

Abstract
Resident microbial communities living on amphibian skin can have significant effects on host health, yet the basic ecology of the host-microbiome relationship of many amphibian taxa is poorly understood. We characterized intraspecific variation in the skin microbiome of the salamander Ensatina eschscholtzii xanthoptica, a subspecies composed of four genetically distinct populations distributed throughout the San Francisco Bay Area and the Sierra Nevada mountains in California, USA. We found that salamanders from four geographically and genetically isolated populations harbor similar skin microbial communities, which are dominated by a common core set of bacterial taxa. Additionally, within a population, the skin microbiome does not appear to differ between salamanders of different ages or sexes. In all cases, the salamander skin microbiomes were significantly different from those of the surrounding terrestrial environment. These results suggest that the relationship between E. e. xanthoptica salamanders and their resident skin microbiomes is conserved, possibly indicating a stable mutualism between the host and microbiome.
Keywords: amphibian, microbiome, symbiosis, Ensatina eschscholtzii
Introduction
Communities of microbes living on and within other organisms play a significant role in many aspects of host life history, from development and physiology to health and behavior [1– 3]. The composition of these resident microbial communities (termed microbiomes) is influenced by many factors, including the host immune system, the environment, and interactions with nonresident microbes [2, 4–6]. However, the drivers of species assemblage and maintenance of host-associated microbiomes in natural systems remain unclear [7, 8].
Amphibian skin has recently emerged as a unique model system in which to study host-associated microbiomes. As amphibians lack protective structures found in other vertebrates such as scales, fur, or feathers, the skin plays a disproportionately large role in physiological processes such as respiration and osmotic balance [7, 9]. Additionally, microbial communities living on amphibian skin have been shown to significantly affect health and disease resistance [10, 11]. As a result, these host microbial communities are an important target for conservation applications, such as efforts aimed at mitigation of a deadly skin disease [12–14]. However, before such conservation strategies can be effectively employed, it is necessary to understand the structure and dynamics of amphibian skin microbial communities in natural systems [15].
Findings from recent studies characterizing amphibian skin microbiomes in natural systems have not been entirely congruous. For example, some studies have shown that the skin microbiome assemblage is primarily determined by host species identity [7, 8, 16], while others show that the host's habitat is a primary determinant of the skin microbiome assemblage [5, 17]. Such seemingly conflicting evidence may be due to the fact that these studies have focused on different taxa, ranging from terrestrial forest salamanders to montane aquatic frogs. To tease apart the effects of host-associated and environmental factors on the skin microbiome, focusing on microbiome variation within a species may be an effective strategy.
Studies of intraspecific variation in microbiome composition provide a model that minimizes differences in physiology, behavior, and ecology which are present in any study of multiple host species. Comparisons across host populations that are similar in all regards except geographic distribution can test whether factors like habitat and environmental microbes influence the skin microbiome. Such an approach can thus provide insight into how conserved the host-microbiome relationship is across the host range.
In addition, intraspecific studies can allow researchers to explore variation in skin microbiome composition across host traits such as sex and age. While sex-specific microbiome variation has not yet been investigated in amphibians, such variation has been observed in the gut and skin of humans and mice [18–20]. It is possible that sex-based differences in hormone and pheromone production present in amphibians may impact the composition of the amphibian skin microbiome as well [21–23]. The effects of age on the amphibian microbiome have been more thoroughly studied, but research has primarily focused on species with aquatic larval stages. In several such species, results indicate a shift in skin microbiome composition between larval and adult life stages [7, 24–26]. To our knowledge, only one study has investigated a direct-developing amphibian (the frog Eleutherodactylus coqui), and it showed conflicting results: some analyses suggested that the microbiome differed between adults and juveniles, but other analyses showed no difference [27]. Thus it remains unclear whether the microbiome shifts through growth and development in direct developing species.
In this study, we investigated intraspecific variation in the skin microbiome of the yellow-eyed ensatina (Ensatina eschscholtzii xanthoptica Gray 1850), a terrestrial direct-developing plethodontid salamander. This subspecies is composed of four genetically and geographically distinct populations distributed in the San Francisco Bay Area and the Sierra Nevada mountains [28]. We used 16S amplicon sequencing to characterize and compare skin microbial communities between populations, age classes, and sexes to explore factors that may be driving skin microbiome assemblage in this salamander.
Our study goals were threefold. First, we tested whether individuals from different populations within this subspecies harbor microbiomes that are similar or distinct from one another. Second, we tested whether the microbiome varies significantly between demographic groups, specifically age classes (juvenile or adult) and sexes. Lastly, we compared skin microbiome composition to that of the surrounding environment (soil) to determine whether salamanders harbor a microbial community that is significantly different from that of their habitat.
Methods
Field sampling
The subspecies Ensatina eschscholtzii xanthoptica is found throughout the San Francisco Bay Area and the Sierra Nevada mountains in California, USA. Individuals can be found under fallen logs and other debris in forested areas [29]. The subspecies is composed of four genetically and geographically distinct populations: North Bay, East Bay, South Bay, and Sierra Nevada (Fig. 1) [28]. For this study, we sampled 10-11 adult salamanders from each of the four populations. We sampled salamanders opportunistically, and distances between salamanders sampled varied. It is therefore possible that some individuals in a population are more closely related than others; however, we did not perform genetic analysis of the host salamanders, so we are unable to determine relatedness.
Fig. 1.
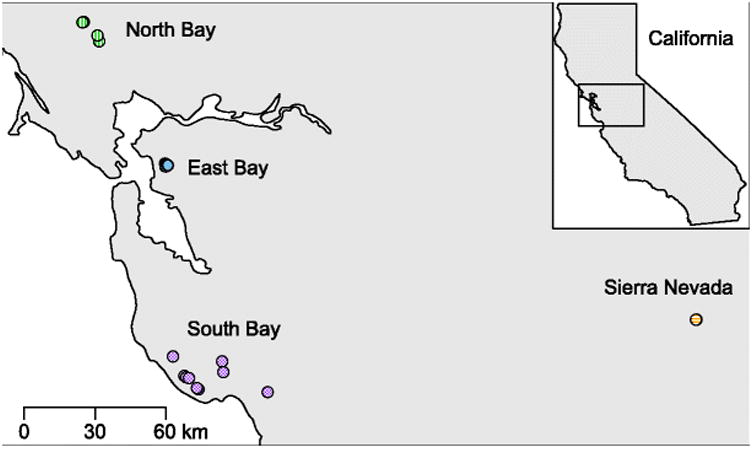
Map of sampling locations. Each point represents an individual salamander sampled; many overlap due to close proximity (East Bay n=11, North Bay n=10, Sierra Nevada n=11, South Bay n=10). Colors correspond to populations.
To compare differences between sexes and age classes, we expanded our sampling of the North Bay population to include equal numbers of males, females, and juveniles (n=10, n=11, n=10 respectively). We chose to use the North Bay population for this comparison because it had the highest salamander abundance; we were unable to find sufficient numbers for age and sex comparisons in the other populations. Nonetheless, we have no reason to expect patterns to differ between populations.
