imaginal disk (Figure 1A and Supplemental Figure S1A). When overexpressed in the wing imaginal disk using the apterous-GAL4 driver (ap-GAL4), Gliotactin spreads away from the TCJ and is found around the cell and along the lateral membrane. As Padash-Barmchi et al. (2010) showed, Gliotactin overexpression leads to overproliferation, apoptosis, and cell delamination, resulting in extrusion of Gliotactin-expressing cells into the wild-type nonapterous side of the imaginal disk (Figure 1, B, F′, and G). Gliotactin is regulated by phosphorylation of two highly conserved tyrosines, leading to endocytosis and lysosome-mediated degradation (Padash-Barmchi et al., 2010). To identify the kinase(s) responsible for controlling Gliotactin, we carried out a screen using RNAi from the Vienna Drosophila RNAi Center (VDRC) collection to the majority of Drosophila kinases. Details of the screen will be published elsewhere (unpublished data). However, in the course of this screen, we identified C-terminal Src kinase (Csk) as a potential kinase for controlling Gliotactin overexpression-induced phenotypes, Gliotactin endocytosis, and localization to the TCJ. When Csk-RNAi was expressed along with Gliotactin (ap>Gli-WT, Csk-RNAi), the Gliotactin overexpression phenotypes were greatly enhanced. Two independent RNAi lines to Csk were tested, and results were the same for both, confirming the lack of off-target effects (Supplemental Figure S1, B and C). The wing imaginal disks were smaller (Figure 1, D and I, and Supplemental Figure S1, B and C) than both control (ap>NLS-GFP) (Figure 1A) or Gliotactin overexpression alone (ap>Gli-WT) (Figure 1B).
FIGURE 1:

Csk levels affect the Gliotactin overexpression phenotypes. Third-instar wing imaginal disks with transgenes or RNAi lines expressed by the apterous-GAL4 (ap>) driver. The dashed lines marked the apterous boundary between the dorsal and ventral compartments with the apterous side (dorsal compartment) on the left. Arrows indicate the leading edge of the Gliotactin positive cells as they move into the control side (posterior compartment). Each panel represents a single Z slice. Scale bars: 30 µm. (A–E) Wing disks immunolabeled for Gliotactin (green) and Dlg (red). DAPI and NLS-GFP in blue. n = 15 disks except n = 8 disks in E. Arrows indicate the leading edge of the Gliotactin expressing cells. (A) Wild-type w1118 wing disks with ap>NLS-GFP. The pouch area of wing imaginal disk is shown. There was no cell migration or ectopic folds in the wild-type wing disk. (B) Gliotactin overexpression (ap>Gli-WT) leads to distinct phenotypes including migration of Gli overexpressing cells into the wild-type (ventral) side of the disk (B′), extra folds due to increased proliferation on the apterous side (B-–B″ and G–G″′). (C) Rescue of Gli overexpression phenotypes by coexpression of Csk-WT. Cell migration to the wild-type side was suppressed (C′) and fewer extra folds were formed (C–C″ and H–H″). (D) Enhancement of Gliotactin overexpression phenotypes by coexpression of Csk- RNAi. Cell migration was enhanced (arrow) and the apterous side was smaller (D′). (E) Gliotactin overexpression in the Csk[J1D8] heterozygous mutant phenocopied the coexpression of Csk-RNAi (E–E′) giving an enhanced cell migration and a smaller dorsal side. (F) Schematic of a Drosophila wing imaginal disk. The region of apterous expression is marked in green and the boundary between dorsal (apterous) and ventral (wild-type) compartments within the pouch region marked in yellow. (F′) Schematic of the migration of Gliotactin overexpressing cells. Migration distances were measured for cells from the apterous boundary into the wild-type/ventral compartment (red arrow) compared with the total distance from the apterous boundary to the distal edge of the disk (black arrow). (G–I) Wing disks immunolabeled for Gliotactin (green), activated Caspase-3 (red), and DAPI (blue). Stars indicate the leading edge of the cells positive for activated Caspase-3 (Cas3). (G) Gliotactin overexpression (ap>Gli-WT) increased levels of activated Caspase-3 with strong expression in migrating cells (G″). (H) Coexpression with Csk-WT (ap>Gli-WT, Csk-WT) reduced the levels of activated Caspase-3 (H″). (I) Coexpression with Csk-RNAi (ap>Gli-WT, Csk-RNAi) led to increased immunolabeling for activated Caspase-3 (I″). (J) Quantification of the cell migration ratios with Gli overexpressed alone (ap>Gli-WT), coexpressed with Csk-WT (ap>Gli-WT, Csk-WT) and coexpressed with Csk-RNAi (ap>Gli-WT, Csk-RNAi) or in the Csk heterozygous mutant (ap>Gli-WT, Csk[J1D8]/+). All showed a significant difference in the cell migration ratios (****p < 0.0001). The difference in cell migration ratios between (ap>Gli-WT, Csk-RNAi) and (ap>Gli-WT, Csk[J1D8]/+) was not significant (NS) (n = 15 disks for each). Bars represent the SD.
With the knockdown of Csk (ap> Gli-WT, Csk-RNAi), there was a high degree of apoptosis as measured by cleaved Caspase-3 immunolabeling (Song, 1997) (Figure 1I and Supplemental Figure S1D). The apterous region was greatly reduced in size with increased extrusion/migration of the Gliotactin expressing cells to the nonapterous side of the disk (Figure 1, D′ and I′, and Supplemental Figure S1, B and C). Similar results were observed when Gliotactin was overexpressed in a heterozygous Csk mutant background (ap>Gli-WT, Csk[J1D8])/+) (Figure 1E). The down-regulation of Csk increased the Gliotactin overexpression phenotypes, suggesting that Csk regulates Gliotactin. To test this hypothesis, we coexpressed Csk (Pedraza et al., 2004) with Gliotactin (ap>Gli-WT, Csk-WT) and found that the Gliotactin overexpression phenotypes were suppressed (Figure 1, C and H). Specifically, while Gliotactin still spread around the cell, the extrusion/migration of the Gli-WT cells was significantly suppressed (Figure 1, C′, H′, and J). The degree of ectopic folds in the Gli-WT side of the disk was reduced, suggesting that the overproliferation phenotype was also suppressed. Further confirming the suppression, there was a reduction in apoptosis as measured by cleaved Caspase-3 immunolabeling (Figure 1H″ and Supplemental Figure S1D).
Dlg plays a role in mediating the deleterious consequences of Gliotactin overexpression, and Dlg is normally down-regulated when Gliotactin is overexpressed (Padash-Barmchi et al., 2013) (Figures 1B″ and 2A″). The down-regulation of Dlg when Csk was coexpressed with Gli-WT was comparable to that in Gliotactin overexpression alone (Figures 1C″ and 2B″ and Supplemental Figure S1E). These results suggest that Csk has a role in regulating Gliotactin, and this effect is independent of changes to Dlg levels.
FIGURE 2:

Coexpression of Csk increases internalization of overexpressed Gliotactin. High-magnification images (60×) from the pouch region of third-instar wing imaginal disks with transgenes or RNAi lines expressed using the apterous-GAL4 (ap>) driver. Disks were immunolabeled for Gliotactin (Gli, green), Dlg (red), and DAPI (blue). The dashed lines represent the apterous boundary with the dorsal (apterous) compartment on the left. Boxed regions were digitally magnified (28×) and shown on the far right with vesicles indicated with arrows. Each en face panel represents a single Z slice taken at the septate junction level. Scale bars: 15 µm, except 5 µm in digitally magnified panels. n = 10 disks in each genotype. (A) ap>Gli-WT. On the apterous side, Gli is found around the membrane and in internal vesicles. Not all the cells contained large vesicles (A′″). Dlg was down-regulated in the apterous side (A″). (B) ap>Gli-WT, Csk-WT. Coexpression of Csk-WT led to an increase cells with large vesicles. Dlg was down-regulated in the apterous side (B″). (C) ap>Gli-WT, Csk-RNAi. Coexpression of Csk-RNAi led to the presence of small vesicles in some cells while many cells did not contain vesicles. Dlg down-regulation in the apterous side was not as extensive (C″ compared with A″ and B″). (D) A digitally magnified area representing the three types of vesicles quantified. Immunolabeling of Gliotactin marked the internal vesicles, and Dlg marked the cell membrane. (E) Analysis of vesicle number and size in (ap>Gli-WT), (ap>Gli-WT, Csk-WT), and (ap>Gli-WT, Csk-RNAi), respectively. Vesicles were placed into three groups based on maximum diameter (type 1: d ≥ 2.5 µm; type 2: 1 µm < d < 2.5 µm; type 3: d ≤ 1 µm. In each n = 10 disks.
Coexpression of Csk increases internalization of overexpressed Gliotactin
As the coexpression of Csk suppressed the Gliotactin phenotypes, we compared the changes to cells overexpressing Gliotactin at the cellular level. A consistent phenotype we observed was changes to cell size. Gliotactin overexpression led to an increase in cell size (Figures 2A″ and 5A″ and Supplemental Figure S5A″) on both the apterous and nonapterous sides. The suppression of Gliotactin phenotypes by Csk (ap>Gli-WT, Csk-WT) also included a suppression of the increased cell size (Figures 2B″ and 5C″ and Supplemental Figure S5C″), whereas the knockdown of Csk (ap>Gli-WT, Csk-RNAi) appeared to further increase cell size (Figures 2C″ and 5E″). The mechanism underlying this effect is not clear, as both sides of the disk were affected, suggesting a change in tissue morphology.
