

Abstract
Mutations in genes that encode for muscle sarcomeric proteins have been identified in humans and two breeds of domestic cats with hypertrophic cardiomyopathy (HCM). This article reviews the history, genetics, and pathogenesis of HCM in the two species in order to give veterinarians a perspective on the genetics of HCM.
Keywords: Feline, Myosin binding protein C, Mutation
Hypertrophic cardiomyopathy (HCM) in people is a genetic disease that has been called a disease of the sarcomere because the preponderance of mutations identified that cause HCM are in genes that encode for sarcomeric proteins.1 Sarcomeres are the basic contractile units of muscle and thus sarcomeric proteins are responsible for the strength, speed, and extent of muscle contraction. In people with HCM, the two most common genes affected by HCM mutations are the myosin heavy chain gene (MYH7), the gene that encodes for the motor protein β-myosin heavy chain (the sarcomeric protein that splits ATP to generate force), and the cardiac myosin binding protein-C gene (MYBPC3), a gene that encodes for the closely related structural and regulatory protein, cardiac myosin binding protein-C (cMyBP-C). To date, the two mutations linked to HCM in domestic cats (one each in Maine Coon and Ragdoll breeds) also occur in MYBPC3.2, 3 This is a review of the genetics of HCM in both humans and domestic cats that focuses on the aspects of human genetics that are germane to veterinarians and on all aspects of feline HCM genetics.
HUMAN HCM
Definition and Etiology
The definition of HCM has evolved as the clinical understanding and the genetic underpinnings of the disease have unfolded. In general terms, HCM is a primary myocardial disease that most commonly causes thickening of the left ventricular (LV) myocardium (either regional asymmetric thickening or concentric thickening/hypertrophy) in the absence of other cardiovascular or systemic causes.4 Wall thickening is thus not secondary to pressure overload (e.g., hypertension, aortic stenosis) or hormonal stimuli (e.g., thyroid disease), but is a primary abnormality of the heart muscle itself. HCM is also distinct from other genetic diseases that can cause LV hypertrophy in humans, especially in childhood, such as Noonan syndrome or Danon’s disease.5,6,7
HCM in humans is most commonly caused by mutations in genes that encode for the proteins that make up the cardiac muscle sarcomere or in proteins closely associated with sarcomere function (Figure 1). Mutations in 11 sarcomeric genes have thus far been linked to HCM and account for about 70% of the disease seen in human patients with HCM.8 That leaves a cause to be identified in approximately 30% of human patients with unexplained LV wall thickening (i.e., HCM not attributable to other systemic causes).4 In these cases the disease may be attributable to novel mutations in either non-coding gene sequences or in other proteins closely associated with sarcomere function such as Ca2+ handling proteins.9 Most mutations are “private” meaning that they are unique to a single family or individual and >1400 distinct gene variants have been identified in patients with HCM.1 Most of the mutations are missense mutations in which one highly conserved DNA nucleotide is replaced by a different nucleotide resulting in a different codon. A different codon may or may not produce a different amino acid but if it does, inclusion of that altered amino acid can disrupt function of that protein which may lead to a disease such as HCM. When a mutation causes a disease (e.g., HCM) it does produce a different amino acid, which forms an abnormal protein.8
Figure 1.
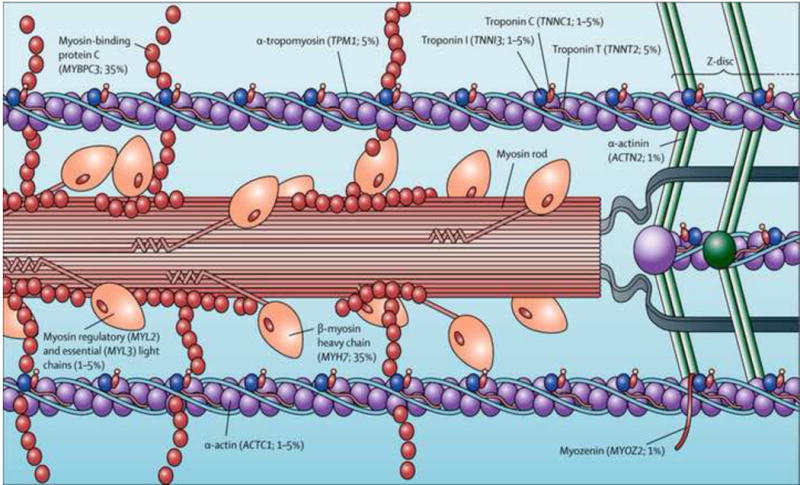
Locations of genes within the cardiac sarcomere known to cause hypertrophic cardiomyopathy in humans. Prevalence of every gene (derived from data of unrelated hypertrophic cardiomyopathy probands with positive genotyping) is shown in parentheses. (From Maron et al. Lancet 2013;381: 242–55 with permission).
Humans with sarcomeric gene mutations express a wide range of phenotypes from no apparent disease to massive wall thickening and a wide range of clinical outcomes from no disease sequelae to sudden death and heart failure. The vast intergenetic and intragenic heterogeneity seen in humans with HCM likely accounts for much of the breadth of these clinical outcomes and probably also accounts for at least some of the variability in the amount of hypertrophy from patient to patient. However, left ventricular hypertrophy is not the only important manifestation of the disease. It is now clear that some sarcomeric mutations cause little or no LV hypertrophy per se, but nonetheless lead to other common sequelae of the disease including diastolic dysfunction, arrhythmia, and sudden cardiac death.10 For instance, mutations in the cardiac troponin T gene commonly predispose patients to sudden death, even if only mild hypertrophy is present.11
In addition to the sarcomeric gene mutations, mutations in other genes occasionally have been suggested as causative for HCM in humans. They include mutations in genes that encode for proteins in the Z-disc (e.g., muscle LIM protein, telethonin, vinculin, LIM domain binding 3, α-actinin 2, myozenin 2, cardiac ankyrin repeat protein, nexilin), and calcium handling protein genes (e.g., phospholamban, calsequestrin 2, calreticulin 3, junctophilin 2).8,12 Many of these have been identified in only one or a few families and thus a solid causal relationship has not been established in all.
Despite the diversity of underlying genetic causes and outcomes, patients with HCM have several clinical features in common. Besides wall thickening, another hallmark of the disease is impaired diastolic function due to the hypertrophy itself, myocardial fibrosis and altered calcium kinetics in the cell.13 HCM in humans (and germane to this discussion, also in Maine Coon cats) is also usually associated with histological changes such as myocyte disarray, a diagnostic feature of the disease, where individual myocytes adopt bizarre shapes and relationships to one another.14 Small coronary vessel disease and myocardial fibrosis are also usually present.15 These histological changes may contribute to the increased susceptibility to arrhythmias and sudden cardiac events in patients with HCM.16 Dynamic left ventricular outflow tract obstruction (LVOTO) due to systolic anterior motion (SAM) of the mitral valve is a common but not consistent feature of HCM in people. It is also labile from time to time. Estimates of prevalence of SAM in people range from 25% to 70% with the presence of SAM being a negative prognostic indicator and associated with increased incidence of sudden death.17,18
Nomenclature
HCM has been known by numerous different names (over 80 different designations) over the years such as idiopathic hypertrophic subaortic stenosis (IHSS), asymmetric septal hypertrophy (ASH), idiopathic cardiomyopathy (ICM), familial hypertrophic cardiomyopathy (FHC), and hypertrophic obstructive cardiomyopathy (HOCM).5 The diversity of names reflects various attempts to describe the range of clinical presentations and their causes. However, the current consensus in human medicine is that these syndromes are collectively referred to as hypertrophic cardiomyopathy (HCM). This is designed to exclude any designation of whether outflow tract obstruction is present or not because obstruction of the LV outflow tract is labile and so can be present on one examination and not on another. It is also meant to include the numerous variations in LV wall thickening that exist (e.g., not just a thick interventricular septum).5,19
History of HCM in Humans
The history of HCM in humans is long and dates back to the late Renaissance period of the 16th and 17th centuries when postmortem anatomic dissection came into being.20 Donald Teare, a pathologist, is generally credited with naming HCM as a distinct disease entity in a paper in 1958 where he described severe LV hypertrophy in a group of young adults that had died suddenly.21 Interestingly, he initially described the disease as asymmetrical hypertrophy of the septum due to a benign tumor of the heart (muscular hamartoma).
In 1960, Hollman, along with Teare, described a family with HCM that included one of the patients from Teare’s original paper along with her sister and brother.22 The pedigree was compatible with an autosomal dominant disease. In 1961, Paré described a family of 77 persons in French Canada of whom 30 had a history of heart disease.23 Many had died suddenly. The family lived in Coaticook, Quebec and because so many family members died of sudden cardiac events, the affliction became known as the Coaticook curse. Much of the family history was related by a 95-year-old family member that described an uncle who died suddenly in 1860 and who was thought to be the first affected family member. The family tree was consistent with an autosomal dominant trait where first degree relatives of affected individuals were at highest risk of developing HCM. Paré continued to follow this family over the next 30 years.
Despite the appearance that HCM was inherited, numerous theories regarding the etiology of HCM were put forth over the ensuing years, including excessive catecholamines, cardiac neural crest cell abnormalities, and, in an infamous (fabricated) paper by Darsee (later retracted), abnormalities in human leukocyte antigens.24,25
The Genetics of Human HCM
In the 1980s, echocardiography became affordable enough to become a common noninvasive tool for reliably diagnosing HCM and so became a tool to establish the phenotype in families where a proband was identified.26 In the same decade, the ability to locate disease-causing mutations in the human genome started to come of age. It was discovered that there were characteristic DNA sequences throughout the genome where specific restriction enzymes cleaved DNA. Slight sequence variations in these sites among individuals resulted in so-called restriction fragment length polymorphisms (RFLPs). By painstakingly comparing RFLP maps in affected and non-affected members of a family it was possible to link a disease to a specific chromosome or region on a chromosome.27 In 1983 this linkage analysis technique was first used successfully to map Huntington’s disease to chromosome 4 in humans.27
It followed shortly thereafter that HCM was the first myocardial disease for which a genetic cause was identified using the same linkage approach. In 1987 Christine Seidman and Bill McKenna teamed up to study the French Canadian family that Paré had discovered almost 3 decades earlier.28 The investigators contacted family members and set up clinics where they performed physical examinations, electrocardiograms, and echocardiograms, and collected blood samples for DNA from over 100 family members over 16 years of age. In the end, 78 family members were included in the study. Of these, 64 were closely related to an affected person and 14 were offspring of unaffected parents. A total of 20 living and 24 deceased family members were diagnosed with HCM.