To explore the effect of environmental microbes on skin microbiota, we also analyzed five soil samples from each population (20 soil samples total). A total of 64 salamander skin swabs and 20 soil samples were used in this study.
Due to drought conditions, populations were sampled at different times of the year as follows: East Bay: March 2014, South Bay: April-May and September 2014, North Bay: December 2014; Sierra Nevada: March 2015.
Salamanders were captured by hand using new nitrile gloves, rinsed with 18 MΩ/cm MilliQ water (25ml for juveniles, 50ml for adults), and swabbed using a sterile synthetic cotton swab, stroking 30 times (10 ventral, 10 dorsal, 5 each side). Soil samples were collected from the soil surface directly under the cover object (e.g. rock or log) of each salamander within approximately 15 cm of an individual's point of capture. Soil was collected in sterile 1.5 mL Eppendorf tubes. Approximately 0.25g of each soil sample was used for DNA extraction. Animals were released within approximately 30 minutes of capture to their original cover objects. Swabs and soil samples were immediately placed on dry ice and then frozen at -80°C until DNA extraction.
Microbiome DNA extraction, amplification, and sequencing
Total genomic DNA from skin swabs and soil samples was extracted using the PowerSoil DNA Isolation Kit according to the manufacturer protocol (MoBio Laboratories, Carlsbad, CA, USA). Five soil samples were randomly chosen from each population for microbiome analysis. We PCR-amplified the V3-V4 region of the bacterial 16S rRNA gene using Illumina primers (see Supplementary Table 1) according to the recommended Illumina protocol, with a slight modification to maximize PCR product (specifically, we increased the number of cycles for Index PCR from 8 to 12) (Illumina Inc, San Diego, CA, USA). Samples were individually barcoded during the index PCR step using sample-specific pairs of dual-indexing primers from the Illumina Nextera Index Kit (see Supplementary Table 1). All samples were run in triplicate to limit PCR bias and produce sufficient quantities of PCR product. Amplified product was quantified using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA, USA). Samples were then pooled at equimolar concentrations and run on the Illumina MiSeq at the San Francisco State University Genomics and Transcriptomics Analysis Core. 16S amplicon libraries are inherently low-diversity, so we added 25% PhiX to the solution before loading the MiSeq to ensure proper cluster density. We sequenced 300bp paired-end reads, which were demultiplexed to produce final reads of ∼460bp for the region of interest.
Bioinformatics and Statistical Analysis
The bioinformatics pipeline QIIME [30] was used for all sequence analyses unless otherwise stated. Sequences were demultiplexed and quality filtered using default protocol (q20 threshold), and the resulting reads were clustered into operational taxonomic units (OTUs) based on 97% similarity, using the cluster seed as the reference sequence. OTUs were then assigned taxonomy using the Greengenes reference database (Greengenes Database Consortium, 2015). For sequences that did not match the reference database, OTUs were clustered de novo, and the centroid of the cluster was chosen as the representative sequence. Any OTUs that were not identified as bacterial were removed. OTUs present in only one sample overall or with less than 100 reads were excluded from our analysis (0.05% threshold) [31].
We used three separate approaches to address our three experimental goals. First, to compare microbiome variation between salamander populations, we analyzed alpha and beta diversity measures across our four populations (n=42). For the North Bay, we randomly selected 5 male and 5 female adults for this analysis. Second, to test for microbiome differences between sexes and age groups, we analyzed microbial diversity within the North Bay population (n=31). Third, to test whether the microbiome of salamanders is significantly distinct from that of their habitat (soil), we compared salamander samples from each population (n=42) to five soil samples from each population (n=20). We rarefied the number of reads per sample for each analysis separately (populations: 31,025; sexes/ages: 24,155; salamanders/soil: 33,855). We also describe a core E. e. xanthoptica microbiome of bacterial OTUs that appeared in 90% or more of salamander samples. For each analysis, all samples were rarefied to include the same number of sequences to ensure equivalent sampling throughout. Patterns of diversity were then analyzed using the core_diversity_analyses.py script in QIIME [30]. The following alpha diversity metrics were calculated for all samples: OTU richness, Chao1, Faith's phylogenetic diversity (PD), and McIntosh evenness. We assessed whether alpha-diversity metrics were normally distributed using Shapiro-Wilk tests. Alpha diversity was compared among groups using analysis of variance (ANOVA) or Kruskal-Wallis tests in R (R Core Team, 2015). Beta diversity was calculated using weighted and unweighted UniFrac distances and Bray-Curtis dissimilarity in QIIME. Bacterial community composition between groups was compared using adonis in QIIME and plotted using principal coordinates analysis (PCoA) in R (R Core Team, 2015). We identified a core microbiome of OTUs that were present on at least 90% of all salamanders sampled.
We used the Antifungal Isolates Database developed by Woodhams et al. (2015) to determine whether any OTUs present in the core microbiome matched bacterial species that have previously been isolated from amphibian skin and shown to have anti-fungal properties. To do so, we filtered our list of core microbiome OTUs by OTU ID to include only those that matched OTUs in the Woodhams (2015) database.
Results
Sequencing
Sequencing resulted in 11,512,105 reads representing 203,784 OTUs, most of which were represented by a single sequence. After filtering of all non-bacterial and low-abundance reads, the final dataset included 4,912,877 reads representing 4398 OTUs across a total of 64 salamander and 20 soil samples.
Alpha Diversity
Our survey of 64 salamanders shows a large degree of variation in skin microbiome diversity across individuals. The number of OTUs (OTU richness) on individual salamanders ranged from 427 to 2,423, while measures of McIntosh evenness ranged from 0.06 (community dominated by few taxa) to 0.94 (highly even community).
Measures of alpha diversity varied somewhat between populations, with East Bay generally exhibiting higher values than the other three populations. OTU richness, phylogenetic diversity (PD), and Chao1 were all normally distributed (Shapiro-Wilk p>0.05), and McIntosh evenness was not (Shapiro-Wilk p<0.001). Analyses of variance (ANOVA) showed significant differences between the four populations in three measures of alpha diversity (OTU richness: F=7.03, df=3, p<0.001; Chao1: F=5.897, df=3, p=0.002; PD: F=5.658, df=3, p=0.003). However, all four populations had similarly values of McIntosh evenness (Kruskal-Wallis p=0.868). Tukey post-hoc tests revealed that OTU richness was higher in the East Bay than all other populations (North Bay: p=0.044; Sierra Nevada: p<0.001; South Bay: p=0.011). The East Bay population also had higher values of Chao1 and PD than the South Bay (Chao1: p=0.018; PD: p=0.024) and Sierra Nevada populations (Chao1: p=0.002; PD: p=0.002), but the North Bay population was not different (Chao1: p=0.112; PD: p=0.080).
