

STAT3 binds the collagen 1α2 enhancer and cooperates with JunB to activate it. We also show that fibroblasts have high basal levels of STAT3 activation, so activation of the enhancer by STAT3 is not affected by IL6. However, IL6 results in protein-level changes. Inhibition of STAT3 blocks collagen production and matrix remodeling.
Abstract
Fibroblast differentiation is a key cellular process that underlies the process of fibrosis, a deadly complication of fibrotic diseases like scleroderma (SSc). This transition coincides with the overproduction of collagen type I (COL1) and other extracellular matrix proteins. High-level expression of the collagen type 1α2 subunit (COL1A2), requires the engagement of a far-upstream enhancer, whose activation is strongly dependent on the AP1 factor JunB. We now report that STAT3 also binds the COL1A2 enhancer and is essential for RNA polymerase recruitment, without affecting JunB binding. STAT3 is required for the increased COL1A2 expression observed in myofibroblasts. We also report that TGFβ partially activates STAT3 and show that inhibiting STAT3 potently blocks TGFβ signaling, matrix remodeling, and TGFβ-induced myofibroblast differentiation. Activation of STAT3 with IL6 transsignaling alone, however, only increased COL1A2 protein expression, leaving COL1A2 mRNA levels unchanged. Our results suggest that activated STAT3 is not the limiting factor for collagen enhancer activation in human lung fibroblasts. Yet, a certain threshold level of STAT3 activity is essential to support activation of the COL1A2 enhancer and TGFβ signaling in fibroblasts. We propose that STAT3 operates at the posttranscriptional as well as the transcriptional level.
INTRODUCTION
Fibrosis is a major complication in many inflammatory diseases. Scleroderma (SSc) is a complex autoimmune disease, characterized by widespread organ fibrosis and thus serves as a model for pathological fibrosis (Varga and Abraham, 2007). At the heart of the fibrotic process lies the differentiation of normal quiescent fibroblasts to myofibroblasts. Myofibroblasts are contractile, biosynthetic cells that actively produce large amounts of extracellular matrix (ECM) and remodel it by contraction. In normal wound healing this is an essential process, allowing wound closure and reformation of lost connective tissue to support reepithelization (Leask and Abraham, 2004; Varga and Abraham, 2007). In pathologic, fibrotic conditions, persistent myofibroblast activation sets off an unremitting, vicious cycle of ECM production and contraction that eventually remodels connective tissue into dense scar tissue (Leask and Abraham, 2004; Varga and Abraham, 2007). Transforming growth factor β (TGFβ) is the master regulator of wound healing and the gene expression program drives myofibroblast differentiation and function (e.g., CTGF, COL1, α-SMA, EDA-fibronectin; Leask and Abraham, 2004). It is strongly implicated in the pathogenesis fibrotic diseases, including SSc (Leask and Abraham, 2004).
To support this differentiation and remodeling process, fibroblasts must increase production of collagen type I (COL1), a major component of the extracellular matrix, which is composed of two chains, α1 (COL1A1) and α2 (COL1A2; Ponticos et al., 2009, 2015). Functional COL1 consists of two α1 chains and one α2 chain tightly coiled around each other in a triple helix. The COL1 triple helices polymerize and become covalently cross-linked forming long fibrils. These fibrils provide a structural support for the attachment of cells and other extracellular matrix components.
High expression of COL1A2 requires engagement of the COL1A2 far-upstream enhancer (Ponticos et al., 2009), whose activation is a hallmark of myofibroblasts (Ponticos et al., 2009). Typically, it is active in scleroderma fibroblasts, which have a high proportion of myofibroblasts but it is weakly active or inactive in healthy control fibroblasts (Ponticos et al., 2009, 2015). There are three DNAse hypersensitive regions (HS3–5) in the COL1A2 enhancer (Figure 1). These regions are attachment sites for transcription factors and specific deletion of HS4 appears to abolish function (Antoniv et al., 2001; Ponticos et al., 2009, 2015). Previously, we have shown that the balance between AP-1 members JunB and c-Jun is critical for enhancer activation (Ponticos et al., 2015). TGFβ activates the enhancer by stabilizing JunB and thus shifting the balance away from c-Jun. JunB then replaces c-Jun in the AP-1 site and fully activates the enhancer (Ponticos et al., 2015).
FIGURE 1:

(A) The structure of the COL1A2 far-upstream enhancer (FUE) showing the proximal promoter (PP), three of the five DNAse hypersensitive sites (HS3–5), the critical AP-1 element in HS4, and the two putative STAT3 sites (arrows indicate DNA strand). (B) Reporter gene analysis of enhancer activation in normal (n = 3) and SSc (n = 3) lung fibroblasts treated with the JAK/STAT inhibitor AG490. The structure of the two DNA constructs used is shown at the top. The WT construct contains all elements of the FUE and PP fused to the firefly luciferase gene, while in ΔHS4 the HS4 region has been deleted to inactivate the enhancer. The results are normalized using a Renilla luciferase transfection control. (C) DNA gel showing the results of a ChIP assay for phosphoSTAT3 near the HS4 region in SSc lung fibroblasts treated with IL6 plus the soluble receptor α(IL6xRα) and SD1029. (D) Western blot of nuclear extracts from the same cells used in C, showing nuclear phosphoSTAT3 levels. TATA binding protein (TBP) was used as the house keeping gene. (E) ChIP analysis using qPCR for the binding of STAT3, JunB, and RNA polymerase II, in the HS4 region of SSc fibroblasts after treatment with SD1029, STAT3 siRNA (siSTAT3), or nontargeting siRNA (siNT). * = p < 0.05.
