

Abstract
Phospholipase D (PLD) activity has been proposed to facilitate multiple steps in cancer progression including growth, metabolism, angiogenesis, and mobility. The canonical enzymes PLD1 and PLD2 enact their diverse effects through hydrolyzing the membrane lipid phosphatidylcholine to generate the second messenger and signaling lipid phosphatidic acid (PA). However, the widespread expression of PLD1 and PLD2 in normal tissues and the additional distinct enzymatic mechanisms through which PA can be generated have produced uncertainty regarding the optimal settings in which PLD inhibition might ameliorate cancer. Recent studies in mouse model systems have demonstrated that inhibition or elimination of PLD activity reduces tumor growth and metastasis. One mechanism proposed for this outcome involves proliferative signaling mediated by receptor tyrosine kinases (RTK) and G protein-coupled receptors (GPCR), which is attenuated when downstream PLD signal propagation is suppressed. The reduced proliferative signaling has been reported to be compounded by dysfunctional energetic metabolism in the tumor cells under conditions of nutrient deprivation. Moreover, cancer cells lacking PLD activity display inefficiencies across multiple steps of the metastatic cascade, limiting the tumor’s lethal spread. Using PLD isoform knockout mice, recent studies have reported on the net effects of inhibition and ablation in multiple cancer models through examining the role of PLD in the non-tumor cells comprising the stroma and microenvironment. The promising results of such in vivo studies, combined with the apparent low toxicity of highly-specific and potent inhibitors, highlights PLD as an attractive target for therapeutic inhibition in cancer. We discuss here the array of anti-tumor effects produced by PLD inhibition and ablation in cancer models with a focus on animal studies.
Keywords: Phospholipase D, Lipid signaling, cancer, angiogenesis, metastasis, metabolism
Introduction
The Phospholipase D (PLD) family exists in essentially all organisms and isoforms have been characterized in viruses, bacteria, yeast, plants, and animals (Jenkins and Frohman, 2005). Humans have six isoforms. PLD1– PLD5 each encode a pair of conserved catalytic motifs of histidine, lysine, and aspartic acid (formally, HxKx4Dx6GSxN) that fold together to form the intact catalytic site (Frohman, 2015). PLD6 encodes only one HKD motif and dimerizes to form the catalytic site. The canonical isoforms are PLD1 (Hammond et al., 1995) and PLD2 (Colley et al., 1997), as they exhibit the classical activity associated with this enzyme family (Selvy et al., 2011), whereas PLD3, PLD4, and PLD5 are thought to be catalytically inactive. PLD1/2 catalyze the transphosphatidylation of the phosphodiester bond found in phosphatidylcholine (Figure 1). This typically involves hydrolysis of phosphatidylcholine to yield phosphatidic acid (PA) and the head-group choline (Yang et al., 1967), although headgroup exchange can also occur, for example with primary alcohols(Sung et al., 1997). The mitochondrially-localized PLD6 is unique for its production of PA from cardiolipin (Choi et al., 2006) and for possessing a phosphodiester bond-hydrolyzing endonuclease activity that generates piwi-interacting RNAs (piRNAs) (Huang et al., 2011). PLD1/2 exert an array of effects using three primary mechanisms: enzymatic production of PA, protein-protein interactions, and GEF (Mahankali et al., 2011) and GAP activities (Huang et al., 2011). PA serves as a second messenger lipid signaling molecule that transduces downstream regulation through binding and activating other proteins, serving as a lipid anchor, and/or inducing negative curvature in cellular membranes (Ammar et al., 2014).
Figure 1.