Within the North Bay population, Student's t-test showed that all alpha diversity metrics were similar between sexes (OTU richness: t(17.48)=0.394, p=0.698; Chao1: t(16.64)=0.514, p=0.614; PD: t(18.4)=0.045, p=0.965; McIntosh evenness: t(13.53)=-1.878, p=0.082). Three of the four alpha diversity metrics were also similar between adults and juveniles (OTU richness: t(19.83)=1.753, p=0.096; PD: t(17.19)=1.2875, p=0.215; McIntosh evenness: t(25.06)=-0.817, p=0.421), but adults had higher values of Chao1 than juveniles (t(20.32)=2.238, p=0.036).
Community composition (beta diversity)
Using both unweighted UniFrac and Bray-Curtis dissimilarity metrics, we found that skin microbial communities differed between the four sampled populations (Fig. 2a, adonis unweighted UniFrac p<0.001, R2=0.16547; Bray-Curtis p<0.001, R2=0.19448). Individuals from the North Bay and Sierra Nevada populations harbored similar skin microbiomes to one another, while those of the East Bay population were significantly different. Individuals in the South Bay population showed variable microbiome community compositions; some were similar to the East Bay, and some were similar to the North Bay/ Sierra Nevada group (Fig 2a). In contrast, the weighted UniFrac analyses showed no significant differences between any of the four populations (Fig 2b, adonis p=0.258).
Fig. 2.
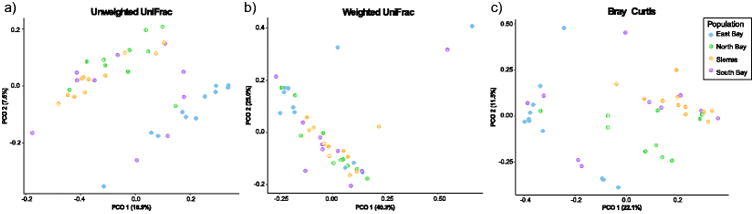
Principal coordinates plots showing patterns of beta diversity across populations. Each point represents the microbiome of an individual salamander. Using a) unweighted UniFrac and b) Bray-Curtis, the skin microbial communities of the different populations are significantly distinct from one another. Using c) weighted UniFrac, the skin microbiomes of the four populations are indistinguishable.
Each population harbored a small set of bacterial taxa that were absent from the other populations, but these unique OTUs were all present at very low abundance (<0.01%, Supplemental Table 3). The East Bay had the most unique OTUs (112), while the other populations had few unique OTUs (14-20). None of these population-specific OTUs matched to any known amphibian mutualists present in the Woodhams et al. (2015) database of bacteria with anti-fungal properties.
For the within-population analyses (North Bay), we found no difference in microbiome composition between males and females (adonis unweighted UniFrac p=0.492; weighted UniFrac p=0.649; Bray-Curtis 0.575). However, two of our diversity metrics revealed slight differences between adults and juveniles (adonis unweighted UniFrac p=0.084 R2=0.04409, Bray-Curtis 0.047 R2=0.06281), while the third did not (weighted UniFrac p=0.451) (Fig. 3).
Fig. 3.
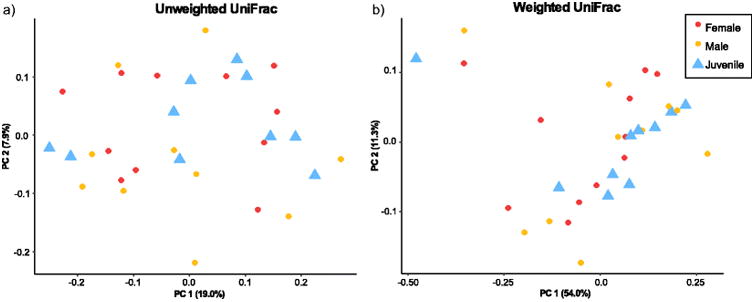
Principal coordinates showing patterns of beta diversity between life history stages. a) Weighted UniFrac and b) Unweighted UniFrac. Unweighted UniFrac and Bray-Curtis analyses showed similar patterns, so Bray-Curtis are not shown.
In all populations, salamander skin microbiomes were highly distinct from microbial communities present in the surrounding habitat (soil) samples (adonis unweighted UniFrac p<0.001, R2=0.10499; weighted UniFrac p<0.001, R2=0.32; Bray-Curtis p<0.001, R2=0.14331) (Fig 4). A number of OTUs were unique to either the soil or salamander datasets, but most were present in both soil and salamander samples (83% of OTUs identified).
Fig. 4.
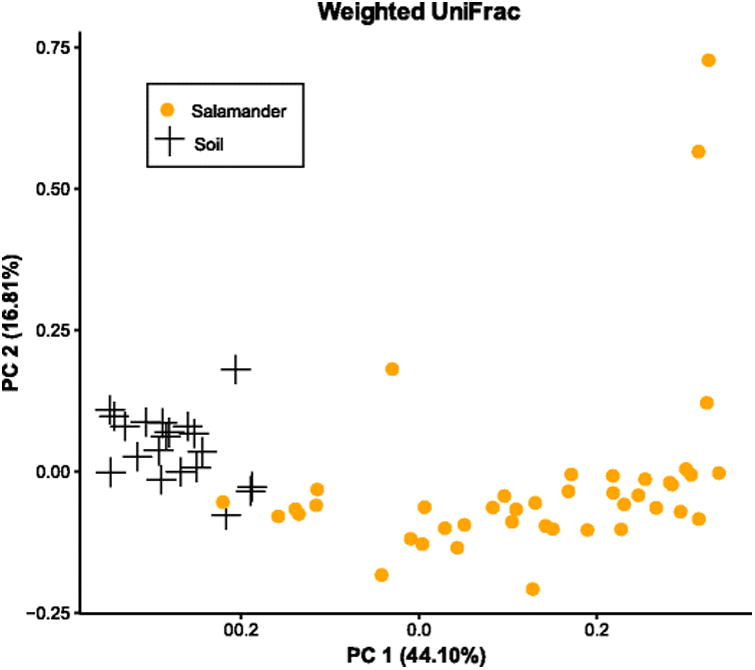
Principal coordinates showing patterns of beta diversity between salamanders and habitat samples (soil). Unweighted UniFrac, weighted UniFrac, and Bray-Curtis analyses provided similar patterns, so only weighted are shown. Soil samples were taken beneath logs where salamanders were found. Salamander skin microbial communities were significantly different from soil microbial communities.
Core microbiome
We identified a core community of 90 OTUs that were present in 90% or more of our salamander samples (Table 1). The core community dominated the skin microbiomes of most individuals, with an average relative abundance of 0.44 (minimum 0.03, maximum 0.82). Two individuals with low core community abundance (0.03 and 0.185, respectively) were dominated by OTUs of the family Chlamydiaceae.
Table 1.