The signal transducer and activator of transcription (STAT) family of transcription factors is important for cell growth and differentiation (Rawlings et al., 2004). STATs are activated in response to growth factor and cytokine through phosphorylation by the JAK kinases (JAK1-3 and TYK2). In addition, JAK-independent activation of STATs by Src and MAPK family members has been described (Rawlings et al., 2004). STAT3 signaling activates a negative feedback loop by up-regulating SOCS3 (Croker et al., 2008). STAT3 signaling has been implicated in fibrosis (particularly in the lung; Pechkovsky et al., 2012; Ray et al., 2013; Pedroza et al., 2016), ECM regulation (Zhang et al., 2003; Ray et al., 2013), and TGFβ signaling (O’Reilly et al., 2014; Pedroza et al., 2016). Fibroblasts do not express the IL6 receptor (Ray et al., 2013), so the soluble IL6 receptor alpha (sIL6Rα) must be supplied in trans. A recent study (Ray et al., 2013), found that trans-IL6 signaling (IL6xRα) through STAT3 increased transcription of the matrix components type I collagen and fibronectin in hypertrophic scar fibroblasts, but not in normal skin fibroblasts. In addition to regulating the ECM and growth factor signaling, STAT 3 can contribute to fibrosis through its links to survival and proliferation (Hirano et al., 2000). A key feature of fibroproliferative disorders is the abnormal persistence of myofibroblasts and their excessive proliferation leading to inappropriate connective tissue expansion (Hinz et al., 2007). STAT3 may thus be contributing to myofibroblast persistence through resistance to apoptosis, so that the self-limiting wound healing response becomes self-perpetuating instead (Shen et al., 2001; Moodley et al., 2003; Pechkovsky et al., 2012). This process of apoptotic resistance and indeed the activation of profibrotic pathways, may be in part dependent on the balance between profibrotic STAT3 and anti-fibrotic STAT1 (Shen et al., 2001; Walters et al., 2005). Finally, O’Donoghue et al. (2012) report that deletion of STAT3 protects mice from bleomycin-induced fibrosis. In this study, we investigate specifically the role of STAT3 in the regulation of COL1 and other extracellular matrix proteins as well as TGFβ signaling in lung fibroblasts explanted from scleroderma patients and healthy volunteers. We report that STAT3 binds the COL1A2 enhancer in the HS4 region and directs activation cooperatively with JunB. We also report that STAT3 signaling is required for the production and remodeling of the extracellular matrix by fibroblasts.
RESULTS
STAT3 activates the COL1A2 far-upstream enhancer
The structure of the COL1A2 far-upstream enhancer and the HS4 region is shown in Figure 1A. An in silico search for STAT3 binding sites identified two sites, 304 and 603 base pairs downstream from the critical AP-1 site, where JunB binds. Both sites are highly conserved. Additional sites were found near the proximal COL1A2 promoter (unpublished data).
To investigate the interaction between STAT3 and the COL1A2 enhancer, we took advantage of the fact that fibroblasts explanted from scleroderma patients, typically contain a high proportion of myofibroblasts, with the COL1A2 enhancer active, even when they are deprived of serum or growth factor stimulation (Ponticos et al., 2009, 2015). We created two different luciferase reporter gene constructs under the control of the COL1A2 enhancer and proximal promoter (Antoniv et al., 2001; Ponticos et al., 2009, 2015): a construct with the -ype enhancer (WT) and a mutant version (ΔHS4), missing the HS4 region containing the STAT3 and JunB sites (Figure 1B). Normal and SSc lung fibroblasts were transfected with the constructs and treated with 100 µM AG490, an inhibitor of JAK2 and JAK3 (also EGFR, HER2), which reduces STAT signaling. Enhancer activation was determined by measuring luciferase expression (Figure 1B). Scleroderma cells, as expected, showed markedly higher luciferase signaling compared with the normal cells but only with the wild-type construct. This increase was eliminated by AG490. In contrast, both cell lines produced the same luminescence with ΔHS4 and AG490 had no effect. The results confirmed that STAT signaling is required and the HS4 region must be present to activate the COL1A2 enhancer, suggesting the STAT3 sites we found in the enhancer are functional.
We assayed the physical binding of STAT3 to the HS4 region by chromatin immunoprecipitation (ChIP), using the more potent and selective JAK family inhibitor SD1029 (Duan et al., 2006). Scleroderma lung fibroblasts were serum starved and treated with 10 µM SD1029 or vehicle for 30 min before stimulation with IL6xRα (as described in Methods and Materials) for a further 30 min. Following IL6xRα treatment, chromatin was immunoprecipitated with a monoclonal antibody (mAb) against phosphorylated STAT3 (pSTAT3). End-point, PCR analysis of the precipitated chromatin DNA clearly detected the presence of the HS4 sequence, irrespective of IL6xRα treatment (Figure 1C), showing that pSTAT3 is indeed bound to the HS4 region. Treatment with SD1029 strongly reduced binding (Figure 1C). Surprisingly, IL6xRα treatment made little difference to the binding of STAT3 to the enhancer suggesting that in myofibroblasts there is already sufficient STAT3 activation to ensure high occupancy of the STAT3 binding sites in the enhancer. The semiquantitative approach was carefully set up to ensure saturation does not obscure the difference between the untreated and IL6 treated cells. We normalized the amount of DNA used in the assay based on an input sample collected before immunoprecipitation and limited the number of PCR cycles to prevent the reaction from getting too close to the plateau phase. Western blotting analysis of nuclear extracts from the treated cells confirmed that untreated cells contain nuclear pSTAT3, which was strongly increased by IL6xRα (Figure 1D). SD1029 blocked the increase in STAT3 phosphorylation by IL6xRα (Figure 1D). SD1029 without IL6xRα eliminated STAT3 phosphorylation (Figure 1D). These findings are in agreement with the ChIP results.
We investigated the functional consequences of STAT3 binding on promoter engagement by assaying JunB binding and RNA polymerase recruitment. When the enhancer is engaged, it contacts the RNA polymerase complex forming at the collagen promoter. Thus, an RNA polymerase ChIP signal is a direct measure of enhancer engagement. Chromatin harvested from scleroderma lung fibroblasts treated overnight with SD1029 or vehicle was immunoprecipitated with antibodies against nonphosphorylated STAT3, JunB, and RNA polymerase II (RNApol II; Figure 1E). The amount of HS4 region DNA in the chromatin precipitated by each antibody was quantified by real-time PCR. The results confirmed binding of all three proteins (Figure 1E). Treatment with SD1029 reduced STAT3 and RNApol II binding, but had no effect on JunB binding. The results show that loss of STAT3 leads to enhancer deactivation and disengagement, even though JunB remains bound. Essentially the same results were obtained when STAT3 was knocked down with small interfering RNA (siRNA).