Initial assessments from clinical data confirmed that the disease was inherited in an autosomal dominant fashion and that penetrance was age-related, meaning that the disease was more prevalent in middle-aged and older family members than it was in younger family members. At the time the study was performed only several hundred RFLP markers were known as opposed to the thousands that are known today. Thus there was no guarantee that a disease locus could be successfully mapped or even that the disease could be attributed to a change (mutation) in a single gene. Nonetheless, 78 RFLPs were followed in a laborious process involving hundreds of person hours to complete the Southern (DNA sequence) blot analyses necessary to identify the RFLPs. In 1989 the investigators identified a single linkage to a region on chromosome 14q1.29 The LOD (logarithm of the odds) score for the locus, which compares the probability that two loci are linked to the probability that they are not, indicated that there was a billion to one chance that the mutation did not reside in this region. Fortuitously, in 1987 the β-myosin heavy chain gene (MYH7) had also been localized to chromosome 14q1 and in 1989 the gene was further sublocalized to chromosome 14q11.2-13.30,31 Consequently, when the list of genes that resided within this region was examined, MYH7 became a gene of interest along with the nearby α-myosin heavy chain gene (MYH6). In 1990, again fortuitously, the complete sequence of MYH7 was published.32,33 Instead of sequencing the entire gene from each family member the investigators fine-mapped the locus they identified with restriction enzymes and sequenced the smaller region of interest they found.
In 1990 the team of researchers first reported a single missense mutation in MYH7 in the large French Canadian family first studied by Paré (family A).34 The mutation occurred in exon 13 and affected codon 403 of MYH7 changing a highly conserved arginine to glutamine (Arg403Glu or R403Q). Importantly, the same mutation was present in all affected family members. That same year the same investigators also identified what was initially thought to be a mutation that resulted in a hybrid α/β myosin heavy chain gene in a different family (family B).35 However, it was subsequently learned that the mutation was actually another point mutation in MYH7 (Arg453Cys or R453C).36 Over the next several years, other families with HCM were identified that had different mutations in MYH7. However, exceptions to the rule soon became evident in that other families with HCM did not have mutations in MYH7.37,38 These studies thus established that while mutations in MYH7 were a prevalent genetic cause of HCM, other genes must also contribute to the development of HCM. This was subsequently proven. For example, an HCM locus was identified on chromosome 15q2 that was later shown to contain mutations in the α-tropomyosin gene.39 Other mutations were subsequently identified in genes encoding troponin T, MyBP-C, myosin essential and regulatory light chains, troponin I and C, and actin.40,41,42,43,44,45 More recently mutations in genes that code for actinin and myozenin 2, two Z-disc (the ends of a sarcomere) proteins, have been linked to HCM in humans.46,47 In addition to genes that encode for sarcomeric proteins, mutations in genes encoding the calcium handling proteins, calsequestrin and junctophilin-2, have been identified in humans with HCM.9,48,49
Since the first report of a genetic cause of HCM in 1990, the identification of new sequence variants in known or novel genes in HCM patients has been exponential. Estimates indicate that over 1400 different gene variants have been identified thus far with the majority of these being in the MYH7 and MYBPC3 genes.8 However, with whole exome data from the NHLBI GO Exome Sequencing Project (ESP) revealing an astonishing amount of genetic variation in the general population (Exome Variant Server. Available at http://evs.gs.washington.edu/EVS/), it remains to be shown that a given variant is actually a causative mutation responsible for HCM rather than a benign polymorphism.50,51 Causal relationships can be demonstrated either through painstaking linkage analyses in large families, as described above, or by other direct demonstrations that a sequence variant causes aberrant protein function or hypertrophy (e.g., in a genetically engineered mouse model).
As suggested by the sheer number of HCM variants detected, it is common for individual families to have a unique or “private” causative mutation that arises independently and spontaneously. However, founder mutations also exist where the same mutation is found in multiple families usually within a similar geographic region.52,53,54 Most founder mutations occur in the MYBPC3 gene, consistent with the general observations that mutations in MYBPC3 cause either more benign disease or disease that occurs later in life. Because these mutations do not confer a selective disadvantage during reproductive years they can be passed on and retained in the population. One prevalent founder mutation is a 25 bp deletion in MYBPC3 that is carried by ~4% of individuals in Southeast India that is thought to have originated in an individual approximately 33,000 years ago.55 Today this mutation affects 20–40 million people and confers a 7-fold increased risk of heart failure, usually after the third decade of life.
Variability in Disease Penetrance and Expression
In classic terms, HCM due to sarcomeric mutations is inherited as an autosomal dominant (Mendelian) trait. This means that children of a parent with a causal mutation and a parent without the mutation will have a 50/50 chance of inheriting the mutation (will be heterozygous) and thus will be at risk of developing HCM. However, it should be emphasized that not all individuals that inherit a causative mutation will go on to develop HCM (i.e., the mutation is not 100% penetrant). In fact, so-called genotype positive, phenotype negative individuals (i.e., G+/LVH−), typically identified by DNA sequencing the relatives of an affected proband, are increasingly common and pose special challenges for clinical disease management.56 This is because while many G+/LVH− individuals will remain disease free for their entire lives, others will go on to develop HCM with age and may have symptoms ranging in severity from mild to severe, including heart failure severe enough to require transplant. The clinical challenge is in differentiating between these disparate outcomes. Thus, identification of “preclinical” signs such as the emergence of diastolic dysfunction that can herald disease progression prior to the emergence of other clinical symptoms (e.g., dyspnea, heart failure) has become a focus of disease surveillance strategies in people.57,58,59
As mentioned, the heterogeneity in HCM disease outcomes is due in part to the vast diversity in the underlying causative gene mutations. But this is combined with differences in gene dosage and other environmental and genetic modifiers that affect disease expression in a given individual. For instance, individuals that are homozygous for a mutation typically develop more severe disease or develop disease sooner than individuals that are heterozygous for a mutation.60 In addition, a small percentage (5–7%) of genotyped individuals carry 2 or more mutations in the same or different sarcomeric genes. In these so-called compound heterozygous cases, disease is also often evident at an earlier age and is more severe.61 In other cases, polymorphisms in other genes such as those associated with the renin-angiotensin-aldosterone system, angiotensin II receptor, and non-coding RNAs have all been proposed to act as modifiers that influence disease expression and severity. An example is the gene that encodes for angiotensin converting enzyme (ACE) where people with the DD genotype have more hypertrophy than those with the ID and II genotypes.62,63
In most cases HCM is not evident at birth but becomes apparent at some point later in life, often during adolescence or later in middle age (age-related penetrance)64. However, infants can be affected depending on the specific mutation, gene dosage (i.e., homozygous or compound heterozygous state) and the gene modifiers involved.65,66 For instance, in one case a novel mutation in myosin light chain 3 (MYL3) caused severe HCM in a 3-month-old infant. In an Amish population, individuals homozygous for mutations in MYBPC3 also develop severe infantile HCM.67,68
As discussed above, the correlation between genotype and phenotype is not well understood. However, as a general rule mutations in the MYH7 (especially R403Q, R453C, and R719W) tend to produce more severe disease than mutations in the MYBPC3 gene.17 Importantly, with regard to a comparison with feline HCM, MYBPC3 gene mutations also more commonly produce HCM later in life. However, there are also individuals with MYBPC3 gene mutations that develop severe HCM early in life. To illustrate the heterogeneity of HCM in family members with the same mutation, one study examined a founder mutation in MYBPC3 (R502W) in 9 families.54 Of the 25 individuals that carried this mutation, 15 had HCM at the time of the study (overall disease penetrance of 60%). However, in one family, all 3 individuals with the mutation had HCM (100% penetrance). Age at the time of diagnosis in these families ranged from 5 years of age in a child with heart failure to an 80 year old woman with only an abnormal ECG. With regard to LVH phenotype, 10 had no hypertrophy, 10 had septal hypertrophy, 3 had concentric hypertrophy, 1 had apical hypertrophy, and 1 had eccentric hypertrophy.
Gene sequence variants can cause poison polypeptides, haploinsufficiency, or disrupt cell homeostasis
Most mutations identified in genes linked to HCM in humans are missense mutations caused by substitution of a single DNA nucleotide with an incorrect nucleotide. Because a sequence of three nucleotides is read as a single codon that directs insertion of a specific amino acid into a nascent polypeptide chain, an incorrect nucleotide can cause incorporation of an incorrect amino acid that directly disrupts the function of the protein formed by the mutant gene. Abnormal proteins produced by mutant genes may be incorporated into sarcomeres and so can produce altered sarcomere function. These so-called “poison polypeptides” can thereby affect heart muscle function and trigger the development of hypertrophy. Alternatively, incorporation of an incorrect amino acid into a protein can also disrupt the overall synthesis, folding, or trafficking of that protein to its correct destination in the cell, thereby causing either less of the protein to be made or less of the protein to be directed to its correct location within the cell. Thus, haploinsufficiency, (i.e. a reduced total amount of the affected protein) can be deleterious if either not enough protein is made or the protein is not available in the right place to perform its function.
Frameshift mutations result from insertion or deletion of a number of DNA nucleotides not divisible by three. These mutations shift the DNA reading frame at the point of the insertion/deletion and thus encode proteins that start with a correct sequence of amino acids but then end with a completely new sequence of amino acids starting from the point of the insertion/deletion mutation. Because novel stop codons (TGA, TAA, TAG) are also often introduced by chance, new sequences often lead to prematurely shortened proteins. If synthesized, the activity of these abnormal proteins can range from deleterious (poison polypeptide) effects on the function of the sarcomere to non-functional proteins that cause haploinsufficiency. However, most commonly these proteins are not produced at all because cell quality control mechanisms efficiently recognize and degrade mutant mRNAs with premature stop codons (e.g., through non-sense mediated mRNA decay) or degrade aberrant or misfolded proteins (e.g., through the ubiquitin-proteasome system [UPS]). Such quality control mechanisms are believed to account for the lack of truncated cardiac myosin binding C protein (cMyBP-C) in myocardium from patients with HCM affected with frameshift mutations in MYBPC3. Instead, reduced amounts of total cMyBP-C protein have been reported in myocardium from patients affected with MYBPC3 truncation mutations as well as in some with missense mutations.69,70 However, it is still not resolved whether modest decreases in the total amount of cMyBP-C protein (haploinsufficiency) per se can lead to functional impairments that cause disease or whether an increased burden on cellular quality control mechanisms disrupt normal cell homeostasis. For instance, truncated cMyBP-C proteins have been shown to be preferred substrates for the UPS that ultimately displace normal substrates for cell protein degradation and thereby disrupt cell homeostasis.71,72
Mutations that affect exon/intron splice sites also occur when either single nucleotide substitutions or insertions/deletions alter the consensus sequences necessary for the proper editing of an mRNA. In these cases, inappropriate exon splicing can lead to a protein with an altered function (by inclusion/exclusion of exons/introns) or to prevention of appropriate regulation of alternatively spliced proteins.
Pathogenesis
Abnormal genes and so abnormal gene processing or gene products (e.g., proteins) result in clinical disease. As discussed above, two straightforward mechanisms by which an abnormal protein can cause disease are 1) dominant negative effects (the abnormal protein has a negative effect on the function of a structure and its effect dominates) caused by impaired function of the abnormal proteins (e.g., poison polypeptide effects) or 2) by a reduction in the amount of protein (haploinsufficiency). Either scenario would be expected to disrupt the ability of sarcomeres to function properly and thereby directly alter the strength, extent, and speed of contraction.