Relative abundances of core OTUs for each population and life history stage. Average relative abundances of the 15 most abundant core taxa in order from highest to lowest abundance. The most abundant OTUs belonged to the genus Pseudomonas.
| Population | Life History Group | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| OTU_IDs | Taxonomy | East Bay | North Bay | Sierras | South Bay | Female | Male | Juvenile | Total |
| 295031, 333782, 750018, 106985*, 1566691*, 552449, 831401*, 277094*, 633252, 589597*, 541859*, 646549, 398604, 108909, 280459, 350105, 202466*, 780555, 669356, 4308187, 284090, 795272, 287032, 256834*, 561294, 279231, 817734, 813617, 142419, 764682, 563273, 560886, 143131, 557974*, 961783*, New.ReferenceOTU5461, New.ReferenceOTU2542, New.ReferenceOTU5608, New.ReferenceOTU152, New.ReferenceOTU3451, New.CleanUp.ReferenceOTU4174133 | Pseudomonas spp. | 0.214503 | 0.350475 | 0.322958 | 0.238365 | 0.360858 | 0.338676 | 0.393682 | 0.317074 |
| 4378239, 624310, 1090029, 732609, New.ReferenceOTU1497 | Sanguibacter spp. | 0.018133 | 0.044374 | 0.055253 | 0.05018 | 0.035045 | 0.036346 | 0.060483 | 0.042831 |
| 537279, 160115*, 544847 | Family Xanthomonadaceae | 0.034513 | 0.039326 | 0.025951 | 0.027777 | 0.030439 | 0.034771 | 0.052622 | 0.035057 |
| 338140, 544177, 829851*, 818602* | Family Pseudomonadaceae | 0.003024 | 0.015187 | 0.01669 | 0.008952 | 0.012721 | 0.015384 | 0.027199 | 0.014165 |
| 580625, 1105814 | Family Bradyrhizobiaceae | 0.007129 | 0.008705 | 0.008677 | 0.008268 | 0.010103 | 0.00941 | 0.00559 | 0.008269 |
| 884655, 1052559, 965129, 1003206 | Family Enterobacteriaceae | 0.00195 | 0.016151 | 0.006479 | 0.003776 | 0.00913 | 0.013283 | 0.003391 | 0.007737 |
| 1110763, 759061, New.ReferenceOTU3114 | Sphingomonas spp. | 0.012255 | 0.007255 | 0.00447 | 0.007523 | 0.007555 | 0.008393 | 0.006429 | 0.007697 |
| 459107*, 348790, 814909, 1108350 | Microbacterium spp. | 0.004407 | 0.006815 | 0.00197 | 0.00415 | 0.005389 | 0.005936 | 0.008436 | 0.0053 |
| 281220, 196652, New.ReferenceOTU2127 | Burkholderia spp. | 0.006198 | 0.002566 | 0.00769 | 0.002298 | 0.005364 | 0.002915 | 0.002531 | 0.004223 |
| 546342, 848824 | Family Sphingobacteriaceae | 0.003176 | 0.003793 | 0.004861 | 0.001178 | 0.005711 | 0.004923 | 0.004803 | 0.004064 |
| 533999*, 250626 | Luteibacter rhizovicinus | 0.004508 | 0.002413 | 0.00411 | 0.003469 | 0.005083 | 0.003806 | 0.002578 | 0.00371 |
| 876170, 279325, 1074625 | Family Microbacteriaceae | 0.006308 | 0.002527 | 0.003386 | 0.004333 | 0.003649 | 0.002999 | 0.0025 | 0.003672 |
| 391286 | Stenotrophomonas spp. | 0.000102 | 0.001893 | 0.006765 | 0.005451 | 0.001185 | 0.00115 | 0.001649 | 0.002599 |
| 1088265 | Erwinia spp. | 0.000729 | 0.002187 | 0.000463 | 0.002115 | 0.001834 | 0.008645 | 0.000482 | 0.00235 |
| 581021 | Propionibacterium acnes | 0.004875 | 0.002233 | 0.001343 | 0.004118 | 0.002147 | 0.000796 | 0.00043 | 0.002277 |
indicates an OTU with previously-documented antifungal properties. Complete taxonomy and remaining OTUs are presented in Supplementary Table 2.
Core OTUs were identified as belonging to the phyla Acidobacteria, Actinobacteria, Bacteroidetes, and Proteobacteria. The majority of core OTUs are members of the genus Pseudomonas, which account for an average relative abundance of 0.31 per salamander. One core OTU was identified as a member of the genus Stenotrophomonas, which has been shown to reside on amphibian skin and exhibit anti-fungal activity as well [33, 34].
Core community abundance did not differ significantly between populations (ANOVA p=0.1656). Fifteen of the 90 core OTUs we identified matched bacterial isolates from the Antifungal Isolates Database [32]. Of these 15 OTUs, ten were identified as Pseudomonas species and two more were unidentified species in the family Pseudomonaceae, one was a Microbacterium species, and two were in the family Xanthomonadaceae (including Luteibacter rhizovicinus) (Table 1). Together, the 15 presumptive anti-fungal OTUs made up an average of 21.3% percent of the core microbiome across all individuals. Abundances of these OTUs were similar across populations (Kruskal-Wallis test p=0.6713).
Discussion
In this study, we examined variation in skin microbiome community of the terrestrial salamander Ensatina eschscholztii xanthoptica, a subspecies composed of four genetically and geographically distinct populations [35]. We characterized variation between populations, ages, and sexes and compared salamanders to their habitats (soil) to determine if any of these factors are correlated with skin microbiome composition in this subspecies. We found that overall, individuals from the four populations of E. e. xanthoptica exhibit similar skin microbial communities, although each population also harbors a set of unique bacterial OTUs. Using two diversity metrics (unweighted UniFrac and Bray-Curtis), we found that the different salamander populations exhibited different microbiomes, but using the weighted UniFrac metric, these patterns disappeared (Fig. 2). Bray-Curtis and unweighted UniFrac distances are based on presence-absence of bacterial OTUs in each sample, while weighted UniFrac distances incorporate presence-absence as well as abundance of bacterial OTUs in each sample. Our results therefore suggest that the most abundant OTUs were similar across populations, but each population also harbored its own characteristic community, including a set of unique OTUs that were present at low abundances (<0.01%), the roles of which remain unknown (Supplemental Table 3). Microbiome communities have been shown to be highly dynamic, shifting composition between seasons, weeks, and even days [30, 36, 37]. It is thus possible for low abundance taxa to become abundant in response to changing environmental conditions or provide important functions even at low densities [38]. In the present study, it is unclear what role these rare or unique bacteria are playing in each population because none of the unique OTUs matched known amphibian mutualists.
Taken together, our analyses suggest that the dominant microbial taxa are relatively conserved across the four populations, but the overall communities are nonetheless distinct. The overall similarity of the dominant microbes between the four sampled populations is striking, and suggests that these taxa may play a role in host processes. These populations are separated by large distances (60 – 250 km), and have presumably been isolated from one another for hundreds to thousands of years [28]. If the skin microbiome was not tightly linked to host processes, we would expect to see differences arising over time in disjunct populations [39], as well as a higher degree of colonization from environmental microbes – yet we found the opposite. In fact, the two salamander populations that had the most similar microbiomes were those that were furthest apart geographically – the North Bay and Sierra Nevada populations (Fig. 2a). Such a high level of similarity between the resident skin microbial communities in these populations suggests that the host-microbiome relationship has been conserved.
