

Abstract
Leflunomide, an anti-inflammatory drug used for the treatment of rheumatoid arthritis, has been marked with a black box warning regarding an increased risk of liver injury. The active metabolite of leflunomide, A771726, which also carries a boxed warning about potential hepatotoxicity, has been marketed as teriflunomide for the treatment of relapsing multiple sclerosis. Thus far, however, the mechanism of liver injury associated with the two drugs has remained elusive. In this study, cytotoxicity assays showed that ATP depletion and subsequent LDH release were induced in a time- and concentration-dependent manner by leflunomide in HepG2 cells, and to a lesser extent, by A77 1726. The decline of cellular ATP levels caused by leflunomide was dramatically exacerbated when galactose was substituted for glucose as the sugar source, indicating a potential mitochondrial liability of leflunomide. By measuring the activities of immuno-captured mitochondrial oxidative phosphorylation (OXPHOS) complexes, we found that leflunomide and A77 1726 preferentially targeted complex V (F1FO ATP synthase), with IC50 values of 35.0 and 63.7 μM, respectively. Bongkrekic acid, a mitochondrial permeability transition pore blocker that targets adenine nucleotide translocase, profoundly attenuated mitochondrial membrane depolarization, ATP depletion, and LDH leakage induced by leflunomide and A77 1726. Substantial alterations of mitochondrial function at the transcript level were observed in leflunomide-treated HepG2 cells, whereas the effects of A77 1726 on the cellular transcriptome were much less profound. Our results suggest that mitochondrial dysfunction may be implicated in the hepatotoxicity associated with leflunomide and A77 1726, with the former exhibiting higher toxicity potency.
Keywords: Drug-induced liver injury, Leflunomide, Oxidative phosphorylation, ATP synthase, ANT
1. Introduction
Leflunomide is an isoxazole derivative that was approved by the U.S. Food and Drug Administration (FDA) in 1998 for the treatment of rheumatoid arthritis (Goldenberg, 1999). As an immunomodulatory agent, leflunomide has been available in over 70 countries worldwide and is one of the most frequently used disease-modifying antirheumatic drugs (Kunkel and Cannon, 2006). Despite the proven efficacy of leflunomide in the management of rheumatoid arthritis, safety concerns have arisen from post-marketing reports of severe liver injury and other adverse events (Aithal, 2011; Alcorn et al., 2009; Keen et al., 2013). In 2010, FDA added a boxed warning to the drug label regarding the risk of severe liver injury from leflunomide, based on a review that identified 49 cases of severe liver injury, including 14 cases of fatal liver failure, in the preceding seven years (U.S. FDA, 2010). The reported adverse events highlight the risk of hepatotoxicity in patients with pre-existing liver disease or elevated liver enzymes and the necessity of monitoring liver functions during leflunomide treatment. Considering the sustained and widespread use of leflunomide, investigations on the mechanism of liver toxicity are needed for the safer use of this drug.
Leflunomide has been shown to undergo a conversion to form its major metabolite A77 1726 and other unidentified metabolites. The pathways and enzymes involved in the formation of A77 1726 have not well-characterized. Some studies suggest that the conversion is mediated by cytochrome P450 (CYP) enzymes and also mediated by a non-enzymatic reaction. A77 1726 can be further metabolized to an oxanilic acid derivative and then excreted (Kalgutkar et al., 2003; Ren et al., 2017; Rozman, 2002; Seah et al., 2008; Shi et al., 2011). The pharmacological activity of leflunomide is primarily mediated by its major metabolite, A77 1726, also known as teriflunomide, which acts through inhibition of dihydroorotate dehydrogenase, a key mitochondrial enzyme in the de novo pyrimidine biosynthetic pathway (Fox et al., 1999). The mean steady-state plasma concentration of A77 1726 can reach 125–230 μM in patients administered the standard dose of leflunomide (Bohanec Grabar et al., 2009; Rozman, 2002; Schmidt et al., 2003). Teriflunomide was approved by the U.S. FDA in 2012 for the treatment of relapsing forms of multiple sclerosis (Miller, 2015). There is a boxed warning in the prescribing information for teriflunomide indicating a possible similar risk of hepatotoxicity as leflunomide, considering that the two drugs produce a similar range of plasma concentrations of teriflunomide at recommended doses (U.S. FDA, 2012). Thus far, there is no evidence that teriflunomide has higher or lower risk of liver injury compared with leflunomide. Nonetheless, there are in vitro data demonstrating that A77 1726 is less cytotoxic than its parent compound and that CYP metabolism of leflunomide is a detoxification process (Shi et al., 2011).
Mitochondria are vital organelles involved in cellular bioenergetics, metabolism, and signaling processes (Grattagliano et al., 2011; Mishra and Chan, 2016; Vakifahmetoglu-Norberg et al., 2017). The primary function of mitochondria is to generate cellular energy in the form of adenosine triphosphate (ATP) through the oxidative phosphorylation (OXPHOS) system. Mitochondria also play a critical role in the regulation of cell death, and the disturbance of mitochondrial function can lead to cell necrosis or apoptosis through mitochondrial membrane permeabilization (Halestrap et al., 2000; Kroemer et al., 2007). Mitochondrial dysfunction has been recognized as a major mechanism of drug-induced liver injury, which is a leading cause of premature termination of clinical trials and post-market drug withdrawals (Pessayre et al., 2012; Ribeiro et al., 2014; Vuda and Kamath, 2016). There has been increased awareness of the necessity of screening for drug-induced mitochondrial dysfunction during the preclinical phase of drug development in the pharmaceutical industry (Nadanaciva and Will 2011a, 2011b). In the current study, we investigated whether leflunomide and its active metabolite exhibit mitochondrial toxicity in human hepatic HepG2 cells to understand better the mechanism of liver injury induced by these agents. Because we demonstrated that endoplasmic reticulum (ER) stress is one of the mechanisms underlies leflunomide-induced toxicity in our previous study (Ren et al., 2017), the interplay between ER stress and mitochondrial impairment was also investigated in this study.
2. Material and methods
2.1. Chemicals and reagents
Leflunomide (≥98% purity) and A77 1726 (≥98% purity) were purchased from Enzo Life Sciences (Farmingdale, NY). Bongkrekic acid, cyclosporine A, and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO). Cell culture media and supplements were purchased from Life Technology (Grand Island, NY) and Atlanta Biologicals (Lawrenceville, GA).
2.2. Cell culture
The human hepatocellular carcinoma cell line HepG2 was purchased from American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2. HepG2 cells were seeded at a density of 3–5 × 105 cells/ml in 96-well tissue culture plates or 60-mm tissue culture dishes, and allowed to adhere for 24 h prior to exposure to DMSO vehicle or test compounds at the indicated concentrations for specified time periods in serum-free medium. The final DMSO concentration in the medium was 0.5% (v/v).
2.3. Cellular adenosine triphosphate content measurement
Total cellular adenosine triphosphate (ATP) levels were determined using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI). The luminescence was measured with a Synergy 2 multi-mode microplate reader (BioTek, Winooski, VT). The percentage of cell viability at each compound concentration was calculated using the equation: % cell viability = (luminescence of compound-treated cells/luminescence of vehicle-treated cells) × 100.
2.4. Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) leakage from cells was used as a measure of cell death. Briefly, at the end of treatment, 10 μl of medium (cell-free supernatant) was collected from each well and Triton-X 100 was added at a final concentration of 1% to lyse the cells. After 1 h, 10 μl of cell lysates combined with medium was harvested. Corresponding samples of supernatants and lysates were transferred to a clear 96-well plate, and 240 μl reaction buffer (81 mM Tris, 204 mM NaCl, 0.2 mM NADH, and 1.7 mM monosodium pyruvate, pH 7.2) was added to each well. The absorption at 340 nm was immediately measured for 5 min at 60-s intervals with a microplate reader (BioTek). LDH activity was determined by the rate of decrease in absorbance at 340 nm due to oxidation of NADH. The percentage of LDH leakage was calculated using the equation: % LDH leakage = [LDH activity in supernatant/LDH activity in (supernatant + cells)] × 100.
2.5. Glucose/galactose assay
HepG2 cells were grown in high-glucose medium or galactose medium (Marroquin et al., 2007). High-glucose medium: DMEM-high glucose (Life Technologies, 11995-065) containing 25 mM glucose and 1 mM sodium pyruvate, supplemented with 5 mM N-2-hydro-xyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Galactose medium: DMEM-no glucose (Life Technologies, 11966-025) supplemented with 10 mM galactose, 2 mM glutamine, 5 mM HEPES, 1 mM sodium pyruvate, 10% FBS, and penicillin/streptomycin as above. Cells were seeded at a density of 5 × 105 cells/ml in 96-well plates and incubated for approximately 24 h prior to treatment with DMSO vehicle or various concentrations of test compounds. After a 24 h treatment, cell viability was assessed by measuring ATP depletion as described above. The EC50 values, defined as the drug concentrations producing a 50% reduction in cellular ATP content, were calculated by fitting the data to the log (inhibitor) versus normalized response-variable slope equation.
2.6. Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP, ΔΨm) was measured using the dual fluorescence dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide). HepG2 cells, cultured in 96-well black wall/clear bottom plates, were treated with DMSO or test compounds for 6 h. After treatment, the supernatant was removed, and cells in each well were incubated with 100 μl of 2.5 μg/ml JC-1 in medium for 20 min at 37°C. After washing twice with PBS, the fluorescence intensities of JC-1 monomers and aggregates were quantified on a microplate reader (BioTek). JC-1 monomers were detected at wavelengths of 485 nm (excitation) and 535 nm (emission), and JC-1 aggregates were detected at the wavelengths of 530 nm (excitation) and 590 nm (emission), respectively. The ratio of JC-1 aggregate (red fluorescence) to monomer (green fluorescence) was used as a measure of MMP. A reduction in the ratio of red/green fluorescence intensity indicates mitochondrial membrane depolarization.
