

Abstract
Monoclonal antibody therapeutics have revolutionized the treatment of diseases such as cancer and autoimmune disorders, and also serve as research reagents for diverse and unparalleled applications. To extend their utility in both contexts, we have begun development of tunable antibodies, whose activity can be controlled by addition of a small molecule. Conceptually, we envision that incorporating cavity-forming mutations into an antibody can disrupt its structure, thereby reducing its affinity for antigen; addition of a small molecule may then restore the active structure, and thus rescue antigen binding. As a first proof of concept towards implementing this strategy, we have incorporated individual tryptophan to glycine mutations into FITC-E2, an anti-fluorescein single-chain variable fragment (scFv). We find that these can disrupt the protein structure and diminish antigen binding, and further that both structure and function can be rescued by addition of indole to complement the deleted side chain. While the magnitude of the affinity difference triggered by indole is modest in this first model system, it nonetheless provides a framework for future mutation/ligand pairs that may induce more dramatic responses. Disrupting and subsequently rescuing antibody activity, as exemplified by this first example, may represent a new approach to “design in” fine-tuned control of antibody activity for a variety of future applications.
Keywords: Designed protein switch, antibody binding domains, single chain variable fragment (scFv), chemical rescue of structure, small molecule modulators, structure-function relationship
Graphical Abstract
INTRODUCTION
Monoclonal antibodies have had transformative impact on biology and medicine, both as tools for scientific discovery and as precisely targeted therapeutic agents. Upwards of 60 antibody-derived therapeutic agents have already reached the clinic, with hundreds more in ongoing clinical trials [2]. Their ability to precisely inhibit or activate some biological target of interest, coupled with dramatic engineering successes to allow antibody humanization and enhanced effector functions [3], antibody-drug conjugates [4], and bi-specific antibodies [5], together provide ample room for antibodies to continue growing as tools for therapeutic intervention.
Their strengths notwithstanding, however, antibody-based therapeutics can still have serious adverse reactions resulting from aberrant modulation of their intended target [6–8]. Thus, a critical but unmet need remains for better control of antibody activity: only then can we fully realize the potential benefits of antibody-based therapeutics while reducing negative side effects. Furthermore, methods for modulating the function of antibodies give us the chance to better understand target biology, including correlations between target engagement and downstream biological effects.
Engineered protein switches that can be selectively activated by small molecules have been used to interrogate a variety of biological processes [9,10]. These switches are typically designed by inserting an “input” domain for ligand sensing into an “output” domain that provides some useful readout [11]. Starting with a maltose-dependent hydrolysis of ampicillin, achieved by fusing maltose-binding protein into beta-lactamase [12], strong interest in this approach has led to a number of successful switches designed in this manner. Modularity amongst “input” and “output” domains has proven difficult to achieve, however, because such designs typically prove very sensitive to the precise location, length, and composition of the linkers connecting the two functional domains [13,14].
An alternative method for introducing small-molecule control into enzymes is “chemical rescue”: in this case, a protein variant is produced in which a sidechain known to be critical for catalysis – often histidine – is deleted, leading to loss of activity. Upon replacement of this functional group, either through a modified substrate [15], or as a complementary exogenous ligand such as imidazole [16], activity can in some cases be restored. Inspired by this work, we recently developed a complementary approach by instead designing control sites directly into the functional protein domain, through a strategy termed “chemical rescue of structure” [17–19]. By introducing a cavity-forming mutation at a site that structurally buttresses the protein active site, the active site geometry is disrupted and protein function is lost; the subsequent addition of an exogenous compound that replaces the deleted atoms can then restore the original active site architecture and rescue protein activity. In the case of a model β-glycosidase, incorporating a tryptophan-to-glycine (W-to-G) mutation near the active site led to an deactivating conformational change; indole binding at the mutation site then reverted this conformational change, and rescued enzyme activity [17]. The indole-bound crystal structure of the mutated enzyme confirmed that indole mimicked the precise interactions that the native tryptophan was making previously, reminiscent in some ways of small molecules that can recapitulate interactions between protein sidechains [20]. Subsequent studies of W-to-G mutations in other systems showed that enhanced protein fluctuations and local unfolding could also mediate the (reversible) loss of activity induced by these cavity-forming mutations [18].
Here we apply the “chemical rescue of structure” approach to antibody binding domains, and we show that it can used as a means to modulate their structure and function. Specifically, we demonstrate this using FITC-E2, a single-chain variable fragment (scFv) that recognizes fluorescein as its antigen [1,21,22]; already, others have designed a variant of a closely-related scFv by incorporating specific peptide segments into the linker connecting the immunoglobulin VH and VL domains, such that fluorescein-binding is dependent on temperature and salt [23]. Here, we find that introducing a W-to-G mutation at the interface between the two immunoglobulin domains leads to loss of activity that can be subsequently restored by indole. In contrast, a W-to-G mutation in the antigen-binding site of either immunoglobulin domain leads to loss of activity that cannot be rescued. Taken together, our results provide improved understanding of the effects of structural perturbation of an antibody, and they provide further insight into how and where one can introduce pharmacological control into antibodies.
RESULTS
Indole rescue of FITC-E2 VH W118G activity
FITC-E2 is an anti-fluorescein scFv that was generated by phage display [1,21,22]. This fusion protein contains the immunoglobulin VH and VL domains connected with a short (Gly4Ser)3 linker. There are a total of five tryptophan residues in the protein (Figure 1A), and each of these was individually mutated to glycine in this study. Two of these, VH W41 and VL W41 (IMGT numbering [24] used throughout), are located at deeply buried positions in the interior of the immunoglobulin domains; mutation of either one to glycine precluded production of soluble protein by expression in E. coli. Two other tryptophan residues, VH W52 and VL W107, are located in the antigen-binding site and make direct contact with fluorescein via hydrogen bonding (VH W52) and pi-pi stacking (VL W107) interactions. The fifth and final tryptophan is the highly-conserved residue VH W118, positioned at the VH-VL domain-domain interface. We expressed and purified the latter three W-to-G mutants, and we tested the fluorescein-binding activity of each one.
Figure 1. Evaluating indole rescue for W-to-G mutants in FITC-E2.