By contrast, the results of diversity-based analysis (Bray-Curtis and unweighted UniFrac) that showed differences in microbiome community across populations suggest that the patterns of similarity in dominant microbes do not fully capture the microbiome community dynamics. The microbiome communities of different populations are significantly different from one another (Fig 2b,c), likely reflecting a combination of factors. Each population differs from the others in environment, host evolution/genetic divergence, and stochastic processes, each of which could impact the microbiome community. For example, due to limitations caused by the significant drought in California throughout our sampling, individuals were found in different times of the year, and experienced different climates, which might have influenced their microbiome communities. In addition, sampling for some populations was more concentrated geographically than other populations (Fig 1). However, microbiome composition does not appear to be related to geographic spread of the individuals sampled; for example, the East Bay was sampled over a relatively small geographic area, but had the highest OTU richness.
Such differences between populations could instead be potentially attributed to habitat differences. While all individuals were found in wooded areas, the vegetative communities were nonetheless different across populations, with East Bay individuals found primarily in suburban oak forest, South Bay populations in redwood forest, and North Bay and Sierra Nevada populations in mixed forests. While the limitations of our data set do not allow us to distinguish between these factors potentially impacting microbial community assemblage, it is nonetheless clear that microbial communities vary across populations, but are still dominated by similar taxa, reflecting a combination of conservation of the host-microbiome relationship and variation in microbiome diversity across conspecific populations separated by significant geographic and temporal distances. These data therefore suggest that both host and environmental processes play a role in the maintenance of microbiome communities in this terrestrial salamander.
Core microbiome
Identifying a core bacterial community is a key step towards understanding the relationship between host and microbiome. Core community taxa are shared across all members of the host group, and are thought to perform ecological and physiological functions that influence host health and physiology [40].
We identified a core microbial community of 90 OTUs that were present in the 90% or more of our salamander samples (Table 1). The most abundant core OTUs belonged to the genus Pseudomonas, an ecologically diverse genus whose members include common species found in the environment, pathogens, and mutualists [41]. Pseudomonas is also one of the most common bacterial taxa in skin microbiomes of other amphibian species (e.g. [7, 42, 43]). Several other core OTUs in our dataset have also been found in other amphibian skin microbiomes, including Sanguibacter and Microbacterium [16, 41, 44], Sphingomonas [45], Luteibacter rhizovicinus [46], Burkholderia [42, 47], and members of the family Xanthomonadaceae (e.g. [24, 48, 49]). In addition, several of the core OTUs in our dataset are known from non-amphibian environments, such as soil and freshwater (Achromobacter, Xanthomonadaceae [50]) or plants and rhizospheres (Luteibacter rhizovicinus; [51]. Others are extremely widespread, and have been found in a variety of environments (e.g. Bradyrhizobiaceae, Enterobacteriaceae, and Sphingobacteriaceae, Burkholderia, Stenotrophomonas) [50]. It is unclear whether these bacterial taxa have any direct effects on the host or are simply commensal species that colonize amphibian skin. In either case, their prevalence and abundance across the salamanders in our study suggest that they are important members of the skin microbiome community.
In addition to common environmental bacteria, we also identified a number of bacterial taxa in the core community that may be amphibian mutualists. For example, previous work has shown that many Pseudomonas species present on amphibian skin exhibit antifungal properties, and likely aid in host defense against fungal pathogens [10, 12, 34, 52, 53]. Twelve Pseudomonas OTUs in our dataset matched bacteria that have previously been isolated from amphibian skin and tested for antifungal activity [32]. We also found one Microbacterium OTU and two OTUs in the family Xanthomonadaceae that matched anti-fungal isolates. We conclude that these taxa are therefore likely to be amphibian mutualists, aiding the host in combatting fungal pathogens such as Batrachochytrium dendrobatidis, the globally distributed agent of the disease chytridiomycosis [54]. The rest of the OTUs present in the core community did not match known anti-fungal species, but their prevalence and abundance in the majority of our samples suggests that they are likely selected for on salamander skin, forming either mutualistic or stable commensal relationships with the amphibian host. Even if these core bacterial taxa do not actively produce anti-fungal compounds on amphibian skin, they may nonetheless be playing a role in host health via alternative ecological mechanisms. For example, diverse communities are often better able to resist pathogens due to the dilution effect, in which the spread and maintenance of pathogens is inhibited by the diverse immune mechanisms and physiological properties of diverse hosts [55]. In addition, OTUs may generally be under negative-frequency-dependent selection, such that OTUs reaching high abundance are easily attacked by pathogens. In such a scenario, diverse communities would be more resistant to pathogens than low-diversity communities dominated by few species [56]. Thus, the diversity within the core community may aid in host pathogen resistance via any (or all) of the above mechanisms.
However, these results should be interpreted with caution. As described above, sequences were assigned to OTUs based on 97% similarity, but bacterial taxa with 97% or more similarity may exhibit different functional and ecological traits [57]. Thus, while sequences in our dataset did match sequences of previously-identified anti-fungal bacteria, it does not necessarily mean the bacteria we sampled have the same capabilities. Conversely, bacteria that were not previously known to be anti-fungal may nonetheless have beneficial properties or functions for the host that have yet to be discovered [58]. Because neither Ensatina nor any other plethodontid salamander from the western United States has been surveyed before, it is possible that OTUs that did not match the anti-fungal database (for example, the OTUs we identified as unique to individual populations) are local mutualists that have not previously been detected. Further research into geographic variation in community composition and assessment of antifungal properties would shed light on this possibility.
While the skin microbiomes of most salamanders in the study were dominated by members of the core community, two individuals with low core community abundance were instead dominated by OTUs of the Chlamydiaceae family. To date, all known members of Chlamydiaceae are intracellular parasites, and are known to cause disease in many animals [59]. Yet Chlamydia has rarely been recorded in amphibians, and its pathogenicity is unclear [60]. In any case, such low core community abundance and dominance of Chlamydiaceae may be an example of dysbiosis, or disruption of a normal, healthy microbiome. Microbial dysbiosis is often associated with poor health or disease [41, 61], although it is not often clear whether dysbiosis leads to disease or is instead a symptom. Alternatively, it is possible that the Chlamydiaceae OTUs we observed represent minimally- or non-pathogen variants that have not previously been documented. The individuals in our study that were dominated by Chlamydiaceae did not show any recognizable signs of disease, but observation time was limited (30min. per individual). In any case, we believe it is important to further explore the prevalence and effects of bacteria in the family Chlamydiaceae on amphibians to determine whether it is a potential threat to amphibian health.
Variation across life history stages
Several studies have shown that skin microbiome composition shifts significantly during the transition from larval stage to adult in amphibians. It is hypothesized that such changes in the microbial community are caused by significant host physiological changes that coincide with a shift from aquatic to terrestrial environments, which themselves harbor significantly different microbial communities [7, 62]. Thus changes in host physiology, environment, or both may contribute to shifts in skin microbiome composition, but it is not easy to tease these effects apart. E. e. xanthoptica is a direct-developing species, and young emerge from the egg as terrestrial hatchlings, bypassing the larval stage [29]. Additionally, hatchlings live in the same habitats as adults. We therefore believe that this species provides an ideal system in which to test the influence of host age on skin microbiome composition, independent of significant metamorphic or ecological changes.
