

SUMMARY
ECSIT is a mitochondrial complex I (CI)-associated protein that has been shown to regulate the production of mitochondrial reactive oxygen species (mROS) following engagement of Toll-like receptors (TLRs). We have generated an Ecsit conditional knockout (CKO) mouse strain to study the in vivo role of ECSIT. ECSIT deletion results in profound alteration of macrophage metabolism, leading to a striking shift to reliance on glycolysis, complete disruption of CI activity, and loss of the CI holoenzyme and multiple subassemblies. An increase in constitutive mROS production in ECSIT-deleted macrophages prevents further TLR-induced mROS production. Surprisingly, ECSIT-deleted cells accumulate damaged mitochondria because of defective mitophagy. ECSIT associates with the mitophagy regulator PINK1 and exhibits Parkin-dependent ubiquitination. However, upon ECSIT deletion, we observed increased mitochondrial Parkin without the expected increase in mitophagy. Taken together, these results demonstrate a key role of ECSIT in CI function, mROS production, and mitophagy-dependent mitochondrial quality control.
In Brief
Macrophages rely on fine-tuning their metabolism to fulfill their anti-bacterial functions. Carneiro et al. show that the complex I assembly factor ECSIT is an essential regulator of the balance between mitochondrial respiration and glycolysis and the maintenance of a healthy mitochondrial pool through mitophagy.
INTRODUCTION
ECSIT (evolutionarily conserved signaling intermediate in Toll pathways) was originally identified as a highly conserved TRAF6-binding protein (Kopp et al., 1999; Xiao et al., 2003). Further studies showed that ECSIT is ubiquitinated by TRAF6 and has a role in intracellular bacterial clearance (West et al., 2011). ECSIT was shown to localize to mitochondria (Vogel et al., 2007) and regulate mitochondrial reactive oxygen species (mROS) production that occurs following engagement of Toll-like receptors (TLRs) (West et al., 2011). Phagocytosis of bacteria by macrophages results in ECSIT-dependent recruitment of mitochondria to the phagosomal membrane (Geng et al., 2015). There, ECSIT-dependent mROS production promotes activation of the phagosomal nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system and ROS-dependent killing of engulfed microbes (West et al., 2011). ECSIT has also been described as a mitochondrial oxidative phosphorylation (OXPHOS) complex I (CI)-associated protein (Vogel et al., 2007). However, the link between ECSIT function as a CI accessory protein and innate immune signaling has remained unclear.
CI is the largest of the five OXPHOS complexes in the mitochondrial inner membrane. CI consists of 44 core and accessory subunits encoded in the mitochondrial and nuclear genomes. Its assembly is a highly complicated process that involves at least 14 additional assembly factors and stepwise assembly of intermediate subcomplexes (Guerrero-Castillo et al., 2017). CI and complex II (CII) are the entry points for electrons from NADH and FADH2, respectively. Electrons are then transferred to complex III (CIII) and complex IV, generating proton translocation from the mitochondrial matrix into the mitochondrial intermembrane space and establishing a proton motive force that drives generation of ATP through F1F0-ATP synthase (complex V) (reviewed by Sazanov, 2015).
The most commonly observed mitochondrial disorders are associated with CI dysfunction. Although CI disorders can result in variable and severe phenotypes, so far, few CI subunits have been targeted for deletion or knockdown in mice. Importantly, a number of CI deficiencies result from mutations in CI subunits critical for CI assembly (Alston et al., 2016; Bénit et al., 2003; Kirby et al., 1999; Koene et al., 2012; Loeffen et al., 2000; Mckenzie and Ryan, 2010; Nouws et al., 2010; Petruzzella and Papa, 2002). ECSIT interacts with the CI assembly factors NDUFAF1 and ACAD9 and was shown, using RNAi knockdown approaches, to be important for CI assembly and NDUFAF1 and ACAD9 stability in mitochondria in the HeLa and HEK293 cell lines (Nouws et al., 2010; Vogel et al., 2007). Furthermore, it was observed that CI levels were reduced, and a 500-kDa CI intermediate accumulated in ECSIT knockdown cells (Vogel et al., 2007). These results suggest that assembly or stability of the CI 1-MDa holoenzyme requires ECSIT. Importantly, although the levels of NDUFAF1 were severely impaired in these studies, the levels of NDUFS3 and ND1, two other CI subunits, remained unchanged (Vogel et al., 2007). Given the important role of NDUFAF1 and ACAD9 in CI assembly, it seemed that ECSIT might predominantly function in concert with these CI assembly factors as part of a central CI assembly complex, the mitochondrial CI assembly (MCIA) (Guerrero-Castillo et al., 2017; Heide et al., 2012; Nouws et al., 2010; Vogel et al., 2007). The function of the MCIA complex is supported by the existence of CI deficiency in patients harboring mutations in genes encoding components of this complex (Alston et al., 2016; Fassone et al., 2011; Mimaki et al., 2012; Nouws et al., 2010). So far, only one ECSIT mutation has been identified in human disease, the V140A variant that triggers hyperinflammation and promotes hyperphagocytic syndrome in extranodal natural killer/T cell lymphoma (ENKTL) (Wen et al., 2018). Furthermore, ECSIT function has only been examined using downregulation in cell lines.
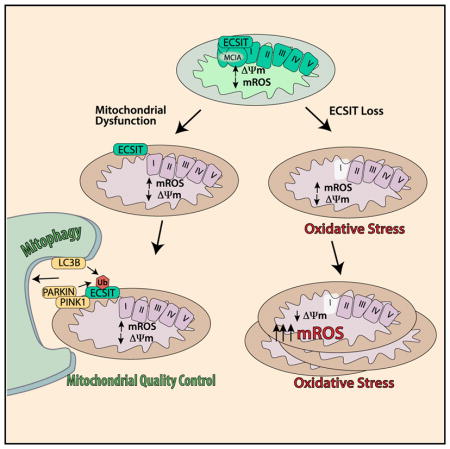
Interestingly, organization of the respiratory chain in macrophages, in particular adaptations in CI and complex II, has recently been shown to be instrumental for resistance to bacterial infection (Garaude et al., 2016; Kelly et al., 2015). Therefore, we decided to examine CI assembly and the expression of CI proteins upon ECSIT deletion in macrophages. Traditional Ecsit knockout (KO) mice have been generated; however, ECSIT deletion results in embryonic lethality, preventing the derivation of KO macrophages (Xiao et al., 2003). To overcome this difficulty, we have generated a conditionally targeted Ecsit knockout (CKO) mouse strain. Here we show that ECSIT can be efficiently deleted in macrophages derived from this mouse strain. ECSIT deletion leads to a metabolic shift in macrophages, accompanied by a complete loss of CI function and an increase in baseline mROS production. Strikingly, we also observed that, despite loss of mitochondrial membrane potential, ECSIT-deleted macrophages exhibit increased mitochondrial mass, suggesting a defect in the mitochondrial quality control pathway involving selective autophagy of damaged mitochondria, or mitophagy. Further analysis revealed that ECSIT interacts with several components of the PINK1/Parkin-dependent mitophagy pathway. Our studies therefore identify ECSIT as a crucial regulator of not only mitochondrial OXPHOS but also of inducible mROS production and mitophagy-dependent mitochondrial quality control.
