

Conventional nonhomologous end joining–based gene knockouts can lead to the production of C-terminally truncated proteins with potentially residual or dominant negative functions. Combining CRISPR/Cas9 with gene traps targeting the first intron (CRISPR-Trap), however, completely prevents the expression of the open reading frame, resulting in clean gene knockouts.
Abstract
CRISPR/Cas9-based genome editing offers the possibility to knock out almost any gene of interest in an affordable and simple manner. The most common strategy is the introduction of a frameshift into the open reading frame (ORF) of the target gene which truncates the coding sequence (CDS) and targets the corresponding transcript for degradation by nonsense-mediated mRNA decay (NMD). However, we show that transcripts containing premature termination codons (PTCs) are not always degraded efficiently and can generate C-terminally truncated proteins which might have residual or dominant negative functions. Therefore, we recommend an alternative approach for knocking out genes, which combines CRISPR/Cas9 with gene traps (CRISPR-Trap) and is applicable to ∼50% of all spliced human protein-coding genes and a large subset of lncRNAs. CRISPR-Trap completely prevents the expression of the ORF and avoids expression of C-terminal truncated proteins. We demonstrate the feasibility of CRISPR-Trap by utilizing it to knock out several genes in different human cell lines. Finally, we also show that this approach can be used to efficiently generate gene replacements allowing for modulation of protein levels for otherwise lethal knockouts (KOs). Thus, CRISPR-Trap offers several advantages over conventional KO approaches and allows for generation of clean CRISPR/Cas9-based KOs.
INTRODUCTION
A decade ago, zinc-fingers, the first customizable site-directed endonucleases, were shown to allow for targeted genome editing in human cells (Urnov et al., 2005). During the past 10 yr, genome editing approaches have rapidly evolved, leading to the development of TALENs (Christian et al., 2010) and the CRISPR/Cas9 technology (Cong et al., 2013; Mali et al., 2013). Among all gene-targeting strategies, CRISPR/Cas9 is the most versatile tool. Owing to its modularity—changing only the single guide RNA (sgRNA) sequence is sufficient to target the Cas9 endonuclease to any desired sequence—CRISPR/Cas9 has become one of the most popular technologies within today’s biotechnology (Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013; Du et al., 2016).
A commonly used strategy to generate gene knockouts (KOs) is to target the coding sequence (CDS) of the gene of interest with CRISPR/Cas9. Upon cleavage by Cas9, one of the two major DNA damage repair pathways usually repairs the double-strand break (DSB); either the accurate homology-directed repair (HDR) pathway or the error-prone nonhomologous end joining (NHEJ). NHEJ typically leads to small insertions/deletions in the vicinity of the DSB, often resulting in a frameshift and generation of a premature termination codon (PTC; Ran et al., 2013). Owing to the truncated CDS, the corresponding mRNA is predicted to be recognized by the nonsense-mediated mRNA decay (NMD) pathway, which degrades transcripts harboring PTCs in a translation-dependent manner (Karousis et al., 2016) and therefore should result in a de facto KO. However, NMD does not target all PTC-containing transcripts with the same efficacy for degradation. The position of the PTC within the transcript (Eberle et al., 2008; Neu-Yilik et al., 2011) and the sequence composition around the termination codon (Loughran et al., 2014) strongly influence the sensitivity of a transcript to NMD. Indeed, different transcripts predicted to be degraded by NMD, some of which were implicated in disease, were shown to escape NMD and in some cases the corresponding truncated protein is detectable (Green et al., 2003; Littink et al., 2009; Malfatti et al., 2015). Consequently, transcripts harboring frameshift mutations mediated by CRISPR/Cas9 may give rise to C-terminally truncated proteins, which could have residual or dominant negative functions. Even though it remains to be elucidated whether C-terminally truncated proteins affect functional studies in cell culture and/or animal models, their occurrence and physiological impact should be considered, especially in the context of clinical trials. Indeed, the first clinical trial testing CRISPR/Cas9 to knock out PDCD1 (programmed cell death 1) in immune cells of cancer patients started last year in China and similar trials are planned in the United States (Cyranoski, 2016).
Here, we propose a clean approach to knock out genes in human cell lines by combining gene traps with CRISPR/Cas9 technology targeting the first intron of genes of interest. Gene traps have been used for almost 20 yr for the generation of transgenic animals (Stanford et al., 1998) but rely on spontaneously occurring HDR, a very rare event. CRISPR-Trap, however, bypasses this limitation and allows flexible and efficient generation of gene KOs. Most important, however, CRISPR-Trap results in the complete abrogation of the corresponding full-length transcript, thereby preventing C-terminally truncated proteins and is directly applicable to ≈50% of all spliced human protein-coding genes and potentially all spliced RNA Pol II transcripts, if the first exon is directly targeted. Furthermore, the approach also allows for gene replacements using the same sgRNA. Additionally, we investigate the dependency of protein and RNA levels on the position of a PTC within a transcript using HEK-293 Flp-In T-REx cells stably expressing human β-globin (HBB) reporter constructs as well as HeLa cells transiently expressing a mini-µ reporter construct. Our results highlight the importance of a well-considered design and thorough analysis of CRISPR/Cas9-created gene KOs.
RESULTS AND DISCUSSION
Residual mRNA and truncated protein expression in KO cell lines with frameshift mutations
While establishing a human-induced pluripotent stem cell (hiPSC) line harboring a missense mutation in the fifth exon of the fused in sarcoma (FUS) gene, we obtained different clones with frameshift mutations in the targeted exon (Supplemental Figure S1). Based on the position of the PTCs within these transcripts, they were predicted to be ideal targets for NMD. The PTCs were positioned >50–55 nucleotides upstream of the 3′ most exon-exon junction, which is a strong trigger of NMD (Schweingruber et al., 2013), and they were located neither close to the 5′ nor to the 3′ end of the transcripts, a feature that was shown to stabilize PTC-containing transcripts (Eberle et al., 2008). Hence, we assumed to have created FUS KO cell lines. Intriguingly, we could detect mRNA, as well as C-terminally truncated FUS protein in some of the clones, whereas there was hardly any mRNA expression and no C-terminally truncated FUS detectable in other clones (Figure 1, A and B). To ensure that this behavior was restricted neither to transcripts of the FUS gene nor to human cell lines, we analyzed available data sets where gene KOs were obtained by introduction of frameshift mutations into the CDS, followed by a whole transcriptome analysis (Huang et al., 2013; Richter et al., 2013; Kabir et al., 2014; McCleland et al., 2015; Sadic et al., 2015; Kim et al., 2016; Lebedeva et al., 2016). In line with our observation, 12–73% mRNA of the KO targets were still detectable in these studies (Supplemental Table S1).