2.7. Measurement of individual OXPHOS complex activity
Direct inhibitory effects of the test compounds on the five OXPHOS complexes were measured using MitoTox™ Complete OXPHOS Activity Assay Panel (Abcam), according to the manufacturer’s protocol. Briefly, leflunomide and A77 1726 were diluted at various concentrations in appropriate assay solution and directly added to each OXPHOS complex: Complexes I, II, IV, and V were immunocaptured from bovine heart mitochondria in a functionally active form by specific monoclonal antibodies immobilized in 96-well microplates; Complex II + III activity was assayed in mitochondrial suspension. The specific inhibitors of each of the five complexes were used as positive controls: rotenone (Complex I), thenoyltrifluoroacetone (TTFA, Complex II), antimycin A (Complex III), potassium cyanide (KCN, Complex IV), and oligomycin (Complex V). Microplate wells coated with a null capture antibody (Complexes I, II, IV, and V) or wells without mitochondria (Complex III) were used as background controls. After the addition of test compounds with assay solution to 96-well microplates, the absorption of each well was immediately measured using a microplate reader (BioTek) at wavelengths of 340 nm (Complex I and V), 600 nm (Complex II), and 550 nm (Complex III and IV) at intervals of 60 s for 2 h (Complex I) or 1 h (Complex II, IV, and V) and 20 s for 5 min (Complex III). The activity of each complex was determined by the rate of change in absorbance after background subtraction. The final DMSO concentration in all the activity assays was 1.5% (vol/vol), which had no inhibitory effect on the enzymes. The IC50 values, defined as the drug concentrations producing a 50% inhibition of OXPHOS activities, were determined using a four-parameter logistic nonlinear regression analysis.
2.8. RNA isolation, sequencing and gene expression analysis
Total RNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA) from four biological replicates of HepG2 cells treated with DMSO (vehicle control), leflunomide (62.5, 125, and 250 μM), or A77 1726 (62.5, 125, and 250 μM) for 6 h. The quantity and purity of RNA were determined using a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, DE). The quality of RNA was assessed on an Agilent 2100 Bioanalyzer using a RNA 6000 Nano kit (Agilent Technologies, Palo Alto, CA). High-quality RNA, with an RNA Integrity Number ≥9.0, was submitted to the Genomics and Microarray Core Facility at the University of Texas Southwestern Medical Center (Dallas, TX) where RNA sequencing (RNA-seq) was performed on an Illumina HiSeq 2000 System. The raw sequencing data were subjected to adapter trimming and quality filtering using Trimmomatic version 0.32 (Bolger et al., 2014). The cleaned reads were mapped to the human reference genome (GRCh37) from the Ensembl database using TopHat version 2.0.8b (Trapnell et al., 2009). Mapped reads were assembled into transcripts, and the expression values were calculated based on the number of fragments per kilobase of transcript per million mapped reads (FPKM) using Cuffiinks version 2.0.2 (Trapnell et al., 2010). Pairwise differential expression analysis was performed using the Bioconductor R package edgeR (Robinson et al., 2010). P-values were adjusted to control false discovery rate due to multiple testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). The differentially expressed genes (DEGs) were determined by absolute fold change > 1.5 and adjusted P < 0.05. A Venn diagram was constructed to depict commonality of DEGs derived from pairwise comparisons using the R package VennDiagram (Chen and Boutros, 2011).
2.9. Western blot analysis
HepG2 cells were grown and treated with leflunomide in 60 mm tissue culture plates. Standard Western blot analyses were performed using antibodies against CHOP (Thermo Fisher Scientific), ATF-4, and GAPDH (as an internal control, Santa Cruz Biotechnology, Santa Cruz, CA), followed by a secondary antibody conjugated with horseradish peroxidase (HRP) (Santa Cruz Biotechnology). The bands were detected by FluorChem E and M Imaging System (ProteinSimple, San Jose, CA).
2.10. Statistical analysis
Statistical comparisons were performed by two-tailed unpaired Student’s t-test or one-way variance-of-analysis (ANOVA) followed by Dunnett’s multiple comparison test. Data analyses were carried out using GraphPad Prism 5 (GraphPad Software, San Diego, CA). The statistical significance level was set at P < 0.05.
3. Results
3.1. Time- and dose-dependent cytotoxicity in HepG2Cells
In our previous study, we compared the cytotoxicity of leflunomide in HepG2 cells with that in the metabolically competent cells HepaRG and primary human hepatocytes. Our results showed that leflunomide was more toxic in HepG2 cells compared to HepaRG cells and the cytotoxicity induced in HepG2 cells was similar to that in primary human hepatocytes (Ren et al., 2017). A study by Shi et al. demonstrated that cytotoxicity of A77 1726 in primary rat hepatocytes can be attenuated by CYP enzymes (Shi et al., 2011), suggesting that metabolites of leflunomide and A77 1726 are less toxic. In addition, it has been reported that the conversion of leflunomide to A77 1726 can occur in a non-enzymatic manner (Kalgutkar et al., 2003; Rozman, 2002). Taken together, the metabolism capacity, which is important for the evaluation of metabolite-mediated toxicity, is of the less concern in the current investigation. Owing to the ease-of-use and some specific features of HepG2 cells in assessing mitochondrial damage susceptibility (Marroquin et al., 2007), we chose HepG2 cells to study the cytotoxicity and mitochondrial dysfunction of leflunomide and A77 1726.
To assess the cytotoxicity of leflunomide and A77 1726, cellular ATP content and LDH release were measured in parallel in HepG2 cells exposed to leflunomide or A77 1726 at various concentrations from 31.25 to 500 μM for 2, 6, and 24 h. As shown in Fig. 1A and B, both drugs caused ATP levels to decline and the release of LDH to increase in time- and concentration-dependent manners. At all the time points, no significant LDH release was observed at 62.5 μM for both drugs. LDH release, an indicator of cell membrane damage, began to increase significantly with 250 μM leflunomide treatment and 500 μM A77 1726 treatment at 2 h. At 6 and 24 h, 125 μM leflunomide induced significant cell membrane disruption with 15.1% and 19.3% of LDH released into the culture medium compared to 9.5% and 10.3% of LDH released from vehicle-treated cells, respectively. By comparison, A77 1726 only caused a slight increase in LDH release at the concentration of 125 μM at 24 h.
Fig. 1.
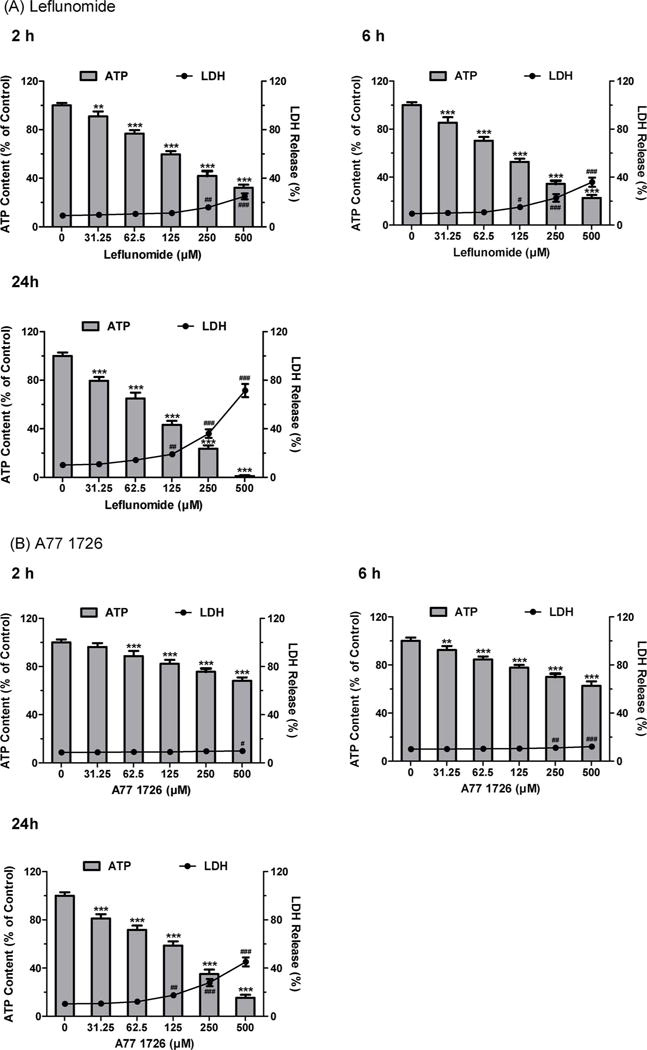
Cytotoxicity of leflunomide (A) and A77 1726 (B) in human hepatoma HepG2 cells. HepG2 cells were treated with DMSO as vehicle control and leflunomide or A77 1726 for 2, 6, and 24 h. Cytotoxicity was assessed by cellular ATP depletion (bars) and LDH release (lines). Data were analyzed for statistical significance using one-way ANOVA followed by Dunnett’s test. Data are expressed as mean ± SD (n = 3). Statistical significance compared with control: **, p < 0.01 and ***, p < 0.001 (ATP); #, p < 0.05, ##, p < 0.01, and ###, p < 0.001 (LDH).
ATP levels were measured to determine whether the cytotoxicity caused by the two drugs was associated with impaired cellular energy metabolism. Both drugs caused significant ATP depletion at 31.25 and 62.5 μM, concentrations at which no or negligible cell death was observed as determined by LDH leakage. At the concentration of 31.25 μM, leflunomide caused a 9.0, 14.6, and 20.5% reduction in ATP levels at 2, 6, and 24 h, respectively, while A77 1726 induced significant ATP depletion of 7.6% and 18.9% at 6 and 24 h, respectively. Prior to the increase of LDH release at 2 h, ATP levels were decreased by 40.3% and 24.3% in cells treated with 125 μM leflunomide and 250 μM A77 1726, respectively. These results show that ATP depletion is more sensitive to leflunomide and A77 1726 than the loss of cell membrane integrity, which implies that the impaired mitochondrial function induced by these drugs may contribute to the increase of LDH release observed at higher concentrations. Overall, A77 1726 exerted lower cytotoxicity in HepG2 cells compared to leflunomide.