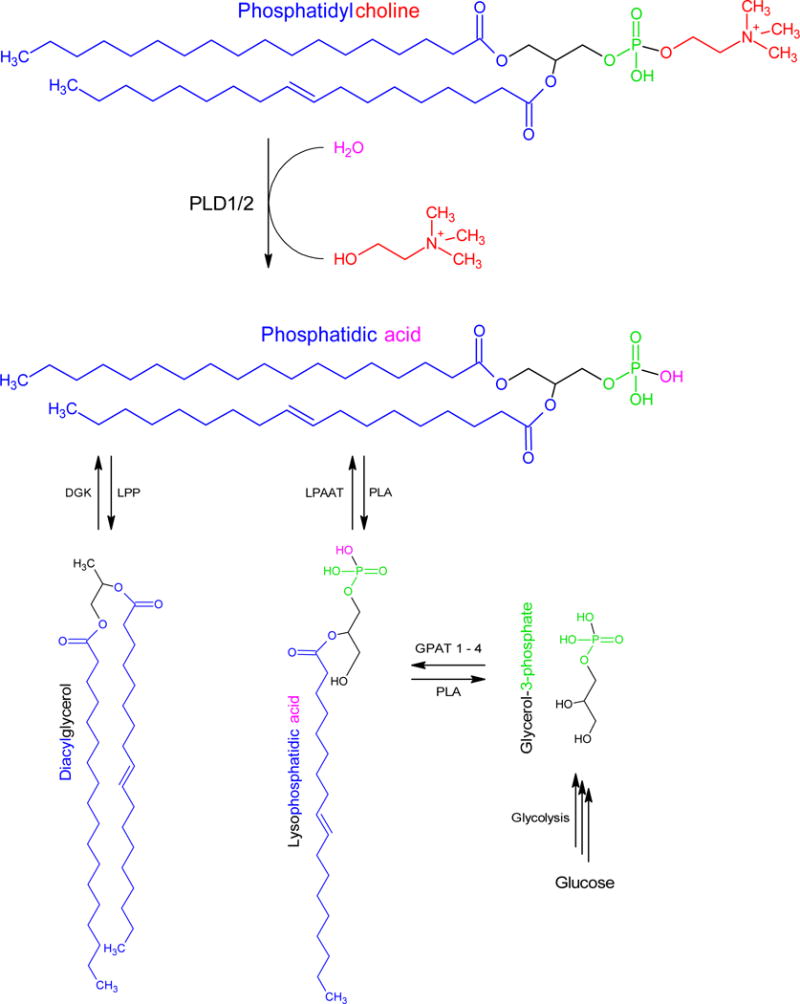
Diagram of the three mechanisms for enzymatic generation of phosphatidic acid (PA). The hydrolysis of phosphatidylcholine by phospholipase D (PLD) releases the headgroup choline to generate PA. Additionally, diacylglycerol can be phosphorylated by diacylglycerol kinase (DGK), whereas LysoPA can be acetylated by LysoPA acetyltransferases (LPAAT). LysoPA can be generated from the glycolytic metabolite glycerol-3-phosphate. PA can be dephosphorylated by lipin to generate diacylglycerol or deacetylation by phospholipases (PLA) to generate LysoPA.
For over 20 years, PLD and its generation of PA have been studied to elucidate a wide range of cellular effects including on cell growth, survival, metabolism, migration, and membrane fusion (Zhang and Frohman, 2014). These roles for PLD activity contribute to multiple disease states including cancer (Bruntz et al., 2014, Gomez-Cambronero, 2014), inflammation (Kang et al., 2014), and neurodegeneration (Brown et al., 2017). PLD is upregulated in a wide range of neoplasms including breast, renal, colorectal, gastric, brain, and thyroid, and expression is correlated with several prognostic measures (Kang et al., 2014). Post-transcriptional regulatory mechanisms, in additional to transcriptional ones, many contribute to the elevation of levels of protein expression and enzymatic activity seen in cancer (Gomez-Cambronero et al., 2017).
Although PLD1 and PLD2 share 50% sequence homology and both enzymatically produce PA, the isoforms have many unique functions. PLD1 and PLD2 differ in normal tissue expression (Kim et al., 2007)(Lopez et al., 1998) and subcellular localization (Du et al., 2004, 2003). As a result, each isoform regulates many unique effects in addition to exhibiting redundancy in some settings (Thielmann et al., 2012) (Ali et al., 2013) (Sanematsu et al., 2013). This review will cover multiple roles of PLD in cancer with a focus on animal models and the effects of specific inhibitors in limiting tumor progression. The discussion will begin with proliferative effects of PLD on tumor growth and angiogenesis. Next, the effects of PLD on metabolism and autophagy will be reviewed. Lastly, metastatic effects and the use of specific and small molecule inhibitors will be compared.