RESULTS
Conditional Deletion of ECSIT in Macrophages
To study the role of ECSIT in mitochondria, and avoid the difficulties posed by early embryonic lethality of the traditional KO mice, we generated an Ecsit CKO mouse (Figure S1). Exon 3 in the Ecsit gene is shared among all three splice variants described in mice, and, therefore, deletion of exon 3 ensures the inactivation of all isoforms of Ecsit (Figure S1; Supplemental Experimental Procedures). Mice with the floxed (f) Ecsit allele were crossed either to Cre-ERT2 or LysM-Cre mice. Use of tamoxifen (Tam) resulted in complete deletion of Ecsit in cultured macrophages from ECSITf/f/Cre-ERT2+ mice compared with macrophages from ECSIT+/+/Cre-ERT2+ mice (Figures 1A, left, and S1D), whereas generation of ECSITf/−/LysM-Cre+ mice (cKO), carrying one floxed allele and one traditional KO allele of ECSIT, resulted in efficient deletion of ECSIT in macrophages both in vitro (bone marrow-derived macrophages [BMDMs]) and in vivo (peritoneal macrophages [PMs]) (Figure 1B). Surprisingly, given the early embryonic lethality resulting from germline Ecsit deletion, deletion of ECSIT only in macrophages had no significant effect on macrophage or monocyte numbers in various tissues (Figure 1C). Therefore, we were able to proceed with the use of conditional ablation of ECSIT to study the role of ECSIT in macrophage metabolism and function. For convenience, we have designated ECSITf/−/LysM-Cre+ BMDMs as cKO macrophages. In addition, we immortalized BMDMs (IBMMs) from the ECSITf/f/Cre-ERT2+ mouse to use in future experiments. IBMMs were treated with 500 nM of tamoxifen (induced knockout [iKO]) or vehicle (wild-type [WT]) for 48 h and washed and cultured for 5–12 days prior to examining ECSIT levels by western blot (Figure 1A, right).
Figure 1. Conditional Deletion of ECSIT in Macrophages Leads to Metabolic Alterations.

(A) Immunoblotting for ECSIT in lysates from ECSIT+/+/Cre-ERT2+ and ECSITf/f/Cre-ERT2+ bone-marrow-derived macrophages (BMDMs) 7 days after deletion induction with tamoxifen (Tam) or vehicle (−) (left) and ECSITf/f/Cre-ERT2+ immortalized BMDMs (iBMMs) treated with vehicle (WT) or tamoxifen (iKO) for 48 hr and cultured for 5 days (right). For subsequent figures, IBMMs were assessed 5–12 days after induction of deletion.
(B) ECSIT levels in ECSIT+/+/LysM-Cre+ (WT) and ECSITf/−/LysM-Cre+ (cKO) BMDMs and peritoneal macrophages (PMs).
(C) Cellularity and proportion in live cells of macrophages (CD11b+ F4/80+) and monocytes (CD11b+ F4/80−) from the spleen (Spln), lymph nodes (LNs), bone marrow (BM), peritoneal cavity (Perit), and lungs of WT and cKO mice (n = 3).
(D) Phenol red-containing cell culture media from ECSIT iKO and WT IBMMs.
(E) Lactate in supernatants from WT or cKO BMDMs, unstimulated (NS) or LPS-stimulated (100 ng/mL) (n = 2).
Shown are means ± SD of n experiments. *p < 0.05 in t test. See also Figure S1.
After ECSIT deletion, the iKO IBMM and cKO BMDM culture supernatants displayed a pronounced change of the phenol red pH indicator in the cell culture medium, suggesting that ECSIT-deleted cells were producing more acidic molecules than WT cells (Figure 1D and data not shown), which is reflective of a dramatic shift in metabolic activity. Genetic defects in CI can be accompanied by lactic acidosis resulting from a metabolic shift toward glycolysis (Bet et al., 1990; Houshmand et al., 1996). Consistent with this observation, there were higher levels of lactate in culture supernatants of cKO BMDMs (Figure 1E) and iKO IBMMs (data not shown), suggesting increased glycolytic activity in both BMDMs and IBMMs. Furthermore, although there was a significant increase in lactate production in lipopolysaccharide (LPS)-stimulated macrophages, consistent with the known glycolytic shift accompanying TLR stimulation (Kelly et al., 2015), there was no significant increase in lactate release in LPS-stimulated ECSIT cKO cells compared with unstimulated cKO cells (Figure 1E). Taken together, these results suggest a significant metabolic shift upon ECSIT deletion in macrophages.
ECSIT Deletion in Macrophages Leads to Changes in Cellular Energetics
To further investigate the metabolic effects of ECSIT deletion in macrophages, we used extracellular flux analysis to measure the extracellular acidification rate (ECAR), indicative of aerobic glycolysis, and oxygen consumption rate (OCR), indicative of mitochondrial respiration. ECSIT-deleted cells displayed a higher ECAR, consistent with increased lactate release and enhanced glycolysis (Figure 2A, left). Although the OCR was not significantly different between WT and iKO cells, there was a significant difference in the ratio between OCR and ECAR (OCR/ECAR), confirming the substantial metabolic shift seen upon ECSIT deletion (Figure 2A, right).
Figure 2. Increased Glycolysis in ECSIT-Deleted Macrophages.

(A) Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) measured in WT and iKO cells (left) and OCR/ECAR ratio (right) (n = 3).
(B) Cell growth measured by crystal violet staining of IBMMs maintained for 48 hr in DMEM without glucose with or without galactose, 5 days after deletion induction (n = 5).
(C) The ECAR was assessed after addition of glucose, oligomycin (oligo), and 2-deoxyglucose (2DG). Left: time course of a representative experiment. Right: determination of glycolysis rate, glycolytic capacity, and glycolytic reserve (n = 2).
Means ± SD of n experiments are shown. *p < 0.05 in t test. See also Figures S2 and S3.
To better understand the macrophage adaptations resulting from ECSIT deletion, we followed cell survival and proliferation in restricted substrate conditions. As expected, ECSIT deletion impaired growth in medium without glucose (Figure 2B). Similar growth impairment was observed when ECSIT-deleted cells were cultured with galactose alone, which does not allow the net production of ATP from glycolysis (Figure 2B). To confirm the increased reliance of macrophages lacking ECSIT on glycolysis, we cultured macrophages in the presence of oxamate, an LDH inhibitor required for lactic acid release and completion of glycolysis. ECSIT iKO macrophages were more sensitive to oxamate than control macrophages (Figure S2A). Consistent with the importance of pyruvate metabolism for lactate production, ECSIT-deleted macrophages benefited more from pyruvate supplementation than WT macrophages (Figure S2B). Furthermore, iKO macrophages were more sensitive to replacement of glucose with glutamine, suggesting that glutamine metabolism and the tricarboxylic acid (TCA) cycle are impaired (Figure S2C).