FIGURE 1:
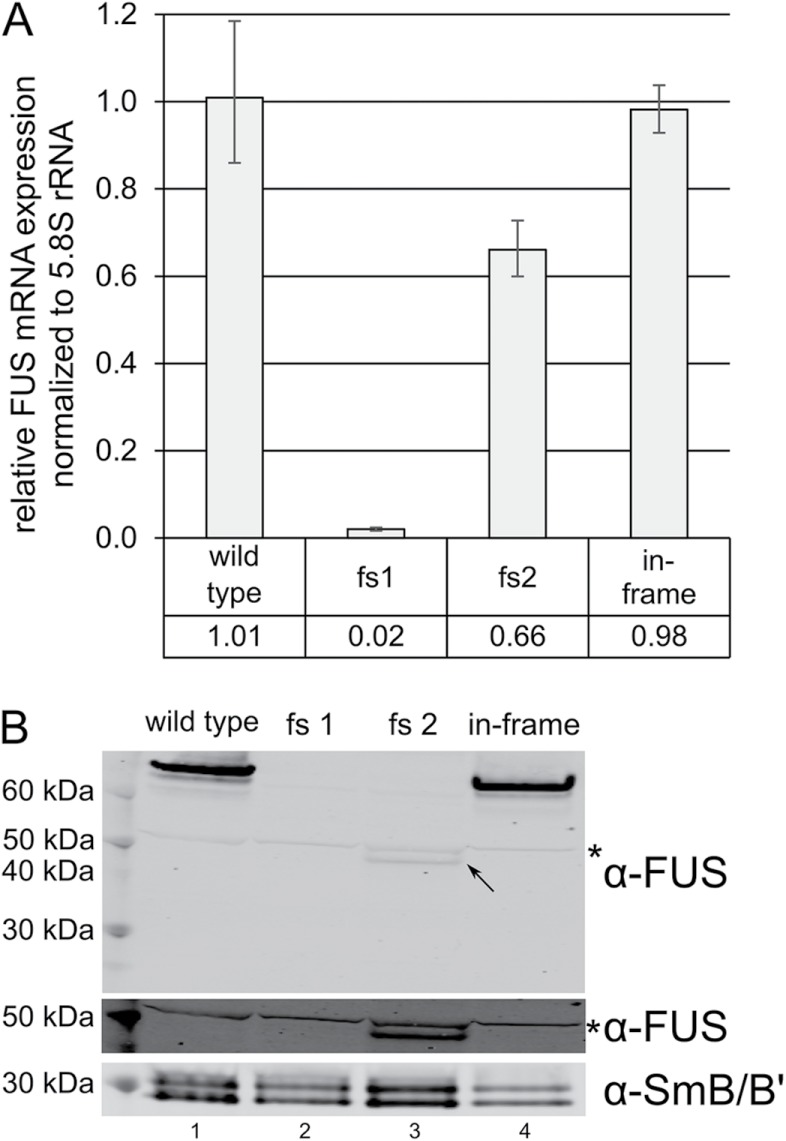
Residual mRNA and C-terminally truncated FUS protein expression in cell lines with FUS frameshift mutations. (A) FUS mRNA levels of two frameshift clones (fs1 and fs2) and one hiPSC line with a homozygous in-frame deletion in exon 5 of the FUS gene relative to wild-type FUS mRNA expression as determined by RT-qPCR. Average values and standard deviations of three biological replicates are shown. (B) Western blot confirming the absence of FUS protein in clone fs1 and showing the C-terminally truncated FUS in clone fs2 (indicated with a black arrow). For better visualization of the band indicated with the black arrow, this section of the Western blot is displayed with a higher exposure (lower membrane part labeled with α-FUS). The asterisk indicates an unspecific band recognized by the α-FUS antibodies equally present in all samples. SmB/B′ served as a loading control.
RNA and protein levels of predicted NMD targets
To address the dependency of mRNA and protein levels of predicted NMD targets on the position of the stop codon within the transcript, we generated a series of reporter constructs (Figure 2A) based on the immunoglobulin (Ig) µ minigene (mini-µ) reporter system (Buhler et al., 2004). Every reporter plasmid codes for Ig µ (including intronic sequences) under a cytomegalovirus promoter with a termination codon at a specific position of the open reading frame (referred to as TER[number] indicating the amino acid position of the TERmination codon). To enable immunodetection, a 2xHA-tag was cloned in front of the CDS, replacing the variable leader exon of the Ig µ minigene. Reporter plasmids harboring PTCs at different positions within the open reading frame (ORF) were transiently transfected into HeLa cells. Protein and mRNA levels were assessed by slot blotting and real-time quantitative PCR (RT-qPCR), respectively (Figure 2, A and B). In agreement with previous data (Eberle et al., 2008), stop codons proximal to the 5′ and the 3′ ends of the transcript only moderately reduced its stability, whereas stop codons more distal to the ends of the mRNA induced its degradation (Figure 2A, black line). Proteins produced from transcripts harboring a stop codon closer to the 3′ end of CDS correlated well with the corresponding mRNA level. On the other hand, only a low level of protein could be detected for transcripts with a very short ORF (Figure 2A, green line). This discrepancy is likely because the peptides produced from the TER30 reporter construct are rapidly degraded after their synthesis or because they are too short to be efficiently detected using slot blots. Notably, it was previously reported that short peptides are difficult to detect by classical Western blotting (Schagger, 2006; Tomisawa et al., 2013). Indeed, already the TER100 construct was undetectable (Supplemental Figure S2A). We therefore reasoned that the TER30 peptide is most likely synthesized, but too small for detection by slot blotting. In support of this, we could detect expression of the TER30 constructs by immunofluorescence microscopy (Supplemental Figure S2B). Collectively, these data indicate that short peptides produced from PTC-containing mRNAs are not necessarily degraded. Consequently, their absence on Western blots, the standard method to analyze protein abundances, is rather explained by a detection problem than by the physical absence of the peptide.