3.2. Cytotoxicity in glucose- and galactose-grown HepG2Cells
HepG2 cells grown in galactose-containing media rely mainly on mitochondrial OXPHOS for ATP generation. It has been reported that galactose-grown HepG2 cells are more sensitive to mitochondrial toxicants than glucose-grown HepG2 cells (Marroquin et al., 2007). To examine the potential mitochondrial toxicity of leflunomide and A77 1726, we treated glucose- and galactose-grown HepG2 cells with various concentrations of the two drugs for 24 h. Leflunomide exhibited markedly different toxicity at concentrations of 62.5, 125, and 250 μM under two culture conditions. Leflunomide was much more toxic when the galactose medium was used (Fig. 2A). The EC50 values were 109.5 ± 4.1 μM and 55.4 ± 2.3 μM for glucose- and galactose-grown HepG2 cells, respectively. By comparison, A77 1726 showed increased toxicity in galactose-grown HepG2 cells only at a much higher concentration of 500 μM (Fig. 2B). The EC50 values were 154.2 ± 6.6 μM and 144.9 ± 8.3 μM under glucose and galactose growth conditions, respectively.
Fig. 2.
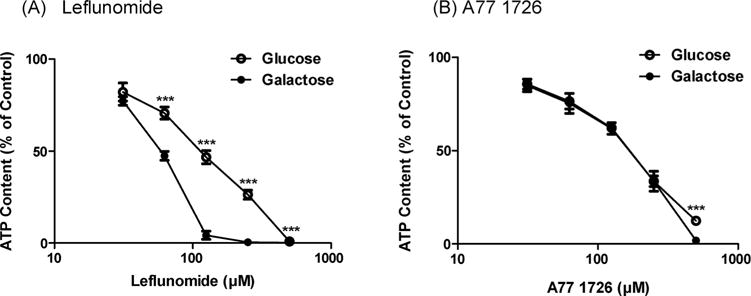
Effects of leflunomide (A) and A77 1726 (B) on mitochondrial function assessed by glucose/galactose assay. Dose responses at 24 h for glucose-grown (25 mM) (open symbol) and galactose-grown (10 mM) (filled symbol) HepG2 cells treated with leflunomide (A) and A77 1726 (B). Data were analyzed for statistical significance using two-tailed unpaired Student’s t-test. Data are expressed as mean ± SD (n = 3). Statistical significance compared between two culture conditions: ***p < 0.001.
3.3. Cytoprotective effects of MPTP blockage
The mitochondrial permeability transition pore (MPTP) is a non-selective channel that mediates the mitochondrial inner membrane permeability. Inappropriate MPTP opening causes mitochondrial membrane depolarization and uncoupling of oxidative phosphorylation, leading to ATP depletion and cell death (Halestrap, 2009; Paradies et al., 2013). To explore the underlying mechanisms of leflunomide- and A77 1726-induced mitochondrial toxicity, bongkrekic acid and cyclosporine A, two MPTP blockers which target adenine nucleotide translocase (ANT) and cyclophilin D (CyPD), respectively, were assessed for their protective effects against the toxicity caused by the two drugs. A marked decrease of mitochondrial membrane potential (MMP) was observed in HepG2 cells treated with 125, 250, and 500 μM of leflunomide, as indicated by a 10.6, 20.9, and 42.6% reduction in the ratio of JC-1 red/green fluorescence intensity, respectively (Fig. 3A1). A77 1726 also caused a loss of MMP, but to a lesser extent than leflunomide, with the ratio of JC-1 red/green fluorescence intensity decreased by 7.9, 13.5, and 29.2% at concentrations of 125, 250, and 500 μM, respectively (Fig. 3B1). The decline of MMP induced by leflunomide or A77 1726 was greatly attenuated by addition of 10 μM bongkrekic acid (Fig. 3A1, B1). Bongkrekic acid also exerted significant protective effects on ATP depletion and LDH leakage caused by the two drugs (Fig. 3A2-3, 3B2-3), indicating a relationship between mitochondrial membrane depolarization and cell injury. By comparison, only moderate protective effects were observed with 1 μM cyclosporine A (Fig. 3A1-3, 3B1-3), suggesting that ANT rather than CypD may be the primary subunit of MPTP involved in the regulation of MMP loss induced by leflunomide and A77 1726.
Fig. 3.
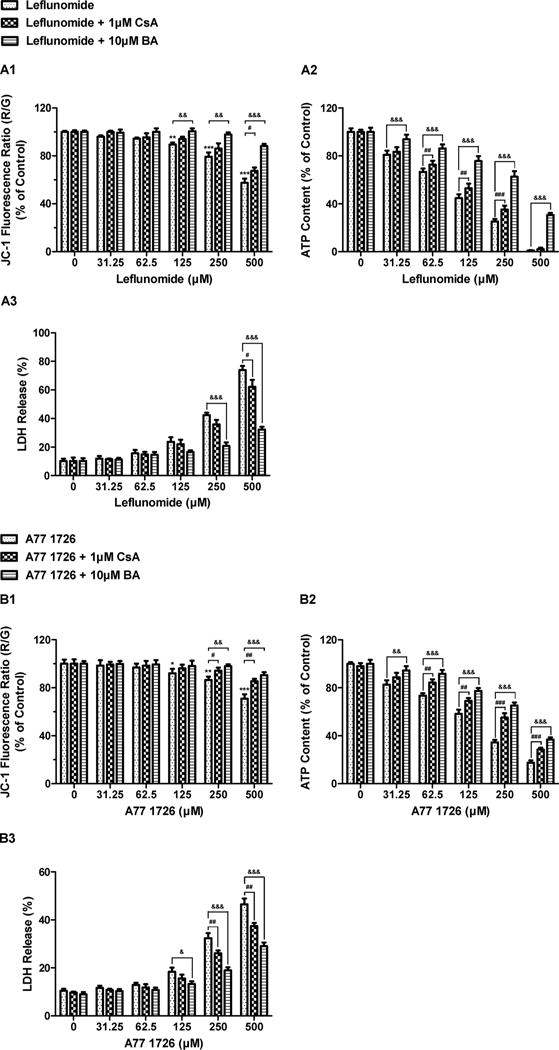
Effects of mitochondrial permeability transition (MPT) inhibitors on ATP depletion, LDH release and mitochondrial membrane potential (MMP) induced by leflunomide (A) and A771726 (B). HepG2 cells were pre-treated with 1 μM cyclosporine A (CsA) or 10 μM bongkrekic acid (BA) followed by a 24-h exposure to leflunomide or A77 1726. MMP was assessed by the JC-1 fluorescent probe. Data were analyzed for statistical significance using one-way ANOVA followed by Dunnett’s test. Data are expressed as mean ± SD (n = 3). Statistical significance compared with control: **, p < 0.01 and ***, p < 0.001 (MMP). Statistical significance compared between treatment in the presence and absence of MPT inhibitors: #, p < 0.05, ##, p < 0.01, and ###, p < 0.001 (CsA); &, p < 0.05, &&, p < 0.01, and &&&, p < 0.001 (BA).
3.4. Inhibitory effects on OXPHOS complexes
Direct inhibition of the activity of each of the five OXPHOS complexes by leflunomide and A77 1726 was measured using commercially available immunocapture-based OXPHOS activity assays. The immunocapture-based OXPHOS activity assays have been demonstrated to provide high specificity and low intra- and inter-assay variations for the detection of mitochondrial inhibitors (Nadanaciva et al., 2007a). These assays have been evaluated using known hepatic mitotoxicants (Nadanaciva et al., 2007a, 2007b). Compared with conventional methods that use isolated intact mitochondria, such assays can produce accurate activity measurements for OXPHOS complexes, especially for Complexes I and V, by eliminating competing enzymes. In this study, the assays were performed in a concentration-response format using serial two-fold dilutions of the two drugs to determine the 50% inhibitory concentration (IC50). The two drugs exhibited different inhibitory potencies on OXPHOS complex activities. Leflunomide potently inhibited Complex V, which catalyzes ATP synthesis, with an IC50 of 35.0 μM (Fig. 4A5). In contrast, leflunomide showed mild inhibition of Complex I with an IC50 of 199.6 μM (Fig. 4A1), and poor inhibition of Complex II, Complex II + III, and Complex IV, with IC50 values > 300 μM (Fig. 4A2-4). A77 1726 also showed preferential inhibition of Complex V, but with lesser potency (IC50 = 63.7 μM), as compared to leflunomide (Fig. 4B5). A77 1726 caused slight inhibition of Complex I with an IC50 of 228.8 μM (Fig. 4B1). Although A77 1726 displayed higher inhibitory potency on Complex II and Complex II + III than leflunomide, the inhibitory effects were negligible due to high IC50 values (Fig. 4B2-3).
Fig. 4.