FIGURE 5:
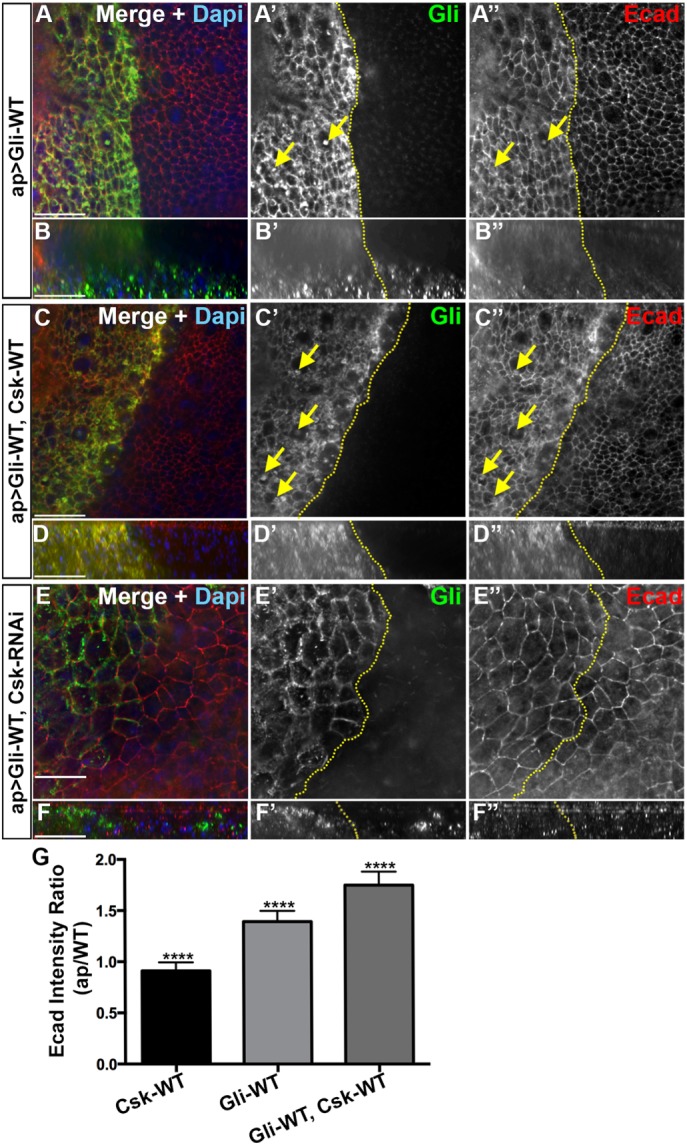
Coexpression of Csk with Gliotactin increases the endocytosis of Ecad. High-magnification images (60×) from the pouch area of third-instar wing imaginal disks with transgenes or RNAi lines expressed by apterous-GAL4 (ap>) driver. Disks were immunolabeled for Gliotactin (green), Ecad (red), and DAPI (blue). The dashed lines represent the boundary between dorsal and ventral compartments of wing disks where the apterous side (dorsal compartment) is on the left. Arrows pointing to colocalization of Ecad with Gliotactin filled vesicles. Each en face panel represents a single Z slice taken at the septate junction level. Scale bars: 15 µm. n = 15 disks in each genotype. (A) ap>Gli-WT. On the apterous side, Gliotactin was found around the membrane and in internal vesicles (A and A′, arrows). Ecad was upregulated in the Gliotactin overexpressing apterous side (A″). Some of the Gliotactin vesicles were colocalized with Ecad in the cytoplasm (A′ and A″). (B) Corresponding side projections of the above panels. (B′) The basolateral spreading of Gliotactin along the lateral membrane and migration of Gliotactin-positive cells to the wild-type side. (B″) Up-regulation of Ecad in the apterous side in both apical and lateral cell membranes. (C) ap>Gli-WT, Csk-WT. Coexpression of Csk-WT led to an increased abundance of large vesicles in the apterous side (C and C′). There was an up-regulation of Ecad level in the cell membranes of cell in apterous side (C″). In the cytoplasm, Ecad was colocalized with the majority of Gliotactin-positive vesicles (C′ and C″, arrows). (D) Corresponding side projections of the above panels. (D′) Suppression of cell migration of Gliotactin positive cells to the nonapterous side. (D″) Up-regulation of Ecad in both apical and lateral cell membranes. (E) ap>Gli-WT, Csk-RNAi. Gliotactin was found around the membrane and formed only small vesicles (E′ and E′″). Changes to Ecad levels or Ecad endocytosis on the apterous side did not appear different from the control side (A″ or C″). (F) Corresponding side projections of the above panels. Ecad levels were the same on the apterous and control sides. (G) Statistical analysis of Ecad immunolabeling. For each genotype, the mean ratios of Ecad intensities in the apterous side vs. WT side are shown. n = 10 disks (****p < 0.0001 between each genotype). Error bars represent SD.
In cells coexpressing Csk and Gli-WT, there was increased levels of Gliotactin endocytosis, and the majority of cells had large internal vesicles filled with Gliotactin (Figure 2, B and B″′). This was compared with Gli-WT expression alone where many but not all cells had large vesicles (Figure 2, A and A″′). In contrast, when Csk was knocked down using RNAi, fewer Gli-WT expressing cells had vesicles, and the vesicle size appeared smaller (Figure 2, C and C″′) compared with Gli-WT alone or Gli-WT plus Csk (Figure 2, A″′ and B″′). To quantify these effects, we measured the number and size of vesicles in the different genotypes. Each vesicle-like structure that was positive for Gliotactin immunolabeling was defined as a vesicle for our analysis. However, as multivesicular bodies (MVBs) are reported to have large diameters (Altick et al., 2009), any larger vesicle may represent a MVB and consist of many small vesicles that cannot be distinguished with light microscopy. The size of vesicles was placed into one of three separate groups based on maximum diameter (type 1: d ≥ 2.5 µm; type 2: 1 µm < d < 2.5 µm; type 3: d ≤ 1 µm) (Figure 2D) and then quantified (Figure 2E and Table 1). In all three backgrounds, the endosome counting was done in a constant area to avoid the effect of cell size changes on endosome counting. To increase the accuracy, a large area encompassing the entire wing pouch starting from the apterous boundary was examined for both apterous and control sides in each disk. There was no significant difference to the total number of vesicles in Gli-WT (ap>Gli-WT) expression versus Gli-WT, Csk-WT coexpression (ap>Gli-WT, Csk-WT) (Figure 2E and Table 1). However, the numbers of larger vesicles (type 1 and 2) were significantly higher in Gli-WT and Csk-WT coexpression, suggesting an increased amount of Gliotactin endocytosis (Table 1). In contrast, the total number of vesicles as well as the numbers in each type when Csk-RNAi was coexpressed with Gli-WT (ap>Gli-WT, Csk-RNAi) was significantly lower, indicating that Gliotactin endocytosis was reduced (Table 1). As Gliotactin endocytosis depends on phosphorylation of conserved tyrosine residues (Y760, Y799) in the intracellular domain of Gliotactin (Padash-Barmchi et al., 2010), our results suggest that Csk controls the endocytosis of Gliotactin likely through increased phosphorylation and thus leads to removal of Gliotactin from the membrane. These results phenocopied previous observations with the overexpression of the GliDD transgene, which mimics phosphorylation of Gliotactin and results in large internal endocytic vesicles (Padash-Barmchi et al., 2010).
TABLE 1:
Comparison of numbers of each type and total number of vesicles in three different genotypes: ap>Gli-WT, (ap>Gli-WT, Csk-WT), and (ap>Gli-WT, Csk-RNAi).
| Type of vesicles | Comparison of number of vesicles | Mean diff. | Significance | p Value | SEM |
|---|---|---|---|---|---|
| Type 1 (d ≥ 2.5 µm) | Gli-WT vs. Gli-WT, Csk-WT | −3.9 | ** | 0.0033 | 0.2981 |
| Gli-WT vs. Gli-WT, Csk-RNAi | 1 | NS | 0.6279 | 1.286 | |
| Gli-WT,Csk-WT vs Gli-WT,Csk-RNAi | 4.9 | *** | 0.0003 | 0 | |
| Type 2 (1 < d < 2.5 µm) | Gli-WT vs. Gli-WT, Csk-WT | −18.7 | * | 0.0374 | 7.237 |
| Gli-WT vs. Gli-WT, Csk-RNAi | 32.6 | **** | 0.0003 | 4.9 | |
| Gli-WT,Csk-WT vs. Gli-WT,Csk-RNAi | 51.3 | **** | <0.0001 | 0.7024 | |
| Type 3 (d ≤ 1 µm) | Gli-WT vs. Gli-WT, Csk-WT | 33.1 | **** | 0.0002 | 7.022 |
| Gli-WT vs. Gli-WT, Csk-RNAi | 65.6 | **** | <0.0001 | 4.899 | |
| Gli-WT,Csk-WT vs. Gli-WT,Csk-RNAi | 32.5 | **** | 0.0003 | 1.638 | |
| All | Gli-WT vs. Gli-WT, Csk-WT | 10 | NS | 0.6772 | 11.01 |
| Gli-WT vs. Gli-WT, Csk-RNAi | 98.7 | **** | <0.0001 | 9.235 | |
| Gli-WT,Csk-WT vs. Gli-WT,Csk-RNAi | 88.7 | **** | <0.0001 | 1.533 |
Summary results of one-way ANOVAs with Tukey post-hoc tests. Mean difference (Mean Diff), Significance, Adjusted p value for multiple comparisons (p value) and SEM are shown. The total number of vesicles in Gliotactin overexpression (ap>Gli-WT) was not significantly different from that in coexpression with Csk-WT (ap>Gli-WT, Csk-WT). The number of largest group (d ≥ 2.5 µm) vesicles was not significantly different in Gliotactin overexpression (ap>Gli-WT) compared with coexpression with Csk-RNAi (ap>GliWT, Csk-RNAi). n = 10 in each genotype.
We confirmed that the type of vesicles that were increased in ap>Gli-WT, Csk-WT were endosomes. Gliotactin vesicles were also positive for both the Rab5 effector protein, Rabenosyn-5 (Rbsn-5) (Tanaka and Nakamura, 2008) (Supplemental Figure S2, A–D) and the late endosome marker Rab7 (Tanaka and Nakamura, 2008; Vanlandingham and Ceresa, 2009) (Supplemental Figure S2, E and F). In Gliotactin overexpression alone and in coexpression with Csk, the majority of the vesicles colocalized with Rbsn-5 (Supplemental Figure S2, A–D), which is present in early to late endosomes (Mottola et al., 2010; Lo˝rincz et al., 2016) and with Rab7 (Supplemental Figure S2, E and F). The size of Rbsn-5 or Rab7 positive vesicles in ap>Gli-WT (Supplemental Figure S2, A″ and E″) was smaller compared with the Gliotactin coexpression with Csk-WT (ap>Gli-WT, Csk-WT) (Supplemental Figure S2, C″ and F″). Of note, the nonapterous wild-type side also showed a strong localization of Rbsn-5 at the level of septate junctions (Supplemental Figure S2, B″ and D″), and there was no apparent change to the level and/or the localization of Rbsn-5 in the Gliotactin overexpressing apterous side compared with the control side. The Gliotactin filled vesicles were not positive for the recycling endosome marker, Rab11 (Supplemental Figure S2, G and H).