While it is intuitive that mutations that disrupt sarcomere function could cause impaired contractile function (e.g., a decrease in contractile force), many mutations that cause HCM surprisingly cause an increase in contractile activity. For instance, the original R408Q mutation in the β-myosin heavy chain has been found consistently to enhance force generation in numerous studies ranging from single molecule studies to force measurements in human myocardial fibers.73,74,75 These and other studies have led to the general idea that myocardial hypercontractility may be one trigger for hypertrophy. Consistent with this, systolic contractile function is often preserved in patients with HCM and the ability of the sarcomeres to generate force in response to a given level of Ca2+ (i.e., the Ca2+ sensitivity of tension) is often increased.76 An increase in the Ca2+ sensitivity of tension per se has been proposed to be a common denominator leading to hypertrophy.77 In support of this idea, normalization of Ca2+ sensitivity of tension in a mouse model of HCM prevented the development of HCM in one study.78 The authors concluded that myofilament desensitization to Ca2+ could be a therapeutic target for treatment of HCM. While this approach could be widely applicable in principle, it is still likely to be a vast oversimplification considering the heterogeneity that underlies HCM. For instance, there are mouse models of severe hypertrophy where the Ca2+ sensitivity of tension is either unchanged or reduced and others where there is a persistent increase in the Ca2+ sensitivity of tension without the development of hypertrophy.79,80 Energetic imbalances caused by inefficient energy consumption during hypercontractility may be one stimulus for the development of hypertrophy.76 Germane to the focus of this review, in one study of HCM in cats, myocardial strain (i.e., shortening of myocardial segments using tissue Doppler imaging echocardiography [TDI]) was decreased despite the usual increase in LV shortening fraction.81 This suggests that the increase in global LV function in cats is due to the decrease in afterload produced by the hypertrophy and that myocardial function is actually decreased.
Mouse models of HCM
The cellular effects of the vast majority of individual sequence variants associated with HCM in humans have not been studied. Of the few that have been investigated, most have been examined primarily using in vitro systems where the biochemistry of the affected protein can be directly investigated. However, even when an abnormal protein is associated with an aberrant function, proof that a given variant causes disease requires either detailed analyses of large family pedigrees (the ‘gold standard’) or recapitulation of the disease phenotype in an animal model. Usually this means expression of the variant gene in transgenic mice to assess the impact of the mutation on cardiac structure and function. While there are inherent limitations to using mice as models of cardiac structure and function in humans, mouse models of human HCM mutations have nonetheless proven tremendously useful in identifying the most proximal causes of the disease, especially in cases where an abnormal protein directly causes altered muscle function (i.e., a so-called “poison polypeptide”) such as the case with the original R403Q mutation first identified in MYH7.82
Mouse models of human HCM have been created for nearly every gene (but not every mutation) thus far associated with HCM, but this section of the review will focus only on MYBPC3 since, at the time of this writing, only mutations in MYBPC3 have been linked to HCM in domestic cats.
The first transgenic mouse model of a mutation in MYBPC3 (known as “t/t” mice) used homologous recombination (genetic recombination in which nucleotide sequences are exchanged between two similar or identical molecules of DNA) to target the precise chromosomal gene locus of MYBC3 and replace one of the two normal (wild-type) alleles with either of two different mutant alleles encoding truncated proteins that lack the C-terminal exons 30 and 31 of the gene.83 This strategy was chosen to mimic human HCM mutations predicted to cause premature truncations of the cMyBP-C protein leading to the production of otherwise normal proteins that lacked amino acids near the C-terminus of the protein. Because these C-terminal amino acids encode the binding sites responsible for anchoring cMyBP-C to myosin and titin, the proteins produced by mutant genes were assumed to be otherwise normal but lacking in these capabilities. Instead, the surprising finding was that these proteins were not made at all, with at most 10% of expected protein detected in homozygous mutant mice. Cell surveillance mechanisms described above that detect and destroy mutant mRNAs and proteins likely account for the observed lack of expression of the mutant truncated proteins.
The findings in the t/t mice thus agree with results from several studies looking at human biopsies showing that truncated proteins were not found in patients.84,85 However, an apparent paradox was that heterozygous mice carrying the t/t mutation, in contrast to human patients that also typically carry only one mutant allele, generally displayed only very mild phenotypes.86,87 Similar results were also reported for two other MYBPC3 knockout models in which the entire gene product was targeted for ablation.88,89 In these models, only very mild deficits in cardiac function were noted in heterozygous mice.89,90 Interestingly, protein expression in the heterozygous mice was normal or near normal, although a study by Stelzer et al found reduced expression of cMyBP-C in heterozygous knockout mice.90 However, the consistent findings that protein levels are ~80–100% of normal (rather than 50%) in the heterozygous mice indicates that gene expression from the remaining unaffected wild-type allele can compensate for the loss of a knockout allele.88,89,90 These results thus argue against haploinsufficiency alone as being a primary cause of HCM, at least in mice. Consistent with this, transgenic expression of as little as 40% of the normal amount of cMyBP-C rescued the disease phenotype in t/t mice.91 It thus remains to be shown that the modest reductions of cMyBP-C observed in some patients with HCM are sufficient to cause disease via haploinsufficiency in people.
Other transgenic MYBPC3 mouse models have been created that do not use homologous recombination to directly target the MYBC3 gene but instead use pro-nuclear injection techniques to add an exogenous mutant cDNA randomly into the genome of a fertilized zygote. The advantage of this method is that it is less expensive and time consuming because transgenic animals can be made relatively quickly using only coding cDNA sequences without the need for extensive knowledge of gene intron or flanking sequences necessary for homologous recombination in embryonic stem cells. Results from a number of transgenic mouse models expressing cMyBP-C mutant proteins have shown various outcomes ranging from mild to severe HCM with differing effects on myocyte force-producing abilities and on myofilament Ca2+ sensitivity of tension.92,93,94,95 Transgenic models have thus been essential in providing “in principal” proof that cMyBP-C proteins created from a mutated MYBPC gene can act as poison polypeptides to disrupt cardiac function in studies designed to test mechanistic hypotheses of cMyBP-C function. However, a disadvantage of these models is that because they lack the normal gene sequences and normal chromosomal localization of the mutation, transgenic cDNAs can bypass normal gene, mRNA, and protein processing which may be essential for ultimately understanding and treating disease pathology.
Aside from gene targeted MYBPC3 knockout mice and the t/t mice, few mouse models exist where single point mutations are targeted directly in the MYBC3 gene at the level of genomic DNA and are therefore subject to normal cell surveillance mRNA and protein processing mechanisms. An exception is a mouse model in which a single point mutation in MYBPC3 is targeted at an intron/exon splice site junction to mimic a human HCM mutation.96 In this model, the gene-targeted mutation resulted in complex expression of gene products ranging from very low expression of the full-length protein with the single amino acid substitution to altered splice variants. While this mouse model reveals the complex ways in which a single point mutation can affect protein expression via distinct pathways, the broader lack of targeted missense models of HCM mutations in MYBPC3 has left unresolved basic questions of whether MYBPC3 mutations disrupt cardiac function through poison polypeptide effects, haploinsufficiency, or other cellular effects. Given the sheer number of variants reported in the MYBC3 gene it is likely that all of these scenarios will contribute to disease etiology depending on the specific mutation. Additional natural or targeted animal models are thus necessary to understand basic mechanisms by which mutations in MYBPC3 lead to disease.
FELINE HCM
Additional insights into the mechanism(s) by which missense mutations in MYBPC3 cause disease that are relevant to both human and veterinary medicine may be found in understanding the genetic causes of feline HCM. By analogy with human HCM, the first genetic causes of feline HCM were identified using sequence analysis of genes encoding sarcomeric proteins. Thus far, two separate single nucleotide substitutions have been identified in MYBPC3, the A31P and R820W mutations, found in the Maine Coon and Ragdoll breeds, respectively.2,3 While these mutations account for a significant proportion of HCM in each of these breeds, as for human HCM, there are likely to be other variants not yet identified because HCM still occurs in cats in these breeds that do not carry either the A31P or R820W mutations.97,98 Nonetheless, because cats with A31P and R820W mutations are the first non-rodent animals that develop HCM as a result of a defined, natural genetic cause, understanding HCM in cats offers an extraordinary opportunity for better understanding the natural history of the disease and for distinguishing the triggers and modifiers that result in progression of occult preclinical disease to heart failure. Conversely the continuing advancement in knowledge regarding HCM in humans is beneficial to veterinary medicine, especially now that at least one cause has been identified in domestic cats.
History of Feline HCM
In August, 1990, Marcia Munro, a cat fancier, found out that her two-year-old female Maine Coon cat had a heart murmur when examined by a veterinary cardiologist (Dr. Joel Edwards) at a cat show. A subsequent echocardiogram revealed a large papillary muscle and systolic anterior motion (SAM) of the mitral valve. While echocardiographic findings were equivocal for HCM at that time, HCM was definitely present on a subsequent echocardiographic exam in December, 1990 and had progressed to more severe HCM in February 1991. Ms. Munro lived close to the Yale School of Medicine library and utilized it to investigate causes of HCM. By happenstance, the same day she went to the library was the same day that Seidman’s landmark article in Cell was published (Sept 7, 1990).34 Based in part on that article and on other information she found in researching the causes of HCM, she telephoned the breeder who had sold her the cat and asked if she knew whether or not any related cats had HCM. The breeder denied any knowledge of this. Ms. Munro subsequently went to several cat shows, talked to other Maine Coon cat breeders on this subject, and was able to identify 6 related cats that had been diagnosed with or had died of HCM. Armed with this knowledge she telephoned the first author with the information. Based on this information the original breeder was contacted by the first author and agreed to have echocardiograms performed on the cats in her colony. The first author and Dr. Paul Pion flew to the destination with a portable ultrasound machine and performed echocardiograms on all of the cats in the cattery as well as a number of cats co-owned by other individuals in the area (July 1991). Seven cats were found to have HCM. The breeder agreed to breed some of these affected cats and to sell the kittens to the first author. Approximately 4 months later, 7 kittens were flown to the University of California, Davis (UCD) to start a research colony dedicated to understanding the cause of and developing treatments for feline HCM.
Maine Coon cat Research Colony
Over the ensuing years, selective breedings of the cats in this colony were performed to identify the pattern of inheritance and provide a detailed family pedigree necessary for establishing the genetic cause of the disease. Echocardiography was performed on cats starting at 3–6 months of age and then every 6 months. Cats identified with HCM (affected) were bred to cats without HCM (unaffected) and to other cats with HCM. This was started by breeding one affected male to 2 affected sisters and one unaffected cousin. Cardiac ultrasound examinations were performed on all offspring to document whether or not they had HCM and when they developed HCM. Cats that died spontaneously were necropsied and their heart morphology noted and heart weight measured (Figures 2 and 3).