Given the increase in lactate production and reliance on glucose for proliferation, we further characterized glycolysis in WT and ECSIT iKO macrophages by measuring the ECAR under different metabolic conditions: after addition of glucose to enable glycolysis; after addition of oligomycin (Oligo), which inhibits mitochondrial ATP synthase, thereby forcing glycolysis; and after addition of the glycolysis inhibitor 2-deoxyglucose (2DG) to confirm the specificity of glycolysis induction. Glycolysis was enhanced after addition of glucose in iKO IBMMs; however, glycolytic capacity (the maximum glycolysis rate in the presence of oligomycin) and glycolytic reserve (the quantitative difference between maximum glycolysis and basal glycolysis) were not significantly different from WT IBMMs (Figure 2C). These changes do not reflect a glycolytic reprogramming at the level of gene expression because mRNA levels of main glycolytic regulators and enzymes, including lactate dehydrogenase A (LDHA) and MCT1, were unchanged in iKO IBMMs (Figure S3). Taken together, these results suggest that CI impairment results in enhanced glycolytic flux in ECSIT-deleted IBMMs.
ECSIT Is Required for CI Function and Mitochondrial OXPHOS
Consistent with the increased reliance of ECSIT-deleted macrophages on glucose, we found that inhibition of glycolysis with 2DG resulted in a more profound decrease in ATP levels in ECSIT iKO and cKO macrophages compared with WT controls (Figure 3A and data not shown). Thus, ECSIT KO cells were more dependent on glycolysis for ATP production, suggesting that increased glycolysis might be a mechanism to compensate for the defect in mitochondrial respiration. We measured the OCR in WT and iKO IBMMs after sequential addition of oligomycin to abrogate the use of OXPHOS and oxygen for ATP production, the protonophore carbonyl cyanide-4-(trifluoromethoxy)phenyl-hydrazone (FCCP) to dissipate the proton gradient leading to oxygen reduction independent of ATP production, and finally the electron transport chain inhibitor rotenone to confirm the specificity of respiratory chain function and identify the level of OXPHOS-independent OCR. ECSIT-deleted IBMMs had a markedly decreased spare respiratory capacity (SRC), as defined by the quantitative difference between the maximal OCR induced by FCCP and the initial basal OCR (Figure 3B). IBMMs lacking ECSIT also exhibited a decreased OCR used for ATP production and proton leak (Figure 3B). Notably, each of these functions depends on establishment of the proton gradient across the mitochondrial inner membrane. This suggests that proton translocation and the mitochondrial membrane potential are impaired in ECSIT-deleted cells, consistent with CI dysfunction. In contrast, coupling (the amount of oxygen consumed to make ATP) was similar between WT and iKO cells, indicating that mitochondrial ATP synthase function is unaffected (Figure 3B, right). Thus, the residual OCR may come from CII function, but it cannot compensate for CI dysfunction in energy-demanding situations, assessed under maximal respiration conditions. Therefore, ECSIT-deleted macrophages are likely to be more sensitive to metabolic stress, as can occur in inflammatory sites where macrophages fulfill their functions.
Figure 3. Loss of CI Activity in the Absence of ECSIT.

(A) ATP levels in iKO and WT IBMMs left untreated (UT) or treated with 50 mM 2DG for 4 hr (n = 3).
(B) OCR in iKO and WT IBMMs, measured after addition of oligomycin, FCCP, and rotenone. Left: time course of a representative experiment. Right: determination of the OCR used for ATP production. SRC, spare respiratory capacity, OCR because of proton leak, and coupling efficiency (n = 2).
(C) CI in-gel activity of mitochondrial complexes from WT and iKO IBMMs; shown is a representative experiment (n = 3). DLDH, dihydrolipoamide dehydrogenase.
(D) CI activity in WT and iKO IBMM mitochondria; CAM, chloramphenicol; shown is a representative experiment (n = 3).
(E) NADH/NAD+ ratio in lysates of iKO IBMMs (n = 2) and cKO BMDMs (n = 3) compared with WT cells.
Shown are means ± SD of n experiments. *p < 0.05 in t test.
To directly examine CI function in the absence of ECSIT, we prepared mitochondria from WT and iKO cells and performed blue native gel electrophoresis (BN-PAGE) followed by in-gel CI activity. A complete lack of CI activity was observed in macrophages lacking ECSIT at the level of the 1-MDa holoenzyme, and no subcomplex with residual CI activity was detected (Figure 3C). Next we measured oxidation of NADH to nicotinamide adenine dinucleotide (NAD+) and observed that CI NADH dehydrogenase activity was fully abolished in mitochondria from ECSIT-deleted macrophages (Figure 3D). Consistent with this finding, the NADH/NAD+ ratio was higher in ECSIT-deleted macrophages (Figure 3E). Taken together with the observed glycolytic shift, these results suggest a complete loss of CI activity and strong impairment of OXPHOS.
ECSIT Is Required for CI Assembly and Stability of CI Proteins
Previously it was observed that proteins in other CI subcomplexes, such as NDUFS3, were minimally affected by small interfering RNA (siRNA)-mediated ECSIT knockdown (Nouws et al., 2010; Vogel et al., 2007). This suggested that, although ECSIT was pivotal for the formation or stability of ECSIT-containing assembly subcomplexes, other CI holoenzyme intermediates were not dependent on ECSIT. Given the profound disruption of CI activity observed in ECSIT-deleted macrophages (Figure 3), we performed BN-PAGE followed by western blotting to examine CI and the ECSIT-containing assembly complexes. Consistent with previous reports, we found that the levels of NDUFAF1 in the ECSIT-associated subcomplexes (≈400 kDa and 700 kDa) in mitochondrial preparations were significantly decreased (Figure 4A), suggesting that function of the MCIA complex might be impaired (Baertling et al., 2017; Guerrero-Castillo et al., 2017). However, in contrast to previous reports, we found that other components of CI, NDUFS3 and ND6, were substantially diminished in ECSIT KO cells and importantly, did not accumulate in detectable assembly intermediates (Figure 4A). CII (succinate dehydrogenase complex, subunit A [SDHA]), complex III (Uqcrc2), and complex V (ATP5F1a and b) were unaffected (Figure 4B). Thus, ECSIT deletion in macrophages resulted in significant disruption of the CI holoenzyme and abrogation of CI subassembly formation without affecting other complexes of the respiratory chain.
Figure 4. Loss of CI Proteins in ECSIT-Deleted Macrophages.

(A and B) BN-PAGE followed by western blot of mitochondrial complexes from WT and iKO IBMMs detected with the indicated proteins from (A) complex I and (B) complexes II, III, V, and VDAC1.
(C and D) Immunoblot of OXPHOS proteins in (C) mitochondrial preparation and total cell lysates of IBMM and (D) total cell lysate of BMDM and PMs.
(E) Immunoblotting of IBMMs transduced with doxycycline-inducible NDUFAF1 (Tet-NDUFAF1) or control (Tet-Empty) and treated with tamoxifen on day 0 and day 1 to induce ECSIT deletion and doxycyclin (Dox) every 24 hr from day 0 or day 1 until analysis on day 3.
Shown are representative experiments (n = 3). See also Figure S4.