We next tested the effect of Csk on a Gliotactin transgene where both tyrosines were converted to phenylalanine to mimic the dephosphorylated state (Gli-FF). Gli-FF is not endocytosed when expressed in a Gli null background and reduces endocytosis when expressed in a wild-type background (Padash-Barmchi et al., 2013) (Figure 3, A and B). It is important to note that Gliotactin forms a dimer or oligomer (Venema et al., 2004), and therefore overexpression of Gli-WT or Gli-FF in a wild-type background is capable of forming oligomers with endogenous Gliotactin (Supplemental Figure S3, C and D). For instance, when Gli-WT was overexpressed in the presence of Gliotactin endogenously tagged with YFP (Gli::YFP, ap>Gli-WT), Gli::YFP spread from the TCJ through the cell membrane in tandem with overexpressed Gli-WT (Supplemental Figure S3E). Therefore, when Gli-FF is expressed, this leads to greatly enhanced overexpression phenotypes (Padash-Barmchi et al., 2013), as the complex of Gli-FF with endogenous Gliotactin reduces the degree of endocytosis, persisting in the membrane longer and enhancing the overexpression phenotypes (Supplemental Figure S3D). Coexpression of Csk-WT with GliFF (ap>Gli-FF, Csk-WT) indicated that Csk is also able to suppress the Gli-FF phenotypes (Figure 3, A and B, and Supplemental Figure S3, AB). Csk expression with Gli-FF also resulted in the formation of large Gliotactin vesicles (Figure 3, B′ and B′″), further confirming the increased level of endocytosis of Gliotactin likely through the increased phosphorylation of the endogenous Gliotactin oligomerized with Gli-FF. Overall, our data indicate that coexpression of Csk leads to an increased endocytosis of Gliotactin through the endocytic/lysosome degradation pathway previously observed for Gliotactin (Padash-Barmchi et al., 2010).
FIGURE 3:

Coexpression of Csk increases the tyrosine phosphorylation associated with overexpressed Gliotactin. Third-instar wing imaginal disks with transgenes or RNAi lines expressed by the apterous-GAL4 (ap>) driver. The dashed lines mark the apterous boundary with the dorsal (apterous) compartment on the left. Areas marked by boxes were digitally magnified 28 times and shown on the far right of each corresponding row with vesicles marked with arrows. Each panel represents a single Z slice at the septate junction level. Scale bars: 15 µm except in C, D, and E (30 µm) and A″′ and B″′ (5 µm). (A, B) High-magnification images (60×) from the wing disk pouch immunolabeled for Gliotactin (Gli, green), Dlg (red), and DAPI (blue). Arrows are pointing to vesicles. n = 6 in A and n = 5 disks in B. (A) ap>GliFF. On the apterous side, Gliotactin was found around the membrane and the occasional small vesicles (A, A″′). (B) ap>GliFF, Csk-WT. Coexpression of Csk-WT led to an increase in vesicle number and size (B′ and B″′). (C–G) PLA signals showing the abundance of Gliotactin and phospho-tyrosine in close (40-nm) proximity. n = 11 in F, n = 9 in G, and n = 8 disks in H. (C′, D′) Higher-resolution images (60×) from the pouch area comparing the levels of PLA in the apterous side vs. WT side. E′: an area showing the maximum level of PLA signals within the apterous side (the area is marked by a star in E). (C, C′) Gliotactin overexpression (ap>Gli-WT) led to increased PLA signals on the apterous side compared with the WT side. (D, D′) Coexpression of Csk-WT further increased the PLA signals on the apterous side. (E, E′) Reduction of PLA signals by coexpression of Csk- RNAi. (F–F″) Side projections showing the increase of PLA (Gli+pY) mostly at the apical side of the Gli-WT overexpression side (arrow). (G–G″) Side projections showing a further increase of amount of PLA (Gli+pY) in Gli-WT, Csk-WT coexpression. PLA signals were mostly at the apical side (arrow). (H) Statistical analysis of PLA signals (between Gli and pY) in ap>Gli-WT and ap>Gli-WT, Csk-WT backgrounds. In each background the mean ratios of PLA signal intensities in apterous side vs. WT side are shown. n = 11 disks in ap>Gli-WT and n = 9 disks in ap>Gli-WT Csk-WT (****p < 0.0001). Bars represent the SD.
Coexpression of Csk increases the tyrosine phosphorylation associated with overexpressed Gliotactin
We next tested the hypothesis that the level of Gliotactin phosphorylation changes with changes to Csk. Gliotactin is phosphorylated at two conserved tyrosine residues (Schulte et al., 2006), where the phospho-mimic form of Gliotactin, Gli-DD, increases endocytosis and the phospho-blocked form Gli-FF reduces endocytosis (Padash-Barmchi et al., 2010). Since antibodies specific to the phosphorylated form of Gliotactin are not available, we used a proximity ligation assay (PLA) (Gajadhar and Guha, 2010) to detect changes in tyrosine phosphorylation within 40 nm of Gliotactin. We compared the degree of phospho-tyrosine and Gliotactin association in three different backgrounds: Gli-WT expression alone (ap>Gli-WT) and coexpressed with Csk (ap>Gli-WT, Csk-WT) or coexpressed with Csk-RNAi (ap>Gli-WT, Csk-RNAi) (Figure 3, F–H). With Gliotactin overexpression alone, there was an elevated level of the PLA signal compared with the nonapterous side, indicating a greater degree of Gliotactin and phospho-tyrosine in close proximity on the apterous side relative to the control side (Figure 3, C and F′). When coexpressed with Csk, the PLA signal level was further increased (Figure 3, D, G, and H). Conversely, when Csk was knocked down, the PLA signal was lower than Gli-WT alone (Figure 3E). Controls for PLA experiments showed a negligible level of PLA when using only one antibody (Supplemental Figure S7, C and D). We observed that the PLA between pTyr and Gliotactin was concentrated in the apical side of the columnar epithelia within the adherens junction domain for both Gli-WT expression alone (Figure 3F) and Gli-WT plus Csk-WT (Figure 3G). Our data suggest that increased Csk levels lead to increased phosphorylation of Gliotactin or a Gliotactin-associated protein and that this interaction appears to be restricted to the apical region of the columnar epithelia.
Csk-mediated regulation of Gliotactin is Src independent
Csk is well established as a negative regulator of Src family kinases (Okada et al., 1991; Okada, 2012). Further, Gliotactin can be phosphorylated on the conserved tyrosine residues by activated Src using an in vitro kinase assay (Padash-Barmchi et al., 2010). This led us to check whether the Csk phenotypes were generated through changes to levels of Src activity. We tested whether the loss of Src in the Gliotactin overexpression could phenocopy the suppression of Gliotactin phenotypes observed with Csk coexpression. Src levels were reduced by using either loss-of-function mutants in either of the two Drosophila Src genes (Src42A and Src64B) (Figure 4) or RNAi lines to each (Supplemental Figure S4, A and B). Reduction of Src levels did not rescue nor enhance the Gliotactin overexpression phenotypes. A 50% reduction in the gene dose of each individual Src had no signficant effect on the Gliotactin overexpression phenotypes: Src64B[D404N], Src64B[KO], or Src42A[myri] heterozygous mutants did not rescue (Figure 4, A–C) and did not phenocopy the Csk rescue of Gli-WT.
FIGURE 4:
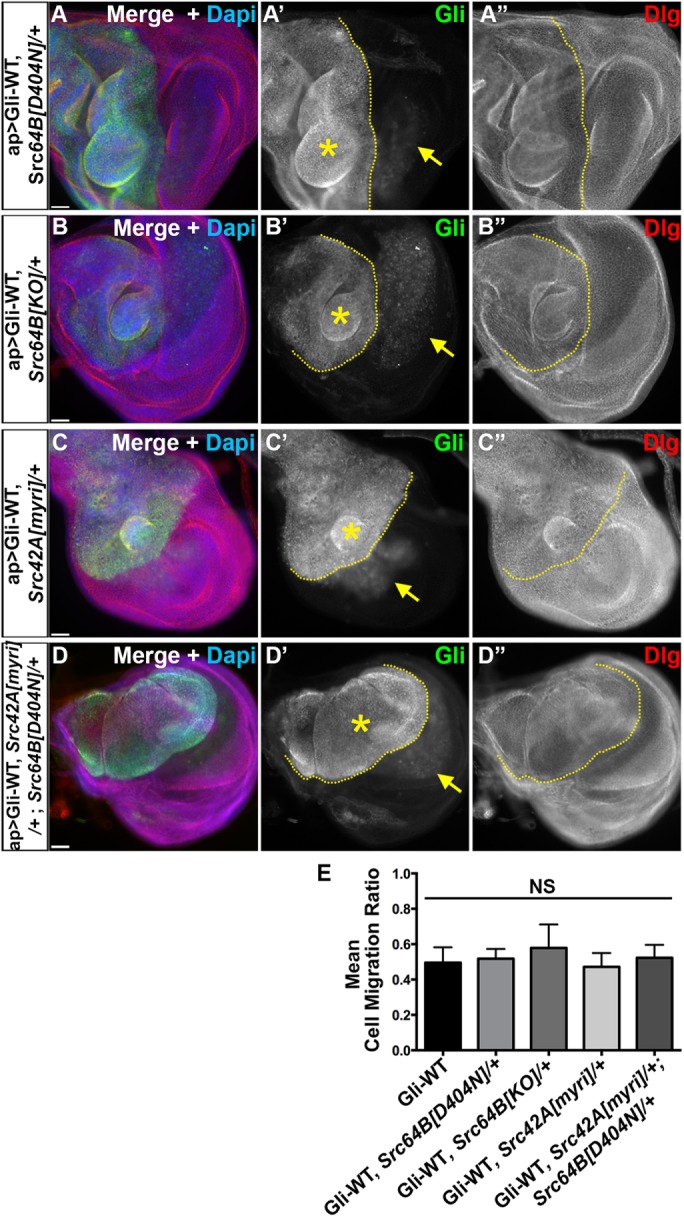
Csk-mediated regulation of Gliotactin is Src independent. Third-instar wing imaginal disks with ap>Gli-WT overexpression in Src -/+ mutants immunolabeled for Gliotactin (Gli, green), Dlg (red), and DAPI (blue). Dashed lines mark the boundary between dorsal and ventral compartments with the apterous side on the left. Each panel represents a single Z slice. Scale bars: 30 µm. n = 10 disks except n = 18 in D. (A–D) Overexpression of Gliotactin in (A) a Src64B[D404N]/+ heterozygous mutant background (ap>Gli-WT, Src64B[D404N]), (B) a Src64B[KO]/+ heterozygous background (ap>Gli-WT, Src64B[KO]/+), (C) a Src42A[myri]/+ heterozygous background (ap>Gli-WT, Src42A[myri]/+), and (D) a Src42A, Src64B heterozygous background (ap>Gli-WT, Src42A[myri]/+; Src64B[KO]/+). Arrows mark the leading edge of the migrating Gli overexpressing cells into the wild-type (ventral) side. Gliotactin overexpression still generated extra folds and overproliferation (stars). (E) Statistical analysis of cell migration ratios in Gliotactin overexpression alone (ap>Gli-WT) compared with expression in Src heterozygous mutant backgrounds. No significant (NS) differences were observed. Bars represent the SD (n = 15 disks).