(A) The crystal structure of the anti-fluorescein scFv FITC-E2, in complex with fluorescein analogue Oregon Green (green spheres) (PDB: 2A9N) [1]. Parts of the scFv corresponding to the heavy and light chains are shown in light grey and dark grey, respectively. Tryptophan residues are highlighted in magenta sticks (IMGT numbering). (B) Relative to the wild-type protein, quenching of fluorescein fluorescence (a readout of scFv binding) was diminished for all three FITC-E2 mutants. Upon addition of 1 mM indole, activity of the VH W118G mutant was partially rescued: from 42% to 86% of the wild-type activity. The other two mutants did not show a statistically significant increase in activity upon addition of indole. Ctrl includes fluorescein and indole, but no scFv. Data are presented as mean ± SEM; n = 3; ****p < 0.0001 for VH W118G with indole vs. no indole.
Binding by FITC-E2 quenches fluorescein’s fluorescence; we used this property to monitor the activity of each FITC-E2 variant. All three mutants showed diminished activity relative to wild-type FITC-E2, with VH W52G and VL W107G showing a larger effect than VH W118G (Figure 1B). Upon addition of 1 mM indole, activity of the VH W118G mutant was restored nearly to the level of wild-type construct; in contrast, there was only a modest increase in activity for VH W52G and VL W107G (not reaching the level of statistical significance).
The rescue of FITC-E2 VH W118G fluorescein-quenching by indole exhibited dose-dependency with a midpoint at 220 μM indole (Figure 2A): this increased protein activity is statistically significant (relative to activity in the absence of indole) at concentrations above 50 μM (i.e. 200X relative to the 250 nM protein concentration) (Figure S1). We next titrated increasing concentrations of protein and correspondingly increasing indole concentrations, with a constant fluorescein concentration (Figure 2B). While there is not a simple binding model that can be used to fit data from two different binding sites with cooperativity between them, it is evident that addition of indole induces tighter antigen binding (shifting the EC50 to lower protein concentrations), and it induces a sharper transition. In contrast, indole does not confer either of these changes to wild-type FITC-E2.
Figure 2. Indole rescues FITC-E2 VH W118G activity.

(A) Addition of indole rescues antigen-binding of FITC-E2 VH W118G in a dose-dependent manner, with EC50 = 220 μM. Data are presented as percent quenching relative to wild-type FITC-E2, using original data collected in Figure S1. (B) Protein titrations with correspondingly increasing indole concentration. Data are presented as mean ± SEM; n= 3. For wild-type FITC-E2, the EC50 values are 23–27 nM regardless of indole concentration. For FITC-E2 VH W118G, the EC50 value is 398 nM in the absence of indole, 241 nM with 241 μM indole (1000X), and 170 nM with 680 μM indole (4000X). Both curves were fit using the Hill equation to facilitate determination of the EC50, but we note that the complexity of the underlying allosteric binding model does not formally allow this equation to necessarily explain the data under all conditions.
Indole enhances binding affinity of FITC-E2 VH W118G for fluorescein
We next sought to confirm the direct interaction of indole with FITC-E2 VH W118G using surface plasmon resonance (SPR). A direct binding assay, in which the protein is immobilized and some combination of fluorescein and indole are flowed over this surface, would be confounded by the fact that the two compounds bind with distinct but interdependent affinities.
Instead, we therefore developed an “inhibition in solution assay” (ISA) [25,26] to monitor the interaction. In ISA experiments, a compound capable of binding the protein is immobilized on the surface of the chip. Measurements are performed under conditions of mass transport limitation, which enable the observed binding response to be derived exclusively from the mass transfer of the protein rather than by association/dissociation kinetics, and should therefore induce a concentration-dependent response. For our experiments, we immobilized a fluorescein analogue (Compound 1, Figure 3A) onto the sensor surface; upon addition of increasing concentrations of FITC-E2 VH W118G we observed a linearly increasing response (Figure S2), confirming the appropriateness of these experimental conditions for further studies [27].
Figure 3. Indole rescues FITC-E2 VH W118G binding affinity for fluorescein.

(A) Structure of the fluorescein analogue immobilized onto the SPR surface for the inhibition in solution assay (ISA). (B) Representative SPR response upon injection of FITC-E2 VH W118G. The presence of increasing fluorescein in solution reduces the observed response, by competing with the immobilized fluorescein for the protein. (C) Fraction of FITC-E2 VH W118G binding immobilized fluorescein, as function of the free fluorescein concentration. The IC50 value shifts from 296 nM in the absence of indole, to 167 nM in the presence of 150 μM indole. Data are presented as mean ± SEM; n = 3.
We next pre-incubated a fixed concentration of FITC-E2 VH W118G protein (150 nM) with varying concentrations of free fluorescein for fifteen minutes, then injected this solution over the surface of the chip (to which our fluorescein analogue was immobilized). The rate of binding was monitored, as a means for reporting on the concentration of free protein in the incubated solution. Our expectation was that pre-incubation with (free) fluorescein would decrease the amount of protein available to bind to the chip, by directly competing for the scFv’s antigen-binding site.
Using this experimental layout, we confirmed that increasing the concentration of free fluorescein in solution diminishes the concentration of free protein available to bind to the chip, as observed though a decreased SPR response (Figure 3B). Here, the amount of protein detected in the SPR response provides an indirect readout of the binding affinity for free fluorescein, by analogy to any other competition assay. Importantly, however, most other competition assays are carried out once equilibrium has been reached between the two competing substrates. By contrast, in this experiment we pre-incubate the protein with free fluorescein and then monitor the initial rate of binding to the immobilized fluorescein: thus, our layout is designed to report on the amount of free protein that was originally present in solution, before a new equilibrium is reached due to competition with the immobilized fluorescein.
We plotted the observed SPR response as a function of the free fluorescein concentration included in the pre-incubation step; as expected, this curve is described by the Hill equation. Upon inclusion of 150 μM indole in the pre-incubation step, we observe that the IC50 value for free fluorescein binding to FITC-E2 VH W118G becomes two-fold tighter (Figure 3C). This result is therefore consistent with our earlier experiments showing enhanced fluorescence quenching in the presence of indole. Through this complementary biophysical assay, then, we have demonstrated that the observed rescue of FITC-E2 VH W118G activity by indole is indeed due to enhanced binding affinity for fluorescein, and not due to some other subtle difference in fluorescence quenching.