STAT3 is required for collagen type 1 expression
We sought to confirm the ChIP results by investigating how modulation of STAT3 affects the transcription of COL1A2 in explanted lung fibroblasts. Cells from healthy controls or scleroderma patients were treated with IL6xRα and 7.5 µM SD1029 (Figure 2A). As expected, scleroderma cells expressed more COL1A2 and this increase was eliminated by SD1029 treatment, while expression in healthy control cells was unaffected (Figure 2A). On the other hand, IL6xRα had no effect on COL1A2 mRNA expression at 24 h (Figure 2A). At the protein level, SD1029 treatment reduced COL1 and fibronectin expression in scleroderma cells in a dose-dependent manner (Figure 2B). Critically, SD1029 treatment reduced both pSTAT3 and STAT3 (Figure 2B). Because SD1029 inhibits all JAKs and may affect other signaling proteins, we verified that STAT3 is the key target using siRNA knockdown. Indeed, knockdown of STAT3 reduced COL1 and fibronectin mRNA expression in scleroderma lung fibroblasts (Figure 2C). This is consistent with the results of the previous section, showing that STAT3 is vital for enhancer activation but in activated fibroblasts there is already sufficient STAT3 for the enhancer to function and additional activation of STAT3 by cytokines does not appreciably affect occupancy of the STAT3 sites in the enhancer.
FIGURE 2:

(A) Normal and scleroderma fibroblasts (n = 3, * = p < 0.05 by Student’s t test) treated with IL6xRα and SD1029. (B) The effect of SD1029 on STAT3 Y705 phosphorylation, FN1 protein, and COL1 protein in serum-starved scleroderma lung fibroblasts. (C) The effect of STAT3 down-regulation with siRNA on COL1A2 and EDA–fibronectin mRNA expression in scleroderma lung fibroblasts (n = 3, * = p < 0.05). (D) Serum-starved scleroderma lung fibroblasts were treated with IL6xRα, TGFβ, and SD1029 for 24 h before assaying COL1A2 mRNA levels (n = 3, * = p < 0.05).
One alternative explanation for the lack of a COL1A2 transcriptional response with IL6xRα is that activation of the enhancer is limited by other components of the transcriptional complex that mediates enhancer activation (e.g., increased JunB/cJun balance), not affected by IL6xRα. If that is the case, then we can expect a synergistic effect between TGFβ signaling and IL6 on COL1 expression. We treated serum-starved SSc lung fibroblasts with both TGFβ and IL6xRα for 24 h and quantified COL1A2 mRNA levels (Figure 2D). We found that IL6 transsignaling did not synergize with TGFβ in up-regulating COL1A2 transcription. On the other hand, SD1029 treatment effectively suppressed the up-regulation of COL1A2 by TGFβ (Figure 2D).
The lack of synergy could also be explained by the differences in signaling between TGFβ and IL6xRα, however. The STAT3 up-regulation produced by IL6xRα is short lived, and therefore it is possible that the increased STAT3 activation does not last long enough to synergize with TGFβ.
Is sustained IL6/sIL6R signaling required for collagen 1 expression?
To examine this possibility, we conducted a series of time-course experiments measuring the intracellular protein expression of STAT3, pSTAT3, COL1A1, and COL1A2, as well as the TGFβ-responsive, myofibroblast-related genes SERPINE1, EDA-FN1, and FN1. We treated serum-starved scleroderma fibroblasts with IL6/sIL6R for 0.5, 2, or 16 h. We used two controls, one at 0.5 h and one at 2 h, to account for the changing basal expression levels. All comparisons were done with the appropriate control (Figure 3A).
FIGURE 3:

(A) Serum-starved lung scleroderma fibroblasts (n = 2) were treated with IL6xRα for 0.5, 2, or 16 h or left untreated. Protein was analyzed by Western blotting and densitometry. A representative blot matching the average is shown. The first histograph shows the normalized density of the bands in the Western shown here. The other histogram on the right shows the average normalized collagen levels from three independent experiments, each with a different fibroblast line (n = 3, * = p < 0.05). (B) Transcriptional analysis of COL1A1, COL1A2, and EDA–FN1 from the cell lines (n = 2) treated as in A. (C) Analysis of COL1 in the media of scleroderma (SSc) and healthy control lung fibroblasts treated with IL6xRα with or without 7.5 µM SD1029 for 16 h. (D) Transcriptional analysis of SOCS3 in response to IL6xRα. * = p < 0.05.
At the protein level, IL6xRα-treated cells at 0.5 h showed a twofold up-regulation of fibronectin/EDA-fibronectin and a 1.5-fold up-regulation of collagen type I (n = 3, both significant, p < 0.05 with the Student’s t test) relative to the untreated control at 0.5 h (Figure 3A). This up-regulation was lost after 2 h (Figure 3A). Real-time PCR analysis of COL1A1, COL1A2, and EDA-fibronectin mRNA levels, showed an upward trend which was not statistically significant (Figure 3B). Collagen is a secreted protein, and therefore we also investigated the effect of IL6 and SD1029 on the secreted COL1 protein levels of scleroderma or normal lung fibroblasts cultured for 16 h (Figure 3C). IL6 resulted in a clear increase in secreted collagen, and this increase was suppressed by 7.5 µM SD1029. Once again there is no transcriptional up-regulation of COL1A2 in response to IL6, but we observed a rapid effect on protein levels, tracking the pSTAT3 levels. In particular, at the 30-min time point, there is not enough time for any transcriptional effect to meaningfully alter protein levels; hence, this must reflect a posttranscriptional effect. It is important to note that collagen protein levels reflect a balance between translation, processing, and secretion, so STAT3 could be affecting any of these processes to produce rapid changes in COL1 protein levels. Using the mRNA extracted from these cells, we then investigated the activation of the STAT3 negative feedback loop by measuring SOCS3 mRNA expression (Figure 3D). We found weak up-regulation of SOCS3 at 30 min, but a dramatic up-regulation at 2 h. The STAT3 negative feedback loop (Croker et al., 2008) is responsible for the rapid shutdown of STAT3 signaling following a single dose of IL6/sIL6R. At 16 h, SOCS3 expression had reverted to background levels.
We reasoned that using two doses of IL6 would result in a more prolonged activation of STAT3 (schematic in Figure 4A) and therefore, if there is any synergy between TGFβ and IL6, it would give us a better chance to observe it. We treated the cells with two doses of IL6xRα, separated by 4 h. TGFβ was only coadministered with the first dose of IL6/sIL6R (Figure 4A). Western blot analysis at 16 h (Figure 4B, lanes 1 and 2) showed a robust phospho-STAT3 signal, confirming that two doses of IL6/sIL6R produce sustained STAT3 activation. Expression of COL1 protein was somewhat higher with IL6/sIL6R, whereas the levels of EDA-FN1, αSMA, and SERPINE1 remained unchanged at 16 h (Figure 4B, lanes 1 and 2). Again, there was no increase in the transcription of these genes with IL6xRα alone (Figure 4C).