To assess whether these results reflect a complete loss of CI proteins versus a failure of mitochondrial import, we performed immunoblotting for several CI components using mitochondrial and total cellular lysates from multiple macrophage types. Upon deletion of ECSIT, we observed a severe reduction in the assembly factor NDUFAF1 and ND6, not only in mitochondrial lysates of IBMMs but also in total cell lysates of IBMMs (Figure 4C), BMDMs, and PMs (Figure 4D). This suggests that ECSIT is essential for maintenance and/or stability of the early 293- and 357-kDa subcomplexes formed by ECSIT and ECSIT binding partners, NDUFAF1, ACAD9, and TMEM126B, components of the MCIA complex, and subsequent subcomplex intermediates, incorporating ND6 (Guerrero-Castillo et al., 2017). We also observed significant loss of another CI subunit, NDUFS3, which has been reported to assemble in an early 170-kDa subcomplex before intervention of ECSIT and the MCIA complex, suggesting broad effects of ECSIT deletion on assembly and abundance of CI subunits.
Given the role of ECSIT in the regulation of multiple transcription factor pathways (Kopp et al., 1999; Xiao et al., 2003), we assessed whether the changes in mitochondrial CI protein levels could be attributable to changes in gene expression. However, despite significant decreases in protein levels, mRNAs encoding these and other mitochondrial respiratory chain components were unchanged in ECSIT-deleted cells (Figure S4A). This suggested that the protein levels of CI subunits were regulated by post-translational mechanisms upon ECSIT loss. Inhibition of proteasomal or lysosomal protein degradation using MG132 or Bafilomycin A1 (BafA1), respectively, did not restore the protein levels of NDUFS3 and NDUFAF1 (Figure S4B), suggesting that other mitochondrial proteases (e.g., LON-ClpP; Pryde et al., 2016), might be responsible for degradation of CI subunits. To test whether the other main assembly factor, NDUFAF1, could rescue CI subunit levels, we reintroduced NDUFAF1 in ECSIT-deleted macrophages. IBMMs were stably transduced with a doxycycline-inducible lentivirus vector expressing NDUFAF1 (Tet-NDUFAF1) or an empty vector (Tet-empty). Cells were treated with tamoxifen to delete ECSIT and with doxycycline to induce NDUFAF1 expression at the same time (day 0), or 24 hr later (day 1). We observed that reintroduction of NDUFAF1 did not restore the protein level of NDUFS3 (Figure 4E), suggesting that the contribution of ECSIT to CI assembly is not simply to stabilize NDUFAF1. Taken together, these results demonstrate broad effects of ECSIT deletion on the composition of CI and a requirement for ECSIT in the maintenance or stability of both the CI holoenzyme and multiple CI subcomplexes.
Dysregulation of mROS Production and Loss of Mitochondrial Membrane Potential in Macrophages Lacking ECSIT
OXPHOS dysfunction is known to lead to ROS production (Breuer et al., 2013; Lin and Beal, 2006). Therefore, we examined the production of cellular ROS in WT and ECSIT-deleted macrophages. We observed a significant increase in total cellular ROS in ECSIT-deleted cells (Figure 5A). We previously showed that ECSIT was required for increased mROS production upon exposure of macrophages to bacterial pathogens and that ECSIT knockdown impaired this inducible mROS production without altering baseline levels (West et al., 2011). However, we found that the basal level of mROS was significantly enhanced in ECSIT cKO BMDMs (Figure 5B), a finding consistent with oxidative stress stemming from CI dysfunction (Leadsham et al., 2013; Pagano et al., 2014; Pitkanen and Robinson, 1996). No further increase in ROS or mROS production was observed when ECSIT-deleted macrophages were treated with the CI inhibitor rotenone or LPS (Figures 5A and 5B). When macrophages were treated with antimycin to inhibit CIII, which, along with CI, is an important source of mROS production, we observed an increase in mROS in both WT and ECSIT-deleted macrophages (Figure 5C). Thus, although ECSIT-deficient cells retain the capacity to increase mROS production, they fail to do so upon LPS stimulation. This is not likely to be due to defects in the TLR signaling pathway because ECSIT-deleted BMDMs exhibit normal induction of the pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) following LPS exposure (Figure S5). These results are consistent with our previous finding that TLR-induced mROS production is dependent on ECSIT (West et al., 2011) and suggest that ECSIT may regulate mROS production during the innate immune response by regulating CI assembly and function.
Figure 5. Mitochondrial Dysfunction in Cells Lacking ECSIT.

(A) IBMMs stimulated with rotenone (1 μM) and LPS (100 ng/mL) for 20 min 7 days after deletion induction, stained with chloromethyl derivative of H2DCFDA (2′,7′-dichlorodihydrofluorescein diacetate) (CM-H2DCFDA) and analyzed by flow cytometry for total ROS (n = 3).
(B) BMDMs stimulated with rotenone and LPS for 20 min, stained with MitoSOX, and analyzed by flow cytometry for mROS (n = 3).
(C) IBMMs stimulated with antimycin A (5 μM) for 20 min and analyzed for mROS like in (B) (n = 3).
(D) Δψm measured by flow cytometry using tetra-methylrhodamine, methyl ester (TMRM) in IBMMs and BMDMs, in the absence (non treated, NT) or presence of CCCP for 1 h (30 μM) (n = 3).
Shown are means ± SD of n experiments. *p < 0.05 in t test. See also Figure S5.
Both direct analysis of CI and changes seen in macrophage metabolism suggest complete loss of CI function upon ECSIT deletion. Maintenance of mitochondrial membrane potential (Δψm) is essential for mitochondrial function. Analysis of Δψm, with uncoupler carbonyl cyanide m-chlorophenyl hydrazine (CCCP) treatment providing a control for collapsed mitochondrial Δψm, revealed that ECSIT-deleted IBMMs and BMDMs have a lower Δψm compared with WT cells (Figure 5D). Thus, consistent with its role in CI assembly and function, loss of ECSIT results in the appearance of indicators of mitochondrial damage, including increased constitutive mROS production and decreased Δψm. These defects also result in loss of LPS-inducible mROS production, which is critical for macrophage bactericidal function.
Defective Mitochondrial Quality Control in ECSIT-Deleted Macrophages
The loss of Δψm in macrophages lacking ECSIT should activate mitochondrial quality control through mitophagy, as occurs upon chemically induced mitochondrial damage (Ashrafi and Schwarz, 2013; Narendra et al., 2008). Therefore, we examined mitochondrial content in WT and ECSIT-deleted macrophages. Surprisingly, and despite decreased Δψm and increased mROS, we found increased mitochondrial DNA content in macrophages lacking ECSIT (Figure 6A). Similarly, we observed increased mitochondrial mass using mitotracker green, which stains mitochondria independently of Δψm (Figure 6B). To assess whether mitophagy is being triggered in ECSIT-deleted cells, we examined mitochondrial recruitment of LC3bII, a late-stage mitophagy marker necessary for lysosomal degradation. We observed a decrease in total and mitochondrial LC3bII protein levels in macrophages lacking ECSIT (Figure 6C). Loss of total LC3bII could indicate an increase in mitophagy or autophagic flux. However, when macrophages were treated with BafA1 to inhibit autophagy, we observed accumulation of LC3bII in WT but not ECSIT-deleted macrophages (Figure 6D). Likewise, the decreased accumulation of VDAC1, TOM20, and Parkin in ECSIT-deleted cells further confirmed that mitophagy was impaired (Figure 6D). These results suggest a role for ECSIT in selective autophagy of damaged mitochondria.
Figure 6. Altered Mitophagy in ECSIT-Deleted Macrophages.