To that end, we examined variation between life history stages within one of our study populations (North Bay, Fig. 1) by comparing the skin microbial communities of equal numbers of juveniles, adult males, and adult females. Two of our community metrics showed that skin microbiomes of juveniles and adults were slightly different from one another (unweighted UniFrac and Bray-Curtis), while one (weighted UniFrac) did not. (Fig. 3). The results of the weighted UniFrac analysis suggest that the dominant members of the skin microbiome community establish early in a salamander's life, and are maintained into adulthood. The results of the unweighted UniFrac and Bray-Curtis analyses, which show slight but significant differences in composition between juveniles and adults, suggest that this process is not immediate, and that either individuals may take time to achieve an optimal adult microbiome community, or the optimal community may be different for juveniles and adults. In addition, the difference between adults and juveniles in our data is much more subtle than those shown by previous studies on metamorphic amphibians [7, 62], which suggests that the temporal stability of the amphibian skin microbiome may vary based on the host's life history strategy (metamorphic vs. direct developing).
We also expected the two sexes to harbor different microbiomes, reflecting sex-based differences in physiology and behavior ([63, 64]). Research in other taxa has shown that the sex of the host may have a strong influence on microbiome composition [21–23], but to our knowledge, no study has yet investigated this in amphibians. In contrast to studies of other taxa, our data showed that male and female salamanders harbor similar microbiomes (Fig. 3), suggesting that host sex is not a determining factor for microbiome community composition in this species.
The similarity of skin microbiome composition across ages and sexes may have several explanations. For example, all individual salamanders within a population occupy similar habitats regardless of age or sex, and are thus exposed to similar environmental conditions, potentially leading to similar skin microbiomes. Additionally, the similarity may be attributable to horizontal or vertical transmission of microbes [42, 65, 66]. Ensatina salamanders are known for their elaborate courtship behaviors, in which males and females engage in prolonged physical contact [29], which may serve to transfer and equalize microbial communities between the sexes. In addition, Ensatina also exhibit maternal care, in which the mother guards their eggs and hatchlings, maintaining nearly constant physical contact for at least several months [29]. This maternal contact may facilitate transfer of microbes to hatchlings, thus establishing the microbiome early in life [42, 67]. In either case, the similarity of microbiomes between ages and sexes suggests that a similar skin microbiome is cultivated and maintained throughout sexes and life stages, supporting the conclusion that the host-microbiome relationship is well conserved.
Impact of environmental microbes
Our final goal was to determine whether salamander skin cultivates a specific microbiome, or is primarily colonized by microbes present in a salamander's habitat. In all four populations, salamander skin microbiomes were distinct from soil microbiomes (Fig. 4), but many of the OTUs present in these communities were not unique to one group or the other. Of the OTUs that were common to both groups, those that were highly abundant on salamander skin were not abundant in the soil, and vice versa (Fig. 5). In agreement with previous studies [5, 7, 17, 58], this pattern suggests that while these different environments (soil and salamander skin) likely exchange bacteria, they each cultivate distinct community compositions.
Fig. 5.
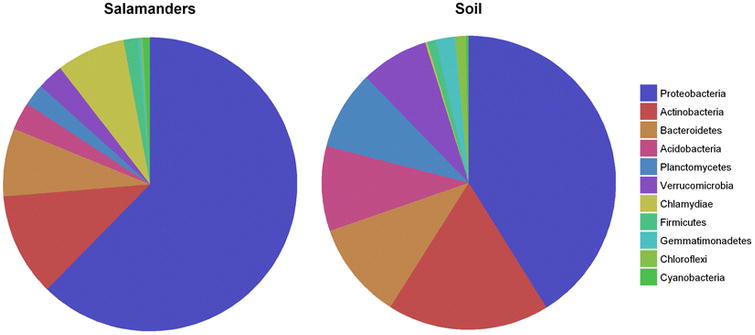
Dominant bacterial phyla present on salamanders and in soil.
Interestingly, we also observed a number of bacterial OTUs that were only present on salamander skin, and were absent from the soil. There are several potential explanations for this finding. First, these OTUs may be environmental microbes that are either present in the soil at extremely low levels such that we were unable to detect them, or they may originate from other areas of the salamander habitat that we did not sample, such as leaf litter or bark surfaces [16]. Alternatively, these OTUs may have coevolved with E. e. xanthoptica, being maintained through generations by horizontal or vertical transmission, thus originating not from the environment but from other salamanders [42, 68]. In either case, it is clear that the salamander skin microbiome does not simply mirror available soil microbes. Rather, it is likely that salamander skin selects for a specific bacterial community composed of environmentally available taxa as well as a possible suite of salamander-specific bacterial lineages.
Conclusions
Our study shows that individuals of four geographically and genetically distinct salamander populations harbor skin microbiomes that are dominated by a common core set of bacterial OTUs, but are nonetheless distinct from one another. Additionally, the skin microbiome appears to shift slightly over age classes, but does not differ between sexes. These results suggest that the relationship between E. e. xanthoptica salamanders and their resident skin microbiomes is significantly influenced by both host and environmental processes.
Supplementary Material
Online Resource 1. Supplementary Table 1. Sequences of Illumina primers used for amplicon sequencing.
Online Resource 2. Supplementary Table 2. Complete list of core bacterial OTUs and average abundances by sample group. (*) indicates an OTU with previously-documented antifungal properties.
Online Resource 3. Supplementary Table 3. List of bacterial OTUs unique to one population. All unique OTUs were present at < 0.01% abundance. The East Bay population had the most unique OTUs.
Acknowledgments
The authors would like to thank S. Ellison for his great help with lab work and troubleshooting. We would also like to thank F. Cipriano and A. Swei for advice on methodology. J. de la Torre provided valuable advice as a member of SPI's Master's thesis committee. SPI would also like to thank her classmates from OEB210 for valuable feedback on the manuscript. We would also like to thank four anonymous reviewers whose comments greatly improved the manuscript. The National Science Foundation provided funding through a research grant (IOS-1258133) awarded to AGZ and VTV as well as a GRFP (DGE-1144152) awarded to SPI. The National Institutes of Health also provided financial support through MBRS-RISE fellowships awarded to SPI and AKB (R25-GM059298).
Footnotes
Ethical approval: All procedures performed with live animals in this study were approved by the Institutional Animal Care and Use Committee at San Francisco State University (Protocol #A12-07). All sampling of wild salamanders was performed with approval from the California Department of Fish and Wildlife (SC-12920) and California State Parks.
