

Abstract
The M-cluster is the [(homocitrate)MoFe7S9C] active site of nitrogenase that is derived from an 8Fe core assembled via coupling and rearrangement of two [Fe4S4] clusters concomitant with the insertion of an interstitial carbon and a ‘9th sulfur’. Combining synthetic [Fe4S4] clusters with an assembly protein template, here we show that sulfite can give rise to the ‘9th sulfur’ that is incorporated in the catalytically important belt region of the cofactor after the radical SAM-dependent carbide insertion and the concurrent 8Fe-core rearrangement have already taken place. Based on the differential reactivity of the formed cluster species, we also propose a new [Fe8S8C] cluster intermediate, the L*-cluster, that is similar to the [Fe8S9C] L- cluster but lacks the ‘9th S’ from sulfite. This work provides a semi-synthetic tool for protein reconstitution that could be widely applicable for the functional analysis of other FeS systems.
Graphical abstract
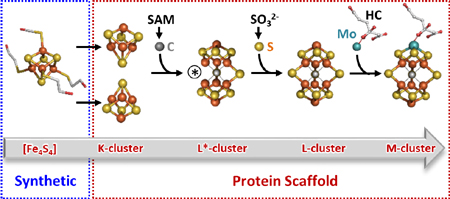
Nitrogenase plays a key role in the global nitrogen cycle, catalyzing the ambient reduction of nitrogen to ammonia at its active cofactor site.1,2 Designated the M-cluster (Fig. 1a, b), the cofactor of the Mo-nitrogenase consists of [MoFe3S3] and [Fe4S3] subclusters that are bridged by three μ2-‘belt’ sulfides (S2−) and one μ6-interstitial carbide (C4−), and it is further coordinated by homocitrate at its Mo end.3–6 Assembly of the M-cluster has attracted considerable attention because it is biologically important and chemically unprecedented. Previously, we have shown that the M-cluster is assembled through (i) formation of an L-cluster ([Fe8S9C]), an 8Fe core of the M-cluster, via coupling and rearrangement of a K-cluster (2x[Fe4S4]) concomitant with incorporation of an interstitial carbide and a ‘9th sulfur (S)’ on NifB, a radical S-adenosyl-L-methionine (SAM) enzyme; (ii) transformation of the L-cluster into a mature M-cluster via insertion of Mo and homocitrate on NifEN, an assembly scaffold; and (iii) transfer of the M-cluster to its binding site in NifDK, the catalytic component of the Mo nitrogenase (Fig. 1a, b).7–12 Further, we have identified early steps along the carbide insertion pathway that involve attachment of the methyl group of SAM to a sulfide atom of the K-cluster, followed by abstraction of a hydrogen atom from this methyl group by a SAM-derived 5′-deoxyadenosyl radical (5′-dA•) and further processing of the resultant carbon-radical species into an interstitial carbide (Fig. 1a, b).7–9
Figure 1. Assembly of the M-cluster.

(a) The assembly of M-cluster involves SAM-dependent conversion of K-cluster to L-cluster on NifB, transfer of L-cluster to NifEN, maturation of L-cluster on NifEN upon insertion of Mo and homocitrate (HC) by NifH, and transfer of the resultant M-cluster to NifDK. (b) Coupling of the 4Fe units of K-cluster (2x[Fe4S4]) into an 8Fe L-cluster ([Fe8S9C]) concomitant with insertion of an interstitial carbide and a ‘9th sulfur’, followed by conversion of the L-cluster to a mature M-cluster ([MoFe7S9C]) via insertion of Mo and HC. Carbide insertion begins with methyl transfer from SAM to the K-cluster and hydrogen abstraction from the SAM-derived methyl group by 5′-dA• and continues with further deprotonation/dehydrogenation of the carbon-radical until a carbide appears in the center of the L-cluster. c. The [Fe4S4]Syn cluster ([Fe4S4(SCH2CH2OH)4]2–) with ligands that are exchangeable for protein-bound cysteines. The hydrogen atoms are omitted for clarity. Color code: Fe, orange; S, yellow; Mo, cyan; C, gray; N, blue; O, red.