Indole stabilizes active FITC-E2 VH W118G
Given our previous observation that modulation of protein stability could lead to inactivation and rescue [18], we next probed the effect of each mutation using differential scanning fluorimetry (DSF). SYPRO Orange dye binds to exposed hydrophobic groups on proteins, leading to enhanced fluorescence intensity; it therefore provides a sensitive readout of protein unfolding [28]. For each protein construct we monitored SYPRO Orange fluorescence intensity as a function of temperature: as expected, each sample underwent an unfolding transition with increasing temperature (Figure S3). The transition temperatures at each set of conditions (as described below) are compiled in Table S1.
In the presence of 5 μM fluorescein (equimolar with the protein concentration), all three W-to-G mutants have lower melting temperature than wild-type FITC-E2, indicating reduced thermal stability (Figure 4A). Unsurprisingly, the magnitude of this effect is largest for the positions that are most deeply buried in the protein structure (VH W118G and VH W52G).
Figure 4. Indole rescues stability of active FITC-E2 VH W118G.
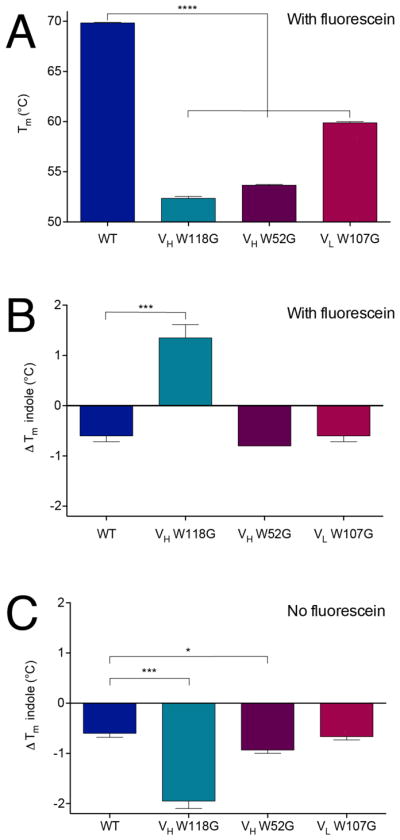
(A) Melting temperatures for all protein constructs show single tryptophan to glycine mutant constructs are less stable than the wild-type protein. (B) In the presence of equimolar fluorescein, addition of 5 mM indole stabilizes the VH W118G construct but not wild type or the other mutants. (C) In the absence of fluorescein, addition of 5 mM indole is not stabilizing for VH W118G or any of the other constructs. Tm = Tm (indole) –Tm (no indole). All data are presented as mean ± SEM; n ≥ 3; *p<0.05, ***p < 0.005, ****p < 0.0001.
Upon addition of 5 mM indole (1000X relative to protein) the melting temperature for VH W118G increases by 1.4 ºC, indicating enhanced thermal stability; in contrast, indole is destabilizing for all other constructs (Figure 4B). The presence of a high fluorescein concentration (far above fluorescein’s dissociation constant), ensures that VH W118G adopts a conformation competent for fluorescein-binding: this conformation presumably matches that of wild-type FITC-E2, which in turn provides the cavity needed for indole binding.
Intriguingly, this effect is not observed in the absence of fluorescein: in this case, addition of indole is now destabilizing for VH W118G (Figure 4C). This observation implies that the introducing the VH W118G mutation has altered the protein conformation, because the indole binding site is no longer available. Instead of the folded and active conformation of wild-type FITC-E2, in the absence of fluorescein and indole the VH W118G mutant must adopt an alternate conformation (or ensemble of conformations) that is at least partially folded, but incapable of binding indole.
Overall, these results highlight the positive cooperativity between fluorescein binding and indole binding. Previously we observed that the presence of indole enhances the interaction of FITC-E2 VH W118G with its antigen; here, we conversely observe that antigen enhances the interaction of the scFv with indole. While the stability of the rescued VH W118G construct is still much less than wild-type, these observations suggest that the inactivation and rescue of FITC-E2 VH W118G derives from a conformational change that is coupled to a change in protein stability.
Indole rescues FITC-E2 VH W118G structure and dynamics
Depending on the length of the linker connecting the two domains as well as the identity of the domains themselves, a number of scFv’s have been observed to form dimers [29–31] or higher-order oligomers [32,33]. These oligomers are typically domain-swapped structures [32,34], and in addition to design of the scFv itself their formation/dissolution can additionally depend on the presence of antigen [35].
To test whether indole was modulating antigen binding by altering the oligomeric state of FITC-E2 VH W118G, we used size-exclusion chromatography multi-angle light scattering (SEC-MALS) [36,37]. Applying this technique to both wild-type FITC-E2 and its VH W118G mutant, both in the absence and presence of indole and antigen (fluorescein), we first noted that in all cases the protein eluted as a single peak. Upon determination of the absolute molar masses of the species present, in all cases we found values consistent with the presence of monomer alone (Table 1).
Table 1. Oligomeric state of protein constructs, as determined by SEC-MALS.
Results confirm that at a protein concentration of 30 μM there is a single species present, with molecular weight matching that of the monomeric protein. This observation holds for WT FITC-E2 and for VH W118G, and applies both in the absence and in the presence of fluorescein and indole.
| Sample | M.W. (buffer alone) | M.W. (buffer with 15 mM indole and 30 μM fluorescein) |
|---|---|---|
| WT (27.165 kDa) | 27.5 kDa ± 5.7% | 27.7 kDa ± 13.8% |
| VH W118G (27.165 kDa) | 26.6 kDa ± 4.5% | 26.8 kDa ± 12.1% |
To investigate the structural and dynamic basis for inactivation and rescue in further detail, we next utilized hydrogen exchange-mass spectrometry (HX-MS). In this experiment, we incubated the protein of interest in a deuterium-containing solution, allowing for exchange of amide hydrogens for deuterium. This exchange is largely dependent on protein structure and dynamics: amide hydrogen atoms involved in strong intramolecular hydrogen bonds will exchange much more slowly than amide hydrogens in unstructured or transiently structured regions [38,39]. Through proteolytic digestion of the protein of interest and mass spectrometry to quantify deuterium uptake in the resulting peptides, we can map these differences to the protein structure. Comparing the exchange rates between different protein variants and conditions enables us to glean information about their conformational preferences.