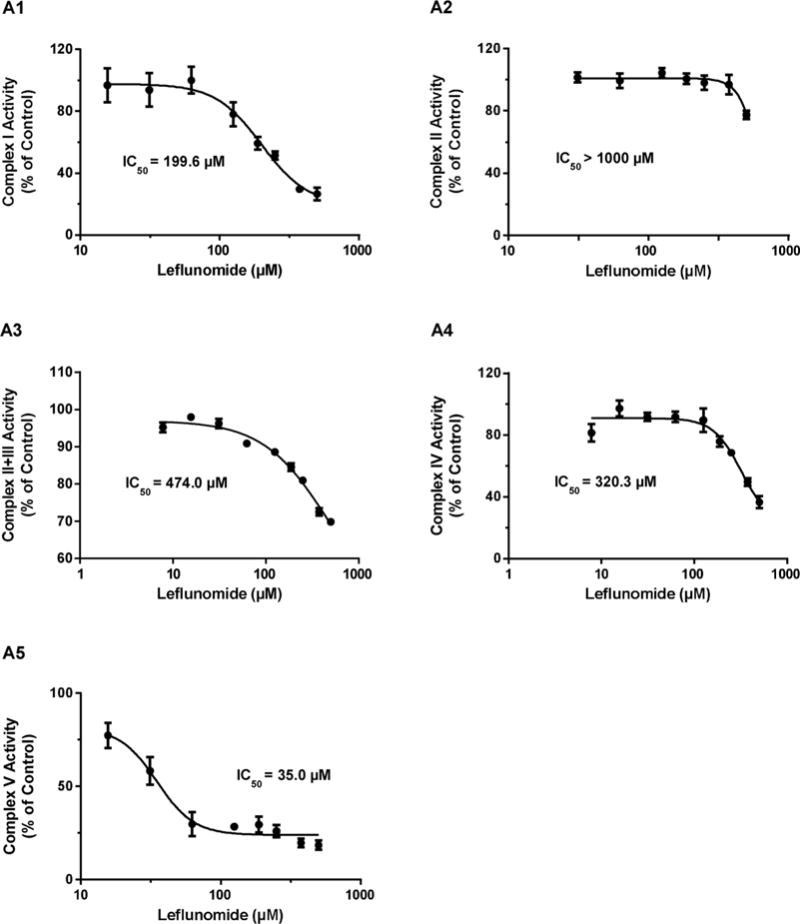
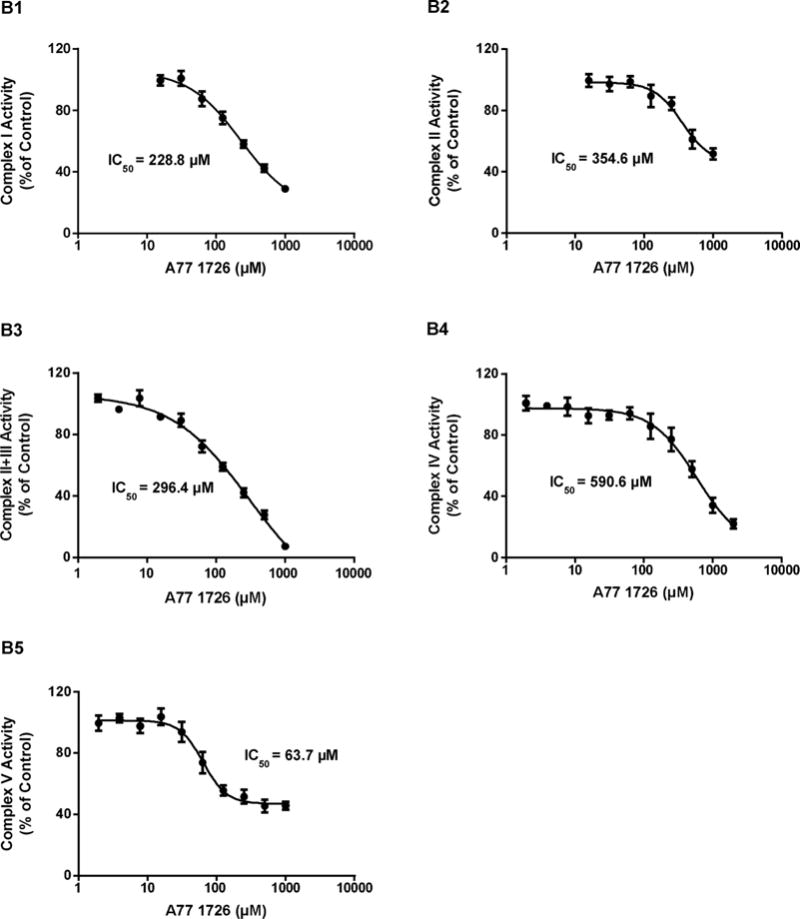
Effects of leflunomide (A) and A77 1726 (B) on the activities of mitochondrial electron transport complexes in isolated bovine heart mitochondria. Data are expressed as mean ± SD (n = 3). The 50% inhibitory concentration (IC50) of leflunomide on Complex I, II, II + III, IV, and V activity were obtained from dose-response curves.
3.5. Transcriptome alterations of mitochondrial function
To assess the effects of leflunomide and A77 1726 on the expression of genes that were related to mitochondrial function, the transcriptomes of HepG2 cells upon a 6-h exposure to DMSO or various concentrations of leflunomide or A77 1726 were quantified by RNA-seq. Pairwise comparisons were conducted among all treatment groups to determine the differential expression of genes in response to leflunomide and A77 1726 exposure. In comparison with leflunomide, A77 1726 exhibited weaker effects on the transcriptome of HepG2 cells. As shown in Fig. 5, at a concentration of 125 μM, leflunomide and A77 1726 induced the differential expression of 2186 and 404 genes, respectively. Seventy-one genes were differentially expressed across all three treatment groups. Among all differentially expressed genes, those associated with mitochondrial function were identified by functional annotations and shown in Table 1, including genes that encode the subunits or assembly factors of the mitochondrial electron transport chain complexes, genes that regulate mitochondrial membrane potential, and genes involved in the mitochondrial energy metabolism.
Fig. 5.
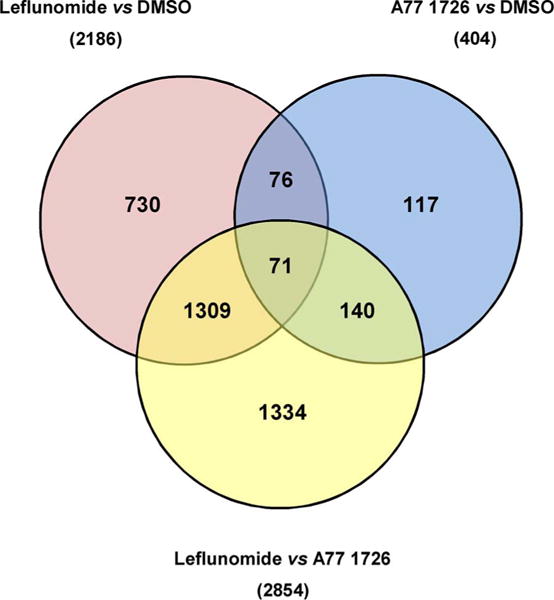
Venn Diagram illustrating the number of differentially expressed genes (DEGs) among HepG2 cells treated with leflunomide (125 μM), A77 1726 (125 μM) and DMSO (vehicle control) for 6 h. Values in parentheses correspond to the total number of DEGs between pairwise comparisons of the three treatment groups. Values in the intersecting regions represent the number of overlapped DEGs between the comparisons, and values in the non-intersecting regions represent the number of DEGs that were unique to the corresponding comparison.
Table 1.
Differentially Expressed Genes Related to Mitochondrial Function.
| Gene | EnsemblID | Leflunomide vs DMSO
|
A77 1726 vs DMSO
|
Leflunomide vs A77 1726
|
|||
|---|---|---|---|---|---|---|---|
| Fold Changea | Adjusted P | Fold Changeb | Adjusted P | Fold Changec | Adjusted P | ||
| ATP5G1 | ENSG00000159199 | −1.62 | 0.021 | 1.07 | 0.57 | −1.74 | 0.009 |
| ABAT | ENSG00000183044 | −1.62 | 0.005 | −1.19 | 0.08 | −1.37 | 0.012 |
| ABCB6 | ENSG00000115657 | −1.59 | 0.048 | 1.21 | 0.30 | −1.92 | 0.016 |
| ABCD3 | ENSG00000117528 | −1.72 | 0.002 | −1.08 | 0.49 | −1.59 | 0.033 |
| ACSS1 | ENSG00000154930 | −1.61 | 0.014 | 1.10 | 0.36 | −1.76 | 0.007 |
| ADPRHL2 | ENSG00000116863 | −1.82 | 0.038 | −1.06 | 0.62 | −1.72 | 0.020 |
| ALDH5A1 | ENSG00000112294 | −1.65 | 0.025 | −1.23 | 0.52 | −1.34 | 0.267 |
| BCL2L1 | ENSG00000171552 | 1.30 | 0.075 | −1.80 | 0.03 | 2.33 | 0.018 |
| BIK | ENSG00000100290 | 19.62 | 0.006 | −1.09 | 0.96 | 21.42 | 0.101 |
| BMF | ENSG00000104081 | 2.62 | 0.011 | −1.02 | 0.63 | 2.68 | 0.012 |
| BNIP3 | ENSG00000176171 | −1.09 | 0.349 | 1.53 | 0.03 | −1.66 | 0.010 |
| CHDH | ENSG00000016391 | −1.56 | 0.019 | −1.83 | 0.04 | 1.17 | 0.067 |
| CLIC1 | ENSG00000213719 | 1.59 | 0.031 | −1.13 | 0.18 | 1.81 | 0.010 |
| CNP | ENSG00000173786 | −1.74 | 0.018 | −1.05 | 0.73 | −1.66 | 0.034 |
| COQ10B | ENSG00000115520 | 2.48 | 0.004 | 1.41 | 0.10 | 1.75 | 0.025 |
| COQ4 | ENSG00000167113 | −1.56 | 0.044 | 1.04 | 0.69 | −1.63 | 0.025 |
| CPT1A | ENSG00000110090 | 4.30 | 0.017 | 1.50 | 0.09 | 2.88 | 0.015 |
| CPT2 | ENSG00000157184 | −1.53 | 0.011 | −1.39 | 0.06 | −1.10 | 0.255 |
| CYP24A | ENSG00000019186 | 7.70 | 0.015 | −1.86 | 0.04 | 14.31 | 0.005 |
| DDX28 | ENSG00000182810 | −1.04 | 0.641 | −1.86 | 0.04 | 1.79 | 0.015 |
| DHODH | ENSG00000102967 | −1.02 | 0.887 | −1.65 | 0.05 | 1.63 | 0.012 |
| DHRS1 | ENSG00000157379 | −1.79 | 0.016 | 1.56 | 0.06 | −2.79 | 0.005 |
| DNAJC19 | ENSG00000205981 | −1.65 | 0.011 | 1.00 | 0.98 | −1.65 | 0.006 |
| DYNLL1 | ENSG00000088986 | −1.83 | 0.010 | −1.01 | 0.74 | −1.81 | 0.005 |
| DYNLL2 | ENSG00000264364 | −1.90 | 0.045 | −1.26 | 0.28 | −1.51 | 0.098 |
| FARS2 | ENSG00000145982 | 1.09 | 0.425 | −1.91 | 0.03 | 2.08 | 0.007 |
| GRPEL2 | ENSG00000164284 | 2.33 | 0.006 | 1.05 | 0.60 | 2.23 | 0.006 |
| HK2 | ENSG00000159399 | 3.24 | 0.009 | −1.04 | 0.65 | 3.36 | 0.004 |
| HRK | ENSG00000135116 | 2.08 | 0.046 | −1.95 | 0.72 | 4.05 | 0.509 |
| HSPA1L | ENSG00000204390 | −2.37 | 0.207 | −2.12 | 0.05 | −1.12 | 0.798 |
| HSPE1 | ENSG00000115541 | −1.59 | 0.023 | −1.03 | 0.65 | −1.55 | 0.009 |
| LGALS3 | ENSG00000131981 | 2.28 | 0.019 | 1.14 | 0.34 | 2.00 | 0.034 |
| MALSU1 | ENSG00000156928 | −1.58 | 0.043 | −1.06 | 0.52 | −1.49 | 0.030 |
| MCL1 | ENSG00000143384 | 2.84 | 0.003 | −1.13 | 0.20 | 3.21 | 0.001 |
| MGEA5 | ENSG00000198408 | 2.58 | 0.044 | 1.26 | 0.09 | 2.06 | 0.038 |
| MIEF2 | ENSG00000177427 | −2.33 | 0.048 | −1.15 | 0.54 | −2.03 | 0.083 |
| MLYCD | ENSG00000103150 | −2.05 | 0.008 | −1.44 | 0.04 | −1.42 | 0.019 |
| MMAB | ENSG00000139428 | −1.59 | 0.038 | −1.11 | 0.54 | −1.44 | 0.044 |