FIGURE 4:

(A) Schematic illustrating the treatment regime. Serum-starved SSc fibroblasts were treated with two doses of IL6xRα 4 h apart. TGFβ, where appropriate, was added together with the first IL6xRα. SD1029 was only added just before the first IL6xRα treatment at zero hours. (B) Protein level analysis of treated cells by Western blotting and densitometry. Transcriptional analysis of (C) COL1A1, COL1a2, and EDA–FN1 and (D) SERPINE1 (PAI-1).
TGFβ treatment modestly increased STAT3 phosphorylation (Figure 4B, lane 3). A combination of TGFβ and two doses of IL6xRα showed the same STAT3 activation as two doses of IL6xRα alone (Figure 4B, lanes 2 and 4). The robust activation of COL1, EDA-FN1, SERPINE1 (mRNA and protein), or αSMA (protein) by TGFβ was the same irrespective of IL6xRα addition (Figure 4B, lanes 3 and 4, and C). Therefore, our data are consistent only with our original hypothesis. Even though TGFβ requires STAT3, there is no obvious synergy on the COL1A2 transcriptional level between IL6 and TGFβ under these experimental conditions because TGFβ activates STAT3 on its own sufficiently to support full activation of the enhancer.
Finally, we examined whether STAT3 is needed for early or late TGFβ signaling. We treated cells with SD1029, TGFβ, and IL6 for 4 h and then a second dose of IL6 was added to relieve the inhibition on STAT3 (schematic in Figure 4A). The second IL6xRα dose restored phosphoSTAT3 levels (Figure 4B, lanes 5 and 7), while in cells treated with TGFβ alone, SD1029 completely blocked any activation of STAT3 (Figure 4B, lane 6). We found the restoration of STAT3 levels after 4 h failed to relieve the inhibition of TGFβ signaling. The effect of TGFβ on the expression of COL1, EDA-FN1, SERPINE1 (mRNA and protein), or αSMA (protein) was still abrogated (Figure 4, B, lanes 5–7, C, and D).
Basal levels of STAT3 in scleroderma
If our hypothesis is correct, then the variation in STAT3 levels between normal and scleroderma fibroblasts should be too small to observe and we can expect constitutive basal STAT3 activation in fibroblasts from both healthy controls (which have few myofibroblasts) and scleroderma patients (which have a lot of myofibroblasts). We measured STAT3 activation after serum starvation in lung fibroblasts explanted from three different healthy controls and three different scleroderma patients (Figure 5, A and B). As expected, we found similar levels of STAT3 phosphorylation (Figure 5B). There was a very small upward trend toward scleroderma, but it was not statistically significant. Individual variation between cell lines was considerable with high STAT3 expression and phosphorylation correlating to higher COL1 and FN1 expression. We confirmed this result by looking at pSTAT3 levels in lung sections obtained from patients and healthy controls using immunohistochemistry (Figure 5C). STAT3 phosphorylation was observed, but the proportion of phosphoSTAT3 positive nuclei did not differ between control healthy and scleroderma fibrotic lung tissue (40.3% control vs. 37.4% for SSc).
FIGURE 5:

(A) Three normal and three scleroderma lung fibroblasts were serum starved overnight and Western blotting used to determine STAT3 phosphorylation. The ratio of pSTAT3 to STAT3 was determined by densitometry (arbitrary units). (B) Levels of STAT3 phosphorylation from A averaged for scleroderma and normal fibroblasts. Statistical analysis was done with the Student’s t test. (C) Immunohistochemistry for STAT3 in lung sections from normal volunteers and scleroderma patients.
JAK/STAT signaling, the extracellular matrix, and myofibroblast function
Given the strong impact of SD1029 on matrix protein synthesis and TGFβ signaling, we investigated the inhibition of key cytoskeletal and ECM rearrangements involved in myofibroblast transition. Using three-color immunofluorescence, we assessed expression of type I procollagen and EDA–fibronectin levels as measures of extracellular matrix biosynthesis (Figure 6A). We used a specific antibody for type I procollagen, which is unprocessed and only found intracellularly, making it a better indicator of the collagen synthesis rate. We also used a specific antibody against EDA-fibronectin, which is an essential component of the ECM laid down specifically by myofibroblasts. Scleroderma lung fibroblasts showed strongly increased procollagen levels and laid down a much more extensive EDA–fibronectin network compared with normal cells (Figure 6A). Treatment with SD1029 had little effect on normal cells, but it normalized the scleroderma cell phenotype (Figure 6A).
FIGURE 6:

(A) Immunofluorescence for type I procollagen (green) and EDA–fibronectin (red) in normal and scleroderma serum-starved lung fibroblasts treated with SD1029. (B) Immunofluorescence for αSMA (green) in normal and scleroderma lung fibroblasts treated with TGFβ and SD1029. The actin cytoskeleton was stained red with phalloidin. (C) Collagen gel contraction assay (n = 3, * = p < 0.05 with a Student’s t test) in scleroderma cells treated with TGFβ, SD1029, and siRNA against STAT3 or nontargeting (NT) siRNA. Normal fibroblasts failed to contract the gel sufficiently to achieve statistical significance.
To assess cytoskeletal rearrangement and stress fiber formation, we stained the fibroblasts with phalloidin and a specific antibody against αSMA (Figure 6B). TGFβ treatment increased the number of αSMA-containing stress fibers. The response of scleroderma fibroblasts was much more pronounced compared with normal fibroblasts (Figure 6B). SD1029 treatment effectively blocked the increased formation of αSMA-containing stress fibers (Figure 6B), showing that cytoskeletal rearrangements in response to TGFβ were blocked.
We used collagen contraction assays to measure the effect of STAT3 inhibition with SD1029 or knockdown with siRNA on contractility, a key feature of the myofibroblast phenotype. Both STAT3 siRNA and SD1029 effectively prevented the fibroblasts from contracting the collagen gels in response to TGFβ (Figure 6C). In contrast, the effect of SD1029 in the scratch wound assay which measures fibroblast migration was not statistically significant (Figure 7).
FIGURE 7:

Scratch wound assay with a scleroderma lung fibroblast line and SD1029. The cells were plated in 24-well plates, allowed to reach confluence, and serum starved overnight with 0.1% BSA in DMEM. A scratch was made with a pipette tip and mitomycin C was added to prevent proliferation. The cells were allowed to migrate overnight with or without 10 µM SD1029. The experiment was carried out in triplicate. The area of the gap was measured with ImageJ and the percentage of closure was calculated as (area at 24 h)/(area at 0 h) × 100. The differences between untreated and treated SD1029 were not statistically significant with a Student’s t test.