FIGURE 2:

RNA and protein levels of predicted NMD targets. (A) mRNA levels and protein levels of mini-µ reporter constructs each containing a PTC at the indicated positions relative to the wild-type construct (stop codon at amino acid position 600). All reporters consist of five exons and four introns (not shown) and encode a 2xHA-tag at their N-terminus. Positions of the stop codons of the different reporters and the exon-exon boundaries (dark lines interrupting the green bar representing the mini-µ reporter gene), identical to all transcripts, are indicated. mRNA and protein levels were determined by RT-qPCR and slot blotting, respectively. Average values and standard deviations of three biological replicates are shown. (B) Representative slot blot displaying the protein expression levels of different reporter constructs detected by anti-HA antibodies (green) and CPSF-100 (red, loading control). A nontransfected control (NTC) was used to estimate the background signal. The last column shows the overlay. (C) HBB PTC−/+ reporter constructs were immunoprecipitated from total cell extracts after 24 h of induction with 250 ng/ml doxycycline. Purified proteins were separated by Nu–PAGE and the gel subjected to silver staining. Both reporter constructs are detectable. (D) Western blot of total cell extracts from C (input) and of the corresponding immunoprecipitations (IP).
As a complementary approach to study protein production from NMD-sensitive transcripts, we generated HEK-293 Flp-In T-REx cells stably expressing 3xFLAG-tagged (N-term) human β-globin (HBB) with (PTC+) or without (PTC−) a PTC at amino acid position 39 of the HBB CDS (Supplemental Figure S2C). The Flp-In T-REx system allows for the introduction of a gene of interest at a specific locus and its inducible expression upon addition of tetracycline (or doxycycline) to the culture medium. Expression of both constructs was induced with doxycycline for 24 h before harvesting. As measured by RT-qPCR, HBB PTC+ mRNA levels were significantly decreased when compared with HBB PTC- mRNA (Supplemental Figure S2D). To monitor protein levels, we performed affinity purification using M2 anti-FLAG antibodies, separated purified proteins by gel electrophoresis, and visualized these by silver staining and Western blotting. Importantly, the same gel system and protein ladder were used for either approach. Intriguingly, both the full-length HBB PTC− and its truncated version HBB PTC+ were detected directly in the gel after silver staining (Figure 2C). Staining intensity, although only semiquantitative, indicates that the HBB PTC+ peptide is less abundant when compared with the truncated HBB PTC− version. In contrast, only the full-length HBB PTC− was detectable by Western blotting (Figure 2D), emphasizing the fact that short peptides are problematic to detect using the latter approach.
CRISPR-Trap; merging CRISPR/Cas9, targeting the first intron with gene traps
In consequence of our findings, we next aimed to establish a gene-targeting approach that would completely abolish expression of full-length transcripts of targeted genes. To this end, we merged CRISPR/Cas9 technology, targeting the first intron of genes of interest, with a gene trap introduced through HDR from a donor plasmid. The gene trap consists of a chimeric intron with a 3′ splice site preceding a customizable cassette (containing, e.g., a selection marker) and a polyadenylation signal (Figure 3A). Our CRISPR-Trap approach can be applied to 1) gene KO, resulting in a premature transcription termination within the first intron of the targeted gene and expression of the selection marker under the promoter of the targeted gene, and 2) gene replacement, allowing for simultaneous expression of a modified gene and a selection marker (Figure 3A). Importantly, CRISPR-Trap is directly applicable to almost 50% of all human protein-coding intron-containing genes because these have a noncoding first exon (Figure 3B). In that case, only the homology arms of the donor plasmid and the sgRNA sequence need to be replaced. Furthermore, cells with off-target integrations of the cassette will not survive the selection, due to the absence of a promoter and hence lack of transcription of the selection marker. Because this approach allows targeting of potentially every spliced transcript, it also allows knocking out or replacing a large subset of long noncoding RNAs (lncRNAs).
FIGURE 3:

CRISPR-Trap. (A) Scheme of the CRISPR-Trap gene-targeting strategy. Cleavage of the first intron of the gene of interest is mediated by CRISPR/Cas9. A donor matrix serves as a template for HDR and is inserted into the first intron. The insert consists of a chimeric intron with a 3′ splice site (dark blue) followed by a customizable cassette (blue) and a SV40 polyadenylation signal (light blue) to terminate transcription in the first intron. The customizable cassette can either mediate a gene KO (left) or a gene replacement (right). Both strategies rely on a selection marker expressed under the promoter of the gene of interest after successful gene targeting. For the latter strategy, the translation of the selection marker depends on an IRES upstream of its CDS. (B) Histogram summarizing the number of amino acids (x-axis) encoded by all known human spliced protein-coding genes (RefSeq release 78). On the y-axis, relative contribution of each bin to the genome is depicted. Note that almost half of all human intron-containing genes have a noncoding first exon.
Following this strategy, we successfully introduced a FUS KO in SH-SY5Y neuroblastoma cells (Reber et al., 2016) as well as in HeLa and hiPSCs (Figure 4, A and B). Although the FUS gene has a coding first exon, our approach is still applicable. The first exon of FUS encodes only four amino acids. Therefore, we designed a cassette that is spliced in frame with the first four codons of FUS, hijacking the endogenous start codon to instead express the selection marker, a zeocin resistance (Reber et al., 2016). Apart from the FUS gene, we applied CRISPR-Trap to knock out the SMG7 gene in HT1080 cells and validated the absence of both the protein (by Western blot; Figure 4C) and the corresponding transcript (by RNA deep sequencing; Figure 4D). Importantly, a direct comparison of SMG7 CRISPR-Trap to frameshift-based KOs exemplifies the advantage of CRISPR-Trap: a frameshift clone in exon 3 expresses a high amount of SMG7 mRNA and frameshifts in exon 15 even result in the expression of a truncated SMG7 protein detectable by Western blot (Supplemental Figure S3).
FIGURE 4:

Generation and characterization of different KO cell lines. (A) RT-qPCR of FUS KO HeLa, SH-SY5Y, and hiPSCs shows the complete absence of the FUS transcript compared with the wild-type cells. Average values and standard deviations of three biological replicates are shown. (B) Western blot confirming the absence of FUS protein in the KO cell lines. Tyrosine tubulin served as a loading control. (C) Western blot showing SMG7 protein levels in WT HT1080 and SMG7 KO clones 11 and 12. The SMG7 blot shows one unspecific band marked with an asterisk and is already documented in Metze et al. (2013) and Colombo et al. (2017). CPSF-73 served as a loading control. (D) SMG7 coverage plot: the coverage of mapped reads from RNA deep sequencing on the SMG7 locus is shown. Introns are reduced in length by a factor of 10 compared with exons for better visualization.