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.McFall-Ngai M, Hadfield MG, Bosch TCG, et al. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A. 2013;110:3229–36. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279–90. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg E, Zilber-Rosenberg I. Symbiosis and development: The hologenome concept. Birth Defects Res Part C - Embryo Today Rev. 2011;93:56–66. doi: 10.1002/bdrc.20196. [DOI] [PubMed] [Google Scholar]
- 4.Naik S, Bouladoux N, Wilhelm C, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–9. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loudon AH, Woodhams DC, Parfrey LW, et al. Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus) ISME J. 2014;8:830–40. doi: 10.1038/ismej.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Küng D, Bigler L, Davis LR, et al. Stability of microbiota facilitated by host immune regulation: informing probiotic strategies to manage amphibian disease. PLoS One. 2014;9:e87101. doi: 10.1371/journal.pone.0087101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kueneman JG, Parfrey LW, Woodhams DC, et al. The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol. 2013;23:1238–1250. doi: 10.1111/mec.12510. [DOI] [PubMed] [Google Scholar]
- 8.McKenzie VJ, Bowers RM, Fierer N, et al. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 2012;6:588–96. doi: 10.1038/ismej.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wardziak T, Luquet E, Plenet S, et al. Impact of both desiccation and exposure to an emergent skin pathogen on transepidermal water exchange in the palmate newt Lissotriton helveticus. Dis Aquat Organ. 2013;104:215–24. doi: 10.3354/dao02602. [DOI] [PubMed] [Google Scholar]
- 10.Harris RN, James TY, Lauer A, et al. Amphibian Pathogen Batrachochytrium dendrobatidis Is Inhibited by the Cutaneous Bacteria of Amphibian Species. Ecohealth. 2006;3:53–56. [Google Scholar]
- 11.Becker MH, Harris RN. Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS One. 2010;5:e10957. doi: 10.1371/journal.pone.0010957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loudon A, Holland J. Interactions between amphibians' symbiotic bacteria cause the production of emergent anti-fungal metabolites. Front Microbiol. 2014;5:1–8. doi: 10.3389/fmicb.2014.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becker MH, Harris RN, Minbiole KPC, et al. Towards a better understanding of the use of probiotics for preventing chytridiomycosis in Panamanian golden frogs. Ecohealth. 2011;8:501–6. doi: 10.1007/s10393-012-0743-0. [DOI] [PubMed] [Google Scholar]
- 14.Becker MH, Richards-Zawacki CL, Gratwicke B, Belden LK. The effect of captivity on the cutaneous bacterial community of the critically endangered Panamanian golden frog (Atelopus zeteki) Biol Conserv. 2014;176:199–206. [Google Scholar]
- 15.Bletz MC, Loudon AH, Becker MH, et al. Mitigating amphibian chytridiomycosis with bioaugmentation: Characteristics of effective probiotics and strategies for their selection and use. Ecol Lett. 2013;16:807–820. doi: 10.1111/ele.12099. [DOI] [PubMed] [Google Scholar]
- 16.Walke JB, Becker MH, Loftus SC, et al. Amphibian skin may select for rare environmental microbes. ISME. 2014:J 1–11. doi: 10.1038/ismej.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzpatrick BM, Allison AL. Similarity and differentiation between bacteria associated with skin of salamanders (Plethodon jordani) and free-living assemblages. FEMS Microbiol Ecol. 2014;88:482–494. doi: 10.1111/1574-6941.12314. [DOI] [PubMed] [Google Scholar]
- 18.Markle JGM, Frank DN, Mortin-toth S, et al. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science (80-) 2013;339:1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 19.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105:17994–9. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez A, Luckey D, Taneja V. The gut microbiome in autoimmunity: Sex matters. Clin Immunol. 2014;159:154–162. doi: 10.1016/j.clim.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore FL, Boyd SK, Kelley DB. Historical perspective: Hormonal regulation of behaviors in amphibians. Horm Behav. 2005;48:373–383. doi: 10.1016/j.yhbeh.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Houck LD, Arnold SJ. Courtship and Mating Behavior. Reprod Biol Phylogeny Urodela. 2003:383–424. [Google Scholar]
- 23.Houck LD. Pheromone communication in amphibians and reptiles. Annu Rev Physiol. 2009;71:161–76. doi: 10.1146/annurev.physiol.010908.163134. [DOI] [PubMed] [Google Scholar]
- 24.Kueneman JG, Woodhams DC, Van Treuren W, et al. Inhibitory bacteria reduce fungi on early life stages of endangered Colorado boreal toads (Anaxyrus boreas) ISME J. 2015;10:1–11. doi: 10.1038/ismej.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carey C, Cohen N, Rollins-Smith L. Amphibian declines: an immunological perspective. Dev Comp Immunol. 1999;23:459–72. doi: 10.1016/s0145-305x(99)00028-2. [DOI] [PubMed] [Google Scholar]
- 26.Rollins-Smith LA. Metamorphosis and the amphibian immune system. Immunol Rev. 1998;166:221–230. doi: 10.1111/j.1600-065x.1998.tb01265.x. [DOI] [PubMed] [Google Scholar]
- 27.Longo AV, Savage AE, Hewson I, Zamudio KR. Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R Soc Open Sci. 2015;2:140377. doi: 10.1098/rsos.140377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuchta SR, Parks DS, Wake DB. Pronounced phylogeographic structure on a small spatial scale: geomorphological evolution and lineage history in the salamander ring species Ensatina eschscholtzii in central coastal California. Mol Phylogenet Evol. 2009;50:240–55. doi: 10.1016/j.ympev.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 29.Stebbins RC. Natural history of the salamanders of the plethodontid genus Ensatina. University of California Press; 1954. [Google Scholar]
- 30.Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bokulich Na, Subramanian S, Faith JJ, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodhams DC, Alford RA, Antwis RE, et al. Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology. 2015;96:595–595. [Google Scholar]
- 33.Flechas SV, Sarmiento C, Cárdenas ME, et al. Surviving chytridiomycosis: differential anti-Batrachochytrium dendrobatidis activity in bacterial isolates from three lowland species of Atelopus. PLoS One. 2012;7:e44832. doi: 10.1371/journal.pone.0044832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker MH, Walke JB, Cikanek S, et al. Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc R Soc B. 2015;282:20142881. doi: 10.1098/rspb.2014.2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuchta SR, Parks DS, Wake DB. Pronounced phylogeographic structure on a small spatial scale: geomorphological evolution and lineage history in the salamander ring species Ensatina eschscholtzii in central coastal California. Mol Phylogenet Evol. 2009;50:240–55. doi: 10.1016/j.ympev.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 36.Davenport ER, Mizrahi-Man O, Michelini K, et al. Seasonal Variation in Human Gut Microbiome Composition. PLoS One. 2014;9:e90731. doi: 10.1371/journal.pone.0090731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer Ea, Cramp RL, Bernal MH, Franklin CE. Changes in cutaneous microbial abundance with sloughing: possible implications for infection and disease in amphibians. Dis Aquat Organ. 2012;101:235–42. doi: 10.3354/dao02523. [DOI] [PubMed] [Google Scholar]