| MRM2 | ENSG00000122687 | −2.00 | 0.024 | −1.37 | 0.17 | −1.46 | 0.047 |
| MRPL49 | ENSG00000149792 | −1.64 | 0.027 | 1.11 | 0.21 | −1.81 | 0.016 |
| MRPL51 | ENSG00000111639 | −1.71 | 0.031 | 1.01 | 0.91 | −1.72 | 0.002 |
| MRPS17 | ENSG00000239789 | −1.65 | 0.027 | −1.23 | 0.13 | −1.35 | 0.007 |
| MRPS18C | ENSG00000163319 | −1.52 | 0.037 | −1.12 | 0.46 | −1.36 | 0.034 |
| MRPS21 | ENSG00000187145 | −1.56 | 0.026 | −1.13 | 0.37 | −1.38 | 0.037 |
| MRPS26 | ENSG00000125901 | −1.51 | 0.014 | −1.01 | 0.87 | −1.50 | 0.013 |
| MRPS30 | ENSG00000112996 | 1.59 | 0.028 | −1.12 | 0.31 | 1.78 | 0.035 |
| MRPS31 | ENSG00000102738 | −1.53 | 0.044 | −1.07 | 0.37 | −1.44 | 0.030 |
| MTERF1 | ENSG00000127989 | −2.55 | 0.036 | 1.19 | 0.42 | −3.02 | 0.008 |
| MTERF2 | ENSG00000120832 | −4.17 | 0.006 | −1.02 | 0.86 | −4.07 | 0.004 |
| MTFR2 | ENSG00000146410 | −1.02 | 0.877 | −2.15 | 0.04 | 2.12 | 0.023 |
| MTX3 | ENSG00000177034 | 1.55 | 0.027 | −1.07 | 0.46 | 1.66 | 0.028 |
| NDUFAF5 | ENSG00000101247 | 1.67 | 0.012 | −1.33 | 0.16 | 2.22 | 0.028 |
| OAT | ENSG00000065154 | 1.17 | 0.026 | 1.63 | 0.03 | −1.40 | 0.024 |
| PDP2 | ENSG00000172840 | −1.66 | 0.018 | 1.34 | 0.10 | −2.23 | 0.013 |
| PIM2 | ENSG00000102096 | −2.92 | 0.034 | 1.56 | 0.09 | −4.55 | 0.005 |
| PLD6 | ENSG00000179598 | 1.65 | 0.026 | −1.05 | 0.68 | 1.74 | 0.015 |
| PMAIP1 | ENSG00000141682 | 2.10 | 0.045 | 1.94 | 0.19 | 1.08 | 0.794 |
| POLG | ENSG00000140521 | −1.57 | 0.013 | 1.04 | 0.65 | −1.62 | 0.020 |
| PPARGC1A | ENSG00000109819 | 2.11 | 0.049 | 1.04 | 0.76 | 2.02 | 0.032 |
| PPP3CC | ENSG00000120910 | 1.82 | 0.048 | −1.10 | 0.81 | 2.01 | 0.191 |
| RAB11FIP5 | ENSG00000135631 | −1.62 | 0.016 | 1.22 | 0.11 | −1.97 | 0.014 |
| RAD51 | ENSG00000051180 | −1.52 | 0.018 | 1.01 | 0.79 | −1.53 | 0.024 |
| RNF144B | ENSG00000137393 | 1.73 | 0.017 | −2.48 | 0.11 | 4.30 | 0.038 |
| SDHAF1 | ENSG00000205138 | −1.99 | 0.047 | 1.04 | 0.86 | −2.08 | 0.023 |
| SDHAF2 | ENSG00000167985 | −1.51 | 0.027 | −1.09 | 0.54 | −1.40 | 0.041 |
| SFXN2 | ENSG00000156398 | −1.84 | 0.016 | 1.16 | 0.31 | −2.12 | 0.007 |
| SFXN3 | ENSG00000107819 | −1.79 | 0.010 | −1.06 | 0.65 | −1.69 | 0.016 |
| SFXN5 | ENSG00000144040 | −1.78 | 0.010 | 1.20 | 0.07 | −2.13 | 0.004 |
| SH3GLB1 | ENSG00000097033 | 1.96 | 0.026 | 1.06 | 0.35 | 1.85 | 0.022 |
| SLC25A14 | ENSG00000102078 | −1.84 | 0.012 | 1.01 | 0.81 | −1.86 | 0.016 |
| SLC25A19 | ENSG00000125454 | −1.92 | 0.012 | −1.25 | 0.13 | −1.54 | 0.051 |
| SLC25A25 | ENSG00000148339 | 9.50 | 0.003 | 1.80 | 0.04 | 5.29 | 0.001 |
| SLC25A29 | ENSG00000197119 | −2.85 | 0.026 | 1.13 | 0.38 | −3.22 | 0.017 |
| SLC25A33 | ENSG00000171612 | 1.98 | 0.006 | 1.35 | 0.13 | 1.47 | 0.031 |
| SLC25A36 | ENSG00000114120 | 1.88 | 0.021 | 1.07 | 0.65 | 1.76 | 0.056 |
| SNN | ENSG00000184602 | −2.89 | 0.006 | 1.39 | 0.06 | −4.01 | 0.004 |
| STARD1 | ENSG00000133121 | 3.63 | 0.008 | −1.62 | 0.06 | 5.90 | 0.003 |
| TFB1M | ENSG00000029639 | −1.13 | 0.444 | −1.65 | 0.02 | 1.46 | 0.098 |
| TFB2M | ENSG00000162851 | −1.66 | 0.006 | −1.25 | 0.05 | −1.33 | 0.015 |
| TIMM10B | ENSG00000132286 | −1.99 | 0.005 | 1.30 | 0.06 | −2.59 | 0.006 |
| TIMM21 | ENSG00000075336 | −2.04 | 0.001 | −1.17 | 0.22 | −1.74 | 0.026 |
| TIMM44 | ENSG00000104980 | −1.51 | 0.048 | −1.02 | 0.73 | −1.48 | 0.065 |
| TIMM8A | ENSG00000126953 | −1.96 | 0.006 | −1.17 | 0.11 | −1.67 | 0.008 |
| TRIAP1 | ENSG00000170855 | −1.68 | 0.007 | 1.09 | 0.24 | −1.82 | 0.004 |
| TUSC2 | ENSG00000114383 | −2.15 | 0.019 | 1.01 | 0.88 | −2.18 | 0.022 |
Fold change was calculated as the ratio of the mean expression levels of the gene between two groups. A positive value of fold change indicates a relatively higher gene expression level while a negative value of fold change indicates a relatively lower gene expression level in
HepG2 cells treated with leflunomide (125 μM) as compared to HepG2 cells treated with DMSO (vehicle control)
HepG2 cells treated with A77 1726 (125 μM) as compared to HepG2 cells treated with DMSO (vehicle control), or
HepG2 cells treated with leflunomide (125 μM) as compared to HepG2 cells treated with A77 1726 (125 μM) for 6 h. Each treatment group contains four replicates. Statistically significant changes (absolute fold change > 1.5; adjusted P < 0.05) were marked in bold.
3.6. Mitochondrial dysfunction associates with ER stress
In our previous study, we demonstrated that endoplasmic reticulum (ER) stress contributes to leflunomide-associated cytotoxicity in hepatic cells (Ren et al., 2017). Mitochondria are both physically and functionally connected with endoplasmic reticulum, the cellular organelle that manufactures, processes, and exports proteins to other intracellular locations. It was of interest to investigate whether leflunomide-dependent mitochondrial toxicity and ER stress are connected. As demonstrated in Fig. 3, the MPTP inhibitor bongkrekic acid significantly reduced the cytotoxicity of leflunomide, whereas little effect was observed for cyclosporine A. Interestingly, we found that the two inhibitors also showed distinct effects on the ER stress proteins (Fig. 6A&B). The presence of bongkrekic acid reduced the induction of two ER stress markers, ATF4 and CHOP, but the addition of cyclosporine A failed to show similar effects.
Fig. 6.