Effects of JAK/STAT3 signaling on translation
Given that our results hinted at a possible posttranscriptional effect on collagen and fibronectin protein production in response to IL6 treatment, we conducted a brief investigation of two initiation factors known to be regulated by STAT3: eIF2α and eIF4e. Recently, a role for STAT3 in autophagy and the control of translation was reported (Shen et al., 2012), with STAT3 enhancing eIF2 activity by preventing α subunit hyperphosphorylation. In addition, it has been reported that IL6 signaling via STAT3 feeds into mTOR/S6/eIF4e pathway (Shi et al., 2002). Activation of eIF4e is accomplished through deactivation of its inhibitor 4EBP1 through phosphorylation.
The results are shown in Figure 8. Surprisingly, there is little eIF2a phosphorylation to begin with and IL6 results in a minor increase rather than a decrease. Conversely, phosphorylation of 4EBP1 was strongly activated by IL6 treatment. Another interesting result was that eIF2α phosphorylation declines later on at 16 h. This offers an interesting explanation for our observation that leaving the cells to recover from serum starvation overnight also results in collagen and fibronectin protein production increases that are not commensurate with the changes observed at the mRNA level.
FIGURE 8:
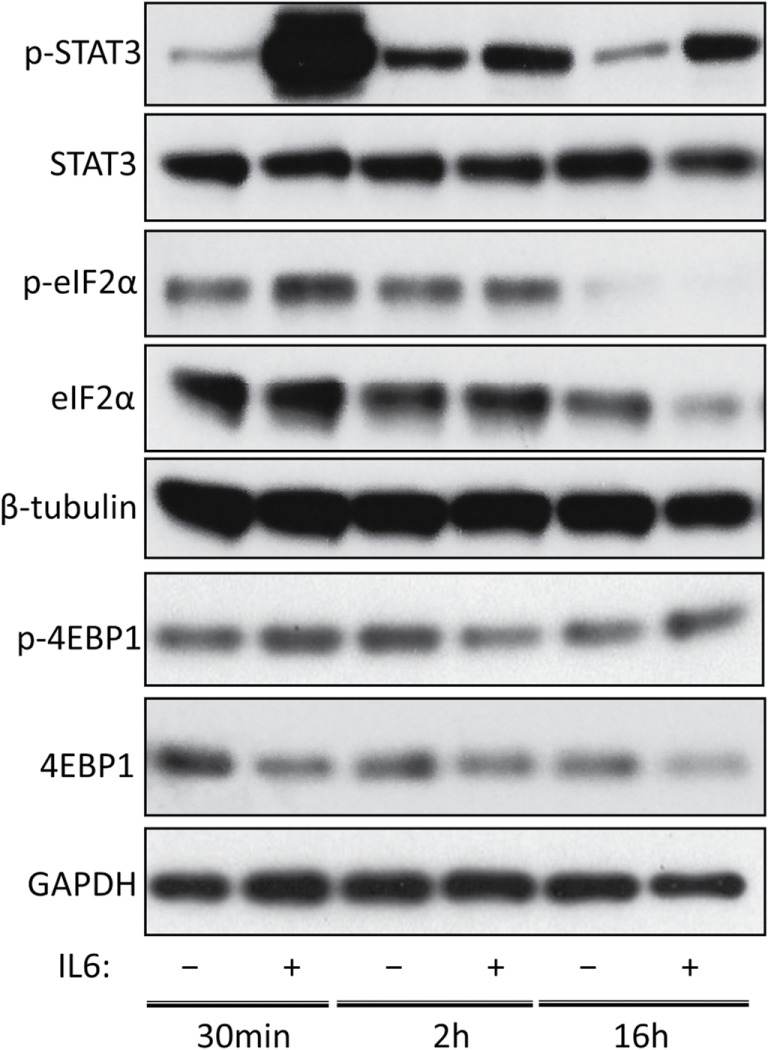
The effect of IL6 on the eIF2α and eIF4e pathways. Primary lung fibroblast cultures from SSc patients were serum starved overnight. The media was replaced with fresh serum-free media containing IL6xRα or the equivalent volume of PBS and protein was harvested after 30 min, 2 h, or 16 h. STAT3 phosphorylation is shown as a positive control for IL6. eIF2α phosphorylation was investigated as a measure of eIF2α deactivation and 4EBP1 expression/phosphorylation was investigated as a measure for eIF4e activation.
DISCUSSION
The molecular control of collagen expression and fibroblast to myofibroblast transition is of considerable medical interest because of their central role in fibrosis. Fibrosis and lung fibrosis specifically is a major cause of death in patients with fibrotic diseases such as SSc (Leask and Abraham, 2004; Varga and Abraham, 2007). Understanding the nature of the dysregulation in the production of COL1 offers vital clues on the dysregulation of extracellular matrix deposition and remodeling in fibrosis. In this report, we show that STAT3, together with JunB, is a key part of the machinery that activates the COL1A2 enhancer and therefore regulates COL1 production. Inhibition of JAK/STAT signaling or STAT3 knockdown prevented enhancer activation and RNA polymerase recruitment without affecting JunB binding. Based on these results, our proposed model for the activation of the COL1A2 enhancer by JunB and STAT3 is shown in Figure 9, A and B.
FIGURE 9:

(A) Interaction between the COL1A2 enhancer and promoter in the presence of STAT3. (B) The enhancer without STAT3. STAT3 acts as a bridge helping to engage the promoter and enhancer, while JunB recruits RNA polymerase. (C) The role of STAT3 in early TGFβ signaling. Basal STAT3 signaling and STAT3 directly induced by TGFβ is sufficient to interact with AP1/JunB and together with SMADs induce early transcriptional activation of matrix proteins. High STAT3 activation enhances translation. The increased expression of profibrotic matrix proteins triggers matrix remodeling, reinforces the TGFβ autocrine loop, and initiates myofibroblast differentiation.