CRISPR-Trap can be used both to create gene KOs and to generate gene replacements. The latter approach is of particular interest when studying essential genes where complete KOs are lethal. As a proof of principle, we targeted the TARDBP gene where conditional KO leads to death in both mice and mouse embryonic stem cells (Chiang et al., 2010). We performed the gene replacement in hiPSCs by introducing a cassette expressing the protein product of the TARDBP gene (TDP-43), N-terminally fused to the destabilizing domain (DD) of mutant human FKBP12 protein (Banaszynski et al., 2006). In addition, the replacement cassette contains an internal ribosomal entry site (IRES) and a puromycin resistance downstream from the DD-TDP-43 ORF allowing selection of successfully edited clones. The DD targets the DD-TDP-43 fusion protein for rapid degradation through the proteasome (Figure 5A). The fusion protein is stable only in the presence of Shield1 ligand bound to the DD. Withdrawal of the ligand from the culture medium leads to the degradation of the fusion protein within a few hours (Banaszynski et al., 2006; Maynard-Smith et al., 2007). We validated the successful introduction of the cassette into hiPSCs by PCR of the corresponding genomic locus (Figure 5B) and demonstrated the efficient degradation of DD-TDP-43 after removal of the ligand (Figure 5C).
FIGURE 5:

Replacing TDP-43 with DD-TDP-43. (A) Scheme of the gene replacement strategy for the TARDBP gene (TDP-43). After successful genome editing, transcription of TARDBP is terminated within its first intron. The first exon is spliced to the cassette which codes for the DD-TDP-43 fusion protein followed by an IRES allowing simultaneous expression of a puromycin resistance marker (PuroR). After translation, DD-TDP-43 is only stable in the presence of Shield1 ligand. In absence of Shield1 ligand, DD-TDP-43 is rapidly degraded by the proteasome. Primers used to analyze the TARDBP genomic locus in B are indicated. (B) PCR of the genomic locus of WT hiPSCs and cells edited on two (homozygous) or one (heterozygous) allele, respectively. Primers aligned to TARDBP in A indicate the expected products. (C) Western blot showing successful replacement of TDP-43 by DD-TDP-43. Wild-type, mono- and biallelic edited cells were grown in the presence or absence (withdrawal 24 h before harvest) of Shield1 ligand. SmB/B′ served as a loading control.
Conclusion
In summary, we show that early truncation of an ORF using gene-targeting approaches often results in inefficient degradation of the corresponding mRNA and production of C-terminally truncated proteins. Moreover, we observe that such peptides are difficult to detect and are most likely missed using classical Western blotting approaches. However, their appearance and potential biological consequences have to be considered, especially in the context of future gene KO approaches using CRISPR/Cas9 for the treatment of human diseases. Additionally, with cellular differences in transcription and protein turnover it is unforeseeable in which cell types and to what extent these peptides could exert unwanted functions (Kristensen et al., 2013; Buszczak et al., 2014). Furthermore, nonsense mutations occurring after structural domains are expected to yield stable truncations and in the cases where the PTC-containing transcripts are not subjected to NMD, these truncations are associated with aggregation and diseases such as coronary artery disease, non-Hodgkin lymphoma, cardiomyopathy, and macular degeneration (Gough et al., 2012). Finally, the mutated mRNA itself can have deleterious side effects, as alterations in mRNA secondary structures caused by indels have been associated with diseases (Zhang et al., 2014).
CRISPR-Trap, however, can bypass these pitfalls. It enables a complete elimination of the full-length transcript, either by hijacking or replacing the endogenous ORF, and consequently prevents the expression of unwanted and potentially problematic genome editing byproducts.
MATERIALS AND METHODS
Oligonucleotides, plasmids, antibodies, computational analysis of publicly available data sets
Used oligonucleotides, plasmids, antibodies, and computational analysis of publicly available data sets are described in the Supplemental Material.
Cell culture
Cells were maintained in DMEM+/+ containing 10% fetal calf serum (FCS), penicillin (100 U/µl), and streptomycin (100 µg/ml) at 37°C and 5% CO2. HeLa, HEK293T, and HT1080 cells were kindly provided by Oliver Mühlemann (University of Bern). The HEK293 Flp-In T-REx cell line was from Thermo Fisher Scientific. The human iPSC line iPS DF6-9-9T.B was purchased from WiCell (Yu et al., 2009). The SH-SY5Y and FUS KO SH-SY5Y cell lines were previously described (Reber et al., 2016). All cell stocks were tested for Mycoplasma contamination and were Mycoplasma-negative. Human iPSCs were maintained on Matrigel-coated plates (Corning) in mTeSR1 (Stemcell Technologies) and grown under hypoxic conditions (5% O2), 5% CO2 and 37°C. 200 nM Shield1 ligand (632189; Takara) was added to the stem cell medium when endogenous TDP-43 was replaced with DD-TDP-43. If not indicated, plasmid DNA transfections were performed using Dogtor (OZ Biosciences) for HeLa and HEK293T, TransIT-LT1 (Mirus Bio) for HT1080, and hiPSCs or Lipofectamine 3000 reagent (Thermo Fisher) for SH-SY5Y and hiPSCs, respectively, according to the manufacturer’s instructions.
RT-qPCR
Cells were harvested by trypsination (HeLa, HEK-293 Flp-In T-REx, SH-SY5Y neuroblastoma, and HT1080) or with 0.5 mM EDTA in phosphate-buffered saline (PBS; hiPSCs). RNA was extracted using TRIzol (Marquis et al., 2009) and reverse transcribed. The RT-qPCR was performed as described in Raczynska et al. (2015). For cells expressing mini-µ reporter constructs, RNA was isolated and DNase treated using the GenElute Mammalian Total RNA Miniprep Kit (RTN350; Sigma-Aldrich) following the manufacturer’s instructions. After reverse transcription, qPCR was performed with Takyon for Probe Assay–No Rox MasterMix (UF-NPCT-B0205; Eurogentec) or with MESA GREEN qPCR MasterMix Plus for SYBR Assay No ROX (05-SY2X-06+NRWOU; Eurogentec) on a Rotor-Gene 6000 R Corbett (Qiagen). Analysis of the qPCR results was conducted as previously described (Metze et al., 2013). Primers and 5′-FAM, 3′-BHQ1 labeled TaqMan probe sequences used in this study for detection of HBB are listed in the Supplemental Materials and Methods.