Since the potential redundancy of the two isoforms of Src cannot be neglected, we also tested a double-mutant line heterozygous for each Src gene (Src42A[myri], Src64B[D404N]) in a Gli-WT overexpression background. Even the double mutant failed to suppress the Gli-WT phenotypes (Figure 4D). The quantification of the Gliotactin cell migration ratios showed that reduction of Src levels either with mutants or RNAi lines plus Gli-WT overexpression was not significantly different from Gli-WT overexpression alone (Figure 4E and Supplemental Figure S4D). In contrast, if Src kinases phosphorylate Gliotactin in vivo, then coexpression with Src-WT should increase the internalization of Gliotactin, leading to a suppression of Gli-WT phenotypes. To test this, we coexpressed Src64B-WT with Gli-WT and found that the phenotypes were not suppressed but rather enhanced (likely due to an additive effect) (Supplemental Figure S4, C and D). Overall, these results suggest that Gliotactin phosphorylation and/or overexpression phenotypes are not generated by Src kinase, and the Csk activity on Gliotactin is Src independent.
Coexpression of Csk with Gliotactin increases the endocytosis of Ecad
Loss of Csk has varying effects depending on whether the reduction is over a broad area or over a discrete region neighboring wild-type cells (Vidal et al., 2006). For instance, the knockdown of Csk within an entire tissue leads to mispatterning and overgrowth while blocking apoptosis. However, Csk knockdown in a discrete region of the wing imaginal disk leads to cell delamination, migration, and cell death via apoptosis (Vidal et al., 2006) in a manner very similar to that of the Gliotactin overexpression phenotypes. This has been attributed to increased Src activity leading to decreased levels of the adhesion junction protein Ecad in Csk down-regulated cells. Vidal et al. (2006) suggested that the relatively high levels of Ecad on the wild-type side trigger the neighboring cells with low Ecad to undergo JNK-mediated apoptosis. Ecad down-regulation as well as up-regulation can lead to cell migration depending on the context (Niewiadomska et al., 1999). As Gliotactin overexpression leads to cell delamination, migration, and JNK-mediated apoptosis (Padash et al., 2010), we tested for changes in Ecad levels at the interface between wild-type and Gliotactin overexpressing cells. The overexpression of Gliotactin does not lead to a down-regulation of Ecad in the apterous side (Padash-Barmchi et al., 2010). Instead, we observed that Ecad levels were elevated on the apterous side (Figure 5, A″ and B″). Interestingly, along with elevated Ecad at the adherens junction (AJ) and throughout the cell, we observed some Gliotactin vesicles that were positive for Ecad (arrows in Figure 5A), suggesting that there is a change to Ecad endocytosis with Gliotactin overexpression. We next tested whether there was a change in Ecad levels when the Gliotactin overexpression phenotypes were suppressed by coexpression of Csk (Figure 5, C, D, and G). In ap>Gli-WT, Csk-WT disks, we observed the increased presence of Ecad within the membrane (Figure 5C) as well as within endocytic vesicles that were also positive for Gliotactin (Figure 5C, arrows). In ap>Gli-WT, Csk-RNAi, there was no noticeable difference in the Ecad levels between the apterous and nonapterous sides (Figure 5, E and F). Expression of Csk alone (ap>Csk-WT) did not lead to changes in Ecad immunolabeling (Figure 5G). The coendocytosis with Gliotactin when Gliotactin is overexpressed, with or without Csk, suggests that Ecad becomes internalized along with Gliotactin when Gliotactin spreads (Figure 5, C″ and D″). Since there was an elevation in Ecad, we tested whether there was a change to another AJ protein Drosophila β-catenin (Armadillo (Arm)) and found that Arm immunolabeling intensities were the same in the apterous side compared with wild-type side in ap>Gli-WT (Supplemental Figure S5, A and B) as well as in ap>Gli-WT, Csk-WT (Supplemental Figure S5, C and D). Csk suppresses the Gliotactin-induced migration phenotypes and yet Ecad levels were significantly elevated (Figure 5G). Therefore, it is unlikely that differences in Ecad levels across the apterous boundary is responsible for the Gliotactin migration phenotypes.
To test whether the changes to Ecad levels might be part of the suppression of the Gliotactin phenotypes, we reduced the gene dosage by 50% of Ecad by using shotgun (shg) loss of function mutants. Specifically, Gliotactin was expressed (ap>Gli-WT) in conjunction with mutant alleles of Ecad (shg[E17B]/+; shg[2]/+) (Supplemental Figure S5E). Conversely, we also increased the Ecad level by coexpressing Ecad (UAS-Shg) with Gli-WT (ap>Gli-WT, Shg-GFP) (Supplemental Figure S5F). Neither loss of Ecad nor coexpression of Ecad lead to a suppression of the Gliotactin triggered phenotypes suggesting that the suppression of Gliotactin phenotypes by Csk is not mediated through changes to Ecad. Overall, these results suggest that the Gliotactin-induced phenotypes and the Csk-mediated suppression of the Gliotactin overexpression phenotypes are independent of changes to both Src and Ecad.
Csk activity on Gliotactin might be indirect
Since our data suggested that the Csk activity on Gliotactin was Src and Ecad independent, we next tested whether Csk directly phosphorylates Gliotactin in vitro. His-tagged fusion proteins of the Gliotactin intracellular domain (Gli-WT) (Padash-Barmchi et al., 2010) were tested in vitro using a Csk kinase assay (Supplemental Figure S6A). While Csk was able to phosphorylate the positive control Src, Gli-WT was not phosphorylated (Supplemental Figure S6A). However, substrate recognition by Csk depends on both the correct three-dimensional structure along with the target amino acids (Ia et al., 2010). As the C-terminal domain used in our assays is natively unfolded in vitro (Zeev-Ben-Mordehai et al., 2003), it may be that Gliotactin does not have the correct structural properties in vitro for recognition by Csk.
To determine whether Gliotactin and Csk can associate in vivo, we tested for the distribution of Csk in wild-type and Gliotactin overexpressing imaginal disks (Figure 6 and Supplemental Figure S6, C–F). In wild type (w1118), Csk immunolabeling was found throughout the cytoplasm with punctalike labeling that occasionally colocalized with Ecad (Figure 6B and Supplemental Figure S6C) but not with Gliotactin (Figure 6, A and B). When Gliotactin was overexpressed, Csk levels at the apical side did not change (Figure 6, C″ and D″), but increased Csk immunolabeling was observed at the basal side of the epithelium in the areas of the delaminated Gliotactin expressing cells under the columnar epithelia on both the apterous and nonapterous side (Figure 6D″). This immunolabeling pattern was different from that in wild-type epithelium (Figure 6B”) and suggested an increase or redistribution of the endogenous Csk into the delaminated cells when Gliotactin was overexpressed. When overexpressed (ap>Csk-WT), Csk levels increased in the cytosol and at the membrane, showing a clear colocalization with Ecad at the AJ (Figure 6, E–H, and Supplemental Figure S6E). The overexpression of Csk did not change the Gliotactin localization at the TCJs (Figure 6E′) but did lead to a spread of Gliotactin down the basolateral membrane (Figure 6F′ and Supplemental Figure S6B). When coexpressed with Gliotactin (ap>Gli-WT, Csk-WT), Csk colocalized with Gliotactin both at the membrane and in the large Gliotactin vesicles (Figure 6, I and J). Overall, the changes to the localization pattern of Csk with changes to Gliotactin and colocalization of overexpressed Csk with Gliotactin suggest that Csk might regulate Gliotactin in vivo.
FIGURE 6:

Csk distribution within the wing imaginal disk. High-magnification images (60×) from the wing pouch from wild type or with transgenes or RNAi lines expressed by apterous-GAL4 (ap>) driver. Disks were immunolabeled for Gliotactin (Gli, green), Csk (red), and Ecad or DAPI (blue). Dashed lines mark the apterous boundary where the apterous side (dorsal compartment) is on the left. Each en face panel represents a single Z slice taken at the septate junction level. Areas that are marked by boxes were digitally magnified by 28 times and placed at the far right of the corresponding row. Arrowheads mark the adherens junction in side projections. Scale bars: 15 µm except in 5-µm digitally magnified panels. n = 10 disks in each genotype. (A) Wild-type disk with Gliotactin labeling at the corners of the cells and Csk puncta throughout the cytoplasm. Csk does not colocalize with Gliotactin (A″′). (B) Corresponding side projections with Gliotactin at the septate junction (B′). Csk labeling was throughout the cytoplasm, less abundant in the septate junction domain, and colocalized with Ecad in the adherens junctions (B″, B″′). (C) ap>Gli-WT wing disks where Csk immunolabeling on the apterous was not changed at the apical level but was increased at the basal side of the epithelium in the areas of the delaminated Gliotactin-expressing cells (arrows). (D) Corresponding side projections with Csk labeling concentrated at the basal side (D″) in the delaminated and migrating Gliotactin-overexpressing cells (arrows) (D′ and D″). There was an increase in Ecad on the apterous side (D″). (E, G) ap>Csk-WT. The apical side of the wing disk with Csk distributed through the cytoplasm and at the membrane. Gliotactin distribution to the TCJ was unchanged (E′). Csk was at the membrane and concentrated at the adherens junctions level (G and G′). There was no change to the Ecad level or localization (G′). (F, H) Corresponding side projections showed an elevated level of Csk through the epithelia with a concentration at the AJ (F and H). In the Csk-overexpressed apterous side, Gliotactin was spread further along the lateral membrane compared with the control side (F′, arrow). There was no difference in Ecad expression or distribution with Csk overexpression (H′). (I) ap>Gli-WT, Csk-WT. Gliotactin was concentrated in large vesicles (I′) along with Csk (arrow) (I″, I″′). (J) Corresponding side projections showed elevated levels of Gliotactin and Csk and the spread from apical to basal in the apterous side (J′ and J″). In the apterous side Ecad was up-regulated (J″′).
We hypothesize that Gliotactin only associates with endogenous and overexpressed Csk when overexpressed (Figure 6C, I). We carried out PLA assays to test whether these two proteins were in close proximity (<40 nm) in vivo. We observed consistent PLA signals between Csk and Gliotactin when Gli-WT was expressed alone (Figure 7A) and the PLA signals were concentrated at the AJ (Figure 7B″). When Gli-WT and Csk-WT were coexpressed, we observed increased PLA signals that were also concentrated at the apical side within the AJ (Figure 7, C″ and D″) and significantly increased compared with Gli-WT alone (Figure 7G). Conversely, when Gli-WT was coexpressed with Csk-RNAi, the PLA signals were reduced (Figure 7, E″ and F″). Controls for PLA experiments showed a negligible level of PLA when using only one antibody (Supplemental Figure S7, C and E). Of interest, the PLA signals were consistently concentrated in the region of the AJs (Figure 7, B″ and D″), even while both Gli-WT and Csk-WT were distributed over a much wider area. This suggests that the close proximity of Gli-WT and Csk-WT was limited to this region possibly due to the presence of an intermediate protein or cofactor. Overall, these in vivo data show that when Gliotactin is overexpressed, Csk is in close proximity to Gliotactin but only in a discrete region of the epithelium and suggests that Csk may phosphorylate Gliotactin or a closely associated protein to Gliotactin within this domain.