Tumor growth and angiogenesis
PLD is activated by phosphorylation, GTPases, and surface receptors including G-protein-coupled receptors (GPCR) and Receptor Tyrosine Kinases (RTK) (McDermott et al., 2004). As such, PLD propagates signaling of many proliferative signals including VEGF and EGF (Chen et al., 2012). The PA generated stimulates downstream targets such as mTOR (Foster et al., 2014) (Yoon et al., 2015) and MAPK (Choi and Han, 2012). However, tumor growth in vivo also depends on the environment in addition to mitogenic stimulation. Factors such as vascularization, metabolism, and autophagy can support or inhibit tumor growth.
Expansion of vasculature is required by a growing tumor to supply nutrients, oxygen, while also providing access to metastatic routes. However, the tumor microenvironment is frequently hypoxic due to irregular vasculature. This chronic hypoxia produces stabilization of the transcription factor hypoxia inducible factor (HIF). HIF can promote angiogenesis, and PLD activity has been reported to promote its translation and stabilization (Toschi et al., 2008). The role of PLD in vertebrate angiogenesis was first shown in the vascular development of zebrafish (Zeng et al., 2009). Pathologically, hypoxia induces PLD2 expression via HIF1α (Liu et al., 2015) and PLD1 through VEGF-stimulated Src kinase to promote neovascularization of the retina (Zhang et al., 2010). Similarly, PLD2 knockout mice have deficient aortic sprouting due to decreased response to VEGF (Lee et al., 2016).
Tumor cell production of VEGF and its stimulation of angiogenesis is important in mouse models of cancer. When either melanoma or Lewis lung carcinoma cells were subcutaneously implanted into PLD1-deficient mice, the resulting tumors grew more slowly than was observed when the tumor cells were implanted into wild-type (WT) mice, and the underlying mechanism was shown to result from greatly decreased neoangiogenesis, which in turn ensued because of a blunted VEGF signaling response by the vascular endothelial cells (Chen et al., 2012). This highlights the role of PLD expression in the tumor environment, as the decrease in neovascularization occurred as a consequence of diminished activation of AKT and MAPK in the VEGF-stimulated endothelial cells. Between propagating proliferative signaling and stimulating vascularization, PLD activity in both the tumor and microenvironment support primary tumor proliferation and growth.
Autophagy and deregulating metabolism
Cancer cells frequently utilize altered metabolic pathways to maintain biosynthetic and bioenergetic demands of rapid proliferation. Historically, cancer metabolism has focused on the observation of increased glycolysis even in the presence of oxygen, known as the Warburg effect. However, a great deal of recent research demonstrates that cancer cells utilize a complex interconversion of multiple metabolites to maintain synthetic and energetic demands (Deberardinis and Chandel, 2016). This flexible network allows cells to maintain adequate levels of lipids, glucose, and amino acids. When nutrient supplies are not sufficient, cells activate macroautophagy (autophagy) to replenish pools. Autophagy is largely repressed by mTOR activity, which is stimulated by PLD-generated PA (Foster et al., 2014). PLD has been suggested to function as a sensor of metabolites including lipid pools, as phosphatidylcholine, the enzymatic substrate of PLD, is one of the most abundant phospholipids of the cell membrane. Glucose activation of mTOR relies on a PLD dependent manor in multiple cell lines (Xu et al., 2011). Furthermore, sufficiency of amino acids activates PLD1 by Vps34 (Yoon et al., 2011). Taken together, PLD is a diverse sensor of biosynthetic pools and a critical regulator of autophagy due to mTOR’s dependency on it.
Consistent with this regulatory pathway, autophagic flux can be stimulated by siRNA knockdown of PLD1 (Jang et al., 2013) or PLD2 (Hwang et al., 2014). However, others have shown a reduction in autophagic flux when either PLD1 or PLD2 is inhibited. In glioblastoma cells, inhibition of PLD1 or PLD2 decreased autophagic flux through Akt phosphorylation of Beclin1 (Ronald C. Bruntz et al., 2014). Similarly, starvation of PLD1 knockout mice resulted in a decrease of LC3+ compartment formation in mouse liver cells and embryonic fibroblasts (Dall’Armi et al., 2010).