While these studies have advanced our understanding of the nitrogenase cofactor assembly, certain crucial details of this process, such as the 4Fe-modular nature of the K-cluster, the origin of the ‘9th S’, and the sequence of events between C and S insertion, have remained unclear. Study of homologously expressed NifB from Azotobacter vinelandii, the organism most studied in nitrogenase research, has historically been hampered by an apparent instability and inactivity of the isolated enzyme, limiting its study to a few reports that used either the NifEN-B fusion protein7–9 or heterologous expression.13,14 Recently, we have established that the NifB protein from Methanosarcina acetivorans (designated MaNifB) is a functional homolog of its counterpart from A. vinelandii that converts the K-cluster to an L-cluster in vitro and subsequently donates the L-cluster to the heterologous biosynthetic machinery of A. vinelandii for further maturation into an M-cluster.13 Identification of a functional methanogen NifB homolog that can be expressed in Escherichia coli circumvents the difficulty of obtaining a stable, active form of the A. vinelandii NifB protein and presents a more simplified system than the A. vinelandii NifEN-B fusion protein.7–9 However, traditional reconstitution of FeS clusters (i.e., using FeCl3 and Na2S) for the as-isolated, cluster-incomplete MaNifB cannot directly facilitate the assessment of whether the K-cluster consists of two 4Fe modules; rather, this method often results in attachment of excess sulfide aggregates to the FeS clusters in the protein, making it impossible to trace the incorporation of the ‘9th S’ or uncouple this event from that of carbide insertion.
Here, we use synthetic [Fe4S4] clusters to reconstitute MaNifB and trace the origin of the ‘9th S’, which allows us to define the sequence of events between carbide- and sulfur-insertion during cofactor assembly while establishing a semi-synthetic approach that could be extended to functional analyses of other FeS systems.
Results & Discussion
In search of an alternative FeS reconstitution approach, we first examined whether a water-soluble, synthetic [Fe4S4] compound ([Fe4S4(SCH2CH2OH)4]2–, designated [Fe4S4]Syn; Fig. 1c) could be used to reconstitute NifH, the obligate reductase component of Mo-nitrogenase. The water-stable [Fe4S4]Syn contains an [Fe4S4] core coordinated by ligands (i.e., -SCH2CH2OH) that are exchangeable with protein-bound cysteines15,16 and, therefore, is a suitable agent for ‘clean’ FeS reconstitutions. Indeed, a dark-brown protein (designated NifH[Fe4S4]) could be re-isolated following incubation of the FeS-depleted apo-NifH (designated NifHapo) with [Fe4S4]Syn clusters (Supplementary Fig. 1). The [Fe4S4] clusters in NifH[Fe4S4] and the as-isolated holo-NifH (designated NifHholo) were indistinguishable from each other, both displaying the same characteristic EPR features in the 0, +1 and +2 oxidation states and undergoing the same line-shape changes of EPR spectra upon nucleotide binding (Supplementary Fig. 2). Moreover, NifH[Fe4S4] and NifHholo displayed nearly identical catalytic profiles when titrated against increasing amounts of the catalytic NifDK component and almost indistinguishable activities in substrate reductions (Supplementary Fig. 2).
Having established a proof-of-concept for the utility of [Fe4S4]Syn clusters in FeS reconstitution, we then generated a [Fe4S4]Syn-reconstituted form of MaNifB (designated MaNifB[Fe4S4]) by re-isolating the protein from an incubation mixture of apo-MaNifB (designated MaNifBapo) and [Fe4S4]Syn clusters. Like the FeCl3/Na2S-reconstituted MaNifB,13 MaNifB[Fe4S4] cleaved SAM into two products: (i) SAH, a product formed upon removal of the methyl group from SAM; and (ii) 5’-dAH, a product formed upon hydrogen abstraction from the SAM-derived methyl group by 5’-dA• (Fig. 2a). Moreover, consistent with the presence of multiple [Fe4S4] clusters (i.e., a SAM cluster and a K-cluster), MaNifB[Fe4S4] displayed a composite, [Fe4S4]1+-specific S=1/2 EPR features in the dithionite (DT)-reduced state (Fig. 2b, trace 1) that became EPR-silent upon indigo disulfonate (IDS)-oxidation to the [Fe4S4]2+ state (Fig. 2b, trace 3). In the presence of SAM, the composite S=1/2 signal of MaNifB[Fe4S4] underwent changes in the DT-reduced state (Fig. 2b, trace 2) concomitant with the appearance of an 8Fe core-specific g=1.92 signal in the IDS-oxidized state (Fig. 2b, trace 4), which was indicative of transformation of a K-cluster (2x[Fe4S4]) into an L-cluster ([Fe8S9C]) via cluster coupling/rearrangement and carbon/sulfur insertion (Fig. 1a, b).7–9,13 Such a transformation was further confirmed by an in vitro cluster maturation assay, wherein the K-cluster on MaNifB[Fe4S4] was converted into an L-cluster upon SAM treatment, followed by the L- to M-cluster maturation on NifEN and the subsequent transfer of the M-cluster to NifDK, resulting in a reconstituted NifDK protein that was active in substrate reduction (Fig. 2c). It should be noted that the maturation protein NifEN was homologously expressed in A. vinelandii and that the successful cluster transfer between this protein and the archaeal MaNifB demonstrates an effective strategy to mix and match nitrogenase proteins from different kingdoms.