One important finding is that although IL6-transsignaling strongly increased STAT3 activation, it did not appreciably change the amount of STAT3 bound to the COL1A2 enhancer or the mRNA level of COL1A2. Indeed, all our results are consistent with the hypothesis that enhancer activation in human lung fibroblasts is not limited by the availability of STAT3, because there is already significant basal STAT3 activation in fibroblasts. STAT3 expression and phosphorylation, as well as binding of COL1A2 to the COL1A2 enhancer are readily detectable in untreated fibroblasts. Similarly, inhibition of STAT3 phosphorylation results in loss of both pSTAT3 and total STAT3 (Figure 2B), in agreement with the well-known fact that STAT3 itself is a well-known target of phosphorylated STAT3 (Yang et al., 2007). However, the impact of STAT3 phosphorylation on STAT3 expression is limited by the induction of SOCS3, which is also a pSTAT3 target. The addition of IL6 does not appreciably increase STAT3 expression despite IL6 treatment, showing that there is already sufficient STAT3 activity to maintain high STAT3 expression and that further increases in phosphorylated STAT3 have no effect due to the induction of SOCS3 (Figure 3D). An important question left unanswered by our study is whether nonphosphorylated STAT3, which is a functional transcription factor (Yang et al., 2007), can also activate the collagen 1A2 enhancer. This may be an additional reason for the lack of a clear transcriptional effect with IL6 on COL1A2. In any case, our results clearly show that STAT3 is naturally activated by TGFβ, so independent activation of STAT3 may not be necessary to support full activation of the enhancer. This explains the apparent lack of synergy between TGFβ and IL6.
Despite the lack of transcriptional activation, we see a consistent increase in COL1 and FN1 protein levels with IL6 transsignaling, strongly suggesting a posttranscriptional effect. We present results that suggest IL6 signaling feeds into the mTOR/S6/eIF4e pathway, offering a very plausible model for the effects of the protein we have observed. IL6 signaling clearly increases 4EBP1 phosphorylation, which relieves its inhibition of eIF4E. This is in agreement with previous results published by our group on skin fibroblasts, where a clear increase in protein expression was observed (Khan et al., 2012)
Our work therefore suggests a dual role for STAT3 in TGFβ signaling: aside from its function as a transcription factor, it also activates the translational machinery. We believe that this explains the clear relationship that we observed between TGFβ and STAT3 signaling and the reason why JAK/STAT inhibition appears so effective at shutting down TGFβ signaling.
TGFβ has long-lasting effects on fibroblasts, which are dependent on early signaling initiating a self-reinforcing process of ECM modification and integrin signaling activation (Thannickal et al., 2003) that culminates in the creation of a high mechanical tension environment and the establishment of a TGFβ autocrine loop (Kojima et al., 2010). This is well illustrated by our data. In Figure 3, it can be seen that after fibroblasts are switched into serum-free media, the expression of matrix proteins—and at the same time STAT3 phosphorylation—increases over time without any treatment, reflecting conditioning of the media by TGFβ and other cytokines/growth factors. Once this process is completed, the myofibroblast phenotype becomes relatively stable. Our results suggest that STAT3 has a direct role in this process. Rather than activate separate pathways that feed into and enhance TGFβ function, STAT3 is actually part of the TGFβ signaling pathway: STAT3 is partially activated by TGFβ and it is directly involved in downstream transcriptional responses. High-level activation of STAT3 by cytokines may enhance sensitivity or speed up the process but the eventual outcome is the same. This is exemplified by the Western blotting results in Figure 3A.
However, inhibition of STAT3 blocks production of matrix protein components both at the transcriptional and posttranscriptional level, preventing TGFβ from initiating the matrix remodeling process, which is an integral part of the myofibroblast transition. This explanation is completely consistent with our observations: STAT3 inhibition potently blocked matrix protein (COL1/FN1) synthesis, the deposition of an EDA–fibronectin network, the appearance of αSMA-containing stress fibers, and consequently fibroblast contractility. The importance of STAT3 in these early events is demonstrated by the fact that overriding STAT3 inhibition 4 h after addition of TGFβ fails to rescue the powerful inhibitory effect of SD1029 on TGFβ signaling as seen in Figure 4. On the basis of these results, we present a model for the interaction between STAT3 and TGFβ in lung fibroblasts in Figure 9C.
Our results offer a new understanding of the avenues of interaction between profibrotic and inflammatory signaling, which underlies the transition of human lung fibroblasts to myofibroblasts. Cytokine signaling appears to potentiate profibrotic signaling rather than synergize with it. They create a permissive effect lowering the threshold at which TGFβ and presumably other profibrotic mediators can initiate the myofibroblast differentiation process. This offers a compelling explanation for our results and helps explain the variability in response between different fibroblast lines.
The differences between fibroblasts of different lineages are particularly relevant. Lung fibroblasts show distinct properties compared with dermal fibroblasts: for example, O’Reilly et al. (2014) and Ray et al. (2013) both report increases in collagen and fibronectin mRNA levels with IL6 treatment in dermal fibroblasts, but the underlying process appears to be completely different. O’Reilly et al. (2013) show a delayed response, which they attribute to IL6 up-regulating gremlin, while Ray et al. (2013) see an instant transcriptional response with IL6. In contrast, Pechkovsky et al. (2012) found no transcriptional effects in lung fibroblasts, in agreement with our results. These differences can be reconciled through variations in basal activation levels of STAT3 and the TGFβ autocrine loop, between different fibroblast lines.
Pedroza et al. (2016) appear at first glance to arrive at a different conclusion. However, upon close inspection of their data, it can be seen that our results are compatible. In their experiments, Pedroza et al. normalize pSTAT3 levels to a house keeping gene, but a closer look at their results shows that there is no obvious change in the level of STAT3 phosphorylation. Although a clear case can be made that an absolute increase in STAT3 means more pSTAT3, if this is driven by increased JAK/STAT signaling, it must necessarily be accompanied by an increase in STAT3 phosphorylation. Yet Pedroza et al. find only increased STAT3 expression and no obvious change in STAT3 phosphorylation. Importantly, there is considerable variation in STAT3 expression even in the normal cells. It is certainly possible that genetic factors leading to increased STAT3 expression may have an important role to play in idiopathic pulmonary fibrosis, but the Pedroza et al. data do not contradict our findings. More interesting is the finding that IL6 signaling activates collagen expression in mouse lung fibroblasts from the bleomycin model. No comparable results are presented with the idiopathic pulmonary fibrosis fibroblasts that the authors clearly have at their disposal. We suspect this may be a critical difference between mouse and human lung fibroblasts.