We employed a continuous labeling approach, in which samples were incubated in deuterium-containing buffer for varying amounts of time, quenched with a low pH buffer, digested with pepsin, and then evaluated via mass spectrometry. A total of 60 unique peptides were unambiguously assigned from pepsin digestion of both wild-type FITC-E2 and the FITC-E2 VH W118G mutant (corresponding to sequence coverage of 80%). To compare samples prepared under different conditions (with or without indole, with or without fluorescein) we calculated the difference in mass for a given peptide, averaged over all time points, and then normalized by the number of exchangeable amide hydrogens in each peptide to yield a quantity termed . We used k-means clustering to classify the strength of effects between samples [40].
Starting with wild-type FITC-E2, we found that addition of 10 μM fluorescein and 10 mM indole led to drastically stronger protection (i.e. less exchange, ) in a single peptide that mapped to the fluorescein binding site (Figure 5A). Enhanced deuterium uptake (i.e. more exchange, ), meanwhile, was not observed for any peptide. This result can be explained solely on the basis of fluorescein binding, which is expected to protect residues in that area from exchange. This observation is further consistent with our earlier observation that binding of wild-type FITC-E2 to fluorescein was unaffected by the presence of indole.
Figure 5. Indole rescues FITC-E2 VH W118G structure and dynamics.

The change in deuterium uptake ( ) for each peptide in the protein of interest, quantified via hydrogen-exchange mass spectrometry. k-means cluster analysis was used to distinguish between groups of peptides with similar extent of protection (horizontal dashed lines). The peptides showing the strongest differences (cluster 3) are then mapped onto the structure of wild-type FITC-E2 (blue/red regions). The position of VH W118 is indicated in orange spheres, for reference. (A) Comparison of deuterium uptake the wild-type protein in the presence of fluorescein and indole, versus deuterium uptake the wild-type protein alone. (B) Comparison of the VH W118G mutant to the wild-type protein (in the absence of fluorescein and indole). (C) Comparison of the VH W118G mutant to the wild-type protein (in the presence of both fluorescein and indole). All data are presented as mean ± SEM; n = 3.
Next, in the absence of fluorescein and indole, we compared wild-type FITC-E2 to its VH W118G mutant. We find that this cavity-forming mutation induces increased deuterium uptake in regions of the VH domain immediately surrounding the mutation site, but also affects the VL domain and remote regions of the VH domain (Figure 5B). As with the W-to-G mutation in an enzyme that we previously studied via HX-MS [18], this observation suggests that introducing the VH W118G mutation induces enhanced conformational fluctuations in the VH domain and its interface with the VL domain, or even dissociation of the domains from one another – though given the scFv format, their linker region must nonetheless keep them flexibly attached to one another.
Finally, we compared wild-type FITC-E2 to its VH W118G mutant, both constructs in the presence of 10 μM fluorescein and 10 mM indole (Figure 5C). Here we observe only a single peptide that exhibits a drastic difference in protection, corresponding to the same peptide that changed previously upon addition of fluorescein and indole to wild-type FITC-E2. This suggests that the structure and dynamics of the rescued mutant are very similar to those of the wild-type, but not identical: the observed difference in deuterium uptake for this peptide may reflect a slight structural difference, which in turn could explain why the rescued mutant does not bind fluorescein with the same binding affinity as wild-type FITC-E2, even at very high indole concentrations.
Collectively then, these observations from hydrogen exchange-mass spectrometry support the model of inactivation and rescue in FITC-E2 VH W118G inferred from our previous experiments. These results also allow the basis of inactivation and rescue to be mapped directly to the protein structure, albeit at low-resolution.
Structure-activity relationship of rescuing ligands
We next explored the structure-activity relationship of rescue activity for the FITC-E2 VH W118G mutant scFv using a series of indole analogues. Given the strategy of replacing tryptophan sidechain with glycine, we expected that the designed cavity would be best rescued by indole, or compounds very closely resembling indole. Starting first with smaller aromatic and heterocyclic analogues (aniline and pyrrole), we confirmed that these do not rescue fluorescein-binding activity; in contrast, an isostere of indole (indazole) shows rescue comparable to that of indole itself (Figure 6A).
Figure 6. Structure-activity relationship for rescue of FITC-E2 VH W118G.

(A) Fluorescein quenching of FITC-E2 VH W118G by several indole analogs. All compounds were tested at a concentration of 250 μM, with 250 nM protein. All data are presented as mean ± SEM; n ≥ 3; ****p < 0.0001 for comparison to DMSO control. (B) The packing environment of Trp118 in the crystal structure of the WT FITC-E2 protein is shown. (C) By recapitulating the location of the sidechain of Trp118, the originally assumed binding mode of indole within the VH W118G binding pocket is shown. The numbering of indole positions for substitution are indicated. (D) Fluorescein quenching of FITC-E2 VH W118G by all methylindoles reveals very similar extent of rescue. All compounds were tested at a concentration of 250 μM, with 250 nM protein. All data are presented as mean ± SEM; n ≥ 3; ****p < 0.0001 for comparison to DMSO control.
In the crystal structure of the wildtype FITC-E2 protein, the sidechain of Trp118 resides in a tightly confined space formed by hydrophobic residues (Figure 6B). From this starting point, our base expectation was that indole would occupy the designed binding site using a position and orientation that exactly recapitulates the original tryptophan sidechain (Figure 6C). If the binding mode can indeed be understood through this simple model, one would expect a strong and distinct structure-activity relationship to emerge: the extent to which additional substituents on indole can be accommodated would be predicted based on the ability of these groups to fit with the surrounding environment. Indeed, our earlier studies showed that the preferences for ligands rescuing the model β-glycosidase mutant could be completely understood on the basis of the wildtype tryptophan’s environment [19]. For FITC-E2 VH W118G, then, one would expect in particular that analogues with small additional substituents at the 2- and 3- positions to rescue activity, but not compounds elaborated at the 4-, 5-, 6-, or 7- positions.
As a test of this model, we evaluated the extent to which all of the seven possible methylindoles rescued activity. In contrast to behavior consistent with this simple model, we observe only mild preferences among this chemical series (Figure 6D). Most notably, all seven compounds rescued activity: there was not a single position of indole at which the additional methyl group led to complete loss of rescue. This observation implies that there must be some degree of malleability in the designed binding site, to accommodate each of these analogues.
While we cannot exclude a model in which the protein undergoes conformational reorganization to fit these different ligands, a more likely explanation is that the indole core adopts alternate binding modes depending on where it harbors substituents. Crystallographic evidence has shown changes in binding mode upon chemical elaboration in a growing number of examples – especially for weakly-binding fragments [41]. Using hydrogen exchange-mass spectrometry we have confirmed that indole rescues the scFv’s interaction with antigen by binding at the designed mutation site, but the relatively “flat” structure-activity relationship suggests that indole may not necessarily be bound in a single pose.