Figure 2.
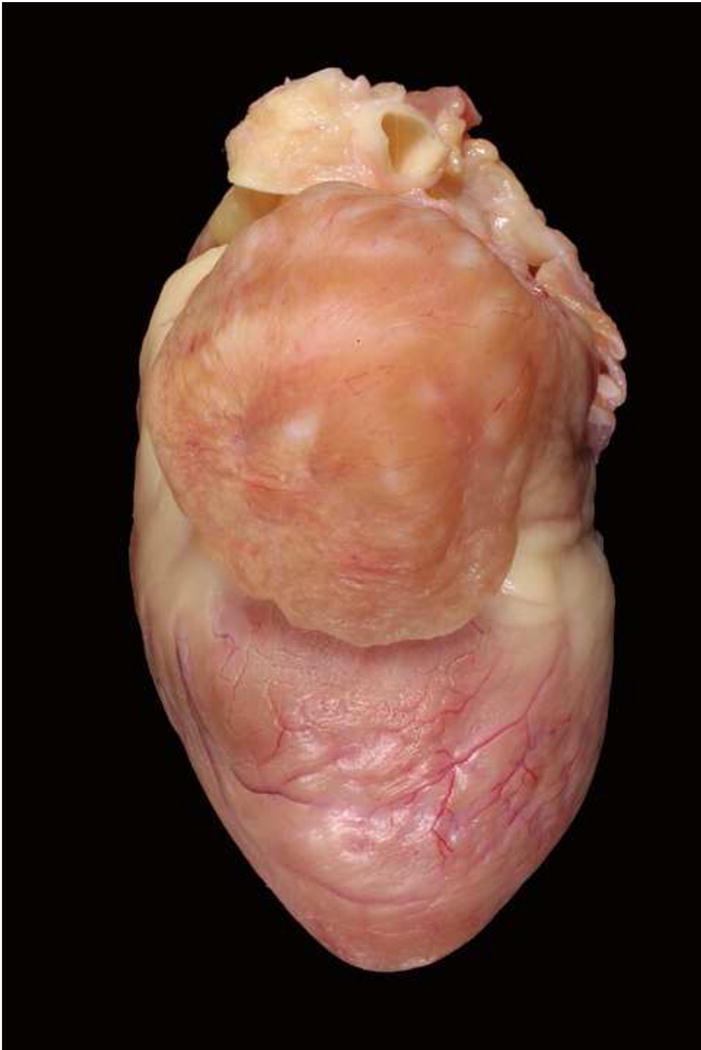
Heart from a Maine Coon cat with HCM and severe left auricular enlargement.
Figure 3.
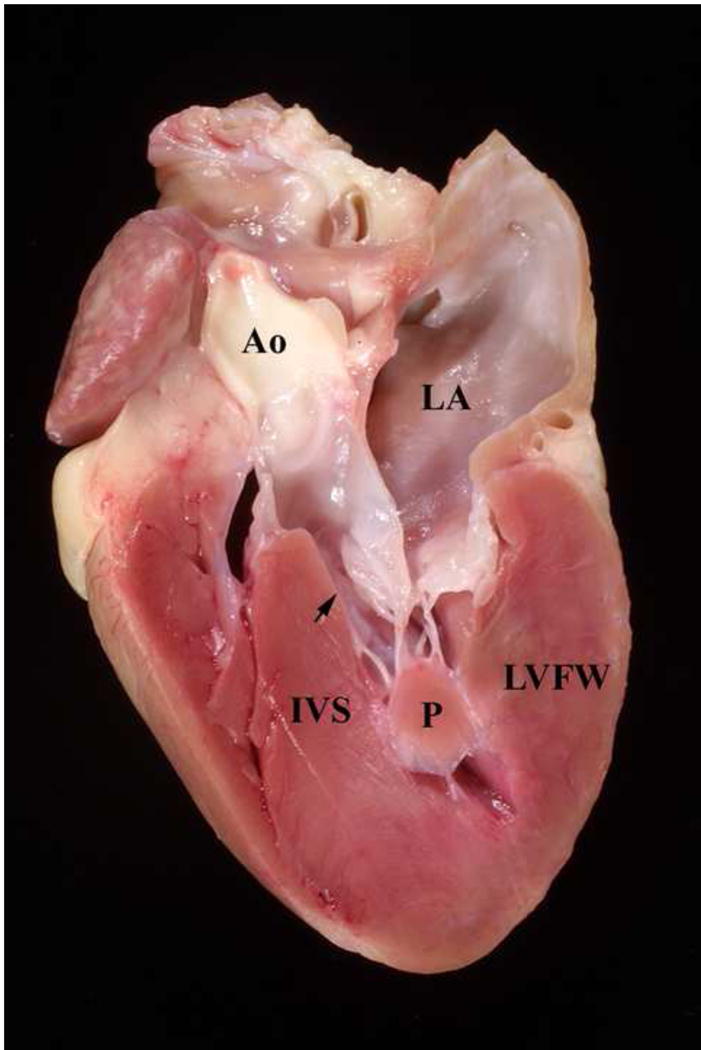
Heart from a Maine Coon cat with HCM. The heart has been sectioned longitudinally. Note the thick interventricular septum (IVS), left ventricular free wall (LVFW), and papillary muscle (P). The arrow points to the narrowed left ventricular outflow tract. Ao = aorta, LA = left atrium
The resultant 4 generation pedigree was consistent with the disease being inherited as an autosomal dominant trait.14 Characteristics were that every generation had affected cats, every affected cat produced at least one affected cat, both males and females were affected, and there was male to male transmission. The disease appeared to be 100% penetrant. When affected cats were bred to unaffected cats approximately 50% (12 of 22) of the cats developed HCM and the other half were normal. In affected cats, HCM was not present before 1 year of age (developed at 13 to 48 months). Males developed more severe disease than females and often developed it earlier (average of 19 months for males vs. 29 months for females). After the disease appeared, it either stabilized or progressed to more severe disease. For example, there were cats that had moderate HCM at the onset that had moderate HCM all of their life and there were cats that progressed from no disease to moderate disease to severe disease while others had no disease for years and then severe disease at the onset. Based on the lack of disease prior to one year of age and the appearance of the disease at a later age, the disease showed clear age-related penetrance.
There were only 2 litters (6 live kittens that survived to adulthood) that were produced from breeding affected cats to affected cats. Four of these 6 cats (both males and 2 of 4 females) developed severe HCM. In these cats the disease was severe by 7–12 months of age in both males and females. Three of these 4 cats died of heart failure or died suddenly between 18 and 25 months of age. Again approximately 50% of the cats in these 2 litters had HCM if one included stillborn kittens in the total count. About 25% were normal and about 25% were stillborn. This was interpreted as possibly meaning that one quarter of the cats were homozygous and homozygosity was lethal. This subsequently proved to be incorrect. Instead, based on current findings, it’s almost certain that these severely affected cats were homozygous for the subsequently identified mutation and the stillborn kittens died for another reason.105
Genetics of Feline HCM
In 2005 the first genetic cause of HCM was identified in the UCD Maine Coon cat research colony by Meurs and colleagues.2 The first clue regarding the identity of the mutation came when myocardial tissue from affected cats in the UCD colony was homogenized and electrophoretically run on SDS-PAGE gels. Analysis of the protein gels identified an apparent decrease in cMyBP-C and myomesin proteins along with anomalous migration of the β-myosin heavy chain protein. Because numerous mutations had already been identified in MYBPC3 in humans it was decided to sequence this gene in these cats. An alteration in the DNA nucleotide sequence was identified in nucleotide 93 in codon 31 where a highly conserved guanine (G) was replaced with cytosine (C). The sequence variant resulted in the codon being changed from GCC (coding for alanine) to CCC (coding for proline). There are two separate ways to denote this mutation. One way is to denote the codon number surrounded by the amino acids altered (A31P or p.A31P). The other is to use the number of the DNA nucleotide that is affected and surround it by the nucleotides that are changed (G91C or c.91G>C). All 4 designations appear in the literature. The mutation was not found in 100 control (non Maine Coon) cats.
Two years later a second mutation in MYBPC3 was identified in Ragdoll cats at a completely separate location (codon 820) where a cytosine (C) was changed to thymine (T) resulting in the codon specifying insertion of a tryptophan (W) instead of arginine (R; R820W) into the protein.3 Interestingly, the R820W mutation along with an R820Q variant have also been identified in human patients with HCM.99,100 Once again the nucleotide sequence in this region of the MYBPC3 gene was highly conserved across mammalian species (human, mouse, rat, dog, cow). Of the 20 cats studied, 9 were homozygous for this mutation.
Once these mutations were identified, genetic tests became available to screen for the mutations in different cat populations. It was subsequently learned that the A31P mutation was very prevalent in Maine Coon cats. In one study, 34% of Maine Coon cats had the mutation.101 Of these, approximately 10% were homozygous for the mutation (~3% of the population). This study also noted that only Maine Coon cats had the A31P mutation although in another study one British Shorthair also had the A31P mutation.102 A very similar percentage of Maine Coon cats (32%) was found to carry the mutation in a subsequent study whereas the percentage was higher in another study (41%).102,103 The high percentage of affected cats was surprising and suggested that the mutation is either more benign than first thought or that it is not 100% penetrant in the general Maine Coon cat population. Otherwise, it would be hard to imagine how the breed could survive.
The low penetrance of the disease in cats carrying the A31P mutation was subsequently confirmed by several studies where testing for the A31P mutation was done during screening clinics for HCM. In a study published in 2010, 83 Maine Coon cats were examined at screening clinics.97 Of these, 18 cats (22%) were positive for the A31P mutation. However, the investigators found that many Maine Coon cats with the A31P mutation did not have echocardiographic evidence of HCM and a few cats without the A31P mutation had HCM. The results suggested that disease penetrance was low in cats with the A31P mutation and confirmed that there is at least one more cause of HCM in Maine Coon cats (this was also noted in the UCD research colony where not all cats with HCM carried the A31P mutation in later years). While many of the cats with the A31P mutation in this study may have been too young (examined prior to the onset of HCM), subsequent studies confirmed that the penetrance of the A31P mutation is low in cats that are heterozygous for the mutation.103,104,105
A similar study looked at the echocardiographic appearance of hearts from Maine Coon cats with the A31P MYBPC3 mutation.104 In this study, echocardiography was performed on 96 Maine Coon cats presented for screening for HCM. Both two-dimensional and TDI echocardiography were performed. Of the 96 cats, 44 had the A31P mutation (38 were heterozygous and 6 were homozygous). Because 45 of the 96 cats were less than 2 years of age many were probably too young to have evidence of HCM if they were heterozygous. Of the 38 heterozygous cats, four had clear evidence of at least moderate HCM. However, only 10 of the 34 heterozygous cats that did not have HCM were over 4 years of age. Of the 44 cats that had the mutation, 13 were male and 31 were female. This potentially introduced an additional bias into the study since female Maine Coon cats appear to get HCM at a later age and get less severe disease.14 Still, this study again clearly showed that the A31P mutation is not nearly 100% penetrant in Maine Coon cats when a young cat is heterozygous for the mutation (11% in this study).