Figure 2. Dithionite (DT)-dependent cluster maturation.
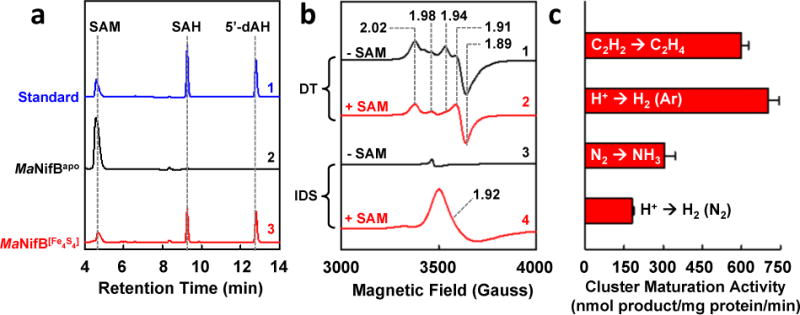
(a) HPLC elution profiles of (1) SAM, SAH and 5’-dAH standards, (2) SAM incubated with MaNifBapo and DT, and (3) SAM incubated with MaNifB[Fe4S4] and DT, showing that contrary to MaNifBapo, MaNifB[Fe4S4] was capable of methyltransfer from one SAM molecule and hydrogen abstraction from this methyl group via a 5’-dA• radical derived from another SAM molecule, generating SAH and 5’-dAH as the respective products of these reactions. (b) EPR spectra of DT-reduced (1, 2) or IDS-oxidized (3, 4) MaNifB[Fe4S4] incubated without (black) or with (red) SAM, demonstrating the formation of an L-cluster on MaNifB[Fe4S4] through a partial disappearance of the composite S=1/2 signal in the DT-reduced state (3) and the concomitant appearance of an L-cluster-specific, g=1.92 signal in the IDS-oxidized state (4). (c) Activities of MaNifB[Fe4S4] upon maturation and transfer of its cluster species to NifDK, showing the competence of MaNifB[Fe4S4] as an M-cluster donor for NifDK upon cluster maturation. Activity data were obtained from three independent experiments (n=6) and presented as mean±s.d. (c).
The observation that an 8Fe L-cluster could be generated from two [Fe4S4]Syn clusters on MaNifB[Fe4S4] provided direct evidence that the K-cluster consists of a [Fe4S4] cluster pair; furthermore, it suggested the possibility of using MaNifB[Fe4S4] to investigate the origin of the ‘9th S’ without complications caused by excess sulfide in the FeCl3/Na2S-reconstituted MaNifB. However, DT (S2O42–), the non-physiological reductant traditionally used in nitrogenase research, is known to break down into a number of S-based products such as sulfide (S2–), sulfite (SO32–) and sulfate (SO42–),17,18 all of which could potentially donate the ‘9th S’ source. To prevent introduction of sulfur species by DT and its breakdown products, we used europium(II) ethyleneglycoltetraacetate (EuII-EGTA, E0’= −0.8 V at pH 8)19 as a reductant in the cluster maturation assay to examine the ability of three physiologically relevant sulfur forms—S2–, SO32–, and SO42–—to serve as the source of the ‘9th S’ for the K- to L-cluster conversion on MaNifB[Fe4S4]. Interestingly, only SO32– supported cluster conversion on MaNifB[Fe4S4] in the presence of SAM, resulting in the formation of an L-cluster that could be further matured into an M-cluster to reconstitute NifDK (Supplementary Fig. 3). More excitingly, upon substitution of 35SO32− for unlabeled SO32− in the same incubation mixture, the 35S radiolabel could be detected in the re-purified MaNifB[Fe4S4] (Fig. 3a, left) and further traced to the L-cluster extracted from MaNifB[Fe4S4] (Fig. 3a, middle). These experiments provide the very first evidence that a DT breakdown product, SO32–, can serve as the external ‘9th S’ source. Notably, when conducted in the presence of DT, the in vitro cluster maturation assays do not require supplementation of SO32− (Fig 2c; Supplementary Fig. 3), which is consistent with an accumulation of SO32− upon decomposition of DT in aqueous solutions.17,18 The extracted L-cluster could then be used in a maturation assay in which it was first converted to an M-cluster on the scaffold protein NifEN and subsequently used for the reconstitution of NifDK (Fig. 3a, right). In the absence of SAM, however, no significant retention of the 35S radiolabel was observed when 35SO32– was supplied (Fig. 3a), suggesting that the insertion of the SAM-derived interstitial carbide likely precedes the incorporation of the ‘9th S’.