FIGURE 7:
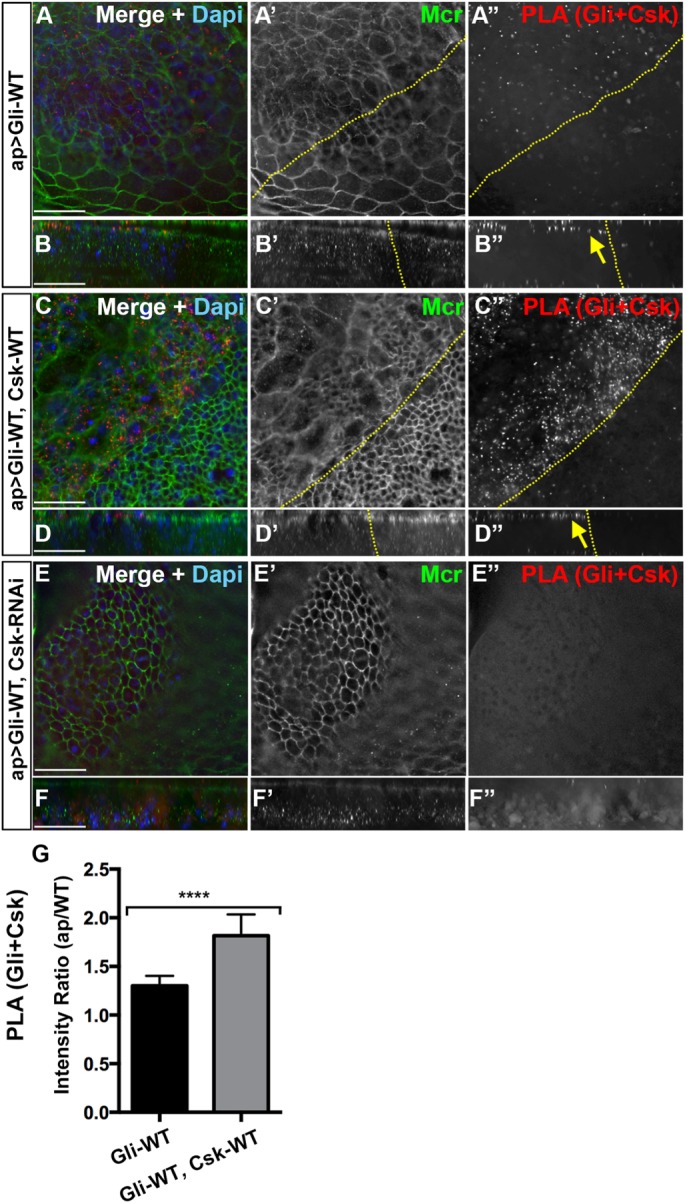
Csk is in close proximity with overexpressed Gliotactin at the AJ. High-magnification images (60×) from the wing pouch with transgenes or RNAi lines expressed by the apterous-GAL4 (ap>) driver. PLA signals between Gliotactin and Csk (red), immunolabeling for Mcr (green), and labeling for DAPI (blue) or Ecad (blue). Dashed lines mark the apterous boundary where the apterous side (dorsal compartment) is on the left. Each panel represents a single Z slice at the septate junction level. Scale bars: 15 µm. n = 10 except n = 12 disks in A. (A) ap>Gli-WT. PLA signals for Gliotactin + Csk were increased compared with the control side (A and A″). A single Z slice at the level of the AJ is shown with some peripodial membrane immunolabeling from apical layers present (asterisk). (B) In the corresponding side projections, the PLA signals were concentrated at the apical side at the level of the AJ (arrow) (B″) with a smaller degree of PLA in the basal side. (C) ap>Gli-WT, Csk-WT. Coexpression of Csk-WT increased the Gliotactin+Csk PLA in the apterous side (C and C″). A single Z slice at the level of the AJ is shown with some peripodial membrane immunolabeling from apical layers present (asterisk). (D) Corresponding side projections. PLA signals were concentrated in the apical side at the level of the AJ (arrow) (D″) with a smaller degree of PLA in the basal side. (E) ap>Gli-WT, Csk-RNAi. PLA signals between Gliotactin+Csk is decreased when the Csk was knocked down (E and E″). The apterous boundary is in these disks is hard to determine and was not marked. Note that this panel is a single Z stack taken from the columnar epithelium at the septate junction level. (F) Corresponding side projections. (G) Statistical analysis of the Gliotactin+Csk PLA signal intensities in ap>Gli-WT and ap>Gli-WT, Csk-WT. The mean ratios of PLA intensities in the apterous side vs. WT side are shown. n = 12 disks in ap>Gli-WT and n = 10 disks in ap>Gli-WT, Csk-WT (****p < 0.0001). Error bars represent the SD.
Down-regulation of Csk affects endogenous Gliotactin
Overall, our results suggest that Csk associates with Gliotactin and triggers endocytosis either by directly or indirectly phosphorylating Gliotactin. If Csk is able to regulate Gliotactin, then we hypothesized that a loss of Csk in an otherwise wild-type background would lead to changes in the distribution of Gliotactin. To test the loss of Csk on endogenous Gliotactin, we expressed Csk-RNAi using the apterous-GAL4 driver (Figure 8). We observed that the knockdown of Csk with apterous-GAL4 (ap>Csk-RNAi) resulted in overproliferation and extra folds of the wing disks without generating a clear cell delamination or migration phenotype (Figure 8B), similarly to prior observations with knockdown of Csk in the wing pouch (Kwon et al., 2015). A small percentage of disks (20%) had low levels of cell migration, but interpretation of these disks was difficult due to the overgrowth of the apterous side, leaving a greatly reduced or absent nonapterous side. To detect whether the endogenous Gliotactin was affected by the loss of Csk, the least-affected areas (those areas still in the apterous side but without extensive folds) were examined. Compared to the Gliotactin localization in the control (ap>NLS GFP) (Figure 8, A′ and F″), the tight localization of Gliotactin to the TCJ was lost when Csk was knocked down (Figure 8, C″ and D′″). Instead, Gliotactin spread away from the TCJ and formed extended ribbonlike structures through the bicellular septate junction (Figure 8, E and E′). In these areas, other septate junction markers, Dlg and Cora, were not mislocalized and remained within the septate junction domain (Figure 8, C″ and D″). Similarly, Ecad was not mislocalized (Supplemental Figure S7, A and B), suggesting that the effect on Gliotactin was not due to loss of either the septate or adherens junctions. These data suggest that Csk has a specific effect on controlling the localization of endogenous Gliotactin. In total, our results suggest that Csk plays a specific role in limiting the expression of Gliotactin to the TCJ likely through phosphorylation and endocytosis of Gliotactin.
FIGURE 8:
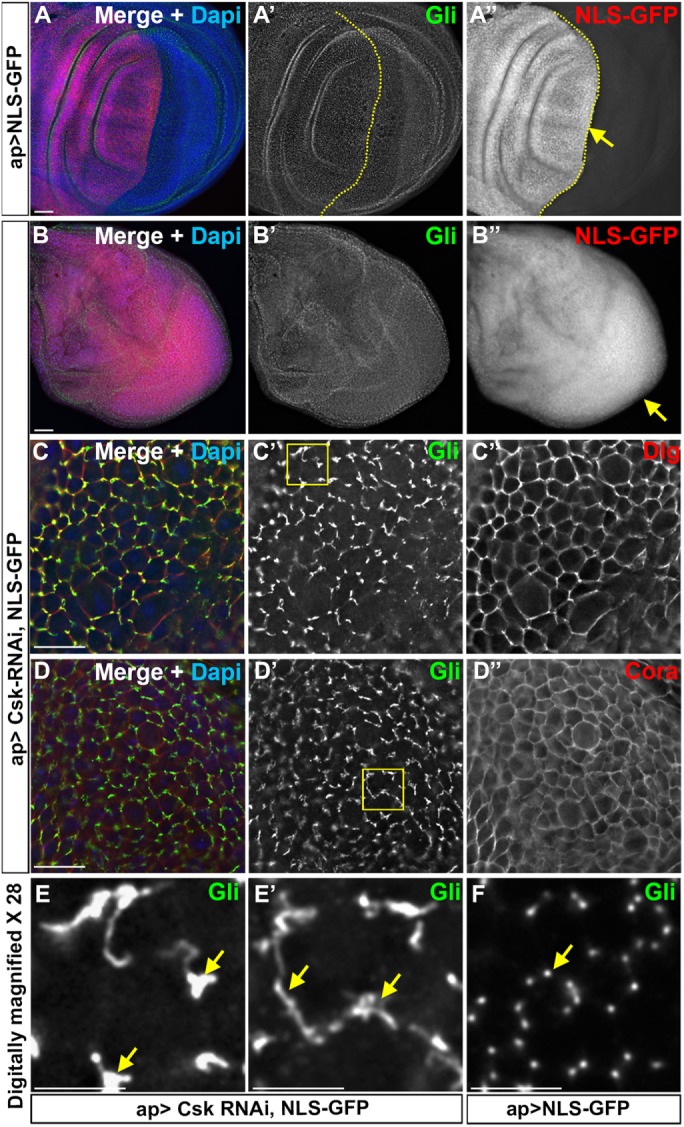
Down-regulation of Csk affects endogenous Gliotactin. Third-instar wing imaginal disks with transgenes or RNAi lines expressed by the apterous-GAL4 (ap>) driver. The apterous boundary is indicated with a dashed line with the apterous side on the left. Each panel represents a single Z slice at the septate junction level. Scale bars: A and B, 30 µm; C and D, 15 µm; and E–E″, 5 µm. n = 10 disks except n = 15 disks in C. (A, B) Wing disks immunolabeled for Gliotactin (Gli, green), NLS-GFP (red), and DAPI (blue). Arrows indicate the leading edge of the migratory cells. (A) ap>NLS-GFP. There was no cell overproliferation or migration of control NLS-GFP-positive cells compared with the nonapterous side. (B) ap>Csk-RNAi, NLS-GFP. Knockdown of Csk resulted in overproliferation resulting in extra folds. The entire disk was filled with NLS-GFP due to overproliferation, making it hard to identify the apterous side. (C, D) Higher magnification images (60×) of ap>Csk-RNAi, NLS-GFP disks immunolabeled for Gliotactin (Green) and Dlg or Cora (red). The images were taken from least-affected areas of least-affected disks. C′ and D′ showed Gliotactin was spreading away from tricellular corners making circular or ribbon like structures. C″ and D″ showed that the Dlg and Cora immunolabeling were not affected, respectively. (E, E′) The 28× digitally magnified images of areas marked by boxes in C′ and D′, respectively. Regions where Gliotactin spread away from the TCJ are indicated (arrows). (F) Wild-type disk 28× digitally magnified. The arrow indicates the punctate-like localization of Gliotactin at the TCJs.