Other than regulating autophagy to maintain metabolism, PLD controls the transportation of multiple metabolites. In response to insulin, PLD activity translocates Glut-4 vesicles to the cell membrane to increase insulin-stimulated glucose uptake (Huang et al., 2005). PLD1 activity also induces the formation of lipid droplets in 3T3 cells (Andersson et al., 2006). When cultured under low glucose conditions, PLD1-inhibited cells display decreased survival after extended deprivation and have dysfunctional transportation of fatty acids to the mitochondria for oxidation (Cai et al., 2016). It is unclear how PLD maintains a dual role by both activating and inhibiting autophagy. Another conflicting question is why PLD seemingly is activated during nutrient satiation, nutrient deprivation, and/or serum starvation. The answer may partially lie in non-standard variations of experimental conditions in defining starvation conditions with respect to glucose, amino acids, glutamine, and serum concentrations. Unique circumstances (or adapted cell lines) may provide a survival or detrimental effect. The role of autophagy as either supportive or destructive to the tumor remains conflicted outside of PLD-specific mechanisms (Levy et al., 2017). Regardless, the involvement of PLD in regulating these multiple aspects of sensing and transporting precursors to maintain biosynthetic and bioenergetic demands emphasizes the importance of defining its role in the rapidly expanding field of cancer metabolism.
Invasion and metastasis
Initiating tumor growth is complicated as cells must first sustain proliferative signals, expand vasculature to receive oxygen and nutrients, and overcome metabolic limitations through autophagy and alternative pathways. To successfully form distant metastases, primary tumor cells must continue to adapt and survive a gauntlet of challenges (Vanharanta and Massagué, 2013). Tumor cells must then (1) detach from adjacent cells, (2) degrade surrounding substrates, (3) invade to vasculature, and (4) intravasate to circulation. Once in the lymphovascular system, the tumor cells are exposed to a barrage of physical, chemical, and immune insults that must be mitigated to (5) survive in circulation. If overcome, these metastatic cells must then (6) extravasate from circulation only to be deposited in an alien environment with different stroma, metabolic substrates, and growth factors further emphasizing the importance of (7) metabolic and proliferative adaptation to successfully expand and leave dormancy. PLD supports each of these steps of the metastatic cascade in addition to growth and vascularization of the primary tumor as previously discussed (Figure 2).
Figure 2.
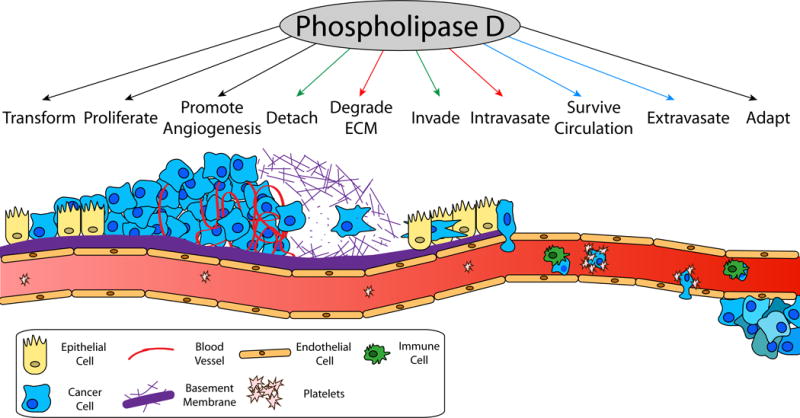
The successful metastasis of a cancer cell from the primary tumor to a distant site requires several steps, each of which necessitates unique cellular characteristics. Expression of PLD has been implicated in supporting each of these processes as indicated by arrows. The effects of PLD in regulating actin polymerization (green arrows), matrix metalloproteinase secretion (red arrows), and platelet activation (blue arrows) are particularly important to the process, although additional mechanisms also contribute. Once initiating tumor cells transform and grow to form the primary tumor, cells must stimulate angiogenesis to maintain nutrients and provide access to the lymphovascular system. Cancer cells must then reduce adhesion to neighboring cells to detach and degrade surrounding extra cellular matrix (ECM). This provides a path for migration cells to invade neighboring stromal cells and reach vasculature. Cancer cells must then intravasate into circulation by opening endothelial cells or crossing at leaky points. When circulating in the blood, cancer cells activate platelets to form protective coverings that prevent immune cell killing and promote trapping in the narrow capillaries of distant organs. The cancer cells must then extravasate and proliferate in the new location, which requires adaptations to new stroma, nutrients, and growth factors.