Overall, we believe our data are instrumental to resolving long-standing questions on the role of cytokine signaling in the fibroblast to myofibroblast transition, posed by the disparities in the literature. JAK/STAT signaling is integrated with profibrotic signals, rather than feeding into them, and STAT3 has both transcriptional and posttranscriptional roles to play. As a transcription factor, it controls COL1A2 expression through binding to its enhancer as a part of a complex that contains AP-1 (e.g., JunB) and presumably other factors. Such transcriptional complexes tend to regulate multiple related genes simultaneously, so we expect that other matrix-related genes are controlled in a similar manner. At the same time, through its role in translation and autophagy, STAT3 appears to directly help regulate the production of various TGFβ-inducible proteins. This combined dual role of STAT3 and the fact that TGFβ itself induces STAT3 phosphorylation, can lead to divergent experimental outcomes depending on the experimental model used and idiopathic factors such as the underlying level of STAT3 expression.
Finally, our report firmly establishes STAT3 as critical component of the fibroblast extracellular matrix remodeling mechanism in the lung and therefore as a target for anti-fibrotic therapy. Although targeting cytokines may be beneficial in ameliorating some aspects of SSc, as evidenced by promising clinical data for toclizumab, an IL6 receptor–blocking antibody, in skin fibrosis (Khanna et al., 2016), we believe that STAT3 also represents a compelling target.
MATERIALS AND METHODS
Patients and cell culture
Fibroblasts were explanted from postmortem scleroderma or healthy control lung biopsy samples. Each fibroblast line used in this study was derived from a distinct individual. They were maintained in DMEM with 10% fetal calf serum and used only between passages 2 and 5.
Reagents
Antibodies to STAT3 and phosphoSTAT3 were obtained from Cell Signaling technologies. Antibodies to JunB and RNA polymerase II as well as rabbit immunoglobulin G (IgG) were obtained from Santacruz. The TATA binding protein (TBP) antibody was obtained from Abcam. The fibronectin antibody was obtained from BD Transduction Laboratories. SD1029 and AG490 were obtained from Calbiochem. All DNA primers were obtained from Integrated DNA Technologies. TGFβ, IL6, and IL6Rα were obtained from R&D Systems.
Transcription factor binding site in silico analysis
Analysis of STAT3 binding to the COL1A2 enhancer was carried out using the Transfac Match program (Matys et al., 2016) and the Jaspar database (Mathelier et al., 2014) using STAT-specific matrices. The degree of conservation for each site was checked using the Galaxy UCSC genome browser (Giardine et al., 2005, Goecks et al., 2010).
DNA constructs and transient transfections
The human collagen far-upstream enhancer (FUE) region and the preparation of the HS1–5 (referred to in the text as WT) and ΔHS4 constructs has been described previously (Antoniv et al., 2001; Ponticos et al., 2015). Transient transfection assays were performed using FuGene 6 transfection reagent according to the manufacturer’s instructions. Briefly, 2 × 105 cells (normal or scleroderma) per transfection were seeded in six-well plates and transfected with 2 μg of total plasmid DNA. A Renilla luciferase plasmid under the control of the Rous sarcoma virus (RSV) promoter was used to control for transfection efficiency. The following day the cells were serum starved overnight with 0.1% bovine serum albumin (BSA) in DMEM, before treatment with 150 µM AG490 or vehicle control (dimethyl sulfoxide) for 24 h. The cells were finally harvested using a dual-light chemiluminescent reporter gene assay system (Tropix) according to the manufacturer’s instructions. The experiment was run in triplicate and repeated three times.
IL6 treatment
IL6 (10 µg/ml) was premixed with an equal volume of the soluble IL6 receptor α (25 µg/ml) in phosphate-buffered saline (PBS), to obtain an equimolar mixture (IL6xRα). IL6xRα was used at 15 ng/ml final IL6 concentration (1.5/1000 dilution).
Chromatin immunoprecipitation
ChIP assays were performed using the EZ ChIP assay kit from Merck Millipore, according to the manufacturer’s instructions. Briefly the cells were seeded in 10-cm plates at 2 × 106 cells per plate and treated as described in the Results section. The chromatin was cross-linked with 1% formaldehyde in media, for 10 min at 37°C. The cells were lysed with SDS–lysis buffer and the chromatin DNA was sheared to 400 base pairs average by sonication. Protein A sepharose beads and specific antibodies (phosphoSTAT3, JunB, RNA polymerase II) were used for immunoprecipitation with rabbit IgG as the negative control. The precipitated DNA was analyzed qualitatively by end-point PCR, using the QIAGEN Fast Cycling PCR kit or quantitatively by real-time PCR (see below) using primers specific for the HS4 region: Fwd – TTCACATGAGCATTTGAGTGTATTG and Rev – TCGTCAGTGTGTAACCCTCATC.
RNA interference
SiRNA against STAT3 was obtained from GE Dharmacon and transfected using Dharmafect 1 according to the manufacturer’s instructions. Fibroblasts were seeded in 25-cm2 flasks at 75% confluence and transfected with 25 nM siRNA. The next day, the fibroblasts were serum starved with 0.1% BSA in DMEM for 24 h and then used for further experiments.
Western blotting
The Thermo Fisher NuPAGE system was used for Western blotting according to the manufacturer’s instructions. Whole cell lysates were prepared in RIPA buffer, and 10–15 µg of protein was loaded per 15-well lane. Nuclear extracts were prepared with the Thermo Fisher NE-PER kit according to the manufacturer’s instructions. Amounts of 2.5–5 µg of protein were loaded per 15-well lane.
Quantitative PCR
Real-time PCR analysis was accomplished using a QIAGEN Rotorgene 6000 thermal cycler with the QIAGEN QuantiFAST RT-PCR kit according to the manufacturer’s instructions. For quantification of ChIP DNA, the reverse transcriptase step was omitted and the results were normalized based on a sample of the chromatin (input) obtained just before the immunoprecipitation step. For the analysis of gene expression, total RNA was isolated using the QIAGEN RNeasy kit according to the manufacturer’s instructions and TBP was used as the house keeping gene. Primer sequences are in Table 1.
TABLE 1:
Primers used for quantitative real-time PCR.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| COL1A2 | TGCTTGCAGTAACCTTATGCCTA | CAGCAAAGTTCCCACCGAGA |
| TBP | AGTGACCCAGCATCACTGTT | GGCAAACCAGAAACCCTTGC |
| EDA–fibronectin | TGCAGTAACCAACATTGATCGC | TCAGGGCTCGAGTAGGTCAC |
| COL1A1 | AATCCTCGAGCACCCTGA | CCCCTGGAAAGAATGGAGAT |
| SERPINE1 | GCTATGGGATTCAAGATTGATGACA | TCTGAAAAGTCCACTTGCTTGAC |
Immunohistochemistry
Lung biopsy specimens obtained from SSc patients or healthy controls were fixed in Formalin and paraffin embedded. Immunohistochemistry was accomplished as described previously (Ponticos et al., 2015), using an antibody specific for phosphorylated STAT3. The cells positive and negative for nuclear phosphoSTAT3 were counted using ImageJ.