- 38.Hajishengallis G, Liang S, Payne MA, et al. Low-Abundance Biofilm Species Orchestrates Inflammatory Periodontal Disease through the Commensal Microbiota and Complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brucker RM, Bordenstein SR. Speciation by symbiosis. Trends Ecol Evol. 2012;27:443–51. doi: 10.1016/j.tree.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 40.Shade A, Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012;14:4–12. doi: 10.1111/j.1462-2920.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 41.Jani AJ, Briggs CJ. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc Natl Acad Sci. 2014;111:E5049–E5058. doi: 10.1073/pnas.1412752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lauer A, Simon MA, Banning JL, et al. Common Cutaneous Bacteria from the Eastern Red-Backed Salamander Can Inhibit Pathogenic Fungi. Copeia. 2007;2007:630–640. [Google Scholar]
- 43.Walke JB, Becker MH, Hughey MC, et al. Most of the dominant members of amphibian skin bacterial communities can be readily cultured. Appl Environ Microbiol. 2015 doi: 10.1128/AEM.01486-15. AEM.01486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krynak KL, Burke DJ, Benard MF. Landscape and water characteristics correlate with immune defense traits across Blanchard's cricket frog (Acris blanchardi) populations. Biol Conserv. 2016;193:153–167. [Google Scholar]
- 45.Walke JB, Becker MH, Hughey MC, et al. Most of the Dominant Members of Amphibian Skin Bacterial Communities Can Be Readily Cultured. Appl Environ Microbiol. 2015;81:6589–6600. doi: 10.1128/AEM.01486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Savage K. Comparative analysis of anti-Bd bacteria from six Malagasy frog species of Ranomafana National Park 2015 [Google Scholar]
- 47.Krynak KL, Burke DJ, Benard MF. Larval Environment Alters Amphibian Immune Defenses Differentially across Life Stages and Populations. PLoS One. 2015;10:e0130383. doi: 10.1371/journal.pone.0130383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belden LK, Hughey MC, Rebollar EA, et al. Panamanian frog species host unique skin bacterial communities. Front Microbiol. 2015;6:1–21. doi: 10.3389/fmicb.2015.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabino-Pinto J, Bletz MC, Islam MM, et al. Composition of the Cutaneous Bacterial Community in Japanese Amphibians: Effects of Captivity, Host Species, and Body Region. Microb Ecol. 2016 doi: 10.1007/s00248-016-0797-6. [DOI] [PubMed] [Google Scholar]
- 50.Murray R, Brenner DJ, Bryant MP. Bergey's Manual of Systematic Bacteriology. Wiliams and Wilkins; 1984. [Google Scholar]
- 51.Johansen JE, Binnerup SJ, Kroer N, Molbak L. Luteibacter rhizovicinus gen. nov., sp. nov., a yellow-pigmented gammaproteobacterium isolated from the rhizosphere of barley (Hordeum vulgare L.) Int J Syst Evol Microbiol. 2005;55:2285–2291. doi: 10.1099/ijs.0.63497-0. [DOI] [PubMed] [Google Scholar]
- 52.Harris RN, Lauer A, Simon MA, et al. Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis Aquat Organ. 2009;83:11–6. doi: 10.3354/dao02004. [DOI] [PubMed] [Google Scholar]
- 53.Harris RN, Brucker RM, Walke JB, et al. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 2009;3:818–24. doi: 10.1038/ismej.2009.27. [DOI] [PubMed] [Google Scholar]
- 54.Wake DB, Vredenburg VT. Colloquium paper: are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc Natl Acad Sci U S A. 2008;(105 Suppl):11466–11473. doi: 10.1073/pnas.0801921105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Civitello DJ, Cohen J, Fatima H, et al. Biodiversity inhibits parasites: Broad evidence for the dilution effect. Proc Natl Acad Sci. 2015;112:8667–8671. doi: 10.1073/pnas.1506279112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winter C, Bouvier T, Weinbauer MG, Thingstad TF. Trade-offs between competition and defense specialists among unicellular planktonic organisms: the “killing the winner” hypothesis revisited. Microbiol Mol Biol Rev. 2010;74:42–57. doi: 10.1128/MMBR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koeppel AF, Wu M. Surprisingly extensive mixed phylogenetic and ecological signals among bacterial Operational Taxonomic Units. Nucleic Acids Res. 2013;41:5175–5188. doi: 10.1093/nar/gkt241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loudon AH, Venkataraman A, Van Treuren W, et al. Vertebrate hosts as Islands: Dynamics of selection, immigration, loss, persistence, and potential function of bacteria on salamander skin. Front Microbiol. 2016;7:1–11. doi: 10.3389/fmicb.2016.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Longbottom D, Coulter LJ. Animal chlamydioses and zoonotic implications. J Comp Pathol. 2003;128:217–244. doi: 10.1053/jcpa.2002.0629. [DOI] [PubMed] [Google Scholar]
- 60.Blumer C, Zimmermann DR, Weilenmann R, et al. Chlamydiae in free-ranging and captive frogs in Switzerland. Vet Pathol. 2007;44:144–150. doi: 10.1354/vp.44-2-144. [DOI] [PubMed] [Google Scholar]
- 61.Hamdi C, Balloi a, Essanaa J, et al. Gut dysbiosis and honeybee health. J Appl Entomol. 2011;135:524–533. [Google Scholar]
- 62.Bresciano JC, Salvador Ca, Paz-y-Miño C, et al. Variation in the Presence of Anti-Batrachochytrium dendrobatidis Bacteria of Amphibians Across Life Stages and Elevations in Ecuador. Ecohealth. 2015 doi: 10.1007/s10393-015-1010-y. [DOI] [PubMed] [Google Scholar]
- 63.Shine RS. Sexual selection and sexual dimorphism in the Amphibia. Copeia. 1979;1979:297–306. [Google Scholar]
- 64.Klein SL. Hormones and mating system affect sex and species differences in immune function among vertebrates. Behav Processes. 2000;51:149–166. doi: 10.1016/s0376-6357(00)00125-x. [DOI] [PubMed] [Google Scholar]
- 65.Walke JB, Harris RN, Reinert LK, et al. Social Immunity in Amphibians: Evidence for Vertical Transmission of Innate Defenses. Biotropica. 2011;43:396–400. [Google Scholar]
- 66.Rebollar EA, Simonetti SJ, Shoemaker WR, Harris RN. Direct and Indirect Horizontal Transmission of the Antifungal Probiotic Bacterium Janthinobacterium lividum on Green Frog (Lithobates clamitans) Tadpoles. Appl Environ Microbiol. 2016 doi: 10.1128/AEM.04147-15. AEM.04147-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Banning JL, Weddle AL, Wahl GW, et al. Antifungal skin bacteria, embryonic survival, and communal nesting in four-toed salamanders, Hemidactylium scutatum. Oecologia. 2008;156:423–9. doi: 10.1007/s00442-008-1002-5. [DOI] [PubMed] [Google Scholar]
- 68.Brucker RM, Bordenstein SR. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution. 2012;66:349–62. doi: 10.1111/j.1558-5646.2011.01454.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Resource 1. Supplementary Table 1. Sequences of Illumina primers used for amplicon sequencing.
Online Resource 2. Supplementary Table 2. Complete list of core bacterial OTUs and average abundances by sample group. (*) indicates an OTU with previously-documented antifungal properties.
Online Resource 3. Supplementary Table 3. List of bacterial OTUs unique to one population. All unique OTUs were present at < 0.01% abundance. The East Bay population had the most unique OTUs.