(A) Ratio of mitochondrial DNA (mtDNA) over nuclear DNA (nucDNA) copies, determined by qPCR in IBMMs 7 days after deletion induction (n = 4).
(B) IBMMs stained with mitotracker green and analyzed by flow cytometry (n = 4). (A and B) Fold change over WT. Shown are means ± SEM of n experiments. *p < 0.05 in t test.
(C and E) Western blot analysis of (C) LC3b and (E) Parkin in total lysate, isolated mitochondria, and cytosol of WT and iKO IBMM (representative experiment of n = 3).
(D) Western blot analysis of cellular lysates of WT and iKO IBMMs treated with 5 nM BafA1 (representative experiment of n = 3).
Upon loss of Δψm, mitophagy is triggered through the PINK1/Parkin system. PINK1 is a ser/thr kinase that is normally imported to the inner mitochondrial membrane and constitutively degraded (Ashrafi and Schwarz, 2013). Upon loss of Δψm, PINK1 import and, therefore, degradation are impaired, and PINK1 accumulates on the outer mitochondrial membrane (OMM) (Narendra et al., 2010). This results in Parkin recruitment and Parkin-dependent ubiquitination of target proteins on the OMM. The ubiquitinated substrates are recognized by adaptor proteins, which mediate recruitment of LC3bII. Therefore, one reason for decreased LC3bII recruitment could be decreased Parkin levels at mitochondria. Instead, when mitochondrial Parkin was examined by western blot, both total and, especially, mitochondrial Parkin protein levels were increased in ECSIT-deficient cells (Figures 6D and 6E). This suggests that the mitochondrial damage that occurs following ECSIT deletion initiates PINK1/Parkin-dependent mitophagy but that Parkin accumulation fails to lead to LC3bII recruitment in the absence of ECSIT. Therefore, selective mitochondrial autophagy is impaired downstream of mitochondrial Parkin accumulation but upstream of LC3bII recruitment.
ECSIT Is a Parkin Substrate
To understand how ECSIT contributes to mitophagy, we investigated the interaction of ECSIT with components of the PINK1/Parkin mitophagy pathway. When co-expressed in 293FT cells, we observed co-immunoprecipitation of ECSIT with PINK1 (Figures 7A and 7B) (Beilina et al., 2005). This interaction was dependent upon mitochondrial localization of ECSIT because it was decreased with a mutant form of ECSIT lacking the mitochondrial localization sequence (Figure 7B; ΔMLS). Furthermore, we observed co-immunoprecipitation of ECSIT with LC3b upon induction of mitophagy with the uncoupling agent CCCP (Figure 7C). When we assessed ECSIT by immunoblotting following induction of mitochondrial damage by CCCP treatment of macrophages, we observed the rapid appearance of a slower-migrating form of ECSIT (Figure 7D, indicated by an arrow), consistent with post-translational modification, such as ubiquitination or stabilization, of full-length ECSIT containing the mitochondrial targeting sequence at the OMM. Interestingly, when immunoprecipitating PINK1, we only recovered the slower-migrating form of ECSIT, whereas the processed form of ECSIT, which co-migrates with ECSITΔMLS, did not interact with PINK1 (Figure 7B). Similarly, we observed interaction between endogenous ECSIT and full-length PINK1 at steady state in BMDMs and IBMMs. This was enhanced by treatment with BafA1 and CCCP, blocking degradation and inducing damage (Figures 7E and 7F). ECSIT also interacted with LC3bII upon mitochondrial damage induction. Furthermore, upon induction of mitochondrial damage and inhibition of autophagy with BafA1, we were able to observe accumulation of multiple slower-migrating forms of ECSIT (Figure 7G). Interestingly, we did not see accumulation of these higher-molecular-weight ECSIT species in the absence of BafA1 treatment (data not shown), suggesting that these species may be normally degraded by autophagic mechanisms. We have previously reported that LPS stimulation leads to accumulation of ECSIT at the OMM (West et al., 2011). Here we see that mitochondrial damage results in accumulation of unprocessed ECSIT (Figures 7C and 7D), ECSIT modification, and association with LC3b.
Figure 7. ECSIT Interacts with Mitophagy Regulators.

(A and B) Coimmunoprecipitation (coIP) of (A) PINK1 with ECSIT and (B) ECSIT with PINK1 and western blot analysis of immunoprecipitated proteins and input lysate after overexpression in 293FT cells (ΔMLS, ECSIT lacking the MLS).
(C) CoIP of LC3b with ECSIT and western blot after overexpression in 293FT cells with or without CCCP for 1 hr (30 μM).
(D) Western blot analysis of ECSIT in lysates of WT IBMMs treated with LPS (100 ng/mL), CCCP (30 μM), or vehicle (DMSO) for the indicated times.
(E and F) Western blot analysis of proteins coimmunoprecipitated with anti-ECSIT or control antibody (immunoglobulin heavy chain [Ig]) in lysates of WT BMDMs (E) or IBMMs (F) treated with 10 nM Bafilomycin A1 (Ba1) for 5 hr and/or 20 μM of CCCP (CC) for 1 hr.
(G) IP of ECSIT and control IP (ctrl) and western blot analysis of immunoprecipitated ECSIT (top, long exposure; bottom, short exposure) and input lysate in WT IBMMs treated with LPS (100 ng/mL), CCCP (30 μM), BafA1 (10 nM), or vehicle (−) for 1 hr.
(H) IP of ubiquitinated proteins and western blot analysis for ECSIT after overexpression of ECSIT, Parkin, PINK1, and VSV-tagged ubiquitin in 293FT.
Given that ECSIT interacts with PINK1 and, upon mitochondrial damage, is likely ubiquitinated and associates with LC3b, we wondered whether ECSIT might be a substrate for Parkin. We therefore tested whether overexpression of Parkin, with or without PINK1, could induce ECSIT ubiquitination (Yoshii et al., 2011). When Parkin was expressed together with vesicular stomatitis virus (VSV)-tagged ubiquitin in 293FT cells, we observed increased ubiquitination of ECSIT (Figure 7H). Parkin-induced ECSIT ubiquitination was further increased by PINK1 co-expression (Figure 7H), consistent with the role of PINK1 in the mitochondrial recruitment of Parkin and activation of Parkin ubiquitin ligase activity. Furthermore, Parkin/PINK1-induced ECSIT ubiquitination was dependent on mitochondrial localization of ECSIT because it was not observed using ECSITΔMLS (Figure 7H). Taken together, these findings suggest that ECSIT may be an important substrate for PINK1/Parkin-mediated autophagy. Consequently, upon loss of ECSIT, both mitochondrial OXPHOS and mitochondrial quality control are impaired, resulting in accumulation of damaged mitochondria with increased mROS production.