Figure 3. Dithionite (DT)-free cluster maturation.
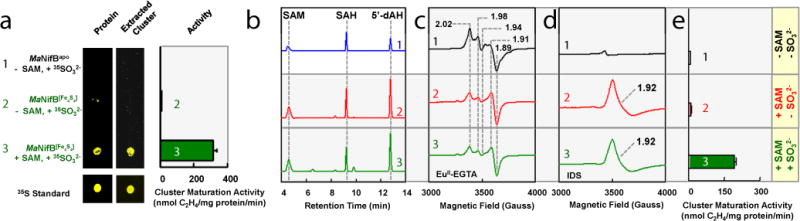
(a) Proteins captured on Ni Sepharose resin after incubation of (1) His-tagged MaNifBapo with 35SO32−, (2) His-tagged MaNifB[Fe4S4] with 35SO32−, and (3) His-tagged MaNifB[Fe4S4] with SAM and 35SO32− (left); clusters extracted from samples a, 2 and a, 3 (middle); and activities of C2H2-reduction by clusters extracted from samples a, 2 and a, 3, after maturation and transfer of clusters to NifDK (right). The 35S label and activity were only detected in SAM/SO32−-treated MaNifB[Fe4S4], suggesting incorporation of S from SO32− into a fully-assembled L-cluster. (b) HPLC elution profiles of (1) SAM, SAH and 5’-dAH standards, (2) SAM incubated with MaNifB[Fe4S4], and (3) SAM incubated with MaNifB[Fe4S4] and SO32−, showing the competence of SAM-treated MaNifB[Fe4S4] in methyltransfer and hydrogen abstraction with or without SO32−-treatment. (c, d) EPR spectra of EuII-EGTA-reduced (c) or IDS-oxidized (d) MaNifB[Fe4S4] incubated without SAM (black), with SAM (red), or with SAM and SO32− (green), suggesting formation of an 8Fe core of the L-cluster in SAM-treated MaNifB[Fe4S4] with or without SO32−. (e) Activities of MaNifB[Fe4S4] treated with (1) no additive, (2) SAM, and (3) SAM and SO32− upon maturation and transfer of clusters to NifDK, showing the sole competence of SAM/SO32−-treated MaNifB[Fe4S4] as an M-cluster donor. Activity data were obtained from three independent experiments (n=6) and presented as mean±s.d. (e).
To further tackle the sequence of events between C and S insertion, we monitored cluster conversion on MaNifB[Fe4S4] in the presence of SAM alone or SAM plus SO32−. When incubated with SAM, MaNifB[Fe4S4] displayed the same SAM cleavage pattern (Fig. 3b) and EPR spectroscopic changes (Fig. 3c, d) with or without SO32− treatment. However, quenching those incubation mixtures with acid to release methanethiol—a technique previously used to detect the SAM-derived methyl group attached to a K-cluster S atom9––liberated substantially less methanethiol in the presence of SO32– (Supplementary Fig. S4), suggesting an improved stability and, consequently, a reduced acid-susceptibility of the cluster species in the SO32−-treated MaNifB[Fe4S4]. Additionally, the SO32−-untreated MaNifB[Fe4S4] was inactive as an L-cluster donor, whereas the SO32−-treated MaNifB[Fe4S4] was active in donating the L-cluster for further maturation into an M-cluster (Fig. 3e). The observation of the 8Fe-core-specific g=1.92 signal in SO32−-untreated MaNifB[Fe4S4] after reaction with SAM (Fig. 3d, trace 2) is particularly intriguing, as it implies that the SAM-dependent carbon insertion and the concurrent 8Fe core rearrangement occur prior to the insertion of the ‘9th S’. Moreover, K-cluster fusion with C in the absence of SO32– leads to the formation of a new intermediate species (designated the L*-cluster) that we tentatively assign as a [Fe8S8C] cluster that closely resembles the L-cluster in architecture but lacks the ‘9th S’ in the catalytically-important belt region of the cluster (Fig. 4). This so-called ‘vacant’ site of the L*-cluster may be occupied by a putative cysteine thiolate or H2O ligand, and studies are underway to assess the exact L*-cluster composition and coordination.