The process of invasion incorporates substrate degradation and migration. Tumors are encapsulated by multiple extra cellular proteins such as collagen. To clear a path for movement, PLD facilitates tumor cells secretion of matrix metalloproteinases to degrade these substrates and rearrangement of the actin cytoskeleton to form invadopodia and mobilize (Wang et al., 2017). After the invading tumor cells reach circulation, survival is immensely difficult. It is estimated that the overwhelming majority of circulating tumor cells (CTC) die due to mechanic sheering, apoptosis, or immune clearance (Stott et al., 2010). Platelets can form aggregations that protect CTC from these elements and may influence a more aggressive phenotype to drive extravasation after arresting in small capillaries (Labelle et al., 2011). PLD activity in platelets is necessary for activation of αIIbβ3 integrins (Elvers et al., 2010) and alpha granule release (Thielmann et al., 2012). When intravenously injected with B16F10 melanoma cells, mice lacking PLD1 developed 50% fewer lung metastatic foci than wild-type mice due to dysfunctional platelet activation and reduced interaction with CTC (Chen et al., 2012).
The occurrence of drug-resistant metastases is the most lethal complication of cancer treatment. Unfortunately, the adaptability of metastatic cells frequently results in formation of secondary tumors that are resistant to therapies previously effective against the primary tumor (Fischer et al., 2015). The ability to sensitize tumor cells to existing therapies offers potential for combinatorial strategies. Radiotherapy is a major approach to cancer, with nearly 50% cancer patients receiving radiation during their treatment (Delaney et al., 2005). In MDA-231 cells, inhibition of PLD1 promotes sensitization to ionizing radiation resulting in increased cell death (Cheol Son et al., 2013). PLD activity also promotes resistance to rapamycin and may serve as an indicator of efficacy of treatment (Chen et al., 2003). Although it was reported that PLD activity down-regulates multidrug resistance-1 gene and decreases sensitivity to vinblastine, the direct mechanism involved in this phenomenon is unclear, as these experiments relied on non-specific stimulators and inhibitors of PLD (Marguerite et al., 2013). To improve long term survival, research is focusing on developing therapies that specifically prevent metastasis, strategically by targeting any step as each is critical for formation. This makes PLD an intriguing target given its multiple points of involvement.
Small molecule inhibitors of PLD
Early research on PLD utilized its high affinity for primary alcohols to out-compete water as reactive nucleophile. Specifically, 1-butanol was frequently used to generate phosphatidylbutanol at the expense of PA production. Interpretation of such studies are complicated, as the concentration of 1-butanol required to fully divert the action of PLD to generate PA is now appreciated to produce a wide range of effects independent of PLD activity (Su et al., 2009). A search for specific small molecule inhibitors by high-throughput screening identified halopemide (Monovich et al., 2007). Halopemide is a psychotropic agent for which multiple clinical trials were conducted without observation of toxicities or adverse effects at plasma concentrations of 100–360 ng/mL (240–860 nM), which was, unknowingly at the time, sufficient to largely inhibit PLD1 and partially inhibit PLD2 (in vivo IC50’s of 21 nM and 300 nM respectively). Optimization then led to the identification of 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), an analog with greatly improved inhibition of PLD1 and PLD2 (1 nM and 44 nM IC50 in vivo, respectively) and lack of activity as a dopamine receptor antagonist (Monovich et al., 2007). This inhibitor has become a commonly used inhibitor of PLD for cell- and mouse-based studies. Brown and Lindsley have since generated and characterized a large family of halopemide-derived inhibitors that may be pharmacologically cleaner, and in some cases are isoform-selective, making them useful for cell culture experiments (Ronald C Bruntz et al., 2014). These compounds though have relatively short half-lives in vivo though (~ 1 hr), making FIPI, with its 5.5 hr half-life, more useful for animal model experiments.
FIPI phenocopies many of the in vivo effects seen in PLD-deficient animal models that were discussed above, including angiogenesis, autophagy, and metabolic deregulation. In cancer mouse models, use of subcutaneously-implanted osmotic pumps to treat xenograft implantation of breast cancer resulted in reduced primary tumor growth and decreased metastasis to the axillary lymph nodes and lungs (Henkels et al., 2016, 2013). Xenografts of hepatocellular carcinoma cells also experienced attenuated tumor growth and EMT when treated with PLD inhibitors (Xiao et al., 2016). A similar reduction in metastasis was seen under acute FIPI inhibition of tail-vein injected melanoma cells (Chen et al., 2012).