Immunoprecipitation of 3xFLAG-tagged HBB
HEK-293 Flp-In T-REx cells (5 × 106) expressing 3xFLAG-tagged HBB were seeded on T150 flasks. The following day, the cells were induced with 250 ng/ml doxycycline hyclate. After 24 h of induction, the cells were harvested by trypsination, washed once in PBS, and resuspended in 500 µl extraction buffer (300 mM NaCl, 0.5% NP-40, 20 mM HEPES, pH 7.4, supplemented with 1× protease inhibitor cocktail (B14001; Biotool). The cells were then sonicated 3 × 5 s, amplitude 45% (Vibracell 75186 sonicator) with cooling on ice in between each cycle. The lysate was centrifuged for 10 min at 16,100 × g at 4°C. Clarified supernatant was incubated with 7.5 µl anti-FLAG beads for 1 h head over tail at 4°C. The beads were then washed once with 1 ml extraction buffer, followed by resuspension in 500 µl extraction buffer supplemented with 10 µl of 20 mg/ml RNase A (R6148; Sigma-Aldrich) and incubated head over tail for 10 min at room temperature. Subsequently, the beads were washed with 2 × 1 ml extraction buffer. Proteins were eluted from the beads by adding 95 µl of 1.5 × LDS sample buffer (NP0008; Thermo Fisher) and incubated for 10 min at 75°C. Before loading the eluates on gel, they were supplemented with 50 mM dithiothreitol (DTT) f.c. and incubated for 10 min at 75°C.
Coupling of anti-FLAG antibodies to Dynabeads M-270 epoxy
M2 anti-FLAG antibodies (F3165; Sigma-Aldrich) were conjugated to Dynabeads M-270 epoxy (14302D; Thermo Fisher Scientific) according to the manufacturer’s instructions. For the coupling reaction, 10 μg of anti-FLAG antibodies per 1 mg (dry weight) of Dynabeads M-270 epoxy were used. Before coupling, magnetic beads were washed twice with 1 ml of 0.1 M sodium phosphate buffer, pH 7.4. After the second wash, the coupling solution (1 M ammonium sulfate, 0.1 M sodium phosphate buffer, pH 7.4, antibodies) was added (20 μl per 1 mg beads). Conjugation was carried out overnight, at 37°C with mixing at 1400 rpm (Eppendorf Thermomixer Comfort). Coupled beads were washed three times with 1 ml PBS, once with 1 ml 0.5% Triton X-100 in PBS, followed by a final wash with 1 ml PBS and then stored at −20°C in 50% glycerol in PBS until use.
Silver staining
Proteins were separated on a NuPAGE 4–12% Bis-Tris Midi Gel (WG1403BOX; Thermo Fisher Scientific) using 1× MES running buffer. After electrophoresis, the gel was incubated overnight in fixing solution (50% MeOH, 12% HAc, 0.05% Formalin). The next day the gel was sequentially washed 3 × 20 min in 35% EtOH, sensitized in 0.02% Na2S2O3 for 2 min, washed 3 × 5 min in H2O, incubated for 20 min in staining solution (0.2% AgNO3, 0.076% Formalin), washed 2 × 1 min in H2O and developed in developing solution (6% Na2CO3, 0.05% Formalin, 0.0004% Na2S2O3) until desired bands intensity. The reaction was stopped by replacing developing solution with stop solution (50% MeOH, 12% HAc) and incubating for 5 min.
Immunoblotting
Cells transfected with mini-µ constructs were harvested by trypsination, counted and lysed either in RIPA buffer (1 ml per 106 cells; 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% Na-deoxycholate, 1 mM EDTA) or gentle hypotonic lysis buffer (1 ml per 106 cells; 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 2 mM EDTA, 0.1% Triton X-100, 1× protease inhibitor cocktail [B14001; Biotool]). Cell lysates were clarified by centrifugation for 15 min at 16,100 × g, 4°C and supernatants were collected for further analysis. Cell equivalents of (1–2) × 105 (8 × 105 for mini-µ reporter constructs) per lane in LDS sample buffer (NP0008; Thermo Fisher) or SDS sample buffer (60 mM Tris-HCl, pH 6.6, 100 mM DTT, 10% glycerol, 2% SDS, 0.01% bromophenol blue), respectively, were loaded. The proteins were separated on a NuPAGE 4–12% Bis-Tris Midi Gel (Thermo Fisher Scientific) or on a 6% or 10% SDS–PAGE. Proteins separated with NuPAGE gels were transferred on a nitrocellulose membrane using the iBlot Gel Transfer System (Thermo Fisher) according to the manufacturer’s instructions. A TE77 ECL Semi-Dry Transfer Unit (Amersham Biosciences) was used to transfer proteins from a SDS–PAGE on a nitrocellulose membrane (Optitran BA-S 85; Whatman). The HBB proteins separated with the NuPAGE gel as well as mini-µ reporter proteins were transferred on a 0.1 µm nitrocellulose membrane (10600105; GE Healthcare) using the TE77 ECL Semi-Dry Transfer Unit. For the slot blot, 8 × 105 cell equivalents were loaded per well of a Bio-Dot SF Microfiltration Apparatus (170-6542; Bio-Rad) and blotted on a 0.1 µm nitrocellulose membrane (10600105; GE Healthcare). Subsequently, membranes were blocked with 5% nonfat dry milk in 0.1% Tween in Tris-buffered saline (TBS), incubated at 4°C overnight with the primary antibodies, washed with 0.1% Tween in TBS, and incubated 1.5 h at room temperature with the fluorescence-labeled secondary antibodies. C-terminal truncated SMG7 protein in clone fs15 (in Supplemental Figure S3C) was detected using the SuperSignal Western Blot Enhancer kit (46640; Thermo Fisher) according to the manufacturer’s instructions. The washed and dried membranes were analyzed using the Odyssey Infrared Imaging System (LI-COR).
Immunofluorescence staining
Twenty-four hours posttransfection, 4 × 104 cells were seeded per well of an eight-chamber slide (7647; Semadeni). Cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed 3 × 5 min with TBS, permeabilized and blocked with 0.5% Triton X-100, 6% bovine serum albumin (BSA) in TBS for 30 min at room temperature. Afterward, cells were incubated with mouse anti-HA (sc-7392; Santa Cruz) diluted 1:1000 in 0.1% Triton X-100, 6% BSA in TBS at 4°C overnight. Subsequently, cells were washed 3 × 5 min with TBS and incubated for 2 h at room temperature with chicken anti-mouse IgG AF488 (A-21200; Thermo Fisher Scientific), diluted 1:500 in 0.1% Triton X-100, 6% BSA, 100 ng/ml 4′,6-diamidin-2-phenylindol in TBS. Before mounting with Vectashield Hardset mounting medium (H-1400; Vector Laboratories), three final washing steps with TBS, 5 min each, were performed. Pictures were acquired with a DFC360 FX monochrome camera (Leica Biosystems) mounted on a Leica DMI6000 B microscope. Analysis was performed with LAS AF software (Leica Biosystems).