Figure 4. Refined model of L-cluster assembly on NifB.
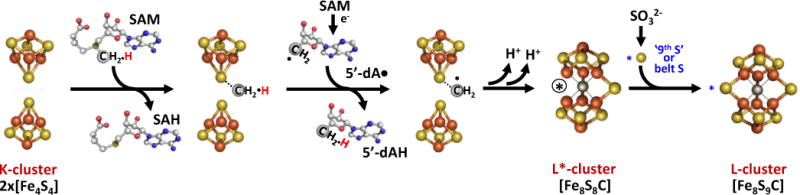
The radical SAM-dependent carbide insertion and the concurrent 8Fe core rearrangement precede the incorporation of the ‘9th S’ at the catalytically important belt region of the nitrogenase cofactor, resulting in an L*-cluster ([Fe8S8C]) that is nearly indistinguishable from the L-cluster ([Fe8S9C]) except for the missing ‘9th S’. The encircled black asterisk represents the ‘vacant’ site of the L*-cluster, which may be occupied by a cysteine thiolate or water molecule. Color code: Fe, orange; Mo, cyan; S, yellow; C, gray; N, blue; O, red.
Our proposal that the ‘9th S’ is inserted when the inorganic core structure of the M-cluster is in place is consistent with the facile exchange of the belt S of the M-cluster with selenium (Se)20 or replacement of a belt S with CO21 under turnover conditions (Supplementary Fig. 5). Interestingly, analogous to the utilization of an oxidized form of S (SO32−) for the incorporation of the ‘9th S’, the belt S of the M-cluster can only exchange with the Se atom in selenocyanate (SeCN−) but not with the more reduced Se in selenide (Se2−) upon turnover.20 It is possible, therefore, that insertion of the ‘9th S’ involves donation of electrons from the electron-rich cluster to the sulfur source, which explains why a more oxidized S species (SO32−)—instead of the fully reduced S (S2−)—is used as a source of the ‘9th S’. In the case of SO42−, however, the S atom is unable to directly interact with the Fe atoms; additionally, the cluster may be poised at a certain redox potential that does not accommodate the redox change required for the conversion of SO42− to a ‘9th S’.
Within the cell, sulfite is one of the central hubs of sulfur metabolism (Supplementary Fig. 6).22–24 A survey of the genomes of 13 nitrogen-fixing microbes,25 including those of A. vinelandii and M. acetivorans, revealed the presence of genes encoding enzymes involved in various sulfite-generating routes; most notably, more than 10 of these organisms possess genes encoding the 3’-phosphoadenosine 5’-phosphosulfate sulfotransferase (PAPS transferase), which converts PAPS to sulfite as part of the assimilatory sulfate reduction pathway (Supplementary Fig. 6). This observation provides support for the physiological relevance of sulfite or related compounds to nitrogenase assembly. Although we cannot rule out the participation of reactive physiological sulfur sources other than SO32– such as persulfides for the in vivo ‘9th S’ delivery,26 it should be noted that persulfides are not stable under reducing in vitro conditions. Nevertheless, our identification of shared sulfite-producing enzymes among multiple diazotrophs suggests that these pathways could be operative for supplying the ‘9th S’ source to NifB. While details of these events are yet to be elucidated, identification of sulfite as the in vitro source of ‘9th S’ that is inserted at a late stage of cofactor assembly suggests the possibility to specifically label this belt S for future mechanistic studies of nitrogenase (Supplementary Fig. 5).