Immunofluorescence
The cells were seeded in an eight-chamber slide at a density of 2 × 104 cells per chamber and allowed to attach overnight. The next day, the fibroblasts were serum starved with 0.1% BSA in DMEM for 24 h, before treatment with TGFβ (2 ng/ml) and SD1029 (10 µM). Immunofluorescence was carried out as described previously (Ponticos et al., 2015), using antibodies against EDA–fibronectin (mouse; Abcam), procollagen1 (rat; Abcam), and αSMA (mouse; DAKO). Phalloidin–Texas Red (Life Technologies), where indicated, was added along with the secondary antibody at a 1:1000 dilution.
Gel contraction assays
A collagen mixture containing one part 0.2 M HEPES, pH 8.0 (Sigma), four parts 2.05 mg/ml rat tail collagen type I (First Link UK), and five parts DMEM was prepared on ice. Fibroblasts were added at 8 × 104 cells/ml and the cell–collagen mixture was added to 24 well plates (1 ml per well). SD1029 was added to the collagen mixture (1–10 µM). The plates were incubated at 37°C for 1 h to allow the collagen to gel and overlaid with 1 ml DMEM. TGFβ, where appropriate, was added to the media at 2 ng/ml. The plates were incubated at 37°C overnight, and the gels were detached and allowed to contract.
Acknowledgments
This research was funded by the Rosetrees Trust, Arthritis Research UK, Royal Free Hospital Charity, and Scleroderma Research UK.
Abbreviations used:
- ChIP
chromatin immunoprecipitation
- COL1
collagen type 1
- ECM
extracellular matrix
- EDA-FN1
extra domain A–fibronectin 1
- FUE
far upstream enhancer for COL1A2
- HS1-5
DNase hypersensitive site 1–5
- IL6xRα
a mixture of interleukin 6 and the interleukin 6 soluble receptor alpha
- PP
proximal promoter
- sIL6Rα
soluble IL6 receptor alpha.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-06-0342) on November 15, 2017.
REFERENCES
- Antoniv TT, De Val S, Wells D, Denton CP, Rabe C, de Crombrugghe B, Ramirez F, Bou-Gharios F. Characterization of an evolutionarily conserved far-upstream enhancer in the human α2(I) collagen (COL1A2) gene. J Biol Chem. 2001;276:21754–21764. doi: 10.1074/jbc.M101397200. [DOI] [PubMed] [Google Scholar]
- Croker BA, Kiu H, Nicholson SE. Socs regulation of the jak/stat signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Bradner JE, Greenberg E, Levine R, Foster R, Mahoney J, Seiden MV. SD-1029 inhibits signal transducer and activator of transcription 3 nuclear translocation. Clin Cancer Res. 2006;12:6844–6852. doi: 10.1158/1078-0432.CCR-06-1330. [DOI] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, The Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat M-L, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- Khan K, Xu S, Nihtyanova S, Derrett-Smith E, Abraham D, Denton CP, Ong VH. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis. 2012;71:1235–1242. doi: 10.1136/annrheumdis-2011-200955. [DOI] [PubMed] [Google Scholar]
- Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, Baron M, Chung L, Fierlbeck G, Lakshminarayanan S, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2 randomised controlled trial. Lancet. 2016;387:2630–2640. doi: 10.1016/S0140-6736(16)00232-4. [DOI] [PubMed] [Google Scholar]
- Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A. Autocrine TGF-βand stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-βsignaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42(Database issue):D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2016;34(Database issue):D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003;29:490–498. doi: 10.1165/rcmb.2002-0262OC. [DOI] [PubMed] [Google Scholar]
- O’Donoghue RJ, Knight DA, Richards CD, Prêle CM, Lau HL, Jarnicki AG, Jones J, Bozinovski S, Vlahos R, Thiem S, et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol Med. 2012;4:939–951. doi: 10.1002/emmm.201100604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. 2014;289:9952–9960. doi: 10.1074/jbc.M113.545822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechkovsky DV, Prêle CM, Wong J, Hogaboam CM, McAnulty RJ, Laurent GJ Zhang SS, Selman M, Mutsaers SE, Knight DA. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol. 2012;180:1398–1412. doi: 10.1016/j.ajpath.2011.12.022. [DOI] [PubMed] [Google Scholar]
- Pedroza M, Le TT, Lewis K, Karmouty-Quintana H, To S, George AT, Blackburn MR, Tweardy DJ, Agarwal SK. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016;30:129–140. doi: 10.1096/fj.15-273953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponticos M, Harvey C, Ikeda T, Abraham D, Bou-Gharios G. JunB mediates enhancer/promoter activity of COL1A2 following TGF-β induction. Nucleic Acids Res. 2009;37:5378–5389. doi: 10.1093/nar/gkp544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponticos M, Papaioannou I, Xu S, Holmes AM, Khan K, Denton CP, Bou-Gharios G, Abraham DJ. Failed degradation of JunB contributes to overproduction of type I collagen and development of dermal fibrosis in patients with systemic sclerosis. Arthritis Rheumatol. 2015;67:243–253. doi: 10.1002/art.38897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J Invest Dermatol. 2013;133:1212–1220. doi: 10.1038/jid.2012.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Devgan G, Darnell JE, Jr, Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci USA. 2001;98:1543–1548. doi: 10.1073/pnas.041588198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Mariño G, Lachkar S, Senovilla L, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–680. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Shi Y, Hsu JH, Hu L, Gera J, Lichtenstein A. Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J Biol Chem. 2002;277:15712–15720. doi: 10.1074/jbc.M200043200. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters DM, Antao-Menezes A, Ingram JL, Rice AB, Nyska A, Tani Y, Kleeberger SR, Bonner JC. Susceptibility of signal transducer and activator of transcription-1-deficient mice to pulmonary fibrogenesis. Am J Pathol. 2005;167:1221–1229. doi: 10.1016/S0002-9440(10)61210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Li C, Halfter H, Liu J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene. 2003;22:894–905. doi: 10.1038/sj.onc.1206158. [DOI] [PubMed] [Google Scholar]