Human iPSC clones with mutations in FUS exon 5
Three 80% confluent hiPSC wells of a six-well plate were transfected using TransIT-2020 (MIR 5404; Mirus Bio) according to the manufacturer’s manual. Each transfection consisted of two plasmids coding for a TALEN binding upstream (left TALEN, pTAL EF1a 023977) and downstream (right TALEN, pTAL EF1a 021443), a donor plasmid (pCLS22315) for HDR, and a FUS exon 5 reporter plasmid according to Flemr and Buhler (2015). The vectors were transfected in a ratio of 2:2:1.25:1 (left TALEN to right TALEN to donor to reporter). A total of 2500, 5000, or 7100 ng of plasmid DNA was transfected on the three wells. Two days after transfection, cells were pooled with Accutase (Thermo Fisher) to generate single cells. Cells were maintained in mTeSR1 containing 10 µM of the ROCK inhibitor Y-27632 (Stemcell Technologies) to enhance single-cell survival. Two days later, puromycin selection (0.5 µg/ml) was started and maintained for 2 d. Colonies originating from single cells were picked and analyzed. RNA and genomic DNA were harvested from TRIzol. FUS expression was analyzed by RT-qPCR and the respective genomic locus was amplified by PCR using the KAPA Taq ReadyMix (KK1006; KAPA Biosystems) according to the manufacturer’s instructions. Primers are presented in the Supplemental Materials and Methods. PCR products were purified over a preparative agarose gel using the Wizard SV Gel and PCR Clean-Up System (A9281; Promega) and sent for Sanger sequencing (Barcode Economy Run Sequencing Service; Microsynth AG). To determine the genomic sequence of clones with different mutations in the two alleles, PCR products were TOPO-TA cloned into the pCRII-TOPO plasmid (K4610-20; Thermo Fisher) according to the manufacturer’s manual. Plasmids were subsequently amplified and purified from single Escherichia coli colonies and sent for sequencing.
Generation of HEK-293 Flp-In T-REx cells expressing 3xFLAG-tagged HBB
The HBB coding sequence (including introns) was PCR-amplified (primer sequenced in the Supplemental Materials and Methods) and cloned into the pcDNA5/FRT/TO 3xFLAG (N) vector using HindIII and NotI restriction sites. The same strategy was applied for the PTC-containing HBB (TER39), resulting in the construct encoding for 3xFLAG HBB with the stop codon at position 64. HEK293 Flp-In T-REx cells expressing 3xFLAG-tagged (N-term) HBB were established according to the manufacturer’s instructions (Flp-In T-Rex; Thermo Fisher Scientific). Briefly, 2 × 106 cells were seeded in 10 ml DMEM (10% FCS, no P/S) on a 100 mm dish. The next day, cells were transfected (20 μl Lipofectamine 2000 in 500 μl Opti-MEM, mixed with 18 μg of pOG44 and 2 μg pcDNA5/FRT/TO in 500 μl Opti-MEM). After 24 h, cells were trypsinized, transferred to a T-175 flask, and cultured in DMEM (10% FCS, no P/S) until attached (3 h) followed by the addition of selection medium (DMEM 10% FCS, 1 × P/S, 100 μg/ml Hygromycin B and 10 μg/ml Blasticidine S). Cells were kept under selection until antibiotic-resistant colonies appeared. The colonies were trypsinized, triturated into a single-cell suspension, transferred to T-75 flask, and cultured in selection medium until 90% confluency. After that, cells were split and kept in culture without any selection before the experiments.
Generation of FUS KO HeLa, SH-SY5Y neuroblastoma, and hiPSC cells
The FUS KO SH-SY5Y cell line was previously described (Reber et al., 2016). FUS KO HeLa and hiPSC cells were generated following the same strategy with some modifications. Briefly, six 80% confluent wells of a six-well plate with HeLa cells were each transfected with 2 µg plasmid coding for the Cas9 endonuclease and the guide RNAs (3 × target A, 3 × target B) targeting the first intron of FUS (pU6gDNA-Cas9-GFP; target sequence A: 5′-TGGATGTCCACCAAGACCTTGG-3′ or target sequence B 5′-TCCAATGGTTAAGGCTTCTGGG-3′ [Sigma-Aldrich]) and 1 µg FUS KO ZeoR donor plasmid (General Biosystems). Four days after transfection, the two conditions were pooled and zeocin selection was started (150 µg/ml) and maintained until analysis of clones originating from single cells for FUS expression by RT-qPCR. Positive clones were expanded and the absence of the protein was verified by Western blotting. Each well (six wells in total in a six-well plate) of 80% confluent hiPSCs was transfected with 2.5 µg plasmid coding for the Cas9 endonuclease and the guide RNAs (3 × target sequence A, 3 × target sequence B) and 5.5 µg FUS KO ZeoR donor plasmid. The transfections were repeated the day after. One day after the second transfection, cells were detached with Accutase (Thermo Fisher Scientific) to generate single cells. Cells were maintained in mTeSR1 containing 10 µM of the ROCK inhibitor Y-27632 (Stemcell Technologies) to enhance single-cell survival. Two days later, zeocin selection (25 µg/ml) was started and maintained for 3 d. Colonies originating from single cells were picked and analyzed for FUS expression by RT-qPCR. Positive clones were expanded, and the absence of the FUS protein was verified by Western blotting.