DISCUSSION
Chemical rescue of structure has been previously applied to build indole-dependent activity into a variety of other proteins; here, we extend this approach to an antibody single-chain variable fragment (scFv). We have shown that adding indole rescues the stability / structure / conformational dynamics of FITC-E2 VH W118G, and these in turn modulate antigen binding. This mechanism of inactivation and rescue, mediated through protein stability, parallels our characterization of a model β-glucuronidase enzyme and of GFP [18]. These examples are in direct contrast to the mechanism initially observed for a β-glycosidase enzyme, which was controlled through a discrete conformational change [17].
Of the three W-to-G mutations in FITC-E2 that allowed for soluble expression, all were found to be inactivating. Among these, however, only one (VH W118G) was rescued by addition of indole; this mutation is located at a position that is close to the antigen binding site, but not in direct contact with the antigen itself. This observation also offers a parallel with our characterization of β-glycosidase mutants [17]: here too we found that a mutated active site residue could not be rescued as effectively as a nearby residue that is not part of the active site. While other studies have focused on active site residues [16,42,43], the distinction between rescuing an active site residue versus rescuing a nearby “buttressing” residue is fundamental to chemical rescue of structure. In this work and in our β-glycosidase studies, we find that – compared to active-site residues, which are by necessity partially exposed – cavity-forming mutations at buried sites offer more potential binding free energy for a rescuing ligand.
From our initial structure-activity relationship (SAR) studies, we observe rescue only from small aromatic heterocyclic compounds that are structurally similar to indole; that said, each of the methylindoles rescued activity to a very similar degree. This observation is in direct contrast to our earlier studies of β-glycosidase: in the latter case there were dramatic differences in activity among the various methylindoles, including some that did not rescue at all, and none rescued activity better than indole itself [19]. This suggests that in the designed β-glycosidase cavity, indole can bind only in a single distinct binding mode; because of this, the presence of additional substituents induces steric clashes in the protein or conformational changes that disrupt the active site geometry. Conversely, the flat SAR profile observed for FITC-E2 VH W118G rescue is consistent with indole adopting many related orientations within the designed binding site. In this scenario, upon chemical elaboration the binding mode(s) that can accommodate the additional substituent is selected, such that most substitutions do not negatively impact rescue.
The binding free energy for a rescuing ligand is a critical consideration for engineering switchable proteins, since it corresponds to the sensitivity of the switch [44]. Even using the largest possible size difference among natural amino acids (W-to-G) and using a buried mutation/rescue site, a relatively high ligand concentration was needed for rescue. Having observed a very similar dynamic range for other indole-based switches, it appears that this binding free energy is simply an intrinsic limitation tied to the small size of the rescuing ligand. Although some of the methylindoles showed slightly enhanced rescue relative to indole itself, all had very similar magnitude. Directed evolution may lead to improved binding affinity, but in essence the cavity has already been subjected to natural evolution in this context of the naturally-occurring tryptophan sidechain – and is therefore likely to be well-optimized already for indole as well.
Moving forward, this highlights the importance of advancing this strategy beyond single cavity-forming mutations: simultaneously incorporating cavity-forming mutations at more than one adjacent site will lead to a larger cavity, which in turn can be rescued by a correspondingly larger ligand. Initial proof-of-concept for this strategy comes from further studies of the β-glycosidase: here a secondary V-to-A mutation was incorporated alongside the previous W-to-G mutation, and the resulting protein variant was rescued using 5-methylindole [19]. Even with this modest increase in the size of the cavity (and thus the size of the rescuing ligand), the sensitivity of the switch was already increased tenfold.
Based on this precedent, we anticipate that incorporating additional cavity-forming mutations adjacent to VH W118G will allow the FITC-E2 antigen-binding to be activated using lower concentrations of the activating ligand. Moreover, we expect that the optimal rescuing ligand for this larger pocket can also be rationally designed, by growing indole into the newly-formed cavity. There are seven framework residues in direct contact with VH W118, and these include both heavy chain positions (42, 50, 103, 105), and light chain positions (42, 50, 118). Among the large set of human antibodies included in the abYsis database [45], the residues in these positions are well conserved (72–99% sequence identity). Based on the strong conservation around the VH W118 side chain, it is likely that the single W-to-G mutation used here will prove transferrable to other antibodies, including those with biological and therapeutic relevance. Expanding the cavity, perhaps into the VL domain, will also allow for rational design of more potent activating ligands, thus decreasing the concentration needed to elicit antigen binding.
In the context of using antibodies to modulate a particular disease-relevant target, developing antibodies whose activity can be controlled by the presence or absence of a small molecule represents a possible new approach to probe the role of a particular target in disease progression. It may become possible for an antibody with attenuated activity to weakly engage its intended target, and subsequently strengthen this interaction through the presence of a small molecule. Thus, the downstream effect of target modulation can also be controlled in a dose-dependent manner by a selectively-distributed small molecule, potentially providing information for target biology and correlation between the amount of target engagement and biological effect. Control of antibody activity in this manner may ultimately even allow for enhanced therapeutic benefit, by diminishing target-related toxicity from the constitutively active form of the antibody.