DISCUSSION
The level and the correct localization of Gliotactin is tightly controlled by phosphorylation followed by endocytosis (Padash-Barmchi et al., 2010) and microRNA-mediated degradation of Gliotactin mRNA (Sharifkhodaei et al., 2016). When overexpressed, the spread of Gliotactin away from the TCJ leads to aberrant phenotypes disrupting the entire epithelial architecture. Tyrosine phosphorylation of Gliotactin is a mechanism to control Gliotactin levels and localization (Padash-Barmchi et al., 2010), yet phosphorylation also leads to the aberrant phenotypes. Here we demonstrate that a tyrosine kinase, C-terminal Src kinase (Csk) regulates Gliotactin through a Src-independent mechanism. Loss of Csk strongly enhanced the Gliotactin overexpression phenotypes, while coexpression of Csk suppressed. Loss of Csk also led to the spread of Gliotactin away from the TCJ when Gliotactin is expressed at normal physiological levels. Overall, our data show that Csk controls both overexpressed and endogenous Gliotactin, suggesting a novel role for Csk in controlling the protein components of the TCJ.
Our results point to a clear role for Csk in regulating Gliotactin phosphorylation, endocytosis, and protein distribution. However, the effect of Csk on Gliotactin could be indirect through the actions of another kinase or direct where Csk itself phosphorylates Gliotactin. Csk is a well-established regulator of Src kinase signaling, but we found that the effects of Csk on Gliotactin were independent of Src. Neither loss nor overexpression of Src suppressed the Gliotactin phenotypes, supporting the conclusion that the in vivo regulation of Gliotactin is not mediated by Src. Our in vitro kinase assay failed to show a direct activity of Csk activity on Gliotactin. However, Csk substrate recognition is dependent on the native three-dimensional structure of the substrate (Ia et al., 2010), such that Src itself is not phosphorylated by Csk when a truncated form of Src is provided, even when the targeted phosphorylation site is present (Ruzzene et al., 1997). Thus Gliotactin may not have the correct structural properties in vitro for recognition by Csk, especially as the intracellular domain of Gliotactin is natively unfolded in vitro (Zeev-Ben-Mordehai et al., 2003).
Our in vivo evidence pointed to an increase in phosphorylation of Gliotactin in the presence of increased Csk. This paired with the PLA signals between Csk and Gliotactin suggested that Csk can associate and perhaps directly phosphorylate Gliotactin but this occurs only when Gliotactin spreads away from the TCJ. In wild-type wing disks, Csk is clearly absent from the septate junction domain and TCJs suggesting that Csk is normally excluded from contact with Gliotactin. The PLA between overexpressed Gli and Csk was concentrated within the adherens junction domain even when both proteins were overexpressed and spread throughout the cell. This suggests that Csk association with Gliotactin requires an intermediate whose distribution is limited to the adherens junction. If Csk directly phosphorylates Gliotactin, then the intermediate(s) could be associated with the adherens junction and be responsible for recruiting Csk and potentially activating Csk at the AJ. Thus the lack of Csk phosphorylation of Gliotactin in vitro may be due to a lack of a specific cofactor for Csk activity. Alternatively, the recruitment of Csk to the adherens junction may activate another tyrosine kinase, which in turn phosphorylates Gliotactin.
Csk is a cytosolic tyrosine protein kinase (Cooper and Howell, 1993) and, unlike Src, Csk is not myristoylated and thus other membrane proteins or lipids recruit Csk to the membrane and cell junctions to activate Csk (Hirao et al., 1997; Kwon et al., 2015). One mechanism is through the SH2 domain of Csk binding to phospho-tyrosine residues of transmembrane protein and membrane-associated proteins. For instance, phospho-tyrosine residues mediate Csk binding to VE-cad (Baumeister et al., 2005) and focal adhesion proteins Pragmin in mammals and Paxillin in Drosophila (Rengifo-Cam et al., 2004; Senda et al., 2016). Junctional adhesion molecule-A acts as a Csk binding protein when phosphorylated and recruits Csk to the integrin-Src complex to suppress Src activity (Naik et al., 2014). Csk is recruited to lipid rafts by binding to PAG1/CBP, which functions to control activation of Src (Okada, 2012). In Drosophila, Ankyrin-repeat, SH3-domain, and proline-rich-region containing protein (dASPP) is located at the adherens junction and functions as a Csk binding protein and as a positive regulator of Csk kinase activity (Langton et al., 2007). Given the localization of Csk to the adherens junction domain, it is possible that Ecad, dASPP, or another adherens junction associated protein could function as a potential intermediate between Csk and Gliotactin.
Another possibility is that Gliotactin can interact with Csk directly but only in the context of a tyrosine kinase that is located at the adherens junction. As outlined above, one means of recruitment of Csk to the membrane involves binding of the Csk SH2 domain to a phospho-tyrosine residue. As Gliotactin has two highly conserved tyrosines (Y760, Y799), phosphorylation of one tyrosine could mediate recruitment Csk and subsequent phosphorylation of the second residue. When overexpressed, Gliotactin is phosphorylated at one or both conserved tyrosine residues (Padash-Barmchi et al., 2010) and it is not known whether these sites are redundant or have different functions. It is possible that there is a second tyrosine kinase associated with the adherens junction that phosphorylates one residue to recruit Csk, which in turn phosphorylates the other. This model would explain the Gliotactin+Csk PLA and the increased Csk recruitment within the Gliotactin positive vesicles when Gliotactin is overexpressed. The increase in Ecad levels with the overexpression of Gliotactin also supports a model where Gliotactin can recruit Csk to the membrane leading to the activation of Csk. The overexpression of Csk alone did not lead to an increase in Ecad, but coexpression of Csk and Gliotactin did. Increased Csk activity would then block Src activity and lead to decreased Ecad endocytosis and increased Ecad (Vidal et al., 2006).
Regardless of whether Csk directly or indirectly phosophorylates Gliotactin, Csk is clearly regulating Gliotactin to control endocytosis and/or localization to the TCJ. As Csk overexpression strongly suppressed the Gliotactin overexpression phenotypes, it is unlikely that Csk is responsible for the phosphorylation-dependent Gliotactin signaling that leads to the overexpression phenotypes. Instead, Csk appears to control Gliotactin endocytosis to ensure tight localization of Gliotactin to the TCJ. When Csk is down-regulated, Gliotactin is found outside the normal domain, suggesting a role in Csk to removing “excess” or mislocalized Gliotactin. It is possible that the effect on endogenous Gliotactin is due to disruption of other proteins known to regulate Gliotactin localization to the TCJ, Dlg (Padash-Barmchi et al., 2013) and Bark/Anakonda (Byri et al., 2015). Loss of either leads to the spread of Gliotactin at the TCJ but also a dramatic down-regulation of Gliotactin on the membrane. With Csk knockdown, Gliotactin levels were not reduced, and spread was not throughout the membrane, suggesting that the mechanism was not mediated by changes to either Dlg or Bark/Anakonda. Instead, the spread of Gliotactin observed with the loss of Csk was similar to that seen when endocytosis was blocked using a dominant-negative Rab5 (Padash-Barmchi et al., 2010).
While the loss of Csk altered the localization of Gliotactin at the TCJ, overexpression of Csk in an otherwise wild-type background only led to a small basolateral spread of Gliotactin that was located to the TCJ. This suggests that Gliotactin at the corners is highly stable and somehow “protected” from phosphorylation by masking the phosphorylation sites or blocking kinase access to Gliotactin. Overall, our data suggest that the normal role of Csk is to control the phosphorylation of Gliotactin and endocytosis to ensure correct localization to the TCJ.
Csk is a cytosolic tyrosine kinase and known as a classic negative regulator of Src, though recently Src-independent roles for Csk have been identified. The roles of Csk can be diverse, as it has both tumor suppressor as well as oncogenic properties (Okada, 2012; Yao et al., 2014). A role for Csk in controlling junctional proteins or signaling events at cell–cell or cell–matrix junctions through regulation of Src activity is well established. However, a prior role for Csk at the tricellular junction or bicellular septate junction was not known. We have uncovered a novel role for Csk as a potential regulator of tricellular junctions independent of Src. It remains to be determined whether Csk activity on Gliotactin is direct or indirect; however, further investigation of the role of Csk in controlling TCJ targets could identify novel targets and functions for Csk. Control of the levels and distribution of tricellular junction proteins is important. For instance, overexpression of the vertebrate tricellular junctional protein Tricellulin leads to tumor formation in some tissues (Kojima and Sawada, 2012). Therefore, Csk regulation of Gliotactin expression could be a useful model to understand the mechanisms associated with control of tricellular junctional proteins that spread beyond their normal distribution.
MATERIALS AND METHODS
Fly stocks
Drosophila fly Stocks were either obtained from the Bloomington Drosophila Stock Center (BDSC), the VDRC, the Kyoto stock center Drosophila Genomics and Genetic Resources Center (DGRC), or from researchers who made them. Fly stocks included w1118 and apterous-GAL4 (BDSC); Gli::YFP (DGRC). UAS transgenes included the following: Gli-WT and Gli-FF (Padash-Barmchi et al., 2010), Csk-WT (Pedraza et al., 2004), Src64B-WT (Dura, 2004), and NLS-GFP (a gift from Douglas Allan, University of British Columbia). RNAi lines used the following: Csk-RNAi 1 (VDRC #109813), Csk-RNAi 2 (VDRC #32877), Csk-RNAi 3 (BDSC #35174), Src64B-RNAi (VDRC #35252), Src64B-RNAi (BDSC #30517), and Src42A- RNAi (VDRC #26019). Mutant alleles include the following: Csk[J1D8] (Stewart et al., 2003), Src64B[D404N] (Strong et al., 2011), Src64B[KO] (Dodson et al., 1998; O’Reilly et al., 2006), and Src42A[myri] (Tateno et al., 2000). Unless specifically stated, all crosses were carried out at 29°C under standard conditions using virgin females from the parental stock: ap-GAL4,GAL80ts;UAS-GliWT#5/SM-TM6,Tb.