Conclusions
Recent chemotherapies have provided meaningful improvements for patient 5-year recurrence-free survival. Unfortunately, the development of chemoresistant metastases is still a common and fatal outcome. As discussed, PLD is a promising target to inhibit multiple aspects of cancer growth and spread due to is multiple roles. Early cell-based studies first highlighted the importance of PLD in propagating proliferative signals and regulating cell mobility, which have been replicated in recent animal studies. These in vivo models have expanded our understanding of how PLD is involved with essentially every step of the metastatic cascade through activity in both cancer and stromal cells. As such, there appear to be several clinical settings in which PLD inhibition might be of significant benefit. Future goals will include generating better inhibitors and interesting pharmaceutical groups in pursuing their development.
Acknowledgments
This work was supported by National Institutes of Health award R01 GM084251 to MAF. ER was supported on National Institutes of Health training grants GM008444 and GM092714.
Footnotes
Competing Interests
The authors have no competing interests to declare.
References
- Ali WH, Chen Q, Delgiorno KE, Su W, Hall JC, Hongu T, Tian H, Kanaho Y, Di Paolo G, Crawford HC, Frohman MA. Deficiencies of the Lipid-Signaling Enzymes Phospholipase D1 and D2 Alter Cytoskeletal Organization, Macrophage Phagocytosis, and Cytokine-Stimulated Neutrophil Recruitment. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammar MR, Kassas N, Bader MF, Vitale N. Phosphatidic acid in neuronal development: A node for membrane and cytoskeleton rearrangements. Biochimie. 2014;107:51–57. doi: 10.1016/j.biochi.2014.07.026. [DOI] [PubMed] [Google Scholar]
- Andersson L, Boström P, Ericson J, Rutberg M, Magnusson B, Marchesan D, Ruiz M, Asp L, Huang P, Frohman Ma, Borén J, Olofsson SO. PLD1 and ERK2 regulate cytosolic lipid droplet formation. J Cell Sci. 2006;119:2246–2257. doi: 10.1242/jcs.02941. [DOI] [PubMed] [Google Scholar]
- Brown HA, Thomas PG, Lindsley CW. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov. 2017;16:351–367. doi: 10.1038/nrd.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Lindsley CW, Brown HA. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev. 2014;66:1033–79. doi: 10.1124/pr.114.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Taylor HE, Lindsley CW, Brown HA. Phospholipase D2 mediates survival signaling through direct regulation of akt in glioblastoma cells. J Biol Chem. 2014;289:600–616. doi: 10.1074/jbc.M113.532978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, He J, Xiong J, Tay LWR, Wang Z, Rog C, Wang J, Xie Y, Wang G, Banno Y, Li F, Zhu M, Du G. Phospholipase D1-regulated autophagy supplies free fatty acids to counter nutrient stress in cancer cells. Cell Death Dis. 2016;7:e2448. doi: 10.1038/cddis.2016.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sato T, Hongu T, Zhang Y, Ali W, Van Der Velden A, Tian H, Di Paolo G, Kanaho Y, Frohman MA. Key Roles for the Lipid Signaling Enzyme Phospholipase D1 in the Tumor Microenvironment During Tumor Angiogenesis and Metastasis. Sci Signal. 2012;5 doi: 10.1126/scisignal.2003257.Key. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zheng Y, Foster Da. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–3942. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- Cheol Son J, Woo Kang D, Mo Yang K, Choi KY, Gen Son T, Min DS. Phospholipase D inhibitor enhances radiosensitivity of breast cancer cells. Exp Mol Med. 2013;45:e38. doi: 10.1038/emm.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Han JS. Overexpression of phospholipase D enhances Bcl-2 expression by activating STAT3 through independent activation of ERK and p38MAPK in HeLa cells. Biochim Biophys Acta-Mol Cell Res. 2012;1823:1082–1091. doi: 10.1016/j.bbamcr.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris aJ, Frohman Ma. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/S0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small Sa, Duff K, Frohman Ma, Wenk MR, Yamamoto A, Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nat Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2 doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment. Cancer. 2005;104:1129–1137. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- Du G, Altshuller YM, Vitale N, Huang P, Chasserot-Golaz S, Morris AJ, Bader MF, Frohman MA. Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J Cell Biol. 2003;162:305–315. doi: 10.1083/jcb.200302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Huang P, Liang BT, Frohman MA. Phospholipase D2 Localizes to the Plasma Membrane and Regulates Angiotensin II Receptor Endocytosis. Mol Biol Cell. 2004;15:1024–1030. doi: 10.1091/mbc.E03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers MEJ, Boesl M, Chen Q, Heemskerk JWM, Stoll G, Frohman MA, Nieswandt B. Impaired αIIb β3 integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal. 2010;3 doi: 10.1126/scisignal.2000551.Impaired. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA, Salloum D, Menon D, Frias MA. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR) J Biol Chem. 2014;289:22583–22588. doi: 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol Sci. 2015;36:137–144. doi: 10.1016/j.tips.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cambronero J. Phosphatidic acid, phospholipase D and tumorigenesis. Adv Biol Regul. 2014;54:197–206. doi: 10.1016/j.jbior.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cambronero J, Fite K, Miller TE. How miRs and mRNA deadenylases could post-transcriptionally regulate expression of tumor-promoting protein PLD. Adv Biol Regul. 2017 doi: 10.1016/j.jbior.2017.08.002. https://doi.org/10.1016/j.jbior.2017.08.002. [DOI] [PMC free article] [PubMed]
- Hammond SM, Altshuller YM, Sung T, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995;270:29640–29643. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene. 2013;32:5551–5562. doi: 10.1038/onc.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels KM, Muppani NR, Gomez-Cambronero J. PLD-specific small-molecule inhibitors decrease tumor-associated macrophages and neutrophils infiltration in breast tumors and lung and liver metastases. PLoS One. 2016;11:1–24. doi: 10.1371/journal.pone.0166553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA. PiRNA-Associated Germline Nuage Formation and Spermatogenesis Require MitoPLD Profusogenic Mitochondrial-Surface Lipid Signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Altshuller YM, Hou JC, Pessin JE, Frohman MA. Insulin-stimulated Plasma Membrane Fusion of Glut4 Glucose Transporter-containing Vesicles Is Regulated by Phospholipase D1. Mol Biol Cell. 2005;16:2614–2623. doi: 10.1091/mbc.E04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WC, Kim MK, Song JH, Choi KY, Min DS. Inhibition of phospholipase D2 induces autophagy in colorectal cancer cells. Exp Mol Med. 2014;46:e124. doi: 10.1038/emm.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YH, Choi KY, Min DS. Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ. 2013;21:1–14. doi: 10.1038/cdd.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Frohman MA. Phospholipase D: A lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DW, Choi KY, Min DS. Functional regulation of phospholipase D expression in cancer and inflammation. J Biol Chem. 2014;289:22575–22582. doi: 10.1074/jbc.R114.569822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee J, Kim S, Shin MK, Min DS, Shin T. Differential expression of phospholipases D1 and D2 in mouse tissues. Cell Biol Int. 2007;31:148–155. doi: 10.1016/j.cellbi.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S, Hynes RO. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Ghim J, Song P, Suh P, Ryu SH. Loss of phospholipase D2 impairs VEGF-induced angiogenesis. BMB Rep. 2016;49:191–196. doi: 10.5483/BMBRep.2016.49.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Du K, Fu Z, Zhang S, Wu X. Hypoxia-inducible factor 1-alpha up-regulates the expression of phospholipase D2 in colon cancer cells under hypoxic conditions. Med Oncol. 2015;32:394. doi: 10.1007/s12032-014-0394-9. [DOI] [PubMed] [Google Scholar]
- Lopez I, Arnold RS, Lambeth JD. Cloning and initial characterization of a human phospholipase D2 (hPLD2): ADP-ribosylation factor regulates hPLD2. J Biol Chem. 1998;273:12846–12852. doi: 10.1074/jbc.273.21.12846. [DOI] [PubMed] [Google Scholar]