DISCUSSION
We have successfully generated a conditional KO mouse for ECSIT, a mitochondrial CI assembly factor. The conditional KO mouse represents a unique model to study the function of ECSIT in different tissues and cell types. Macrophages lacking ECSIT exhibit profound disruption of mitochondrial CI. ECSIT deletion led to increased dependence on glycolysis and mitochondrial respiratory chain dysfunction. Although the observed increase in mROS production and decrease in Δψm would be expected to increase mitophagy, we observed an increase in mitochondrial mass. Activation and recruitment of the mitophagy machinery is disrupted in cells lacking ECSIT. We found that ECSIT undergoes ubiquitination following mitochondrial damage and that ECSIT can interact with PINK1, Parkin, and LC3bII. Finally, we showed that Parkin induces ubiquitination of mitochondrial ECSIT. Upon loss of ECSIT, we observed increased mitochondrial accumulation of Parkin, suggesting initiation of mitophagic pathways. However, LC3bII recruitment is diminished and accumulation of damaged mitochondria occurs. These results suggest that ECSIT is a key mediator of PINK1/Parkin-dependent mitophagy, allowing recruitment of the degradative autophagy machinery downstream of mitochondrial damage signaling. In the future, it will be important to determine which of the selective autophagy receptors mediate ECSIT-dependent LC3bII recruitment to damaged mitochondria or whether ECSIT can act as a mitophagy receptor to more fully elucidate the role of ECSIT in mitophagy.
ECSIT has previously been described as a CI chaperone and was identified in a subcomplex of 370 kDa known as the MCIA complex, containing TMEM126B, NDUFAF1, and ACAD9 (Heide et al., 2012). More recently, it has been reported that TMEM126B and another transmembrane protein, TIMMDC1, have a role in putting together two membrane arm subcomplexes (Andrews et al., 2013; Guarani et al., 2014). TIMMDC1 immunoprecipitated with ECSIT and NDUFAF1, and immunoprecipitation (IP)-mass spectrometry (MS) analysis showed several interactions of this protein with CI subunits, including all proteins of the MCIA complex (Guarani et al., 2014). The MCIA complex likely helps to assemble the membrane arm of CI with TIMMDC1 (Guerrero-Castillo et al., 2017). Previously, Vogel et al. (2007) showed that ECSIT knockdown reduced NDUFAF1 levels and impaired CI enzymatic activity and assembly in HeLa cells. Although the NDUFAF1 protein was completely absent in ECSIT knockdown cells, it appeared that levels of other CI components (i.e., NDUFS3) were unaffected, resulting in accumulation of a 500-kDa CI intermediate (Vogel et al., 2007). Surprisingly, in ECSIT KO macrophages, the 370-kDa subcomplex is not detected, and we observed loss of the NDUFS3 subunit, which is involved in the earliest steps of CI assembly (Guarani et al., 2014; Mckenzie and Ryan, 2010; Mimaki et al., 2012; Vartak et al., 2014). Furthermore, and in contrast to knockdown experiments, no CI intermediates were detected (Figures 3 and 4). ECSIT-deleted macrophages hence exhibit a more severe phenotype than previously observed with ECSIT knockdown approaches. These results suggest an essential role for ECSIT in CI assembly/stability in macrophages. Although NDUFAF1 is a well described CI assembly factor, and ECSIT deletion leads to its disappearance, it is unlikely that the role of ECSIT in CI assembly is only to stabilize NDUFAF1 because NDUFAF1 knockdown leads to impaired CI assembly but not its complete disruption (Nouws et al., 2010; Vogel et al., 2007). In addition, we show that the reintroduction of NDUFAF1 in ECSIT-deleted cells does not restore normal levels of NDUFS3 protein, suggesting that NDUFAF1 alone is not sufficient to restore CI subunits. Moreover, although TIMMDC1 is involved in the membrane anchoring of the Q subcomplex containing NDUFS3, TIMMDC1-depleted cells did not show reduced levels of NDUFS3 or NDUFAF1 (Guarani et al., 2014), suggesting that the role of ECSIT in CI assembly is independent of TIMMDC1 as well. One possibility is that ECSIT has a role in stabilizing an early peripheral arm intermediate containing NDUFS3 by facilitating its anchoring in the membrane by ND1, for example (Guerrero-Castillo et al., 2017). It is important to highlight that ECSIT and NDUFS3 were co-purified by tandem affinity purification (TAP) tag and IP-MS, indicating a putative interaction between these proteins (Guarani et al., 2014; West et al., 2011).
The differences observed in our data and that of Vogel et al. (2007) might be due to the use of different cell types or due to the fact that previous experiments were performed using a knockdown approach. Importantly, nuclear encoded mRNA levels for NDUFAF1 and NDUFS3 and mitochondrially encoded ND6 mRNA levels were unchanged in ECSIT-deleted cells (Figure S4A), indicating that the regulation of CI disappearance is post-transcriptional. Mitochondria have a sophisticated quality control system that repairs or degrades misfolded, oxidized, or unassembled proteins. This system includes several mitochondrial chaperones and proteases besides the ubiquitin-proteasome system (UPS) (Heo and Rutter, 2011; Karbowski and Youle, 2011; Livnat-Levanon and Glickman, 2011). However, the UPS pathway and lysosomal degradation did not seem to be involved in the degradation of the CI subunits NDUFAF1 and NDUFS3 (Figure S4B). It is possible that the CI subunits are translocated and imported to the mitochondria to be degraded by mitochondrial proteases. It is important to note that some non-assembled OXPHOS subunits have been described as substrates for mitochondrial ATPases associated with diverse cellular activities (AAA) proteases (Arlt et al., 1996; Guzélin et al., 1996; Stiburek et al., 2012) and that LON/ClpP proteases have been shown to mediate degradation of CI under conditions of increased mROS production (Pryde et al., 2016).
Mitochondrial alterations in ECSIT-deleted macrophages lead to drastic effects on mitochondrial functions. Surprisingly, ATP levels were unaltered, suggesting that increased glycolysis compensates for lower ATP production by mitochondria. This is supported by a dramatic decrease in ATP levels upon inhibition of glycolysis in cells lacking ECSIT (Figure 3A). However, this metabolic shift is not a reprogramming phenomenon because mRNA levels of key glycolytic regulators and enzymes were not affected (Figure S3), and the mechanism of this increased flux is still unknown. Thus, without an increase in glycolytic capacity, we expect that ECSIT-deleted cells will be more sensitive to ATP-demanding stresses, and the function of cells like macrophages is likely to be altered in vivo, at sites of ongoing inflammation, for example.
We initially deleted ECSIT to confirm the role of ECSIT in the induction of mROS by TLRs upon phagocytosis of bacterial pathogens. Although the induction of mROS by bacterial products was abrogated in macrophages lacking ECSIT, this analysis was complicated by changes in baseline mROS production. Macrophages lacking ECSIT exhibit a significant increase in constitutive mROS production (Figure 5B). This is in agreement with other reports where deficiencies in CI subunits were studied (Jin et al., 2014; Miwa et al., 2014; Vogel et al., 2007). Given that CI is totally absent in ECSIT KO macrophages, it is likely that there might be another source of mROS in these cells. For example, complex III and, to a much lesser extent, CII have also been implicated in ROS production (Bolisetty and Jaimes, 2013; Casteilla et al., 2001; McLennan and Degli Esposti, 2000; Yankovskaya et al., 2003). Another possibility is the generation of ROS through alpha-KGDH (alpha-ketoglutarate dehydrogenase). It was reported that alpha-KGDH can produce ROS as a result of an increased NADH:NAD+ ratio (Starkov et al., 2004; Tretter and Adam-Vizi, 2004), as observed in ECSIT-deleted cells (Figure 3E). Nevertheless, additional studies are necessary to understand the source of increased ROS in ECSIT KO macrophages because other enzymes have been implicated in ROS generation as well (Mrácek et al., 2009; Zorov et al., 2014).