Immunofluorescence labeling
Wing imaginal disks were dissected from wandering third-instar larvae and immunolabeled using standard protocols: larvae were dissected in 1× phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min. All subsequent washes were in PBS plus 0.1% Triton X-100 (PBST), and disks were blocked in PBST with BSA (0.5%) prior to primary antibody incubation overnight at 4°C, secondary antibodies were incubated for 2 h at room temperature. Disks were cleared in 70% glycerol and mounted with Vectashield (Invitrogen). Primary antibodies were used at the following dilutions: mouse anti-Gli IF6.3 at 1:100 (Auld et al., 1995), rabbit anti-Gli at 1:600 (the Gliotactin polyclonal antibody was generated by immunizing rabbits with a peptide corresponding to the amino acid sequence [CQPAAQPRRTHLVEGVPQTS] and subsequent affinity purification, both by YenZym Antibodies, LLC), guinea pig anti-Mcr at 1:800 (Hall et al., 2014), rabbit anti-Csk at 1:1200 (Langton et al., 2007), rabbit anti-phospho-Tyr at 1:100 (Cell Signaling), rabbit anti-Rbsn 5 at 1:6000 (Tanaka and Nakamura, 2008), rabbit anti-Rab7 at 1/2000 (Tanaka and Nakamura, 2008), rabbit anti Rab11 at 1/8000 (Tanaka and Nakamura, 2008), and rabbit anti-cleaved Caspase 3 at 1:200 (Cell Signaling). Mouse anti-Arm at 1:50 (Riggleman et al., 1990), mouse anti-Dlg 4F3 at 1:100 (Parnas et al., 2001), rat anti–DE-cadherin at 1:35 (Oda et al., 1994), and mouse anti-Coracle (9C and C615-16B cocktail) at 1:100 (Fehon et al., 1994) were obtained from the Developmental Studies Hybridoma Bank (University of Iowa), and DAPI (4’,6-diamidino-2-phenylindole) was used at 1:1000 (Thermo Scientific). The following secondary antibodies were used at 1:300: goat anti-rabbit Alexa488, goat anti-rabbit Alexa568, goat anti rabbit Alexa647, goat anti-mouse Alexa488, goat anti-mouse Alexa568, goat anti-mouse Alexa647, goat anti-rat Alexa568, goat anti-rat Alexa647, and goat anti-guinea pig Alexa488.
Proximity ligation assay
PLA were carried according to the manufacturer’s specifications (Duolink, Sigma). Total volumes of the PLA reactions were 80 µl for 15 wing disks. Prior to PLA, the isolation of wing imaginal disks, fixation, and incubation with primary antibodies were done as described above. Disks were incubated at 37°C, and timings were as follows: initial incubation with PLA probe solution for 1.5 h, ligation reaction for 1 h, and amplification reaction for 2 h. Disks were equilibrated and mounted in Vectashield. PLA controls used only one antibody at a time to ensure specificity.
Imaging
For higher magnification images, z-series stacks were collected with 0.2-µm steps using a DeltaVision Spectris microscope (Applied Precision, GE) with a 60× (1.4 NA) oil immersion lens and CoolSnap HQ digital camera. Lower magnification images were collected with a 20× air lens. Deconvolution was done with SoftWorx (Applied Precision) software with 6–10 iterations using a point-spread function calculated with 0.2-µm beads conjugated with Alexa Fluor 568 (Molecular Probes) mounted in Vectashield. Image processing and side projections were done using SoftWorx. Figures were made using Adobe Photoshop 4 or CC 2017. Lower-resolution imagines (20× objective) were also obtained using a Zeiss Axioskop and Northern Eclipse software.
Statistical analysis
The statistical analysis and graphing were done using Prism 6.0. One-way analysis of varience (ANOVA) was done to perform pairwise comparisons among multiple genotypes. Tukey post hoc was done to determine significance. Student’s t test was done when only two genotypes were to be compared. Samples were neither randomized nor blinded. All disks in each sample set were analyzed.
Cell migration.
Using ImageJ (http://imagej.nih.gov/ij/), the migration distance of Gliotactin positive cells from the apterous boundary to the control side was measured for each disk and expressed as a ratio to the total distance from the apterous boundary to the outer edge of the control side. To measure each distance, the line tool and the Analyze/ Measure option in ImageJ were used. For all panels, the dorsal/ventral apterous side was identified based on immunolabeling of the overexpressed Gliotactin at the level of the septate junction domain as the epithelium/boundary is intact at this level.
Counting of vesicles.
For each disk, three Z stacks (at the level of the septate junction domain) from 60× objective images were compressed using the quick projection function in SoftWorx. Endosome counting was done manually marking each endosome with the aid of cell counter tool in ImageJ. For each disk, counting was done in a large and constant area (892,500 pixels2) that extended from the apterous boundary through the wing pouch from digitally magnified images (2×).
Immunolabeling intensity.
Using ImageJ, intensities of immunolabeling or PLA (mean gray values) were measured in two constant size boxes drawn on the apterous and the control side of the pouch area of wing disk and the ratios of the apterous/wild type were calculated.
Biochemistry
Extraction of His-tagged Gliotactin C-terminal constructs.
BL21DE3 pLysS cells carrying pET28a (Novagen) plasmids containing Gliotactin C-terminal constructs (Padash-Barmchi et al., 2010) were cultured for 2 h prior to the induction of protein expression with 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) for 45 min. The cells were lysed in 40 mM imidazole, 300 mM NaCl, 50 mM Tris-HCl, pH 8, 1/100 PhosStop (Roche), and 1/100 Complete (Roche). Nickel NTA-agarose (Qiagen) was used for the purification of His-tagged proteins and eluted with 250 mM imidazole buffer (300 mM NaCl, 50 mM Tris-HCl, pH 8) and dialyzed using Slide-A-Lyzer 7000MWCO cassettes (Pierce) into Kinase Buffer (25 mM MOPS [3-(N-morpholino)propanesulfonic acid], pH 7.2, 12.5 mM glycerol-2-phosphate, 20 mM MgCl2, 25 mM MnCl2, 5 mM EGTA [ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid], 2 mM EDTA, 0.25 mM dithiothreitol [DTT], and 0.1 mM sodium orthovanadate).
Kinase assay.
A 20-µl kinase reaction (0.5 µg His tagged GliCter in pET28, 0.5 µg GST–Csk [Cell Signaling Technologies], 1× Kinase Buffer, 1 mM ATP) was incubated at 30°C for 30 min. In the control experiment 0.5 µg of His tagged human Src (Creative Biomart) was used.
Li-cor Western blot analysis.
Half of the kinase reaction was loaded on 10% SDS–PAGE followed by Western blot on nitrocellulose membranes. Blots were blocked using 50% Odyssey blocking buffer (LI-COR) diluted in PBS and analyzed with LI-COR Odyssey fluorescent detection systems. The primary antibodies mouse anti-Gli IF6.3 at 1/5000 and rabbit anti-phosphoTyr (Cell Signaling) at 1/5000 and secondary antibodies goat anti-rabbit IRDye 680 at 1: 20,000, goat anti-mouse IRDye 800 at 1: 20,000 were used (All IRDye antibodies from Rockland).
Supplementary Material
Acknowledgments
We thank Tian Xu for providing us with Csk-WT transgenic flies, Nick Tapon for Csk antibodies, Makoto Nakamura for Rbsn-5 and Rab7 antibodies, and Ninan Abraham for helpful discussions on the biochemical work and for use of the LI-COR Odyssey fluorescent detection system. We also thank the Vienna Drosophila RNAi center and the Bloomington stock center for providing fly stocks and the Developmental Studies Hybridoma Bank for providing antibodies. We thank Till Matzat for his comments on the article and Auld lab members for support and suggestions throughout this work. This research was funded by grant MOP-123420 from the Canadian Institutes of Health Research.
Abbreviations used:
- ap
apterous
- Csk
C-terminal Src kinase
- Gli
Gliotactin
- SJ
septate junction
- TCJ
tricellular junction.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-04-0251) on November 22, 2017.
REFERENCES
- Altick AL, Baryshnikova LM, Vu TQ, Von Bartheld CS. Quantitative analysis of multivesicular bodies (MVBs) in the hypoglossal nerve: evidence that neurotrophic factors do not use MVBs for retrograde axonal transport. J Comp Neurol. 2009;514:641–657. doi: 10.1002/cne.22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Fetter RD, Broadie K, Goodman CS. Gliotactin, a novel transmembrane protein on peripheral gila, is required to form the blood-nerve barrier in Drosophila. Cell. 1995;81:757–767. doi: 10.1016/0092-8674(95)90537-5. [DOI] [PubMed] [Google Scholar]
- Bätz T, Förster D, Luschnig S. The transmembrane protein Macroglobulin complement-related is essential for septate junction formation and epithelial barrier function in Drosophila. Development. 2014;141:899–908. doi: 10.1242/dev.102160. [DOI] [PubMed] [Google Scholar]
- Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J. 2005;24:1686–1695. doi: 10.1038/sj.emboj.7600647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner S, Littleton JT, Broadie K, Bhat MA, Harbecke R, Lengyel JA, Chiquet-Ehrismann R, Prokop A, Bellen HJ. A Drosophila neurexin is required for septate junction and blood-nerve barrier formation and function. Cell. 1996;87:1059–1068. doi: 10.1016/s0092-8674(00)81800-0. [DOI] [PubMed] [Google Scholar]
- Byri S, Misra T, Syed ZA, Bätz T, Shah J, Boril L, Glashauser J, Aegerter-Wilmsen T, Matzat T, Moussian B, et al. The triple-repeat protein anakonda controls epithelial tricellular junction formation in Drosophila. Dev Cell. 2015;33:535–548. doi: 10.1016/j.devcel.2015.03.023. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Howell B. The when and how of Src regulation. Cell. 1993;73:1051–1054. doi: 10.1016/0092-8674(93)90634-3. [DOI] [PubMed] [Google Scholar]
- Dodson GS, Guarnieri DJ, Simon MA. Src64 is required for ovarian ring canal morphogenesis during Drosophila oogenesis. Development. 1998;125:2883–2892. doi: 10.1242/dev.125.15.2883. [DOI] [PubMed] [Google Scholar]
- Faivre-Sarrailh C, Banerjee S, Li J, Hortsch M, Laval M. Drosophila contactin, a homolog of vertebrate contactin, is required for septate junction organization and paracellular barrier function. Development. 2004;131:4931–4942. doi: 10.1242/dev.01372. [DOI] [PubMed] [Google Scholar]
- Fehon RG, Dawson IA, Artavanis-Tsakonas S. A Drosophila homologue of membrane-skeleton protein 4.1 is associated with septate junctions and is encoded by the coracle gene. Development. 1994;120:545–557. doi: 10.1242/dev.120.3.545. [DOI] [PubMed] [Google Scholar]