MATERIALS AND METHODS
Protein expression and purification
A plasmid encoding the FITC-E2 scFv in the pCANTAB6 vector was provided by MedImmune UK. All of our studies make use of the VH W111.4A variant (which we refer to here as “wild-type”), because this variant provides better yields and shows no difference in antigen binding relative to the originally-reported construct [1,22]. Point mutations and TEV site insertions were made using the QuikChange II Site Directed Mutagenesis Kit (Agilent Technologies), and verified with direct sequencing analysis. Plasmids were transformed into HB2151 E. coli cells. Cultures were grown in 2xYT media at 30ºC, and expression was induced by the addition of 1 mM IPTG at an OD600 of 0.5–0.6. After induction, the culture was grown at 30°C for a further 3 hours. The periplasmic protein fraction was prepared by cold osmotic shock. Cells were harvested, resuspended in TES buffer (200 mM Tris, 0.5 mM EDTA, 0.5 M sucrose pH 8.0, cOmplete Protease Inhibitor Cocktail (Roche)) (1/40th culture volume), and incubated on ice for 10 min. A 1:5 dilution of TES was then added (1.5/40th culture volume) and the solution was incubated on ice for a further 30 min. Solutions were then centrifuged (24,000 x g, 10 min, 4°C). The supernatant was loaded onto a pre-equilibrated 5 mL Ni-NTA column (HisTrap HP, GE Healthcare) and washed with 5 CV of purification buffer (50 mM Tris-HCl, 300 mM sodium chloride, pH 8.0) followed by 5 CV of purification buffer with 40 mM imidazole. The target protein was eluted in purification buffer containing 400 mM imidazole. Fractions were collected and analyzed by SDS-PAGE. Those fractions containing the target protein were digested with TEV protease at 4°C for 36 hours while dialyzing against TEV buffer (150 mM NaCl, 50 mM Tris HCl, 0.5 mM EDTA, pH 8.0). Completeness of cleavage was assessed by SDS-PAGE. Protein was then loaded onto a pre-equilibrated gel filtration column (HiLoad 16/60 Superdex 75, GE Healthcare) and eluted with PBS buffer. Fractions were analyzed by SDS-PAGE, concentrated, and pooled.
Fluorescence Quenching Assay
For indole titrations, samples were prepared in PBS (1X, pH 7.1) with 250 nM protein, 10 nM fluorescein, and varying concentrations of indole (70 μL total volume). For SAR studies with indole analogues, all compounds were stocked at 10 mM in DMSO. Samples were prepared in PBS (1X, pH 7.1) with 250 nM protein, 10 nM fluorescein, and 250 μM compound (70 μL total volume). For protein titrations, samples were prepared in PBS with 10 nM fluorescein, and varying concentrations of protein in the presence or absence of a constant ratio of indole (i.e. the indole was serially diluted in the same fashion as the protein). The samples were added to three wells (20 μL each) of a black 384-well flat bottom low-volume microplate (Greiner). Fluorescence measurements were acquired on a Tecan M100 plate reader with excitation 490 nm, emission 513 nm, gain 167, and Z-position 2500 μm. Blank measurements containing all sample components except fluorescein were subtracted to remove background signal. All samples were normalized to a control in which protein and indole were not included. Samples were run in independent triplicate experiments. Statistical significance was evaluated using a one-tailed Student’s t-test, and results were considered statistically significant at p < 0.05.
Thermal Shift Assay
Samples were prepared in PBS (1X, pH 7.1) with 5 μM protein, 5X SYPRO Orange, 5μM fluorescein, and 5 mM indole in PBS (25 μL total volume). Prepared samples were added to separate wells of a white 96-well PCR plate (ThermoFisher). Fluorescent measurements were carried out a minimum of three times using a Bio-Rad CFX96 RT PCR System instrument (Excitation: 515–535 nm/Emission: 560–580 nm). The plate was heated from 25 to 95°C with a heating rate of 0.5°C/min. The minimum values on a plot of the negative first derivative of relative fluorescence units (RFUs) versus temperature (i.e. -d(RFU)/dT versus T) were used to determine the melting temperature. Samples were run in independent triplicate experiments. Statistical significance was evaluated using a one-tailed Student’s t-test, and results were considered statistically significant at p < 0.05.
Synthesis of Compound 1
To 6-(Fluorescein-5-Carboxamido) hexanoic acid, succinimidyl ester (5 mg, 8.52 μmol, 1.0 equiv.) was added to a solution of tert-butyl (3-aminopropyl)carbamate in DMSO (20 mM in 0.448 mL, 8.95 μmol, 1.05 equiv.) followed by triethylamine (1.3 μL, 9.38 μmol), 1.1 equiv.). The reaction was allowed to stir at room temperature for 16 hours, after which it was injected onto an Atlantis T3 column (19 mm x 100 mm, 5 μm) and partially purified via Gilson HPLC (5% to 60% MeCN in water + 0.1% TFA over 15 minutes) to remove the DMSO. Fractions containing the intermediate were pooled and concentrated. To the intermediate was added HCl (4N in dioxane, 4 mL). The reaction was allowed to stir at room temperature for 2 hours, after which it was concentrated under reduced pressure to afford a crude yellow solid. The crude product was taken up in water (5% DMSO), filtered (0.45 μm), injected onto an Atlantis T3 column (19 mm x 100 mm, 5 μm), and purified via Gilson HPLC (5% to 60% MeCN in water + 0.1% TFA over 15 minutes). Fractions containing the products were combined, frozen, and lyophilized to yield compound 1 as a yellow solid (2.3 mg, 41%). 1H NMR (300 MHz, DMSO-d6) δ 8.79 (t, J = 5.56 Hz, 1H), 8.43 (d, J = 0.75 Hz, 1H), 8.22 (dd, J = 8.1, 1.5 Hz, 1H), 7.94 (s, 1H), 7.68 (br s, 2H), 7.35 (d, J = 7.9 Hz, 1H), 6.68 (d, J = 1.7 Hz, 2H), 6.62–6.44 (m, 4H), 3.35–3.25 (m, 2H), 3.19–3.01 (m, 2H), 2.86–2.65 (m, 2H), 2.18–2.02 (m, 2H), 1.74–1.62 (m, 2H), 1.62–1.47 (m, 4H), 1.40–1.20 (m, 4H). LCMS (ES+) calculated for C30H31N3O7 (M+H) m/z 546.2 Da, found 546.2 Da.
SPR Inhibition in Solution Assay (ISA)
The assay was run on a Biacore T200 (GE Healthcare). Immobilization of Compound 1 (10 mM in DMSO) on a CM5 chip (GE Healthcare) with running buffer (PBS, 0.005% P20) was initiated by an injection of EDC/NHS (400 mM/100 mM) at 5 μL/min for 7 min. The ligand was coupled to the surface of the chip by an injection of 200 μM solution (10 mM NaAc, pH 5.0) at 5 μL/min for 25 min. The surface was deactivated with an injection of 1M ethanolamine at 5 μL/min for 7 min. The final immobilization level was 560 RU. Protein was pre-incubated with fluorescein (and indole, if desired) for 15 minutes, then injected into the instrument at 35 μL/min. Regeneration was achieved by 30 sec injection of 0.5% SDS. The normalized SPR response was determined by the slope of the response measured 8 seconds after the start of the injection relative to a protein only control. All experimental runs were carried out in triplicate.