Homozygous cats appear to be a different story. Of the 6 cats that were homozygous for the A31P MYBPC3 mutation in the aforementioned study, 4 had clear echocardiographic evidence of HCM and the other two had abnormal diastolic function as assessed by TDI, an abnormality often present in cats that go on to develop HCM. Consequently, it appears that all 6 of these cats either had HCM or a preclinical stage of HCM, which is reasonable evidence that the mutation is causal. Two cats in this study without the known mutation also had HCM. This once again documented that there is at least one more cause of HCM in Maine Coon cats.
Another recent study into the penetrance of the A31P mutation looked at 332 cats.103 This study also showed that penetrance for Maine Coon cats heterozygous for the mutation is low (6%), that most but not all cats homozygous for the mutation get HCM (penetrance is high), and that most of the Maine Coon cats that get severe HCM prior to 6 years of age do so because they are homozygous for the mutation. The prevalence of HCM in the study population was 6%. Eighteen cats were homozygous and 89 cats were heterozygous for the mutation. The odds ratio for having HCM for homozygous cats was very high at 21.6 (95% confidence interval 7–66). Overall, 50% of the cats that were homozygous for the mutation had HCM. Only two cats over four years of age were homozygous and both had HCM.
In another recent study, penetrance was estimated to be 0.08 (8 Maine Coon cats in 100 with the A31P mutation will develop HCM) for cats heterozygous for the A31P mutation.105 However, this study again showed that the penetrance is much higher in cats that are homozygous for the A31P mutation (0.58 in this study), which again demonstrates that the mutation is causal rather than simply a benign polymorphism. Consequently, it appears that the majority of the clinically apparent disease seen in young to middle-aged Maine Coon cats is in cats that are homozygous for the mutation.
Although Maine Coon cats that are heterozygous for the A31P mutation often do not show clear evidence of LV wall thickening, cardiac function may still be compromised. In one study, heterozygous cats were examined echocardiographically using both two-dimensional and TDI to look for LV wall thickening and to assess diastolic function.104 Only 4 of the 38 cats heterozygous for the A31P mutation had LV hypertrophy. However, as a group, the remaining cats had reduced early diastolic LV wall velocity suggesting impaired diastolic function, a hallmark of HCM that, as mentioned previously, is often considered one of the pre-clinical manifestations of the disease. In other words, the A31P mutation can cause subclinical diastolic dysfunction that may or may not progress to wall thickening. Similar to human HCM, identifying the early sequelae of the disease and identifying additional environmental and genetic factors that provoke disease progression will be essential to understanding the pathophysiology of HCM and so its prevention or treatment in cats.
Findings appear to be similar in Ragdoll cats. In one study the effect of the R820W mutation was examined in 236 Ragdoll cats that had been screened for the mutation.98 Of these cats, 156 did not have, 68 were heterozygous for, and 12 were homozygous for the mutation. Only 15 cats had died of HCM at the time a questionnaire was sent to each owner. Cardiac death was unusual in the cats that did not have the mutation and in cats that were heterozygous for the mutation whereas cardiac death was common (7 of the 12 died of HCM) in cats that were homozygous for the mutation. The average age of death for the cats that were homozygous for the mutation was 5.65 years but death due to HCM occurred as early as 5 months of age and as late as 10.9 years of age. Consequently, both Maine Coon cats and Ragdoll cats that are homozygous for a mutation in MYBPC3 are at high risk of developing severe HCM and may do so at an early age. Cats that are heterozygous for either mutation, however, are at low risk of developing HCM, at least prior to 4–5 years of age.
HCM is prevalent in many breeds of cats but studies to prove that it is hereditary and to examine its mode of inheritance are few.106,107 After the first two mutations in MYBPC3 were identified, other feline breeds were screened for mutations in sarcomeric genes. In one study, 14 cats with HCM representing 5 different breeds (Sphynx, Norwegian Forest Cat, Siberian, British Shorthair, and Maine Coon [without the A31P mutation]) were examined for mutations in genes encoding cardiac troponin I, troponin T, MYBPC3, essential and regulatory light chains, alpha-tropomyosin, actin, and MYH7. The coding sequences of these genes were sequenced and examined for sequence variants linked to disease.108 However, none were identified. Instead, 33 single nucleotide polymorphisms (SNPs) were found that either did not segregate with disease or did not change an amino acid. Although the number of cats sequenced was small, the flood of causal mutations seen in human patients with HCM has thus far not been borne out in cats with HCM. However, with reduced costs and the widespread availability of next generation and whole genome sequencing methods, it is hoped that additional variants will be identified. Such novel gene approaches have recently been used successfully to identify genetic causes of dilated cardiomyopathy in Doberman Pinschers and arrhythmogenic right ventricular cardiomyopathy in Boxer dogs.109,110
Now that one cause of HCM is known in two breeds, cat breeders have the opportunity to reduce the prevalence of HCM in Maine Coon and Ragdoll cats. Ideally, all breeding cats would be screened for the mutation and cats with a known mutation would not be bred. However, because of the high incidence of these mutations in the breeding populations there is a risk of creating unintended consequences if this were to be done by reducing the size of the gene pool and so increasing the incidence of other genetic diseases in each breed. This risk may not be as high as it appears to be since breeders are already highly selective in their breeding practices. Regardless, the ideal of removing the two mutations from all cat populations probably cannot be achieved because it would require the cooperation of, if not all, at least the majority of breeders, both professional and non-professional. The likelihood of that occurring is negligible. One practical recommendation is for breeders to screen breeding cats and then to try to primarily breed cats that either do not have the mutation or selectively breed cats that are heterozygous for the mutation based on breed standards. We also recommend that two cats that are heterozygous not be bred to each other and try to mate any cat with an identified mutation only once. Homozygous cats should not be bred, primarily because of the risk of producing additional homozygous cats (if bred to a homozygous or heterozygous cat) but also because cats without a mutation are never produced. Kittens produced from normal to heterozygous matings should be tested. There is no reason to test kittens that are the products of mating two normal (non-mutated) cats.
Pathogenesis
The two genetic causes of HCM identified thus far in domestic cats occur in the same gene, MYBPC3, which encodes the protein cMyBP-C. cMyBP-C is a muscle regulatory protein that influences the force and speed of cardiac contraction and contributes to both diastolic and systolic function as well as the heart’s ability to increase contractility in response to inotropic stimuli.111,112 The feline HCM mutations each cause single amino acid substitutions in different domains of the affected proteins, but similar to human MYBPC3 mutations it is currently unknown whether the mutations cause disease by disrupting the normal function of cMyBP-C, by affecting protein stability leading to haploinsufficiency, or by impairment of other cell processes. Additional studies utilizing both purpose bred cats and client-owned cats carrying the mutations should be helpful in distinguishing between these possibilities with the goal of designing effective therapeutic strategies.
The cardiac myosin binding protein C gene is over 21 kb and is made up of 35 exons (4.5 kb) of which 34 code for amino acids. The protein is made up of 1173 amino acids. As shown in Figure 4, cMyBP-C is a member of the immunoglobulin (Ig) superfamily of proteins being composed of 11 modular domains folded into compact β-sheets with homology to either Ig-like or fibronectin (Fn)-like domains. The domains are numbered sequentially C0–C10 beginning at the N-terminus of the protein. The A31P mutation identified in Maine Coon cats places the altered amino acid near the N-terminus of cMyBP-C within C0, the first Ig-like domain of cMyBP-C, whereas the R820W mutation found in Ragdoll cats occurs closer to the center of the molecule in domain C6. Between C0 and C1 there is an unstructured linker that is rich in proline and alanine residues and between C1 and C2 there is a partially structured linker of ~100 residues that is referred to as the M-domain. The M-domain is a key regulatory subunit of cMyBP-C that is phosphorylated by a number of protein kinases (e.g., protein kinase A) following β-adrenergic stimulation.113,114
Figure 4.
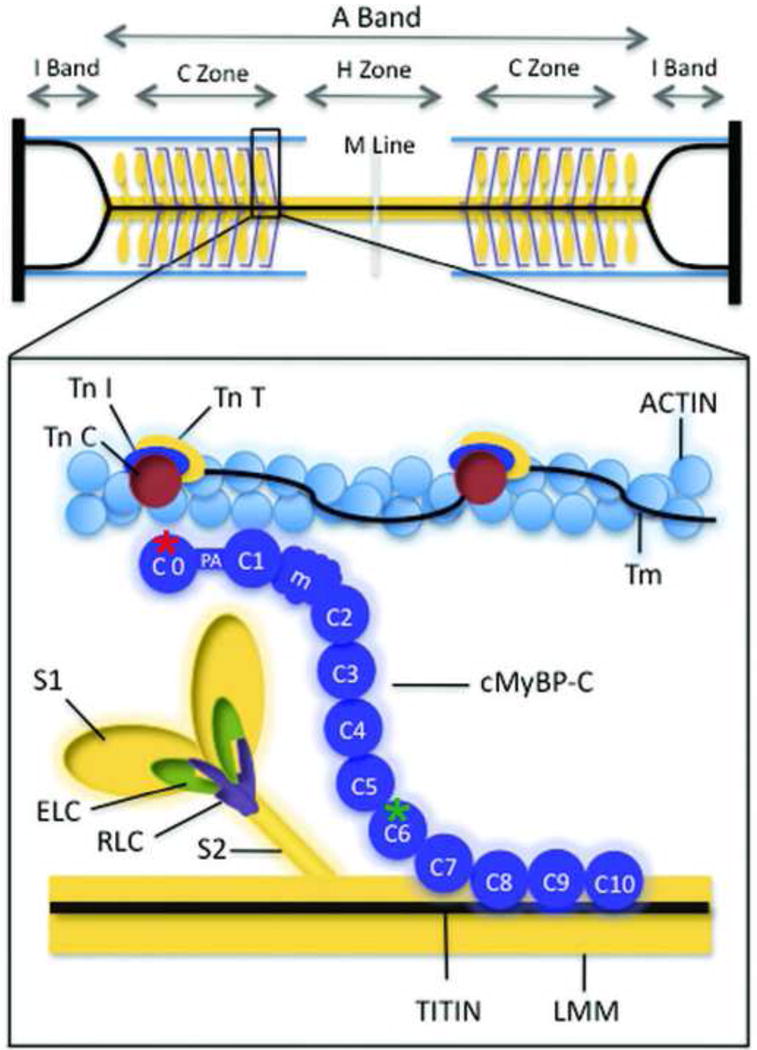
Myosin binding proteins of the thick filament are shown. This simplified diagram depicts major proteins of the thick and thin filaments. Myosin ELC and RLC are shown binding to myosin S1 near the junction with myosin S2. cMyBP-C is shown in a hypothetical arrangement extending from the thick filament to the thin filament, but the precise position of cMyBP-C relative to myosin is not known. Individual cMyBP-C domains are numbered C0 to C10, starting at the N terminus of cMyBP-C. Site of the amino acid sequence abnormalities produced by the Maine Coon and Ragdoll mutations are marked by a red and a green asterisk respectively. LMM indicates light meromyosin; m, the position of the cMyBP-C regulatory motif between domains C1 and C2 that contains PKA phosphorylation sites; PA, a proline/alanine-rich linker sequence between C0 and C1; Tm, tropomyosin; TnC, troponin C; TnI, troponin I; TnT, troponin T. (From Harris et al. Circ Res 2011;108 (6): 751–764 with permission).