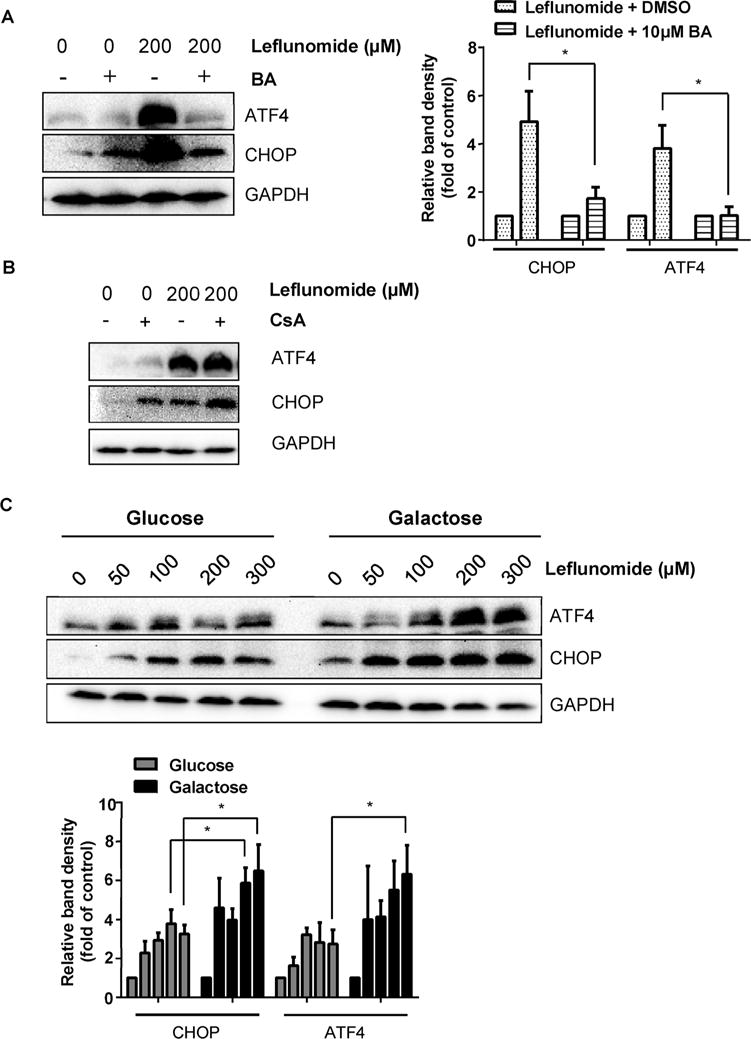
Mitochondrial dysfunction modulates the level of ER stress. (A) HepG2 cells were pre-treated with MPTP inhibitor bongkrekic acid (BA) (10 μM) for 2 h before exposure to 200 μM of leflunomide for 6 h. Western blot showed that the presence of BA attenuated the leflunomide-induced activation of ATF4 and CHOP. (B) HepG2 cells were pre-treated with MPTP inhibitor cyclosporine A (CsA) (1 μM) for 2 h before exposure to 200 μM of leflunomide for 6 h. Representative Western blot showed that CsA failed to alleviate the ER stress induced by leflunomide. (C) Glucose-grown and galactose-grown HepG2 cells were exposed to 50 to 300 μM of leflunomide for 6 h. Western blot showed that galactose-grown HepG2 cells exhibited more prominent increase in both ATF4 and CHOP protein levels. Similar Western results were obtained from three independent experiments. Intensities of bands were normalized to the amount of GAPDH. *p < 0.05 versus treatment of corresponding control.
Moreover, galactose-grown HepG2 cells treated with leflunomide showed higher sensitivity to mitochondrial damage (Fig. 2A&B) and also displayed more profound increases in ATF4 and CHOP compared to leflunomide-treated glucose-grown cells (Fig. 6C). A slight increase in CHOP was also observed in galactose-grown DMSO-treated cells; however, the increase is not statistically significant (Fig. 6). These results indicate that the level of mitochondrial damage correlates positively with the extent of ER stress.
4. Discussion
Drug-induced mitochondrial dysfunction has received growing attention in recent years as a cause of late-stage clinical failures and the withdrawal of marketed drugs. Drugs may disturb mitochondrial functions via a number of different mechanisms, including direct inhibition of OXPHOS complexes, uncoupling of electron transport from ATP production, irreversible opening of MPTP, impairment of mtDNA replication and mtDNA-encoded polypeptide synthesis, inhibition of fatty acid β-oxidation, or diminished efficiency of the tricarboxylic acid cycle (Labbe et al., 2008; Vuda and Kamath, 2016). Despite long-term recognition of leflunomide-induced liver injury, the mechanisms responsible for its toxicity are not fully understood. In the current study, we assessed the hepatotoxicity induced by leflunomide and its active metabolite in HepG2 cells and explored the underlying mechanisms, with mitochondrial dysfunction being the primary focus.
In the clinic, a dose regimen of 20 mg (or 10 mg if 20 mg is not tolerated) leflunomide once daily with or without a 100 mg loading dose for three days is recommended for the treatment of rheumatoid arthritis (Sanofi-Aventis U.S. LLC, 2016). At the standard daily dosage of 20 mg leflunomide, the average steady-state plasma concentration of A77 1726 is approximately 125 μM (Bohanec Grabar et al., 2009; Rozman, 2002). Pharmacokinetic studies have shown that plasma/serum concentrations of A77 1726 are dose proportional following either single (20–100 mg) or repeated doses (5–25 mg daily) of leflunomide (Li et al., 2002; Mladenovic et al., 1995; Rozman, 2002). Moreover, large inter-individual variability has been observed in the steady-state plasma concentrations of A77 1726 among patients taking therapeutic doses of leflunomide, with an 80-fold concentration range observed between the highest and lowest values (Bohanec Grabar et al., 2009; Chan et al., 2005; Keen et al., 2013; van Roon et al., 2005). Therefore, intermediate and clinically relevant concentrations of leflunomide and A77 1726, 125 μM each, were used in this study to investigate the mechanisms involved in drug-induced liver toxicity. In our study, the EC50 values of leflunomide and A77 1726 in HepG2 cells measured with the ATP assay were 109.5 and 154.2 μM, respectively (Fig. 2), indicating that the concentrations that caused cytotoxicity are close to the plasma concentration achievable in patients and are clinically relevant.
To examine the cytotoxicity of leflunomide and A77 1726 in HepG2 cells, we first measured the cellular ATP content following treatment with the two drugs. Cellular ATP levels began to decrease significantly after a 2-h exposure of HepG2 cells to leflunomide and A77 1726 at concentrations of 31.25 and 62.5 μM, respectively, in a time- and concentration-dependent manner. By comparison, LDH leakage, an indicator of cell membrane damage, began at higher concentrations at all time points (Fig. 1). Mitochondria house the enzymatic system that produces the majority of cellular energy in the form of ATP, and necrotic cell death ensues when cellular ATP levels fall profoundly within a short time or when reduced ATP levels persist for a longer time (Kushnareva and Newmeyer, 2010). The early occurrence of ATP depletion prior to cell membrane collapse implicated impairment of mitochondrial function in HepG2 cells treated with the two drugs, and therefore further assays were conducted to explore the cytotoxic mechanism related to the mitochondrial dysfunction.
Increased metabolism of glucose into lactic acid in the presence of oxygen, also known as aerobic glycolysis, is a prominent feature of some highly proliferative cells (Gatenby and Gillies, 2004). These cells do not rely on mitochondrial respiration for energy production due to their high glycolytic activity. However, respiration rates can be increased to maintain ATP levels when these cells are cultured in galactose-containing media (Marroquin et al., 2007). Replacement of glucose with galactose as the sole sugar source causes these cells to redirect their energy metabolism to OXPHOS to maintain ATP levels and therefore become sensitive to mitochondrial toxicants (Rodriguez-Enriquez et al., 2001). An assessment strategy for drug-induced mitochondrial dysfunction has been established based on the distinct effects of mitochondrial toxicants on the viability of HepG2 cells in response to the sugar source (Marroquin et al., 2007). Recently, a similar approach was implemented in a study with primary rat hepatocytes to identify drugs associated with mitochondrial dysfunction (Liu et al., 2016). By using this strategy, we examined the mitochondrial toxicity of leflunomide and A77 1726 in glucose- versus galactose-grown HepG2 cells (Fig. 2). The reduction of ATP content induced by leflunomide was significantly greater in galactose-grown HepG2 cells as compared with glucose-grown HepG2 cells, as indicated by a roughly two-fold difference in EC50 values. In contrast, a difference between glucose- and galactose-grown cells treated with A77 1726 was only apparent at a high concentration of 500 μM, and no substantial difference in EC50 values was observed. Moreover, the EC50 value of leflunomide was approximately 2.6-fold higher than that of A77 1726 in galactose-grown HepG2 cells. These results indicate that leflunomide and A77 1726 may have mitochondrial liabilities, with the former exhibiting higher potency.
Mitochondrial membrane permeability is regulated by MPTP, a non-selective channel that opens in response to calcium overload and various effectors, such as adenine nucleotide depletion, inorganic phosphate elevation, oxidative stress, and mitochondrial depolarization (Halestrap et al., 2002). MPTP opening leads to collapse of MMP and ATP production, and eventually, cell death (Crompton, 1999; Halestrap, 2009). In this study, we observed dose-dependent decreases of MMP in HepG2 cells treated with leflunomide and A77 1726 (Fig. 3). To a large extent, these drug-induced declines in MMP were reversed by bongkrekic acid, an ANT inhibitor. ATP depletion and LDH leakage induced by the two drugs were also dramatically attenuated by bongkrekic acid. In contrast, the protective effects of the CypD inhibitor cyclosporine A were less pronounced. In general, ANT is considered as a structural component of MPTP, but evidence also suggests that ANT can act a regulator of MPTP activity in association with CypD, mitochondrial phosphate carrier (PiC), F1FO ATP synthase or other pore-forming components (Baines, 2009; Gutierrez-Aguilar and Baines, 2015; Karch and Molkentin, 2014). The binding of bongkrekic acid locks ANT in a conformation to facilitate MPTP blockage (Fiore et al., 1998). It has also been proposed that cyclosporine A may modulate the pore-forming activity of F1FO ATP synthase through ANT or PiC that binds with CypD, which may explain the mild MPTP blockage effects of cyclosporine A observed in this study (Gutierrez-Aguilar and Baines, 2015; Karch and Molkentin, 2014). On the other hand, in HepG2 cells, it is possible that CypD is more sensitive than ANT in response to mitotoxicants. Nonetheless, our results indicate that mitochondrial membrane depolarization induced by leflunomide and A77 1726 may be primarily mediated via the modulation of ANT.