Size-Exclusion Chromatography Multi-Angle Light Scattering (SEC-MALS)
SEC-MALS was performed using an Agilent 1200 Series HPLC with a TSKgel G3000SWXL Column (Tosoh Bioscience, King of Prussia, PA) coupled to a downstream DAWN HELEOS II MALS detector (Wyatt Technology, Santa Barbara, CA), followed by an Optilab T-rEX differential refractometer (Wyatt Technology). The WT and VH W118G scFv proteins (20 μl of ~0.8 mg/mL) were analyzed in both PBS buffer alone, and in PBS buffer with 15 mM indole and 30 μM fluorescein. The data were collected and evaluated using the ASTRA 7 software (Wyatt Technology). Molecular weights were calculated from a single elution peak using the Zimm model and a dn/dc value of 0.1850 mL/g.
Hydrogen exchange mass spectrometry
The methods of automated hydrogen exchange-mass spectrometry analysis used here have been described in detail previously [46], except for the following details. Stock solution of wild-type FITC-E2 and FITC-E2 VH W118G were diluted to a final concentration of 10 μM using a 1% (v/v) DMSO 6 mM phosphate buffer, containing 135 mM NaCl with and without 10 μM fluorescein and 10 mM indole at pH 7.1. To initiate the hydrogen exchange process, 5 μL of protein stock was diluted with 36 μL of D2O labeling buffer with and without fluorescein and indole. Thereafter, the reaction was incubated at 25°C for seven exchange time points ranging from 30 seconds to 24 hours. All HX labeling was performed in triplicate for each labeling time. Hydrogen exchange was quenched by 1:1 dilution with 0.2 M phosphate buffer at pH 2.5 containing 4 M guanidine HCl and 0.4 M TCEP in H2O at 1°C. Samples were immediately injected onto a 100 μL loop prior to pepsin digestion, desalting (Poroshell 120 SB-C18 2.1 × 5 mm, 2.7 μm particles, Agilent Technologies, Santa Clara, CA), and peptide separation. A combination of mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) was used to elute the peptides. To minimize carry-over of peptides from previous runs, the desalting trap was washed between each run using an acetonitrile gradient while the pepsin column was back-flushed with 0.1% formic acid.
High-resolution mass measurements in combination with CID tandem mass spectrometry on an Agilent 6530 Q-TOF were used to identify wild-type and VH W118G peptides. A total of 60 unique peptides could be unambiguously assigned from pepsin digestion of both wild-type FITC-E2 and the FITC-E2 VH W118G mutant (corresponding to sequence coverage of 80%).
To compare data from different conditions, the difference in mass for a given peptide, over all time points, was divided by the number of exchangeable amide protons ( ). Experimental error on ( ) was estimated using propagation of the standard errors of the means. To classify the strength of the effect between samples, k-means clustering with k = 3 was employed. Peptides with HX differences ( ) in the first cluster were considered insignificant, peptides in the second cluster were considered to have intermediate differences, and peptides in the third cluster had the most dramatic differences.
Supplementary Material
Acknowledgments
We thank Kate Wickson and James Hunt for their helpful discussions, Ron Tomlinson for synthesis of Compound 1, Trevor Wilkinson at MedImmune UK for providing us with the FITC-E2 vector, and Stefan Geschwindner for help designing the SPR ISA experiment. An equipment loan from Agilent Technologies to the Weis lab is gratefully acknowledged. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health through grant R01GM112736, and by the Human Frontier Science Program (J.K.).
References
- 1.Honegger A, Spinelli S, Cambillau C, Plückthun A. A mutation designed to alter crystal packing permits structural analysis of a tight-binding fluorescein-scFv complex. Protein Sci. 2005;14:2537–49. doi: 10.1110/ps.051520605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. 2008;20:460–70. doi: 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Deonarain MP, Yahioglu G, Stamati I, Marklew J. Emerging formats for next-generation antibody drug conjugates. Expert Opin Drug Discov. 2015;10:463–81. doi: 10.1517/17460441.2015.1025049. [DOI] [PubMed] [Google Scholar]
- 5.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–38. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 7.Muller PY, Milton MN. The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov. 2012;11:751–61. doi: 10.1038/nrd3801. [DOI] [PubMed] [Google Scholar]
- 8.Sathish JG, Sethu S, Bielsky MC, de Haan L, French NS, Govindappa K, Green J, Griffiths CE, Holgate S, Jones D, Kimber I, Moggs J, Naisbitt DJ, Pirmohamed M, Reichmann G, Sims J, Subramanyam M, Todd MD, Van Der Laan JW, Weaver RJ, Park BK. Challenges and approaches for the development of safer immunomodulatory biologics. Nat Rev Drug Discov. 2013;12:306–24. doi: 10.1038/nrd3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buskirk AR, Liu DR. Creating Small-Molecule-Dependent Switches to Modulate Biological Functions. Chemistry & Biology. 2005;12:151–61. doi: 10.1016/j.chembiol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 10.Stein V, Alexandrov K. Synthetic protein switches: design principles and applications. Trends Biotechnol. 2015;33:101–10. doi: 10.1016/j.tibtech.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Ostermeier M. Engineering allosteric protein switches by domain insertion. Protein Eng Des Sel. 2005;18:359–64. doi: 10.1093/protein/gzi048. [DOI] [PubMed] [Google Scholar]
- 12.Guntas G, Ostermeier M. Creation of an allosteric enzyme by domain insertion. J Mol Biol. 2004;336:263–73. doi: 10.1016/j.jmb.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Kim JR, Ostermeier M. Modulation of effector affinity by hinge region mutations also modulates switching activity in an engineered allosteric TEM1 beta-lactamase switch. Arch Biochem Biophys. 2006;446:44–51. doi: 10.1016/j.abb.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Dagliyan O, Shirvanyants D, Karginov AV, Ding F, Fee L, Chandrasekaran SN, Freisinger CM, Smolen GA, Huttenlocher A, Hahn KM, Dokholyan NV. Rational design of a ligand-controlled protein conformational switch. Proc Natl Acad Sci U S A. 2013;110:6800–4. doi: 10.1073/pnas.1218319110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter P, Wells JA. Engineering enzyme specificity by “substrate-assisted catalysis”. Science. 1987;237:394–9. doi: 10.1126/science.3299704. [DOI] [PubMed] [Google Scholar]