Our previous study in macrophages revealed induction of mROS as a consequence of interaction of activated TLR signaling complexes containing TRAF6 with mitochondrial ECSIT (West et al., 2011). We observed a relocalization of ECSIT from the inner mitochondrial membrane to the OMM. Using knockdown approaches, we previously found that ECSIT was important for mitochondrial recruitment of TRAF6 and for inducible production of mROS in LPS-stimulated macrophages. The results obtained with ECSIT deletion in macrophages support an important role in the regulation of CI-dependent ROS (Figure 5) and suggest that this role may be secondary to regulation of CI assembly and protein stability. Therefore, these findings provide further support for the assertion that CI is the key source of mROS during bacterial phagocytosis.
It will be critical to explore the physiological relevance of the mechanisms elucidated here by examining the effects of ECSIT loss on innate immunity in in vivo models of infection. Based on our previous work (West et al., 2011), we expect that perturbed metabolism and mROS inducibility in ECSIT-deleted macrophages will impair resistance to bacterial infection. Moreover, multiple studies suggest that crosstalk between anti-bacterial autophagy and mitophagy (Randow and Youle, 2014) contributes to resistance to bacterial infection. Autophagy (Benjamin et al., 2013) and LC3-associated phagocytosis (Sarkar et al., 2017) have been shown to restrict S. typhimurium infection, and Parkin can contribute to resistance against M. tuberculosis and S. typhimurium (Manzanillo et al., 2013). Because we show that ECSIT is involved in mitophagy and that it interacts with LC3b, ECSIT loss might affect either or both of these pathways in addition to its expected effects on ROS production.
In summary, our results present ECSIT as a link between mitochondrial quality control, CI function, and mROS production in macrophages and reveal a unique homeostasis of CI in macrophages, which could be instrumental for macrophage function as suggested by Garaude et al. (2016).
EXPERIMENTAL PROCEDURES
Further details and an outline of resources used in this work can be found in the Supplemental Experimental Procedures.
Mice
Animals used for experiments were age-matched (8–12 weeks old) and sex-matched and were bred and housed under standard conditions in accordance with Columbia University Institutional Animal Care and Use Committee policies. All mouse protocols were approved by Columbia University. The Ecsitf/f mouse was generated following methods explained in the Supplemental Experimental Procedures (Yusa et al., 2011), backcrossed into the C57BL/6 background, and then bred to the Ecsit+/− (Xiao et al., 2003) and LysM-Cre (The Jackson Laboratory) or Rosa26Cre-ERT2 (B. Reizis, personal communication) mouse strains.
Cell Systems
BMDMs were harvested from 8- to 12-week-old ECSITf/−/LysM-Cre+ (WT) and ECSIT+/+/LysM-Cre+ (cKO) or ECSITf/f/Cre-ERT2+ littermates and cultured for a period of 7 days in DMEM containing L929 conditioned medium. BMDMs from ECSITf/f/Cre-ERT2+ mice were treated with 4-hydroxy (4OH)-tamoxifen (500 nM) or vehicle on days 1, 3, and 6 of differentiation and plated for experiments on day 7. IBMMs from ECSITf/f/Cre-ERT2+ mice were treated with 500 nM of 4OH-tamoxifen or vehicle for 48 hr, washed, and cultured for 5–12 days prior to testing. These macrophages are referred to as iKO or WT, respectively. Relevance of the use of IBMMs was tested in assays performed in both BMDMs and IBMMs.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 6 software. All data were tested for normal distribution of variables. Normally distributed data are displayed as means ± SD unless otherwise noted. Comparisons between two groups were performed with Student’s t test when normally distributed or Mann-Whitney test otherwise. Groups of three or more were analyzed by one-way ANOVA or the Kruskal-Wallis test. Values of n for each experiment are reported in the figures and figure legends. p < 0.05 was considered significant. Statistical parameters for each experiment can be found in the figure legends.
Supplementary Material
Highlights.
Loss of ECSIT in macrophages leads to a striking glycolytic shift
ECSIT is essential for complex I assembly and stability in macrophages
Role of ECSIT in mROS production and removal of damaged mitochondria by mitophagy
Acknowledgments
We are grateful to Dr. B. Reizis and Dr. Thomas Ludwig for providing the Rosa26Cre-ERT2 mouse strain; Dr. R. Jaenisch and Dr. D. Trono for providing the plasmids used in lentivirus generation for IBMM transduction; Dr. N. Mizushima for the pMXs-IP HA-Parkin plasmid (Yoshii et al., 2011); Dr. M. Cookson for the pCMVTNT PINK1 C-myc plasmid (Beilina et al., 2005); Drs. P. Chastagner, A. Israel, and C. Brou for the pcDNA3 VSV-Ub plasmid; Dr. T. Meila for the YFP-LC3 plasmid; and Dr. Kosuke Yusa for the pMCS-DTA plasmid (Yusa et al., 2011). We thank Dr. Alexander Tzagoloff from Columbia University and Dr. Malgorzata Rak from Robert-Debré University Hospital for providing ATP synthase F1 alpha and beta subunits and ND6 antibodies, respectively, and Dr. Victor Lin from Columbia University for technical assistance and advice. We also thank Dr. Sujatha Gurunathan for help with writing the manuscript. The study was supported by grants from the NIH (AI33443 and ES025677) and institutional funds from Columbia University. A.L. was partly supported by EMBO Long-Term Fellowship ALTF-390-2012.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.051.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, F.R.G.C., A.L., M.S.H., and S.G.; Methodology, F.R.G.C., A.L., and S.G.; Investigation, F.R.G.C. and A.L.; Resources, J.J.S.; Writing – Original Draft, F.R.G.C., A.L., M.S.H., and S.G.; Writing – Review and Editing, A.L., M.S.H., and S.G.; Funding Acquisition, A.L. and S.G.