Inhibition of mitochondrial electron transport complexes underlies the mechanism of liver toxicity induced by a variety of mitochondrial toxicants (Dykens et al., 2008a, 2008b; Nadanaciva et al., 2007a, 2007b; Will et al., 2008). To investigate further the direct effects of leflunomide and A77 1726 on mitochondrial function, we performed immunocapture-based OXPHOS activity assays to measure the perturbation of the activities of individual electron transport chain components by the two drugs. Such immunocapture-based assays have proven superiority over conventional methods in accurate activity measurements for OXPHOS complexes, especially for Complex I and Complex V, by removing the confounding influence of competing enzymes present in intact mitochondria (Nadanaciva et al., 2007a). Leflunomide and A77 1726 both displayed preferential inhibition of Complex V, with IC50 values of 35.0 and 63.7 μM, respectively (Fig. 4). This is in accordance with the concentrations at which the two drugs induced early ATP depletion in HepG2 cells (Fig. 1). Complex V, known as F1FO ATP synthase, is a large protein complex located in the inner mitochondrial membrane, where it catalyzes the conversion of ADP to ATP using the energy generated by the translocation of protons from the intermembrane space into the matrix (Jonckheere et al., 2012). Notably, the two drugs also targeted ANT, which mediates the exchange of mitochondrial ATP with cytosolic ADP for ATP synthesis (Halestrap and Brenner, 2003). Recent models of MPTP formation proposed that the pore forms from the F1FO ATP synthase and that ANT may modulate MPTP through its interaction with the pore-forming c subunit of the ATP synthase FO complex (Bernardi and Di Lisa, 2015; Bernardi et al., 2015; Bonora et al., 2013; Halestrap, 2014; Karch and Molkentin, 2014). Our results suggested that the inhibitory effects of leflunomide and A77 1726 on both mitochondrial Complex V and ANT may account for the rapid ATP depletion following exposure of HepG2 cells to the two drugs. The association between the two mechanisms needs further investigation.
Our RNA-seq analysis revealed that leflunomide induced alterations in the expression of a number of genes related with mitochondrial function, whereas A77 1726 exerted more modest effects on the transcriptome. An interesting observation of this study is that leflunomide might mainly affect the components of the inner mitochondrial membrane. A number of proteins that exhibited significant changes at gene expression level localize in the inner membrane (Table 1). ATP5G1, a gene encoding the mitochondrial FO ATP synthase subunit C1, a component of the inner membrane (Jonckheere et al., 2012), was significantly down-regulated by leflunomide but not A77 1726. The c-subunit ring of the ATP synthase FO complex, as discussed above, has been implicated as a core component of MPTP. The reduction of ATP5G1 expression induced by leflunomide may be involved in the exacerbated mitochondrial membrane depolarization.
Inner membrane translocases and solute carriers also typically localize to the inner mitochondrial membrane (Schmidt et al., 2010). Our gene expression analysis also showed that several members of SLC25A family, which comprises of mitochondrial solute carriers in the inner membrane, were also altered in expression following leflunomide exposure (Table 1). Particularly, SLC25A25, a gene encoding a calcium-binding ATP-Mg/Pi carrier, was dramatically up-regulated by leflunomide, and to a lesser extent, by A77 1726. This carrier transports ATP-Mg in exchange for phosphate and catalyzes mitochondrial uptake/effiux of adenine nucleotides (del Arco and Satrustegui, 2004; Fiermonte et al., 2004). The up-regulation of SLC25A25 may occur in response to the changes in mitochondrial adenine nucleotide content caused by the two drugs. Moreover, several genes encoding translocases of mitochondrial inner membrane, including TIMM8A, TIMM10B, TIMM21, and TIMM44, were down-regulated by leflunomide but not A77 1726. These translocases mediate the import and insertion of proteins across or into the mitochondrial inner membrane, which require MMP and ATP production as driving forces (Hutu et al., 2008; van der Laan et al., 2006). The reduced expression of the translocases, together with the loss of MMP and depletion of ATP, may contribute to defects in the mitochondrial protein import system, which may also be implicated in the hepatotoxicity induced by leflunomide.
In our study, measurement of the activity of caspase 3/7, an executor of apoptosis, upon leflunomide treatment at 2 h or 6 h time point did not show activation of caspase 3/7 (data not shown), suggesting that the observed cytotoxicity was not caspase-dependent. It could be possible that the depletion of ATP disturbed the function of transmembrane ion-dependent ATPases, which could result in osmotic failure, and thus, cell death. This hypothesis is consistent with the results of LDH release, which indicated the integrity of cell membranes was significantly disrupted at the presence of leflunomide. Nevertheless, other mechanisms could also contribute to this process, including ER stress, a mechanism we discussed in length in our previous paper (Ren et al., 2017).
Mitochondrial dysfunction has been recognized to be a critical contributor to drug-induced liver toxicity, and ER stress has gained more attention recently in the field of liver toxicity (Chen et al., 2014, 2015; Ren et al., 2016; Uzi et al., 2013). Mitochondria and ER share direct contacts via the mitochondrial-associated membranes (MAM) and these physical associations enable a tight functional connection between the two organelles (Kornmann et al., 2009; Vance, 1990). Therefore, it is not surprising that ER stress and mitochondrial dysfunction contribute coordinately to the cytotoxicity. In our studies, we showed that both mitochondrial impairment and ER stress (Ren et al., 2017) are involved in leflunomide’s toxicity and that mitochondrial impairment seems closely related to ER stress. ATP depletion, a result of impaired cellular bioenergetic, primarily leads to imbalanced mitochondrial dynamics and membrane depolarization (Pathak et al., 2013; Valero, 2014) and impacts other cellular functions such as the functions of the ER, including protein folding and calcium level regulation since they are ATP dependent. One of the plausible explanations is that the effect of bioenergetic failure and mitochondrial damage caused by leflunomide exposure could disrupt normal ER function and result in ER stress. Therefore, when ATP depletion was attenuated by the MPTP inhibitor bongkrekic acid, ER stress was also reduced (Fig. 6A), while on the other hand, cells in galactose containing medium suffered more prominent ATP loss and more severe ER stress (Fig. 6C). It is needed to emphasize that major endpoint we used for our studies is ATP content. ATP decrease is considered as an indicator of mitochondrial dysfunction; however, ATP content is commonly used for general cytotoxicity evaluation. In a future study, we will use more specific assays and endpoints to study the interplay between mitochondrial impairment and ER stress.
Lastly, it is worth mentioning that a study by Latchoumycandane et al. reported a protective role of leflunomide and A77 1726 in human hepatic HC-04 cells. Treatment with leflunomide or A77 1726 protected cells from acetaminophen-induced cytotoxicity by inhibiting mitochondrial permeability transition (Latchoumycandane et al., 2006). In addition to the difference in treatment effects between our current study and the study by Latchoumycandane et al., another discrepancy is that our previous study showed that JNK was activated in response to leflunomide exposure (Ren et al., 2017), while leflunomide inhibited JNK activation induced by acetaminophen (Latchoumycandane et al., 2006). Although both of our studies and their study employed hepatic cells, the drug concentrations used were quite different. The maximal concentration of leflunomide was 30 μM to exert the protective effect in the study by Latchoumycandane et al. Their study has been performed under the condition that leflunomide itself was not toxic to the cells; while our investigations have been focused on the toxic effects of leflunomide and subtoxic and toxic concentrations were used. Different stress conditions could attribute to the biphasic effect of leflunomide; the mechanisms for these distinct effects warrant further investigation. Nonetheless, considering there is a concentration shift from the protective effect to the cytotoxic effect, precautions should be taken when using leflunomide for different purposes.
In summary, in the current study, we reported that leflunomide and its active metabolite A77 1726 caused impairment of mitochondrial function, with leflunomide showing higher potency. Our results indicate that inhibition of mitochondrial OXPHOS complexes and collapse of mitochondrial membrane potential via ANT modulation, which caused rapid ATP depletion, leading to cell injury, may be two possible mechanisms underlying the hepatotoxicity of leflunomide and A77 7126.
Acknowledgments
Z.R. was supported by appointments to the Postgraduate Research Program at the National Center for Toxicological Research administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. FDA. This article is not an official guidance or policy statement of the U.S. Food and Drug Administration (FDA). No official support or endorsement by the U.S. FDA is intended or should be inferred.