Moreover, the methodology of using synthetic metalloclusters as reported herein could be applicable to a wide array of FeS-containing enzyme classes. The [Fe4S4]Syn cluster could prove particularly useful for reconstituting other heterologously expressed proteins in the radical SAM superfamily27 as well as other [Fe4S4]-cluster-containing enzymes associated with important processes such as DNA synthesis and gene regulation.28–30 Strategies to incorporate synthetic cofactors within apoproteins have been successfully employed in the hydrogenase field,31–33 to gain mechanistic and biosynthetic insight. These approaches can also be used to generate artificial enzymes with altered reactivities.34–36
Conclusions
In summary, we have demonstrated the utility of a water stable and soluble [Fe4S4] cluster to reconstitute the nitrogenase protein NifB for the determination of the L-cluster ‘9th S’ source in vitro. In the process, we have discovered that SO32–, and not S2– or SO42–, can donate an S atom for this purpose, and that previous successful studies of cluster maturation on NifB appear to have benefited from the presence of the reductant DT, which can potentially provide SO32– to the system. More excitingly, we have identified for the first time a putative [Fe8S8C] intermediate, the L*-cluster, that can be matured to the L-cluster upon addition of SO32–. Further investigation of the L*-cluster on NifB is an ongoing pursuit.
Methods
Methods other than protein reconstitution and 35S-labeling experiments are included in Supplementary Information.
Protein Reconstitution
Purified NifHholo or MaNifB was treated with 20 mM bathophenanthroline disulfonate, an iron chelator, in a buffer containing 5 mM MgATP, 2 mM dithionite (DT; Na2S2O4), 50 mM Tris-HCl (pH 8.0) and 500 mM NaCl, followed by incubation at room temperature for 1 h. The mixture was diluted with a buffer containing 50 mM Tris-HCl (pH 8.0) and loaded on a Q Sepharose column (GE Healthcare). Subsequently, the column was washed with a buffer containing 2 mM DT, 50 mM Tris-HCl (pH 8.0) and 100 mM NaCl prior to elution of apo-NifH (designated NifHapo) or apo-MaNifB (designated MaNifBapo) with a buffer containing 50 mM Tris-HCl (pH 8.0). Reconstitution of NifHapo or MaNifBapo with [Fe4S4]Syn was carried out by adding a dimethylformamide (DMF) solution of [Fe4S4]Syn dropwise to NifHapo or MaNifBapo at molar ratios of 1:1 and 5:1, respectively, in a buffer containing 2 mM DT, 20 mM BME, 50 mM Tris-HCl (pH 8.0) and 500 mM NaCl, with continuous stirring on ice. After incubation on ice for 1 h, the reaction mixture was diluted with a buffer containing 2 mM DT and 50 mM Tris-HCl (pH 8.0) and loaded on a Q Sepharose column. The column was then washed with a buffer containing 2 mM DT, 50 mM Tris-HCl and 100 mM NaCl prior to elution of the reconstituted NifH (designated NifH[Fe4S4]) or MaNifB (designated MaNifB[Fe4S4]) with a buffer containing 2 mM DT, 50 mM Tris-HCl (pH 8.0) and 500 mM NaCl. The DT-free reconstitution of MaNifBapo was carried out by passing MaNifBapo through a Sephadex G-25 desalting column (GE Healthcare) to remove DT, diluting the protein with a buffer containing 20 mM BME, 50 mM Tris-HCl (pH 8.0), and treating the protein solution with a DMF solution of [Fe4S4]Syn as described above. The mixture was directly loaded on a Q Sepharose column after incubation on ice for 30 min, and the column was washed with a buffer containing 0.5 mM EuII-EGTA, 50 mM Tris-HCl and 100 mM NaCl prior to elution of the reconstituted NifH[Fe4S4] or MaNifB[Fe4S4] with a buffer containing 2 mM EuII-EGTA, 50 mM Tris-HCl (pH 8.0) and 500 mM NaCl. All solutions described above contained 10% (v/v) glycerol.