The different domains and linkers of cMyBP-C are thought to mediate different functional effects of cMyBP-C by interacting with various other sarcomeric proteins. For instance, domains C8–C10 anchor cMyBP-C to myosin and titin in the thick filament and these interactions account for the characteristic localization of cMyBP-C under electron microscopy to a series of 7–9 discrete bands (corresponding roughly to 1 cMyBP-C molecule for every 9 myosin molecules) within each half sarcomere.115 Domains C1-M-C2 also bind to myosin but at another segment of the β-myosin heavy chain (i.e., the myosin S2 subfragment) that is closer to the catalytic force generating myosin heads (myosin S1).116,117 These N-terminal domains of cMyBP-C are thus thought to influence the ability of myosin to interact with actin and thereby to inhibit or enhance contraction. In addition, the same N-terminal domains along with C0 (a domain unique to cardiac isoforms of MyBP-C) also interact directly with actin and can promote activation of contraction through a novel mechanism that shifts tropomyosin on the thin filament.118 Activating effects of the N terminal domains of cMyBP-C could thus account for the ability of cMyBP-C to maintain force during systole and prolong the ejection phase of systole.119
Because the A31P substitution occurs in the C0 domain, it is possible that the mutation disrupts the ability of C0 to interact with actin and/or with the thin filament and thereby functions as a poison polypeptide to affect systolic or diastolic function. Consistent with this idea, 3 mutations in humans that cause HCM that occur in the M-domain of cMyBP-C (which also binds to actin) reduce the binding affinity of cMyBP-C for actin, whereas another mutation that causes HCM increases actin binding affinity.120 Alternatively, because C0 also interacts with the regulatory light chain (RLC) of myosin, the A31P mutation could disrupt interactions with RLC and thereby alter myosin force generating capabilities.121 Interestingly, very little is known regarding the “middle” domains of cMyBP-C (C3 through C7).122 Thus, the functional significance of C6 which harbors the abnormal amino acid created by the R820W mutation in Ragdoll cats and humans is currently unknown.
The idea that the A31P gene mutation could create a protein that functions as a poison polypeptide is further supported by the observation that the mutant A31P cMyBP-C is properly incorporated in muscle sarcomeres. As shown in Figure 5, an antibody specific for the A31P mutation recognized the abnormal A31P protein in myofibrils from heterozygous and homozygous affected cats.123 Furthermore, this A31P protein showed proper localization, appearing as two fluorescent (green) bands in each sarcomere where each band represents the merged fluorescence from the 7–9 cMyBP-C stripes in each half sarcomere. However, in the original study describing the A31P mutation Meurs et al found that the total amount of cMyBP-C in heterozygous and homozygous affected cats was reduced, suggesting that haploinsufficiency or other cellular effects could play a role.2 Consistent with this idea in silico algorithms predicted that the A31P mutation could disrupt folding of the C0 Ig domain and Ratti et al reported that, unlike wild-type recombinant C0, C0 could not be expressed as a soluble protein when the A31P mutation was included in the sequence.121 Other mutations in the Ig domains of cMyBP-C and titin also appear to reduce domain stability, potentially leading to increased susceptibility to proteolysis or degradation by the UPS.122,124
Figure 5.
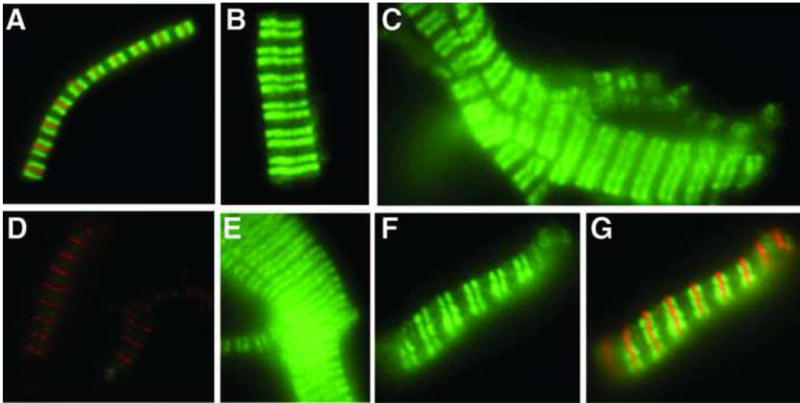
Incorporation of the A31P cMyBP-C missense mutation in sarcomeres of A31P-affected cats. Top Row, Immunofluorescent localization of cMyBP-C (green) was performed using a polyclonal antibody against cMyBPC. The antibody recognized cMyBP-C in myofibrils from unaffected wild-type (A), heterozygous (B), and homozygous A31P cats (C). Counterstaining with an antibody to myomesin (red) in A indicates M-line position. Bottom Row, Immunofluorescent localization of A31P mutant cMyBP-C (green) was performed using an affinity-purified antibody specific for the A31P mutation. The antibody did not cross-react with wild-type cMyBP-C in myofibrils from unaffected wild-type cats (D) but recognized A31P cMyBP-C in myofibrils from heterozygous (E) and homozygous (F) A31P cats. Counterstaining with an antibody to myomesin (red) indicates M-line position in D. G, Merged image of the same myofibril as shown in F showing counterstaining with an antibody to myomesin (red) to indicate M-line position. (From Harris et al. Circ Res 2011;108 (6): 751–764 with permission).
Additional studies are thus needed in cats with the A31P mutation to resolve the molecular basis of disease and to hopefully formulate effective therapies where none currently exist.125,126 For instance, by analogy with treatment of other human diseases, strategies can be used that seek to stabilize misfolded but otherwise functional proteins such as the case for mutations in the cystic fibrosis chloride channel gene or that target the UPS using drugs under development for cancer and neurodegenerative disorders.127,128 While it is true that genetic screening can remove the A31P and R820W alleles from cat populations by selective breeding, HCM remains the most common cause of heart failure in cats. By analogy with human HCM, feline HCM is likely to have multiple causes due to mutations in multiple genes. By understanding the molecular basis for HCM in cats carrying A31P and R820W mutations new therapeutic options with potentially broader applicability may emerge for use in both veterinary and human medicine.
Conclusion
HCM in cats and humans is a complex, multifactorial disease often resulting in heart failure, thromboembolism, arrhythmia, and sudden cardiac death. Following the discovery of the R403Q mutation in the β-myosin heavy chain as the first genetic cause of HCM in humans, enormous progress has been made in uncovering other genetic mutations linked to the disease and in understanding the molecular basis for the disease.34 Progress in understanding the genetic basis of the disease in humans led directly to the identification of two causative mutations (A31P and R820W) identified so far in domestic cats.2,3 By analogy with the human disease, it is hoped that additional genes and mutations in those genes will be identified as causative in cats using next generation sequencing methods. However, for both cats and humans, identification of the genetic causes of HCM is still only a first step toward the goals of preventing and treating HCM. The vast heterogeneity of underlying causes combined with other genetic and environmental modifiers that lead to diverse disease outcomes presents a formidable challenge to the idea that HCM can be treated as a single disease or with a single therapy. Instead, it is more likely that multiple final common pathways will emerge that can be identified and selectively targeted with therapeutics. Critical to discovery of these pathways will be a better understanding of the molecular and cellular consequences of different mutations and a better understanding of the natural history of the disease, including its earliest preclinical stages. Insights into these processes gained from understanding HCM in people, rodent models, and cats should therefore advance both human and veterinary medicine.