References
- Aithal GP. Hepatotoxicity related to antirheumatic drugs. Nat Rev Rheumatol. 2011;7:139–150. doi: 10.1038/nrrheum.2010.214. [DOI] [PubMed] [Google Scholar]
- Alcorn N, Saunders S, Madhok R. Benefit-risk assessment of leflunomide: an appraisal of leflunomide in rheumatoid arthritis 10 years after licensing. Drug Saf. 2009;32:1123–1134. doi: 10.2165/11316650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:850–857. doi: 10.1016/j.yjmcc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995;57:289–300. [Google Scholar]
- Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore channel formation by F-ATP synthase, integration in signal transduction and role in pathophysiology. Physiol Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohanec Grabar P, Grabnar I, Rozman B, Logar D, Tomsic M, Suput D, Trdan T, Peterlin Masic L, Mrhar A, Dolzan V. Investigation of the influence of CYP1A2 and CYP2C19 genetic polymorphism on 2-Cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide (A77 1726) pharmacokinetics in leflunomide-treated patients with rheumatoid arthritis. Drug Metab Dispos. 2009;37:2061–2068. doi: 10.1124/dmd.109.027482. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. ABBV Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan V, Charles BG, Tett SE. Population pharmacokinetics and association between A77 1726 plasma concentrations and disease activity measures following administration of leflunomide to people with rheumatoid arthritis. Br J Clin Pharmacol. 2005;60:257–264. doi: 10.1111/j.1365-2125.2005.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xuan J, Couch L, Iyer A, Wu Y, Li QZ, Guo L. Sertraline induces endoplasmic reticulum stress in hepatic cells. Toxicology. 2014;322:78–88. doi: 10.1016/j.tox.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang Z, Wu Y, Shi Q, Yan H, Mei N, Tolleson WH, Guo L. Endoplasmic reticulum stress and store-operated calcium entry contribute to usnic acid-Induced toxicity in hepatic cells. Toxicol Sci. 2015;146:116–126. doi: 10.1093/toxsci/kfv075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt. 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- del Arco A, Satrustegui J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J Biol Chem. 2004;279:24701–24713. doi: 10.1074/jbc.M401417200. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol Appl Pharmacol. 2008a;233:203–210. doi: 10.1016/j.taap.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Jamieson JD, Marroquin LD, Nadanaciva S, Xu JJ, Dunn MC, Smith AR, Will Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol Sci. 2008b;103:335–345. doi: 10.1093/toxsci/kfn056. [DOI] [PubMed] [Google Scholar]
- Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722–30730. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- Fiore C, Trezeguet V, Le Saux A, Roux P, Schwimmer C, Dianoux AC, Noel F, Lauquin GJ, Brandolin G, Vignais PV. The mitochondrial ADP/ATP carrier: structural, physiological and pathological aspects. Biochimie. 1998;80:137–150. doi: 10.1016/s0300-9084(98)80020-5. [DOI] [PubMed] [Google Scholar]
- Fox RI, Herrmann ML, Frangou CG, Wahl GM, Morris RE, Strand V, Kirschbaum BJ. Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93:198–208. doi: 10.1006/clim.1999.4777. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Goldenberg MM. Leflunomide, a novel immunomodulator for the treatment of active rheumatoid arthritis. Clin Ther. 1999;21:1837–1852. doi: 10.1016/S0149-2918(00)86732-6. (discussion 1821) [DOI] [PubMed] [Google Scholar]
- Grattagliano I, Russmann S, Diogo C, Bonfrate L, Oliveira PJ, Wang DQ, Portincasa P. Mitochondria in chronic liver disease. Curr Drug Targets. 2011;12:879–893. doi: 10.2174/138945011795528877. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2015;1850:2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Doran E, Gillespie JP, O’Toole A. Mitochondria and cell death. Biochem Soc Trans. 2000;28:170–177. doi: 10.1042/bst0280170. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The C ring of the F1Fo ATP synthase forms the mitochondrial permeability transition pore: a critical appraisal. Front Oncol. 2014;4:234. doi: 10.3389/fonc.2014.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutu DP, Guiard B, Chacinska A, Becker D, Pfanner N, Rehling P, van der Laan M. Mitochondrial protein import motor: differential role of Tim44 in the recruitment of Pam17 and J-complex to the presequence translocase. Mol Biol Cell. 2008;19:2642–2649. doi: 10.1091/mbc.E07-12-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar AS, Nguyen HT, Vaz AD, Doan A, Dalvie DK, McLeod DG, Murray JC. In vitro metabolism studies on the isoxazole ring scission in the anti-inflammatory agent lefluonomide to its active alpha-cyanoenol metabolite A771726: mechanistic similarities with the cytochrome P450-catalyzed dehydration of aldoximes. Drug Metab Dispos. 2003;31:1240–1250. doi: 10.1124/dmd.31.10.1240. [DOI] [PubMed] [Google Scholar]
- Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10396–10397. doi: 10.1073/pnas.1410104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen HI, Conaghan PG, Tett SE. Safety evaluation of leflunomide in rheumatoid arthritis. Expert Opin Drug Saf. 2013;12:581–588. doi: 10.1517/14740338.2013.798299. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kunkel G, Cannon G. Leflunomide in the treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2006;2:17–31. doi: 10.1586/1744666X.2.1.17. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann N Y Acad Sci. 2010;1201:50–57. doi: 10.1111/j.1749-6632.2010.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22:335–353. doi: 10.1111/j.1472-8206.2008.00608.x. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Seah QM, Tan RC, Sattabongkot J, Beerheide W, Boelsterli UA. Leflunomide or A77 1726 protect from acetaminophen-induced cell injury through inhibition of JNK-mediated mitochondrial permeability transition in immortalized human hepatocytes. Toxicol Appl Pharmacol. 2006;217:125–133. doi: 10.1016/j.taap.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Li J, Yao HW, Jin Y, Zhang YF, Li CY, Li YH, Xu SY. Pharmacokinetics of leflunomide in Chinese healthy volunteers. Acta Pharmacol Sin. 2002;23:551–555. [PubMed] [Google Scholar]
- Liu C, Sekine S, Ito K. Assessment of mitochondrial dysfunction-related, drug-induced hepatotoxicity in primary rat hepatocytes. Toxicol Appl Pharmacol. 2016;302:23–30. doi: 10.1016/j.taap.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Miller AE. Teriflunomide: a once-daily oral medication for the treatment of relapsing forms of multiple sclerosis. Clin Ther. 2015;37:2366–2380. doi: 10.1016/j.clinthera.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladenovic V, Domljan Z, Rozman B, Jajic I, Mihajlovic D, Dordevic J, Popovic M, Dimitrijevic M, Zivkovic M, Campion G, et al. Safety and effectiveness of leflunomide in the treatment of patients with active rheumatoid arthritis. Results of a randomized, placebo-controlled, phase II study. Arthritis Rheum. 1995;38:1595–1603. doi: 10.1002/art.1780381111. [DOI] [PubMed] [Google Scholar]
- Nadanaciva S, Will Y. Investigating mitochondrial dysfunction to increase drug safety in the pharmaceutical industry. Curr Drug Targets. 2011a;12:774–782. doi: 10.2174/138945011795528985. [DOI] [PubMed] [Google Scholar]
- Nadanaciva S, Will Y. New insights in drug-induced mitochondrial toxicity. Curr Pharm Des. 2011b;17:2100–2112. doi: 10.2174/138161211796904795. [DOI] [PubMed] [Google Scholar]
- Nadanaciva S, Bernal A, Aggeler R, Capaldi R, Will Y. Target identification of drug induced mitochondrial toxicity using immunocapture based OXPHOS activity assays. Toxicol In Vitro. 2007a;21:902–911. doi: 10.1016/j.tiv.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Nadanaciva S, Dykens JA, Bernal A, Capaldi RA, Will Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol Appl Pharmacol. 2007b;223:277–287. doi: 10.1016/j.taap.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Changes in the mitochondrial permeability transition pore in aging and age-associated diseases. Mech Ageing Dev. 2013;134:1–9. doi: 10.1016/j.mad.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Pathak D, Berthet A, Nakamura K. Energy failure: does it contribute to neurodegeneration? Ann Neurol. 2013;74:506–516. doi: 10.1002/ana.24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessayre D, Fromenty B, Berson A, Robin MA, Letteron P, Moreau R, Mansouri A. Central role of mitochondria in drug-induced liver injury. Drug Metab Rev. 2012;44:34–87. doi: 10.3109/03602532.2011.604086. [DOI] [PubMed] [Google Scholar]
- Ren Z, Chen S, Zhang J, Doshi U, Li AP, Guo L. Endoplasmic reticulum stress induction and ERK1/2 activation contribute to nefazodone-Induced toxicity in hepatic cells. Toxicol Sci. 2016;154:368–380. doi: 10.1093/toxsci/kfw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Chen S, Qing T, Xuan J, Couch L, Yu D, Ning B, Shi L, Guo L. Endoplasmic reticulum stress and MAPK signaling pathway activation underlie leflunomide-induced toxicity in HepG2Cells. Toxicology. 2017;392:11–21. doi: 10.1016/j.tox.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MP, Santos AE, Custodio JB. Mitochondria: the gateway for tamoxifen-induced liver injury. Toxicology. 2014;323:10–18. doi: 10.1016/j.tox.2014.05.009. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Juarez O, Rodriguez-Zavala JS, Moreno-Sanchez R. Multisite control of the Crabtree effect in ascites hepatoma cells. Eur J Biochem. 2001;268:2512–2519. doi: 10.1046/j.1432-1327.2001.02140.x. [DOI] [PubMed] [Google Scholar]
- Rozman B. Clinical pharmacokinetics of leflunomide. Clin Pharmacokinet. 2002;41:421–430. doi: 10.2165/00003088-200241060-00003. [DOI] [PubMed] [Google Scholar]
- Sanofi-Aventis U.S. LLC. ARAVA® (leflunomide): Prescribing Information. Bridgewater, NJ: 2016. [Google Scholar]
- Schmidt A, Schwind B, Gillich M, Brune K, Hinz B. Simultaneous determination of leflunomide and its active metabolite, A77 1726, in human plasma by high-performance liquid chromatography. Biomed Chromatogr. 2003;17:276–281. doi: 10.1002/bmc.244. [DOI] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Seah QM, New LS, Chan EC, Boelsterli UA. Oxidative bioactivation and toxicity of leflunomide in immortalized human hepatocytes and kinetics of the non-enzymatic conversion to its major metabolite, A77 1726. Drug Metab Lett. 2008;2:153–157. doi: 10.2174/187231208785425791. [DOI] [PubMed] [Google Scholar]
- Shi Q, Yang X, Greenhaw J, Salminen WF. Hepatic cytochrome P450s attenuate the cytotoxicity induced by leflunomide and its active metabolite A77 1726 in primary cultured rat hepatocytes. Toxicol Sci. 2011;122:579–586. doi: 10.1093/toxsci/kfr106. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. FDA. FDA Drug Safety Communication: New boxed warning for severe liver injury with arthritis drug Arava (leflunomide) 2010 (URL: https://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm218679.htm)
- U.S. FDA. FDA Approved Labeling Text. 2012 (URL: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf)
- Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y, Xavier R, Chung RT, Tirosh B, Shibolet O. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol. 2013;59:495–503. doi: 10.1016/j.jhep.2013.04.024. [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426–431. doi: 10.1016/j.bbrc.2016.11.088. [DOI] [PubMed] [Google Scholar]
- Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des. 2014;20:5507–5509. doi: 10.2174/138161282035140911142118. [DOI] [PubMed] [Google Scholar]
- van Roon EN, Jansen TL, van de Laar MA, Janssen M, Yska JP, Keuper R, Houtman PM, Brouwers JR. Therapeutic drug monitoring of A77 1726, the active metabolite of leflunomide: serum concentrations predict response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:569–574. doi: 10.1136/ard.2004.025205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan M, Wiedemann N, Mick DU, Guiard B, Rehling P, Pfanner N. A role for Tim21 in membrane-potential-dependent preprotein sorting in mitochondria. Curr Biol. 2006;16:2271–2276. doi: 10.1016/j.cub.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Vuda M, Kamath A. Drug induced mitochondrial dysfunction: mechanisms and adverse clinical consequences. Mitochondrion. 2016;31:63–74. doi: 10.1016/j.mito.2016.10.005. [DOI] [PubMed] [Google Scholar]
- Will Y, Dykens JA, Nadanaciva S, Hirakawa B, Jamieson J, Marroquin LD, Hynes J, Patyna S, Jessen BA. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. 2008;106:153–161. doi: 10.1093/toxsci/kfn157. [DOI] [PubMed] [Google Scholar]