Generation of SMG7 KO HT1080 cells by CRISPR-Trap and high-throughput sequencing
Six 90% confluent wells of a six-well plate with HT1080 cells were each transfected with 1.5 µg of the plasmid coding for the Cas9 endonuclease and the guide RNA targeting the first intron of SMG7 (pU6gDNA-Cas9-GFP; target sequence: ACCAAACGTTGCCGGGCACGGGG) and 1.5 µg of the SMG7 KO ZeoR donor plasmid. Three days after transfection, the six wells were pooled and zeocin (500 µg/ml) selection was started and maintained until analysis of SMG7 expression by RT-qPCR in clones originating from single cells. Positive clones were expanded and the absence of the protein was verified by Western blotting. Wild-type (WT) HT1080 cells and two independent SMG7 KO clones were analyzed by high-throughput sequencing. The library was prepared with the Illumina TruSeq Stranded Total RNA Library Prep Kit (chemistry v3), which also includes a ribo-zero depletion step. The reads were produced by means of a HiSeq3000 machine, with a length of 100 base pairs and in single-end mode. The sequencing depth was around 85 million per sample. Every condition was analyzed in triplicate. The reads were mapped to the human genome (hg38) with the program HISAT2 (Kim et al., 2015). The genome index was created with the exon and splice site information taken from the Ensembl transcriptome annotation (release 84). The insert sequence containing the resistance cassette was added to the SMG7 locus. The locus coverage was investigated by means of wig files.
Generation of SMG7 KO HT1080 cells by conventional CRISPR approach
Four 80% confluent wells of a six-well plate with HT1080 cells were each transfected with 2.8 or 4.8 µg of the plasmid coding for the Cas9 endonuclease and the guide RNA targeting either the third (pCRISPR-EF1a-Cas9-GFP Smg7 exon 3 t1; target sequence: 5′-GAAAATGCTAGTTACCGATTTGG-3′) or the 15th exon of SMG7 (pCRISPR-EF1a-Cas9-GFP Smg7 exon 15 t1; target sequence: 5′-GGAGTTACTTGCTTGAGTTGGGG-3′) and 200 ng empty pSUPuro. Two days after transfection, the two wells transfected with the same plasmids were pooled on a 15 cm plate and puromycin (2 µg/ml) selection was conducted for 2 d. Colonies originating from single cells were picked and analyzed. RNA and genomic DNA were harvested from TRIzol. SMG7 expression was analyzed by RT-qPCR and the respective genomic locus was amplified by PCR using the Maximo Taq DNA Polymerase 2X-preMix (S113; GeneON) according to the manufacturer’s instructions. Primers are presented in the Supplemental Materials and Methods. PCR products were purified over a preparative agarose gel using the Wizard SV Gel and PCR Clean-Up System (A9281; Promega) and sent for Sanger sequencing (Barcode Economy Run Sequencing Service; Microsynth AG). To determine the genomic sequence of clones with different mutations in the two alleles, PCR products were TOPO-TA cloned into the pCRII-TOPO plasmid (K4610-20; Thermo Fisher) according to the manufacturer’s manual. Plasmids were subsequently amplified and purified from single E. coli colonies and sent for sequencing.
Gene replacement of TDP-43 by DD-TDP-43 in hiPSCs
The first intron of the TARDBP gene was targeted using pCRISPR-EF1a-SpCas9 TDP KO t2, coding for the sgRNA targeting the sequence 5′-CTGAAGGCTTATGATGCGTCAGG-3′ in the first intron of TARDBP together with a TDP-43 replacement matrix (General Biosystems). A 100 mm dish with 80% confluent hiPSCs was transfected with 5 µg pCRISPR-EF1a-eSpCas9 (1.1) and 25 µg of the donor matrix. Cells were maintained in mTeSR1 containing 10 µM Y-27632 and 200 nM Shield1 ligand to stabilize DD-TDP-43 in case of successful genome editing. The transfection was repeated the next day. After the second transfection, cells were detached using Accutase and transferred onto a 150 mm dish. Positive clones were selected with puromycin (0.25 µg/ml) in mTeSR1 containing 10 µM Y-27632 and 200 nM Shield1 ligand for 3 d. Y-27632 (10 µM) was maintained for four more days until single cells grew to small colonies. Afterward, cells were maintained in mTeSR1 containing 200 nM Shield1 ligand. Colonies originating from single cells were picked. RNA and genomic DNA were extracted using TRIzol. TDP-43 and DD-TDP-43 expression were analyzed by RT-qPCR and the respective genomic locus was analyzed by PCR using the KAPA Taq ReadyMix (KK1006; KAPA Biosystems) according to the manufacturer’s instructions. Used primers are present in the Supplemental Materials and Methods. Positive clones were expanded and grown in the presence or absence of Shield1 ligand (withdrawal of Shield1 ligand 24 h before harvest). Replacement of the endogenous protein was verified by Western blotting.
Supplementary Material
Acknowledgments
We thank Christian Kroun Damgaard (Aarhus University) for providing the pcDNA5/FRT/TO 3xFLAG (N) plasmid, Oliver Mühlemann for providing cell lines and feedback on the manuscript, and Nicole Kleinschmidt and Karin Schranz for their excellent technical support. This research and related results were made possible by the support of the NOMIS Foundation to M.-D.R. and the National Centre of Competence in Research (NCCR) RNA & Disease funded by the Swiss National Science Foundation. We further thank the Swiss National Science Foundation (IZK0Z3_166772) and the Karolinska Institute for the International Short Visit Grants to J.A.B., the Swedish Medical Research Council (2016-02112) to E.H., Christoph Schweingruber for helpful suggestions, and Andrea Eberle for critical comments on the manuscript.
Abbreviations used:
- CDS
coding sequence
- DD
destabilizing domain
- DSB
double-strand break
- FUS
fused in sarcoma
- HBB
human β-globin
- HDR
homology-directed repair
- hiPSC
human induced pluripotent stem cell
- Ig
immunoglobulin
- IP
immunoprecipitation
- IRES
internal ribosomal entry site
- KO
knockout
- lncRNA
long noncoding RNA
- NHEJ
nonhomologous end joining
- NMD
nonsense-mediated mRNA decay
- NTC
nontransfected control
- ORF
open reading frame
- PTC
premature termination codon
- sgRNA
single guide RNA
- TARDBP
TAR DNA-binding protein 43.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-05-0288) on November 22, 2017.