Abbreviations
- cMyBP-C
cardiac myosin binding protein-C
- HCM
hypertrophic cardiomyopathy
- LV
left ventricle
- LVOTO
left ventricular outflow tract obstruction
- MYH7
β-myosin heavy chain gene
- MYBPC3
cardiac myosin binding protein C gene
- RFLP
restriction fragment length polymorphism
- SAM
systolic anterior motion of the mitral valve
- TDI
tissue Doppler imaging
- UPS
ubiquitin-proteasome system
Definitions
- cDNA
Complementary DNA, which is DNA transcribed from messenger RNA and so contains only coding sequences (exons; lacks introns)
- C-terminus
The end of an amino acid chain (protein or polypeptide) terminated by a free carboxyl group (–COOH)
- Codon
A series of 3 DNA nucleotides that encode for an amino acid
- Conserved nucleotide sequence (highly conserved)
Similar or identical nucleotide sequences in a gene that are the same across species
- DNA nucleotide
Deoxyribose (sugar) plus one of four nitrogenous bases - adenine, cytosine, guanine, or thymine
- Haploinsufficiency
A reduced total amount of an affected protein (i.e., protein produced by a mutant gene) in the cell, which results in cellular dysfunction
- Heterozygous
An individual that carries one mutant copy and one normal copy of a gene
- Homozygous
An individual that carries two copies of a mutant gene, with one copy inherited from each parent
- N-terminus
The start of a protein or polypeptide chain that has an amino acid with a free amine group (–NH2)
- Poison polypeptide
An abnormal protein produced by a mutant gene that is incorporated into the cell and results in cellular dysfunction
- Polymorphism
Natural nucleotide variations in a gene that have no adverse effects on the individual and that occur with fairly high frequency in the general population
- Proband
An affected individual that is the basis for starting a genetic study
- Penetrance
The extent to which a particular gene or set of genes is expressed in the phenotypes of individuals carrying it, measured by the proportion of carriers showing the characteristic phenotype
- Phenotype
The composite of an organism’s observable characteristics or traits (e.g., its morphology, development, or biochemical or physiological properties)
References
- 1.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. The Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 2.Meurs KM, Sanchez X, David RM, Bowles NE, Towbin JA, Reiser PJ, Kittleson JA, Munro MJ, Dryburgh K, Macdonald KA, Kittleson MD. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet. 2005;14:3587–3593. doi: 10.1093/hmg/ddi386. [DOI] [PubMed] [Google Scholar]
- 3.Meurs KM, Norgard MM, Ederer MM, Hendrix KP, Kittleson MD. A substitution mutation in the myosin binding protein C gene in Ragdoll hypertrophic cardiomyopathy. Genomics. 2007;90:261–264. doi: 10.1016/j.ygeno.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Writing Committee Members. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–e831. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Seidman CE, Ackerman MJ, Towbin JA, Maron MS, Ommen SR, Nishimura RA, Gersh BJ. How should hypertrophic cardiomyopathy be classified?: What’s in a name? Dilemmas in nomenclature characterizing hypertrophic cardiomyopathy and left ventricular hypertrophy. Circ Cardiovasc Genet. 2009;2:81–86. doi: 10.1161/CIRCGENETICS.108.788703. [DOI] [PubMed] [Google Scholar]
- 6.Colquitt JL, Noonan JA. Cardiac findings in Noonan Syndrome on long-term follow-up. Congenit Heart Dis. 2014;9:144–150. doi: 10.1111/chd.12102. [DOI] [PubMed] [Google Scholar]
- 7.Boustany R-MN. Lysosomal storage diseases—the horizon expands. Nat Rev Neurol. 2013;9:583–598. doi: 10.1038/nrneurol.2013.163. [DOI] [PubMed] [Google Scholar]
- 8.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years. J Am Coll Cardiol. 2012;60:705–715. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 9.Landstrom AP, Ackerman MJ. Beyond the cardiac myofilament: hypertrophic cardiomyopathy-associated mutations in genes that encode calcium-handling proteins. Curr Mol Med. 2012;12:507–518. doi: 10.2174/156652412800620020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efthimiadis GK. Hypertrophic cardiomyopathy in 2013: Current speculations and future perspectives. World J Cardiol. 2014;6:26. doi: 10.4330/wjc.v6.i2.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29:549–555. doi: 10.1016/s0735-1097(96)00530-x. [DOI] [PubMed] [Google Scholar]
- 12.Tian T, Liu Y, Zhou X, Song L. Progress in the molecular genetics of hypertrophic cardiomyopathy: A mini-review. Gerontology. 2013;59:199–205. doi: 10.1159/000346146. [DOI] [PubMed] [Google Scholar]
- 13.Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, Marian AJ. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kittleson MD, Meurs KM, Munro MJ, Kittleson JA, Liu SK, Pion PD, Towbin JA. Familial hypertrophic cardiomyopathy in Maine Coon cats: An animal model of human disease. Circulation. 1999;99:3172–3180. doi: 10.1161/01.cir.99.24.3172. [DOI] [PubMed] [Google Scholar]
- 15.Maron BJ, Wolfson JK, Epstein SE, Roberts WC. Intramural (―small vessel‖) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1986;8:545–557. doi: 10.1016/s0735-1097(86)80181-4. [DOI] [PubMed] [Google Scholar]
- 16.Varnava AM, Elliott PM, Sharma S, McKenna WJ, Davies MJ. Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart Br Card Soc. 2000;84:476–482. doi: 10.1136/heart.84.5.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 18.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–1075. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 19.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, Wigle ED. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 20.Coats CJ, Hollman A. Hypertrophic cardiomyopathy: lessons from history. Heart. 2007;94:1258–1263. doi: 10.1136/hrt.2008.153452. [DOI] [PubMed] [Google Scholar]
- 21.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollman A, Goodwin JF, Teare D, Renwick JW. A family with obstructive cardiomyopathy (asymmetrical hypertrophy) Br Heart J. 1960;22:449–456. doi: 10.1136/hrt.22.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paré JAP, Fraser RG, Pirozynski WJ, Shanks JA, Stubington D. Hereditary cardiovascular dysplasia. Am J Med. 1961;31:37–62. doi: 10.1016/0002-9343(61)90222-4. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin JF. Prospects and predictions for the cardiomyopathies. Circulation. 1974;50:210–219. doi: 10.1161/01.cir.50.2.210. [DOI] [PubMed] [Google Scholar]
- 25.Darsee JR, Heymsfield SB, Nutter DO. Hypertrophic cardiomyopathy and human leukocyte antigen linkage: differentiation of two forms of hypertrophic cardiomyopathy. N Engl J Med. 1960;300:877–882. doi: 10.1056/NEJM197904193001602. [DOI] [PubMed] [Google Scholar]
- 26.Feigenbaum H. Evolution of echocardiography. Circulation. 1996;93:1321–1327. doi: 10.1161/01.cir.93.7.1321. [DOI] [PubMed] [Google Scholar]
- 27.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shoulson I, Bonilla E, Martin JB. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306:234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- 28.Seidman CE, Seidman JG. Identifying Sarcomere Gene Mutations in Hypertrophic Cardiomyopathy: A Personal History. Circ Res. 2011;108:743–750. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 30.Saez LJ, Gianola KM, McNally EM, Feghali R, Eddy R, Shows TB, Leinwand LA. Human cardiac myosin heavy chain genes and their linkage in the genome. Nucleic Acids Res. 1987;15:5443–5459. doi: 10.1093/nar/15.13.5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuoka R, Yoshida MC, Kanda N, Kimura M, Ozasa H, Takao A. Human cardiac myosin heavy-chain gene mapped within chromosome region 14q11.2-q13. Am J Med Genet. 1989;32:279–284. doi: 10.1002/ajmg.1320320234. [DOI] [PubMed] [Google Scholar]
- 32.Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP. The complete sequence of the human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics. 1990;8:194–206. doi: 10.1016/0888-7543(90)90272-v. [DOI] [PubMed] [Google Scholar]
- 33.Liew CC, Sole MJ, Yamauchi-Takihara K, Kellam B, Anderson DH, Lin LP, Liew JC. Complete sequence and organization of the human cardiac beta-myosin heavy chain gene. Nucleic Acids Res. 1990;18:3647–3651. doi: 10.1093/nar/18.12.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 35.Tanigawa G, Jarcho JA, Kass S, Solomon SD, Vosberg HP, Seidman JG, Seidman CE. A molecular basis for familial hypertrophic cardiomyopathy: an alpha/beta cardiac myosin heavy chain hybrid gene. Cell. 1990;62:991–998. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- 36.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy [see comments] N Engl J Med. 1992;326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 37.Marian AJ, Yu QT, Mares A, Jr, Hill R, Roberts R, Perryman MB. Detection of a new mutation in the beta-myosin heavy chain gene in an individual with hypertrophic cardiomyopathy. J Clin Invest. 1992;90:2156–2165. doi: 10.1172/JCI116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dufour C, Dausse E, Fetler L, Dubourg O, Bouhour JB, Vosberg HP, Guicheney P, Komajda M, Schwartz K. Identification of a mutation near a functional site of the beta cardiac myosin heavy chain gene in a family with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 1994;26:1241–1247. doi: 10.1006/jmcc.1994.1142. [DOI] [PubMed] [Google Scholar]
- 39.Thierfelder L, MacRae C, Watkins H, Tomfohrde J, Williams M, McKenna W, Bohm K, Noeske G, Schlepper M, Bowcock A. A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc Natl Acad Sci U S A. 1993;90:6270–6274. doi: 10.1073/pnas.90.13.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 41.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 42.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 43.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 44.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 45.Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 46.Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. 2010;55:1127–1135. doi: 10.1016/j.jacc.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 48.Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, Ikeda A, Kamatani N, Takeshima H, Matsuoka R. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet. 2007;52:543–548. doi: 10.1007/s10038-007-0149-y. [DOI] [PubMed] [Google Scholar]
- 49.Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XHT, Claycomb WC, Ko J-K, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunsø S, Svendsen JH, Olesen MS. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013;21:918–928. doi: 10.1038/ejhg.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapplinger JD, Landstrom AP, Bos JM, Salisbury BA, Callis TE, Ackerman MJ. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res. 2014;7:347–361. doi: 10.1007/s12265-014-9542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christiaans I, Nannenberg EA, Dooijes D, Jongbloed RJE, Michels M, Postema PG, Majoor-Krakauer D, van den Wijngaard A, Mannens MMAM, van Tintelen JP, van Langen IM, Wilde AAM. Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth Heart J. 2010;18:248–254. doi: 10.1007/BF03091771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA. The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet. 1999;65:1308–1320. doi: 10.1086/302623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Page SP, Kounas S, Syrris P, Christiansen M, Frank-Hansen R, Andersen PS, Elliott PM, McKenna WJ. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5:156–166. doi: 10.1161/CIRCGENETICS.111.960831. [DOI] [PubMed] [Google Scholar]
- 55.Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, Narasimhan C, Ayapati DR, Ayub Q, Mehdi SQ, Oppenheimer S, Richards MB, Price AL, Patterson N, Reich D, Singh L, Tyler-Smith C, Thangaraj K. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho CY. Hypertrophic cardiomyopathy in 2012. Circulation. 2012;125:1432–1438. doi: 10.1161/CIRCULATIONAHA.110.017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho CY, López B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, González A, Colan SD, Seidman JG, Díez J, Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Captur G, Mohun TJ, Finocchiaro G, Wilson R, Levine J, Conner L, Lopes L, Patel V, Sado DM, Li C, Bassett P, Herrey A, Tome Esteban M, McKenna WJ, Seidman C, Muthurangu V, Bluemke DA, Ho CY, Elliott PM, Moon JC. Advanced assessment of cardiac morphology and prediction of gene carriage by CMR in hypertrophic cardiomyopathy - The HCMNET/UCL collaboration. Heart. 2014;100:A72–A73. [Google Scholar]
- 59.Ho CY, Abbasi SA, Neilan TG, Shah RV, Chen Y, Heydari B, Cirino AL, Lakdawala NK, Orav EJ, Gonzalez A, Lopez B, Diez J, Jerosch-Herold M, Kwong RY. T1 Measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6:415–422. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation. 2000;102:1950–1955. doi: 10.1161/01.cir.102.16.1950. [DOI] [PubMed] [Google Scholar]