References
- Alston CL, Compton AG, Formosa LE, Strecker V, Oláhová M, Haack TB, Smet J, Stouffs K, Diakumis P, Ciara E, et al. Biallelic Mutations in TMEM126B Cause Severe Complex I Deficiency with a Variable Clinical Phenotype. Am J Hum Genet. 2016;99:217–227. doi: 10.1016/j.ajhg.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE. Assembly factors for the membrane arm of human complex I. Proc Natl Acad Sci USA. 2013;110:18934–18939. doi: 10.1073/pnas.1319247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt H, Tauer R, Feldmann H, Neupert W, Langer T. The YTA10–12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell. 1996;85:875–885. doi: 10.1016/s0092-8674(00)81271-4. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, Sánchez-Caballero L, van den Brand MAM, Wintjes LT, Brink M, van den Brandt FA, Wilson C, Rodenburg RJT, Nijtmans LGJ. NDUFAF4 variants are associated with Leigh syndrome and cause a specific mitochondrial complex I assembly defect. Eur J Hum Genet. 2017;25:1273–1277. doi: 10.1038/ejhg.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rötig A, Munnich A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- Benjamin JL, Sumpter R, Jr, Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013;13:723–734. doi: 10.1016/j.chom.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bet L, Bresolin N, Moggio M, Meola G, Prelle A, Schapira AH, Binzoni T, Chomyn A, Fortunato F, Cerretelli P, et al. A case of mitochondrial myopathy, lactic acidosis and complex I deficiency. J Neurol. 1990;237:399–404. doi: 10.1007/BF00314729. [DOI] [PubMed] [Google Scholar]
- Bolisetty S, Jaimes EA. Mitochondria and reactive oxygen species: physiology and pathophysiology. Int J Mol Sci. 2013;14:6306–6344. doi: 10.3390/ijms14036306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer ME, Koopman WJ, Koene S, Nooteboom M, Rodenburg RJ, Willems PH, Smeitink JA. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol Dis. 2013;51:27–34. doi: 10.1016/j.nbd.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Casteilla L, Rigoulet M, Pénicaud L. Mitochondrial ROS metabolism: modulation by uncoupling proteins. IUBMB Life. 2001;52:181–188. doi: 10.1080/15216540152845984. [DOI] [PubMed] [Google Scholar]
- Fassone E, Taanman JW, Hargreaves IP, Sebire NJ, Cleary MA, Burch M, Rahman S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet. 2011;48:691–697. doi: 10.1136/jmedgenet-2011-100340. [DOI] [PubMed] [Google Scholar]
- Garaude J, Acín-Pérez R, Martínez-Cano S, Enamorado M, Ugolini M, Nistal-Villán E, Hervás-Stubbs S, Pelegrín P, Sander LE, Enríquez JA, Sancho D. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol. 2016;17:1037–1045. doi: 10.1038/ni.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H, Hong L, Xie C, Li X, Zhao H, et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol. 2015;16:1142–1152. doi: 10.1038/ni.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarani V, Paulo J, Zhai B, Huttlin EL, Gygi SP, Harper JW. TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Mol Cell Biol. 2014;34:847–861. doi: 10.1128/MCB.01551-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, Nijtmans L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017;25:128–139. doi: 10.1016/j.cmet.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Guzélin E, Rep M, Grivell LA. Afg3p, a mitochondrial ATP-dependent metalloprotease, is involved in degradation of mitochondrially-encoded Cox1, Cox3, Cob, Su6, Su8 and Su9 subunits of the inner membrane complexes III, IV and V. FEBS Lett. 1996;381:42–46. doi: 10.1016/0014-5793(96)00074-9. [DOI] [PubMed] [Google Scholar]
- Heide H, Bleier L, Steger M, Ackermann J, Dröse S, Schwamb B, Zörnig M, Reichert AS, Koch I, Wittig I, Brandt U. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Heo JM, Rutter J. Ubiquitin-dependent mitochondrial protein degradation. Int J Biochem Cell Biol. 2011;43:1422–1426. doi: 10.1016/j.biocel.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houshmand M, Larsson NG, Oldfors A, Tulinius M, Holme E. Fatal mitochondrial myopathy, lactic acidosis, and complex I deficiency associated with a heteroplasmic A–> G mutation at position 3251 in the mitochondrial tRNALeu(UUR) gne. Hum Genet. 1996;97:269–273. doi: 10.1007/BF02185750. [DOI] [PubMed] [Google Scholar]
- Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014;20:483–498. doi: 10.1016/j.cmet.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011;23:476–482. doi: 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, Tannahill GM, Murphy MP, O’Neill LA. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J Biol Chem. 2015;290:20348–20359. doi: 10.1074/jbc.M115.662114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- Koene S, Rodenburg RJ, van der Knaap MS, Willemsen MA, Sperl W, Laugel V, Ostergaard E, Tarnopolsky M, Martin MA, Nesbitt V, et al. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J Inherit Metab Dis. 2012;35:737–747. doi: 10.1007/s10545-012-9492-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E, Medzhitov R, Carothers J, Xiao C, Douglas I, Janeway CA, Ghosh S. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999;13:2059–2071. doi: 10.1101/gad.13.16.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadsham JE, Sanders G, Giannaki S, Bastow EL, Hutton R, Naeimi WR, Breitenbach M, Gourlay CW. Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 2013;18:279–286. doi: 10.1016/j.cmet.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Livnat-Levanon N, Glickman MH. Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim Biophys Acta. 2011;1809:80–87. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, van den Heuvel LP. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenzie M, Ryan MT. Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life. 2010;62:497–502. doi: 10.1002/iub.335. [DOI] [PubMed] [Google Scholar]
- McLennan HR, Degli Esposti M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr. 2000;32:153–162. doi: 10.1023/a:1005507913372. [DOI] [PubMed] [Google Scholar]
- Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, von Zglinicki T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun. 2014;5:3837. doi: 10.1038/ncomms4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrácek T, Pecinová A, Vrbacký M, Drahota Z, Houstek J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch Biochem Biophys. 2009;481:30–36. doi: 10.1016/j.abb.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Pagano G, Talamanca AA, Castello G, Cordero MD, d’Ischia M, Gadaleta MN, Pallardó FV, Petrovic S, Tiano L, Zatterale A. Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: toward mitochondria-targeted clinical strategies. Oxid Med Cell Longev. 2014;2014:541230. doi: 10.1155/2014/541230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzella V, Papa S. Mutations in human nuclear genes encoding for subunits of mitochondrial respiratory complex I: the NDUFS4 gene. Gene. 2002;286:149–154. doi: 10.1016/s0378-1119(01)00810-1. [DOI] [PubMed] [Google Scholar]
- Pitkanen S, Robinson BH. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of super-oxide dismutase. J Clin Invest. 1996;98:345–351. doi: 10.1172/JCI118798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde KR, Taanman JW, Schapira AH. A LON-ClpP Proteolytic Axis Degrades Complex I to Extinguish ROS Production in Depolarized Mitochondria. Cell Rep. 2016;17:2522–2531. doi: 10.1016/j.celrep.2016.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–411. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Tindle C, Pranadinata RF, Reed S, Eckmann L, Stappenbeck TS, Ernst PB, Das S. ELMO1 Regulates Autophagy Induction and Bacterial Clearance During Enteric Infection. J Infect Dis. 2017;216:1655–1666. doi: 10.1093/infdis/jix528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol. 2015;16:375–388. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, Houstek J, Zeman J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartak RS, Semwal MK, Bai Y. An update on complex I assembly: the assembly of players. J Bioenerg Biomembr. 2014;46:323–328. doi: 10.1007/s10863-014-9564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, Janssen RJ, van den Brand MA, Dieteren CE, Verkaart S, Koopman WJ, Willems PH, Pluk W, van den Heuvel LP, Smeitink JA, Nijtmans LG. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Ma H, Cai Q, Lin S, Lei X, He B, Wu S, Wang Z, Gao Y, Liu W, et al. Recurrent ECSIT mutation encoding V140A triggers hyperinflammation and promotes hemophagocytic syndrome in extranodal NK/T cell lymphoma. Nat Med. 2018;24:154–164. doi: 10.1038/nm.4456. [DOI] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Shim JH, Klüppel M, Zhang SS, Dong C, Flavell RA, Fu XY, Wrana JL, Hogan BL, Ghosh S. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 2003;17:2933–2949. doi: 10.1101/gad.1145603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, Léger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.