REFERENCES
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Paillusson A, Muhlemann O. Efficient downregulation of immunoglobulin mu mRNA with premature translation-termination codons requires the 5’-half of the VDJ exon. Nucleic Acids Res. 2004;32:3304–3315. doi: 10.1093/nar/gkh651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M, Signer RA, Morrison SJ. Cellular differences in protein synthesis regulate tissue homeostasis. Cell. 2014;159:242–251. doi: 10.1016/j.cell.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci USA. 2010;107:16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Muhlemann O. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. 2017;23:189–201. doi: 10.1261/rna.059055.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyranoski D. CRISPR gene-editing tested in a person for the first time. Nature. 2016;539:479. doi: 10.1038/nature.2016.20988. [DOI] [PubMed] [Google Scholar]
- Du QS, Cui J, Zhang CJ, He K. Visualization analysis of CRISPR/Cas9 gene editing technology studies. J Zhejiang Univ Sci B. 2016;17:798–806. doi: 10.1631/jzus.B1601985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Stalder L, Mathys H, Orozco RZ, Muhlemann O. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008;6:e92. doi: 10.1371/journal.pbio.0060092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, Buhler M. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep. 2015;12:709–716. doi: 10.1016/j.celrep.2015.06.051. [DOI] [PubMed] [Google Scholar]
- Gough CA, Homma K, Yamaguchi-Kabata Y, Shimada MK, Chakraborty R, Fujii Y, Iwama H, Minoshima S, Sakamoto S, Sato Y, et al. Prediction of protein-destabilizing polymorphisms by manual curation with protein structure. PLoS One. 2012;7:e50445. doi: 10.1371/journal.pone.0050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Lewis BP, Hillman RT, Blanchette M, Lareau LF, Garnett AT, Rio DC, Brenner SE. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics. 2003;19(suppl 1):i118–i121. doi: 10.1093/bioinformatics/btg1015. [DOI] [PubMed] [Google Scholar]
- Huang ML, Sivagurunathan S, Ting S, Jansson PJ, Austin CJ, Kelly M, Semsarian C, Zhang D, Richardson DR. Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: activation of the integrated stress response, eIF2α phosphorylation, and the induction of downstream targets. Am J Pathol. 2013;183:745–757. doi: 10.1016/j.ajpath.2013.05.032. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir S, Hockemeyer D, de Lange T. TALEN gene knockouts reveal no requirement for the conserved human shelterin protein Rap1 in telomere protection and length regulation. Cell Rep. 2014;9:1273–1280. doi: 10.1016/j.celrep.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis ED, Nasif S, Muhlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA. 2016;7:661–682. doi: 10.1002/wrna.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Lee JH, Park BC, Park TS. Myotube differentiation in CRISPR/Cas9-mediated MyoD knockout quail myoblast cells. Asian-Australas J Anim Sci. 2016;30:1029–1036. doi: 10.5713/ajas.16.0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, Foster LJ. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol Syst Biol. 2013;9:689. doi: 10.1038/msb.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, de Jesus Domingues AM, Butter F, Ketting RF. Characterization of genetic loss-of-function of Fus in zebrafish. RNA Biol. 2016;14:29–35. doi: 10.1080/15476286.2016.1256532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littink KW, van Genderen MM, Collin RW, Roosing S, de Brouwer AP, Riemslag FC, Venselaar H, Thiadens AA, Hoyng CB, Rohrschneider K. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009;50:2344–2350. doi: 10.1167/iovs.08-2553. [DOI] [PubMed] [Google Scholar]
- Loughran G, Chou MY, Ivanov IP, Jungreis I, Kellis M, Kiran AM, Baranov PV, Atkins JF. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 2014;42:8928–8938. doi: 10.1093/nar/gku608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti E, Bohm J, Lacene E, Beuvin M, Romero NB, Laporte J. A premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J Neuromuscul Dis. 2015;2:219–227. doi: 10.3233/JND-150085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis J, Kampfer SS, Angehrn L, Schumperli D. Doxycycline-controlled splicing modulation by regulated antisense U7 snRNA expression cassettes. Gene Ther. 2009;16:70–77. doi: 10.1038/gt.2008.138. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith LA, Chen LC, Banaszynski LA, Ooi AG, Wandless TJ. A directed approach for engineering conditional protein stability using biologically silent small molecules. J Biol Chem. 2007;282:24866–24872. doi: 10.1074/jbc.M703902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Soukup TM, Liu SD, Esensten JH, de Sousa e Melo F, Yaylaoglu M, Warming S, Roose-Girma M, Firestein R. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J Pathol. 2015;237:508–519. doi: 10.1002/path.4596. [DOI] [PubMed] [Google Scholar]
- Metze S, Herzog VA, Ruepp MD, Muhlemann O. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA. 2013;19:1432–1448. doi: 10.1261/rna.038893.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE. Mechanism of escape from nonsense-mediated mRNA decay of human β-globin transcripts with nonsense mutations in the first exon. RNA. 2011;17:843–854. doi: 10.1261/rna.2401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczynska KD, Ruepp MD, Brzek A, Reber S, Romeo V, Rindlisbacher B, Heller M, Szweykowska-Kulinska Z, Jarmolowski A, Schumperli D. FUS/TLS contributes to replication-dependent histone gene expression by interaction with U7 snRNPs and histone-specific transcription factors. Nucleic Acids Res. 2015;43:9711–9728. doi: 10.1093/nar/gkv794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber S, Stettler J, Filosa G, Colombo M, Jutzi D, Lenzken SC, Schweingruber C, Bruggmann R, Bachi A, Barabino SM. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016;35:1504–1521. doi: 10.15252/embj.201593791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter S, Morrison S, Connor T, Su J, Print CG, Ronimus RS, McGee SL, Wilson WR. Zinc finger nuclease mediated knockout of ADP-dependent glucokinase in cancer cell lines: effects on cell survival and mitochondrial oxidative metabolism. PLoS One. 2013;8:e65267. doi: 10.1371/journal.pone.0065267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadic D, Schmidt K, Groh S, Kondofersky I, Ellwart J, Fuchs C, Theis FJ, Schotta G. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 2015;16:836–850. doi: 10.15252/embr.201439937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H. Tricine–SDS–PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829:612–623. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Stanford WL, Caruana G, Vallis KA, Inamdar M, Hidaka M, Bautch VL, Bernstein A. Expression trapping: identification of novel genes expressed in hematopoietic and endothelial lineages by gene trapping in ES cells. Blood. 1998;92:4622–4631. [PubMed] [Google Scholar]
- Tomisawa S, Abe C, Kamiya M, Kikukawa T, Demura M, Kawano K, Aizawa T. A new approach to detect small peptides clearly and sensitively by Western blotting using a vacuum-assisted detection method. Biophysics (Nagoya-shi) 2013;9:79–83. doi: 10.2142/biophysics.9.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lin H, Zhao H, Hao Y, Mort M, Cooper DN, Zhou Y, Liu Y. Impact of human pathogenic micro-insertions and micro-deletions on post-transcriptional regulation. Hum Mol Genet. 2014;23:3024–3034. doi: 10.1093/hmg/ddu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.