- Fristrom DK. Septate junctions in imaginal disks of Drosophila: a model for the redistribution of septa during cell rearrangement. J Cell Biol. 1982;94:77–87. doi: 10.1083/jcb.94.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16:181–188. doi: 10.1016/j.tcb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Gajadhar A, Guha A. A proximity ligation assay using transiently transfected, epitope-tagged proteins: application for in situ detection of dimerized receptor tyrosine kinases. BioTechniques. 2010;48:145–152. doi: 10.2144/000113354. [DOI] [PubMed] [Google Scholar]
- Genova JL, Fehon RG. Neuroglian, Gliotactin, and the Na+/K+ ATPase are essential for septate junction function in Drosophila. J Cell Biol. 2003;161:979–989. doi: 10.1083/jcb.200212054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall S, Bone C, Oshima K, Zhang L, Mcgraw M, Lucas B, Fehon RG, Ward RE. Macroglobulin complement-related encodes a protein required for septate junction organization and paracellular barrier function in Drosophila. Development. 2014;141:889–898. doi: 10.1242/dev.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt A, Pflanz R, Behr M, Tarp T, Riedel D, Schuh R. Bark beetle controls epithelial morphogenesis by septate junction maturation in Drosophila. Dev Biol. 2015;400:237–247. doi: 10.1016/j.ydbio.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Hirao A, Hamaguchi I, Suda T, Yamaguchi N. Translocation of the Csk homologous kinase (Chk/Hyl) controls activity of CD36-anchored Lyn tyrosine kinase in thrombin-stimulated platelets. EMBO J. 1997;16:2342–2351. doi: 10.1093/emboj/16.9.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ia KK, Mills RD, Hossain MI, Chan K-C, Jarasrassamee B, Jorissen RN, Cheng H-C. Structural elements and allosteric mechanisms governing regulation and catalysis of CSK-family kinases and their inhibition of Src-family kinases. Growth Factors. 2010;28:329–350. doi: 10.3109/08977194.2010.484424. [DOI] [PubMed] [Google Scholar]
- Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171:939–945. doi: 10.1083/jcb.200510043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–1124. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- Kojima T, Sawada N. Regulation of tight junctions in human normal pancreatic duct epithelial cells and cancer cells. Ann NY Acad Sci. 2012;1257:85–92. doi: 10.1111/j.1749-6632.2012.06579.x. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, Waghmare I, Verghese S, Singh A, Singh A, Kango-Singh M. Drosophila C-terminal Src kinase regulates growth via the Hippo signaling pathway. Dev Biol. 2015;397:67–76. doi: 10.1016/j.ydbio.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Langton PF, Colombani J, Aerne BL, Tapon N. Drosophila ASPP regulates C-terminal Src kinase activity. Dev Cell. 2007;13:773–782. doi: 10.1016/j.devcel.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Lo˝rincz P, Lakatos Z, Varga Á, Maruzs T, Simon-Vecsei Z, Darula Z, Benko˝ P, Csordás G, Lippai M, Andó I, et al. MiniCORVET is a Vps8-containing early endosomal tether in Drosophila. Elife. 2016;5:e14226. doi: 10.7554/eLife.14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, Akashi M, Nishi E, Furuse M. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J Cell Sci. 2011;124:548–55. doi: 10.1242/jcs.072058. [DOI] [PubMed] [Google Scholar]
- Mottola G, Classen A-K, González-Gaitán M, Eaton S, Zerial M. A novel function for the Rab5 effector Rabenosyn-5 in planar cell polarity. Development. 2010;137:2353–2364. doi: 10.1242/dev.048413. [DOI] [PubMed] [Google Scholar]
- Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule-A suppresses platelet integrin aIIbb3 signaling by recruiting Csk to the integrin-c–Src complex. Blood. 2014;123:1393–1402. doi: 10.1182/blood-2013-04-496232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Meyer EJ, Kroesen A, McKinney SA, Gibson MC. Epithelial junctions maintain tissue architecture by directing planar spindle orientation. Nature. 2013;500:359–362. doi: 10.1038/nature12335. [DOI] [PubMed] [Google Scholar]
- Nayak G, Lee SI, Yousaf R, Edelmann SE, Trincot C, Van Itallie CM, Sinha GP, Rafeeq M, Jones SM, Belyantseva IA, et al. Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J Clin Invest. 2013;123:4036–4049. doi: 10.1172/JCI69031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska P, Godt D, Tepass U. D E-Cadherin is required for intercellular motility during Drosophila oogenesis. J Cell Biol. 1999;144:533–547. doi: 10.1083/jcb.144.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot-Timothee C, Graf F, Noirot CH. The specialization of septate junctions in regions of tricellular junctions. J Ultrastruct Res. 1982;78:152–165. doi: 10.1016/s0022-5320(82)80020-8. [DOI] [PubMed] [Google Scholar]
- Oda H, Uemura T, Harada Y, Iwai Y, Takeichi M. A drosophila homolog of cadherin associated with armadillo and essential for embryonic cell-cell adhesion. Dev Biol. 1994;165:716–726. doi: 10.1006/dbio.1994.1287. [DOI] [PubMed] [Google Scholar]
- O'Reilly AM, Ballew AC, Miyazawa B, Stocker H, Hafen E, Simon MA. Csk differentially regulates Src64 during distinct morphological events in Drosophila germ cells. Development. 2006;133:2627–2638. doi: 10.1242/dev.02423. [DOI] [PubMed] [Google Scholar]
- Okada M. Regulation of the Src family kinases by Csk. Int J Biol Sci. 2012;8:1385–1397. doi: 10.7150/ijbs.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem. 1991;266:24249–24252. [PubMed] [Google Scholar]
- Padash-Barmchi M, Browne K, Sturgeon K, Jusiak B, Auld VJ. Control of Gliotactin localization and levels by tyrosine phosphorylation and endocytosis is necessary for survival of polarized epithelia. J Cell Sci. 2010;123:4052–4062. doi: 10.1242/jcs.066605. [DOI] [PubMed] [Google Scholar]
- Padash-Barmchi M, Charish K, Que J, Auld VJ. Gliotactin and discs large are co-regulated to maintain epithelial integrity. J Cell Sci. 2013;126:1134–1143. doi: 10.1242/jcs.113803. [DOI] [PubMed] [Google Scholar]
- Paul SM, Ternet M, Salvaterra PM, Beitel GJ. The Na+/K+ ATPase is required for septate junction function and epithelial tube-size control in the Drosophila tracheal system. Development. 2003;130:4963–4974. doi: 10.1242/dev.00691. [DOI] [PubMed] [Google Scholar]
- Pedraza LG, Stewart aR, Li D-M, Xu T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene. 2004;23:4754–4762. doi: 10.1038/sj.onc.1207635. [DOI] [PubMed] [Google Scholar]
- Riggleman B, Schedl P, Wieschaus E. Spatial expression of the Drosophila segment polarity gene armadillo is posttranscriptionally regulated by wingless. Cell. 1990;63:549–560. doi: 10.1016/0092-8674(90)90451-j. [DOI] [PubMed] [Google Scholar]
- Ruzzene M, Songyang Z, Marin O, Donella-Deana A, Brunati AM, Guerra B, Agostinis P, Cantley LC, Pinna LA. Sequence specificity of C-terminal Src kinase (CSK) A comparison with Src-related kinases c-Fgr and Lyn. Eur J Biochem. 1997;246:433–439. doi: 10.1111/j.1432-1033.1997.t01-1-00433.x. [DOI] [PubMed] [Google Scholar]
- Schulte J, Charish K, Que J, Ravn S, MacKinnon C, Auld VJ. Gliotactin and Discs large form a protein complex at the tricellular junction of polarized epithelial cells in Drosophila. J Cell Sci. 2006;119:4391–4401. doi: 10.1242/jcs.03208. [DOI] [PubMed] [Google Scholar]
- Schulte J, Tepass U, Auld VJ. Gliotactin, a novel marker of tricellular junctions, is necessary for septate junction development in Drosophila. J Cell Biol. 2003;161:991–1000. doi: 10.1083/jcb.200303192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senda Y, Murata-Kamiya N, Hatakeyama M. Csk-mediated EPIYA phosphorylation of Pragmin creates a feed-forward Csk activation loop that promotes cell motility. Cancer Sci. 2016;107:972–980. doi: 10.1111/cas.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifkhodaei Z, Barmchi MP, Gilbert MM, Samarasekera G, Fulga TA, Van Vactor D, Auld VJ. The tricellular junction protein Gliotactin auto-regulates mRNA levels via BMP signaling induction of miR-184. J Cell Sci. 2016;129:1477–1489. doi: 10.1242/jcs.178608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, McCall K, Steller H. DCP-1, a Drosophila cell death protease essential for development. Science. 1997;275:536–540. doi: 10.1126/science.275.5299.536. [DOI] [PubMed] [Google Scholar]
- Stewart RA, Li D-M, Huang H, Xu T. A genetic screen for modifiers of the lats tumor suppressor gene identifies C-terminal Src kinase as a regulator of cell proliferation in Drosophila. Oncogene. 2003;22:6436–6444. doi: 10.1038/sj.onc.1206820. [DOI] [PubMed] [Google Scholar]
- Strong TC, Kaur G, Thomas JH. Mutations in the catalytic loop HRD motif alter the activity and function of drosophila Src64. PLoS One. 2011;6:e28100. doi: 10.1371/journal.pone.0028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Nakamura A. The endocytic pathway acts downstream of Oskar in Drosophila germ plasm assembly. Development. 2008;135:1107–1117. doi: 10.1242/dev.017293. [DOI] [PubMed] [Google Scholar]
- Tateno M, Nishida Y, Adachi-yamada T. Regulation of JNK by Src during Drosophila development. Science. 2000;287:324–327. doi: 10.1126/science.287.5451.324. [DOI] [PubMed] [Google Scholar]
- Tepass U, Tanentzapf G, Ward R, Fehon R. Epithelial cell polarity and cell junctions in Drosophila. Annu Rev Genet. 2001;35:747–784. doi: 10.1146/annurev.genet.35.102401.091415. [DOI] [PubMed] [Google Scholar]
- Tyler S. Epithelium–the primary building block for metazoan complexity. Integr Comp Biol. 2003;43:55–63. doi: 10.1093/icb/43.1.55. [DOI] [PubMed] [Google Scholar]
- Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284:12110–12124. doi: 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema DR, Zeev-Ben-Mordehai T, Auld VJ. Transient apical polarization of Gliotactin and Coracle is required for parallel alignment of wing hairs in Drosophila. Dev Biol. 2004;275:301–314. doi: 10.1016/j.ydbio.2004.07.040. [DOI] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL. Csk-Deficient boundary cells are eliminated from normal drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell. 2006:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Yao Q, Liu BQ, Li H, McGarrigle D, Xing BW, Zhou MT, Wang Z, Zhang JJ, Huang XY, Guo L. C-terminal src kinase (CSK)-mediated phosphorylation of eukaryotic elongation factor 2 (EEF2) promotes proteolytic cleavage and nuclear translocation of EEF2. J Biol Chem. 2014;289:12666–12678. doi: 10.1074/jbc.M113.546481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeev-Ben-Mordehai T, Rydberg EH, Solomon A, Toker L, Auld VJ, Silman I, Botti S, Sussman JL. The intracellular domain of the drosophila cholinesterase-like neural adhesion protein, Gliotactin, is natively unfolded. Proteins. 2003;53:758–767. doi: 10.1002/prot.10471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.