- 16.Toney MD, Kirsch JF. Direct Bronsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science. 1989 doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- 17.Deckert K, Budiardjo SJ, Brunner LC, Lovell S, Karanicolas J. Designing Allosteric Control into Enzymes by Chemical Rescue of Structure. J Am Chem Soc. 2012;134:10055–60. doi: 10.1021/ja301409g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Y, DiPrimio N, Keppel TR, Vo B, Fraser K, Battaile KP, Egan C, Bystroff C, Lovell S, Weis DD, Anderson JC, Karanicolas J. The Designability of Protein Switches by Chemical Rescue of Structure: Mechanisms of Inactivation and Reactivation. J Am Chem Soc. 2013;135:18840–9. doi: 10.1021/ja407644b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budiardjo SJ, Licknack TJ, Cory MB, Kapros D, Roy A, Lovell S, Douglas J, Karanicolas J. Full and Partial Agonism of a Designed Enzyme Switch. ACS Synth Biol. 2016;5:1475–84. doi: 10.1021/acssynbio.6b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci U S A. 2006;103:15422–7. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–14. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 22.Pedrazzi G, Schwesinger F, Honegger A, Krebber C, Plückthun A. Affinity and folding properties both influence the selection of antibodies with the selectively infective phage (SIP) methodology. FEBS Letters. 1997;415:289–93. doi: 10.1016/s0014-5793(97)01143-5. [DOI] [PubMed] [Google Scholar]
- 23.Blenner MA, Banta S. Characterization of the 4D5Flu single-chain antibody with a stimulus-responsive elastin-like peptide linker: a potential reporter of peptide linker conformation. Protein Sci. 2008;17:527–36. doi: 10.1110/ps.073257308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lefranc MP, Pommie C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27:55–77. doi: 10.1016/s0145-305x(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 25.Geschwindner S, Olsson LL, Albert JS, Deinum J, Edwards PD, de Beer T, Folmer RH. Discovery of a novel warhead against beta-secretase through fragment-based lead generation. J Med Chem. 2007;50:5903–11. doi: 10.1021/jm070825k. [DOI] [PubMed] [Google Scholar]
- 26.Geschwindner S, Dekker N, Horsefield R, Tigerstrom A, Johansson P, Scott CW, Albert JS. Development of a plate-based optical biosensor fragment screening methodology to identify phosphodiesterase 10A inhibitors. J Med Chem. 2013;56:3228–34. doi: 10.1021/jm301665y. [DOI] [PubMed] [Google Scholar]
- 27.Holdgate GA, Anderson M, Edfeldt F, Geschwindner S. Affinity-based, biophysical methods to detect and analyze ligand binding to recombinant proteins: matching high information content with high throughput. J Struct Biol. 2010;172:142–57. doi: 10.1016/j.jsb.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–21. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 29.Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993;90:6444–8. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essig NZ, Wood JF, Howard AJ, Raag R, Whitlow M. Crystallization of single-chain Fv proteins. J Mol Biol. 1993;234:897–901. doi: 10.1006/jmbi.1993.1638. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths AD, Malmqvist M, Marks JD, Bye JM, Embleton MJ, McCafferty J, Baier M, Holliger KP, Gorick BD, Hughes-Jones NC, et al. Human anti-self antibodies with high specificity from phage display libraries. EMBO J. 1993;12:725–34. doi: 10.1002/j.1460-2075.1993.tb05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pei XY, Holliger P, Murzin AG, Williams RL. The 2.0-A resolution crystal structure of a trimeric antibody fragment with noncognate VH-VL domain pairs shows a rearrangement of VH CDR3. Proc Natl Acad Sci U S A. 1997;94:9637–42. doi: 10.1073/pnas.94.18.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iliades P, Kortt AA, Hudson PJ. Triabodies: single chain Fv fragments without a linker form trivalent trimers. FEBS Lett. 1997;409:437–41. doi: 10.1016/s0014-5793(97)00475-4. [DOI] [PubMed] [Google Scholar]
- 34.Perisic O, Webb PA, Holliger P, Winter G, Williams RL. Crystal structure of a diabody, a bivalent antibody fragment. Structure. 1994;2:1217–26. doi: 10.1016/s0969-2126(94)00123-5. [DOI] [PubMed] [Google Scholar]
- 35.Arndt KM, Muller KM, Pluckthun A. Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry. 1998;37:12918–26. doi: 10.1021/bi9810407. [DOI] [PubMed] [Google Scholar]
- 36.Tarazona MP, Saiz E. Combination of SEC/MALS experimental procedures and theoretical analysis for studying the solution properties of macromolecules. J Biochem Biophys Methods. 2003;56:95–116. doi: 10.1016/s0165-022x(03)00075-7. [DOI] [PubMed] [Google Scholar]
- 37.Folta-Stogniew E. Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods Mol Biol. 2006;328:97–112. doi: 10.1385/1-59745-026-X:97. [DOI] [PubMed] [Google Scholar]
- 38.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–34. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 39.Vadas O, Burke JE. Probing the dynamic regulation of peripheral membrane proteins using hydrogen deuterium exchange-MS (HDX-MS) Biochem Soc Trans. 2015;43:773–86. doi: 10.1042/BST20150065. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Song M. Ckmeans.1d.dp: Optimal k-means Clustering in One Dimension by Dynamic Programming. The R journal. 2011;3:29–33. [PMC free article] [PubMed] [Google Scholar]
- 41.Malhotra S, Karanicolas J. When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode? J Med Chem. 2017;60:128–45. doi: 10.1021/acs.jmedchem.6b00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–7. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 43.Lamba V, Yabukarski F, Herschlag D. An Activator-Blocker Pair Provides a Controllable On-Off Switch for a Ketosteroid Isomerase Active Site Mutant. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b03547. [DOI] [PubMed]
- 44.Vallée-Bélisle A, Ricci F, Plaxco KW. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc Natl Acad Sci U S A. 2009;106:13802–7. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin AC. abYsis: Integrated Antibody Sequence and Structure-Management, Analysis, and Prediction. J Mol Biol. 2017;429:356–64. doi: 10.1016/j.jmb.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 46.Toth RTt, Mills BJ, Joshi SB, Esfandiary R, Bishop SM, Middaugh CR, Volkin DB, Weis DD. Empirical Correction for Differences in Chemical Exchange Rates in Hydrogen Exchange-Mass Spectrometry Measurements. Anal Chem. 2017 doi: 10.1021/acs.analchem.7b01396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.