- Mahankali M, Peng HJ, Henkels KM, Dinauer MC, Gomez-Cambronero J. Phospholipase D2 (PLD2) is a guanine nucleotide exchange factor (GEF) for the GTPase Rac2. Proc Natl Acad Sci. 2011;108:19617–19622. doi: 10.1073/pnas.1114692108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguerite V, Gkikopoulou E, Alberto JM, Guéant JL, Merten M. Phospholipase D activation mediates cobalamin-induced downregulation of Multidrug Resistance-1 gene and increase in sensitivity to vinblastine in HepG2 cells. Int J Biochem Cell Biol. 2013;45:213–220. doi: 10.1016/j.biocel.2012.09.018. [DOI] [PubMed] [Google Scholar]
- McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du Z, LaSala D, Boyar W, Steed P. Optimization of Halopemide for Phospholipase D2 inhibition. Bioorganic Med Chem Lett. 2007;17:2310–2311. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- Sanematsu F, Nishikimi A, Watanabe M, Hongu T, Tanaka Y, Kanaho Y, Côté JF, Fukui Y. Phosphatidic acid-dependent recruitment and function of the rac activator DOCK1 during dorsal ruffle formation. J Biol Chem. 2013;288:8092–8100. doi: 10.1074/jbc.M112.410423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvy PE, Lavieri RR, Lindsley CW, Brown HA. Phospholipase D: Enzymology, functionality, and chemical modulation. Chem Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, Inserra EJ, Ulman M, Springer S, Nakamura Z, Moore AL, Tsukrov DI, Kempner ME, Dahl DM, Wu CL, Iafrate AJ, Smith MR, Tompkins RG, Sequist LV, Toner M, Haber DA, Maheswaran S. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2:25ra23. doi: 10.1126/scitranslmed.3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Yeku O, Olepu S, Genna A, Park J, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75:437–446. doi: 10.1124/mol.108.053298.have. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht JA, Frohman MA. Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J. 1997;16:4519–4530. doi: 10.1093/emboj/16.15.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielmann I, Stegner D, Kraft P, Hagedorn I, Krohne G, Kleinschnitz C, Stoll G, Nieswandt B. Redundant functions of phospholipases D1 and D2 in platelet α-granule release. J Thromb Haemost. 2012;10:2361–2372. doi: 10.1111/j.1538-7836.2012.04924.x. [DOI] [PubMed] [Google Scholar]
- Toschi a, Edelstein J, Rockwell P, Ohh M, Foster Da. HIF alpha expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene. 2008;27:2746–2753. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- Vanharanta S, Massagué J. Origins of Metastatic Traits. Cancer Cell. 2013;24:410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang F, He J, Wu P, Tay LWR, Cai M, Nian W, Weng Y, Qin L, Chang JT, McIntire LB, Di Paolo G, Xu J, Peng J, Du G. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev Cell. 2017;43:186–197.e7. doi: 10.1016/j.devcel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Sun Q, Bei Y, Zhang L, Dimitrova-Shumkovska J, Lv D, Yang Y, Cao Y, Zhao Y, Song M, Song Y, Wang F, Yang C. Therapeutic inhibition of phospholipase D1 suppresses hepatocellular carcinoma. Clin Sci (Lond) 2016;130:1125–1136. doi: 10.1042/CS20160087. [DOI] [PubMed] [Google Scholar]
- Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. Phospholipase D Mediates Nutrient Input to Mammalian Target of Rapamycin Complex 1 (mTORC1) *. J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SF, Freer S, Benson AA. Transphosphatidylation by phospholipase D. J. Biol. Chem. 1967;242:477–484. [PubMed] [Google Scholar]
- Yoon M, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid–sensing mTORC1 pathway. J Cell Biol. 2011;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Rosenberger CL, Wu C, Truong N, Sweedler JV, Chen J. Rapid mitogenic regulation of the mTORC1 Inhibitor, DEPTOR, by phosphatidic acid. Mol Cell. 2015;58:549–556. doi: 10.1016/j.molcel.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng XXI, Zheng X, Xiang Y, Cho HP, Jessen JR, Zhong TP, Solnica-Krezel L, Brown HA. Phospholipase D1 is required for angiogenesis of intersegmental blood vessels in zebrafish. Dev Biol. 2009;328:363–376. doi: 10.1016/j.ydbio.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang D, Kundumani-Sridharan V, Gadiparthi L, Johnson DA, Tigyi GJ, Rao GN. PLD1-dependent PKCγ activation downstream to Src is essential for the development of pathologic retinal neovascularization. Blood. 2010;116:1377–1385. doi: 10.1182/blood-2010-02-271478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Frohman MA. Cellular and physiological roles for phospholipase D1 in cancer. J Biol Chem. 2014;289:22567–22574. doi: 10.1074/jbc.R114.576876. [DOI] [PMC free article] [PubMed] [Google Scholar]