- 61.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 62.Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death [see comments] Lancet. 1993;342:1085–1086. doi: 10.1016/0140-6736(93)92064-z. [DOI] [PubMed] [Google Scholar]
- 63.Lechin M, Quiñones MA, Omran A, Hill R, Yu QT, Rakowski H, Wigle D, Liew CC, Sole M, Roberts R. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation. 1995;92:1808–1812. doi: 10.1161/01.cir.92.7.1808. [DOI] [PubMed] [Google Scholar]
- 64.Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med. 1986;315:610–614. doi: 10.1056/NEJM198609043151003. [DOI] [PubMed] [Google Scholar]
- 65.Mongiovì M, Fesslova V, Fazio G, Barbaro G, Pipitone S. Diagnosis and prognosis of fetal cardiomyopathies: a review. Curr Pharm Des. 2010;16:2929–2934. doi: 10.2174/138161210793176428. [DOI] [PubMed] [Google Scholar]
- 66.Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: Importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. doi: 10.1016/j.cardfail.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jay A, Chikarmane R, Poulik J, Misra VK. Infantile hypertrophic cardiomyopathy associated with a novel MYL3 mutation. Cardiology. 2013;124:248–251. doi: 10.1159/000347138. [DOI] [PubMed] [Google Scholar]
- 68.Zahka K, Kalidas K, Simpson MA, Cross H, Keller B, Galambos C, Gurtz K, Patton MA, Crosby AH. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency amongst the Amish. Heart. 2008 doi: 10.1136/hrt.2007.127241. [DOI] [PubMed] [Google Scholar]
- 69.Van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 70.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 71.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T, Zolk O. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 72.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 73.Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–91. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 74.Belus A, Piroddi N, Scellini B, Tesi C, d’ Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008;586:3639–3694. doi: 10.1113/jphysiol.2008.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Witjas-Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJM, Michels M, Ho CY, Kraft T, Poggesi C, van der Velden J. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol. 2014 doi: 10.1113/jphysiol.2014.274571. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011;109:86–96. doi: 10.1161/CIRCRESAHA.111.242974. [DOI] [PubMed] [Google Scholar]
- 77.Spudich JA. Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RT, Sadayappan S, Robbins J, Wieczorek DF, Solaro RJ, Wolska BM. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet. 2014;7:132–143. doi: 10.1161/CIRCGENETICS.113.000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 80.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wess G, Sarkar R, Hartmann K. Assessment of left ventricular systolic function by strain imaging echocardiography in various stages of feline hypertrophic cardiomyopathy. J Vet Intern Med. 2010;24:1375–1382. doi: 10.1111/j.1939-1676.2010.0586.x. [DOI] [PubMed] [Google Scholar]
- 82.Milani-Nejad N, Janssen PML. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol Ther. 2014;141:235–249. doi: 10.1016/j.pharmthera.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rottbauer W, Gautel M, Zehelein J, Labeit S, Franz WM, Fischer C, Vollrath B, Mall G, Dietz R, Kubler W, Katus HA. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest. 1997;100:475–482. doi: 10.1172/JCI119555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, Fischer C, Ochs J, McKenna WJ, Klues H, Vosberg HP. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101:1396–1402. doi: 10.1161/01.cir.101.12.1396. [DOI] [PubMed] [Google Scholar]
- 86.McConnell BK, Fatkin D, Semsarian C, Jones KA, Georgakopoulos D, Maguire CT, Healey MJ, Mudd JO, Moskowitz IP, Conner DA, Giewat M, Wakimoto H, Berul CI, Schoen FJ, Kass DA, Seidman CE, Seidman JG. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ Res. 2001;88:383–389. doi: 10.1161/01.res.88.4.383. [DOI] [PubMed] [Google Scholar]
- 87.Barefield D, Kumar M, de Tombe PP, Sadayappan S. Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. AJP Heart Circ Physiol. 2014;306:H807–H815. doi: 10.1152/ajpheart.00913.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 89.Carrier L, Knöll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine M-L, Fiszman M, Ross J, Schwartz K, Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 90.Cheng Y, Wan X, McElfresh TA, Chen X, Gresham KS, Rosenbaum DS, Chandler MP, Stelzer JE. Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. AJP Heart Circ Physiol. 2013;305:H52–H65. doi: 10.1152/ajpheart.00929.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 94.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang L, Ji X, Barefield D, Sadayappan S, Kawai M. Phosphorylation of cMyBP-C affects contractile mechanisms in a site-specific manner. Biophys J. 2014;106:1112–1122. doi: 10.1016/j.bpj.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- 97.Wess G, Schinner C, Weber K, Küchenhoff H, Hartmann K. Association of A31P and A74T polymorphisms in the myosin binding protein C3 gene and hypertrophic cardiomyopathy in Maine Coon and other breed cats. J Vet Intern Med. 2010;24:527–532. doi: 10.1111/j.1939-1676.2010.0514.x. [DOI] [PubMed] [Google Scholar]
- 98.Borgeat K, Casamian-Sorrosal D, Helps C, Luis Fuentes V, Connolly DJ. Association of the myosin binding protein C3 mutation (MYBPC3 R820W) with cardiac death in a survey of 236 Ragdoll cats. J Vet Cardiol. 2014;16:73–80. doi: 10.1016/j.jvc.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 99.Ripoll Vera T, Monserrat Iglesias L, Hermida Prieto M, Ortiz M, Rodriguez Garcia I, Govea Callizo N, Gómez Navarro C, Rosell Andreo J, Gámez Martínez JM, Pons Lladó G, Cremer Luengos D, Torres Marqués J. The R820W mutation in the MYBPC3 gene, associated with hypertrophic cardiomyopathy in cats, causes hypertrophic cardiomyopathy and left ventricular non-compaction in humans. Int J Cardiol. 2010;145:405–407. doi: 10.1016/j.ijcard.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 100.Konno T, Shimizu M, Ino H, Matsuyama T, Yamaguchi M, Terai H, Hayashi K, Mabuchi T, Kiyama M, Sakata K, Hayashi T, Inoue M, Kaneda T, Mabuchi H. A novel missense mutation in the myosin binding protein-C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J Am Coll Cardiol. 2003;41:781–786. doi: 10.1016/s0735-1097(02)02957-1. [DOI] [PubMed] [Google Scholar]
- 101.Fries R, Heaney AM, Meurs KM. Prevalence of the myosin-binding protein C mutation in Maine Coon cats. J Vet Intern Med. 2008;22:893–896. doi: 10.1111/j.1939-1676.2008.0113.x. [DOI] [PubMed] [Google Scholar]
- 102.Mary J, Chetboul V, Sampedrano CC, Abitbol M, Gouni V, Trehiou-Sechi E, Tissier R, Queney G, Pouchelon JL, Thomas A. Prevalence of the MYBPC3-A31P mutation in a large European feline population and association with hypertrophic cardiomyopathy in the Maine Coon breed. J Vet Cardiol. 2010;12:155–161. doi: 10.1016/j.jvc.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 103.Godiksen MT, Granstrom S, Koch J, Christiansen M. Hypertrophic cardiomyopathy in young Maine Coon cats caused by the p.A31P cMyBP-C mutation–the clinical significance of having the mutation. Acta Vet Scand. 2011;53:7. doi: 10.1186/1751-0147-53-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sampedrano C, Chetboul V, Mary J, Tissier R, Abitbol M, Serres F, Gouni V, Thomas A, Pouchelon JL. Prospective echocardiographic and tissue Doppler imaging screening of a population of Maine Coon cats tested for the A31P mutation in the myosin-binding protein C gene: A specific analysis of the heterozygous status. J Vet Intern Med. 2009;23:91–99. doi: 10.1111/j.1939-1676.2008.0218.x. [DOI] [PubMed] [Google Scholar]
- 105.Longeri M, Ferrari P, Knafelz P, Mezzelani A, Marabotti A, Milanesi L, Pertica G, Polli M, Brambilla PG, Kittleson M, Lyons LA, Porciello F. Myosin-binding protein C DNA variants in domestic cats (A31P, A74T, R820W) and their association with hypertrophic cardiomyopathy. J Vet Intern Med. 2013;27:275–285. doi: 10.1111/jvim.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Silverman SJ, Stern JA, Meurs KM. Hypertrophic cardiomyopathy in the Sphynx cat: A retrospective evaluation of clinical presentation and heritable etiology. J Feline Med Surg. 2012;14:246–249. doi: 10.1177/1098612X11435040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trehiou-Sechi E, Tissier R, Gouni V, Misbach C, Petit AMP, Balouka D, Carlos Sampedrano C, Castaignet M, Pouchelon JL, Chetboul V. Comparative echocardiographic and clinical features of hypertrophic cardiomyopathy in 5 breeds of cats: A retrospective analysis of 344 cases (2001–2011) J Vet Intern Med. 2012;26:532–541. doi: 10.1111/j.1939-1676.2012.00906.x. [DOI] [PubMed] [Google Scholar]
- 108.Meurs KM, Norgard MM, Kuan M, Haggstrom J, Kittleson M. Analysis of 8 sarcomeric candidate genes for feline hypertrophic cardiomyopathy mutations in cats with hypertrophic cardiomyopathy. J Vet Intern Med. 2009;23:840–843. doi: 10.1111/j.1939-1676.2009.0341.x. [DOI] [PubMed] [Google Scholar]
- 109.Meurs KM, Lahmers S, Keene BW, White SN, Oyama MA, Mauceli E, Lindblad-Toh K. A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Hum Genet. 2012;25:25. doi: 10.1007/s00439-012-1158-2. [DOI] [PubMed] [Google Scholar]
- 110.Meurs KM, Mauceli E, Lahmers S, Acland GM, White SN, Lindblad-Toh K. Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet. 2010;128:315–324. doi: 10.1007/s00439-010-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C: redefining its structure and function. Biophys Rev. 2012;4:93–106. doi: 10.1007/s12551-012-0067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tong CW, Nair NA, Doersch KM, Liu Y, Rosas PC. Cardiac myosin-binding protein-C is a critical mediator of diastolic function. Pflüg Arch - Eur J Physiol. 2014;466:451–457. doi: 10.1007/s00424-014-1442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bardswell SC, Cuello F, Kentish JC, Avkiran M. cMyBP-C as a promiscuous substrate: phosphorylation by non-PKA kinases and its potential significance. J Muscle Res Cell Motil. 2012;33:53–60. doi: 10.1007/s10974-011-9276-3. [DOI] [PubMed] [Google Scholar]
- 115.Luther PK, Bennett PM, Knupp C, Craig R, Padrón R, Harris SP, Patel J, Moss RL. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J Mol Biol. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453:254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- 117.Ababou A, Rostkova E, Mistry S, Masurier CL, Gautel M, Pfuhl M. Myosin binding protein C positioned to play a key role in regulation of muscle contraction: Structure and interactions of domain C1. J Mol Biol. 2008;384:615–630. doi: 10.1016/j.jmb.2008.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, Robbins J, Warshaw DM, Craig R. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci. 2014;111:2170–2175. doi: 10.1073/pnas.1316001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palmer BM, Georgakopoulos D, Janssen PM, Wang Y, Alpert NR, Belardi DF, Harris SP, Moss RL, Burgon PG, Seidman CE, Seidman JG, Maughan DW, Kass DA. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circ Res. 2004;94:1249–1255. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 120.Bezold KL, Shaffer JF, Khosa JK, Hoye ER, Harris SP. A gain-of-function mutation in the M-domain of cardiac myosin-binding protein-C increases binding to actin. J Biol Chem. 2013;288:21496–21505. doi: 10.1074/jbc.M113.474346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286:12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Idowu SM, Gautel M, Perkins SJ, Pfuhl M. Structure, stability and dynamics of the central domain of cardiac myosin binding protein C (MyBP-C): implications for multidomain assembly and causes for cardiomyopathy. J Mol Biol. 2003;329:745–761. doi: 10.1016/s0022-2836(03)00425-x. [DOI] [PubMed] [Google Scholar]
- 123.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Anderson BR, Bogomolovas J, Labeit S, Granzier H. Single molecule force spectroscopy on titin implicates immunoglobulin domain stability as a cardiac disease mechanism. J Biol Chem. 2013;288:5303–5315. doi: 10.1074/jbc.M112.401372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abbott JA. Feline hypertrophic cardiomyopathy: An update. Vet Clin North Am Small Anim Pract. 2010;40:685–700. doi: 10.1016/j.cvsm.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 126.Rishniw M, Pion PD. Is treatment of feline hypertrophic cardiomyopathy based in science or faith? J Feline Med Surg. 2011;13:487–497. doi: 10.1016/j.jfms.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol Ther. 2014 doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 128.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]