35S-labeling Experiments
To monitor the event of ‘9th S’ insertion on MaNifB[Fe4S4] (see Fig. 3a, left), three different reactions were assembled, each containing, in a total volume of 40 μL, 2 mM EuII-EGTA, 2 mM Na235SO3, 10% (v/v) glycerol, 50 mM Tris-HCl (pH 8.0) and (i) 22 nmol DT-free MaNifBapo; (ii) 22 nmol DT-free MaNifB[Fe4S4]; and (iii) 22 nmol DT-free MaNifB[Fe4S4] and 5 mM SAM. All reactions were incubated for 30 min at room temperature with intermittent mixing and then run over Ni Sepharose resin (25 μL packed volume; GE Healthcare) pre-equilibrated with a buffer containing 2 mM EuII-EGTA, 500 mM NaCl, 10% (v/v) glycerol and 25 mM Tris-HCl (pH 8.0). The Ni Sepharose resin was washed with 100 μL of the same equilibration buffer before the bound protein was eluted with 80 μL buffer containing 2 mM EuII-EGTA, 250 mM imidazole, 500 mM NaCl, 10% (v/v) glycerol and 25 mM Tris-HCl (pH 8.0). The protein eluent was then diluted with 270 μL equilibration buffer and mixed with Ni Sepharose resin (12.5 μL packed volume; GE Healthcare). Subsequently, the protein-bound Ni Sepharose resin was re-suspended in the equilibration buffer and applied directly onto a Whatman Grade 1 qualitative filter paper (GE Healthcare). The blots were dried and exposed to a GE Healthcare Storage Phosphor Screen GP (20 × 25 cm) for 16 h before imaging was performed on a GE Healthcare Typhoon Trio+ variable mode imager. To trace the 35S radiolabel into the MaNifB-bound cluster species (see Fig. 3a, middle), reactions ii and iii above were scaled up by 6-fold, followed by extraction of cluster species from these reaction mixtures using a protocol adapted from a previously described procedure.8 The extracted clusters were then blotted onto a GE Healthcare Whatman filter paper for imaging as described above.
Data Availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files and from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
This work was supported by NIH-NIGMS grant GM67626 (to M.W.R. and Y.H.), DOE-BES grant DE-DC0014470 (to M.W.R. and Y.H.), a Takeda Science Foundation grant (to Y.O.), and Grant-in-Aids for Scientific Research (No. 23000007, 16H04116) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to K.T. and Y.O.).
Footnotes
Author contributions
K.T. (Irvine), C.C.L., N.S.S., Y.H. and M.W.R. designed and analyzed experiments. K.T. (Irvine) performed experiments. K.T. (Nagoya) and Y.O. provided materials. Y.H. and M.W.R. analyzed experiments and wrote the manuscript with input from all authors.
Competing interests
The authors declare no competing financial and non-financial interests.
References
- 1.Burgess BK, Lowe D. J Mechanism of molybdenum nitrogenase. Chem Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Rees DC, et al. Structural basis of biological nitrogen fixation. Philos Trans A Math Phys Eng Sci. 2005;363:971–984. doi: 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- 3.Chan MK, Kim J, Rees DC. The nitrogenase FeMo-cofactor and P-cluster pair: 2.2 Å resolution structures. Science. 1993;260:792–794. doi: 10.1126/science.8484118. [DOI] [PubMed] [Google Scholar]
- 4.Einsle O, et al. Nitrogenase MoFe-protein at 1.16 Å resolution: a central ligand in the FeMo-cofactor. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 5.Spatzal T, et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancaster KM, et al. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiig JA, Hu Y, Ribbe MW. NifEN-B complex of Azotobacter vinelandii is fully functional in nitrogenase FeMo cofactor assembly. Proc Natl Acad Sci USA. 2011;108:8623–8627. doi: 10.1073/pnas.1102773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiig JA, Hu Y, Lee CC, Ribbe MW. Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science. 2012;337:1672–1675. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiig JA, Hu Y, Ribbe MW. Refining the pathway of carbide insertion into the nitrogenase M-cluster. Nat Commun. 2015;6:8034. doi: 10.1038/ncomms9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, et al. FeMo cofactor maturation on NifEN. Proc Natl Acad Sci USA. 2006;103:17119–17124. doi: 10.1073/pnas.0602647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y, et al. Nitrogenase Fe protein: a molybdate/homocitrate insertase. Proc Natl Acad Sci USA. 2006;103:17125–17230. doi: 10.1073/pnas.0602651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribbe MW, Hu Y, Hodgson KO, Hedman B. Biosynthesis of nitrogenase metalloclusters. Chem Rev. 2014;114:4063–4080. doi: 10.1021/cr400463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fay AW, Wiig JA, Lee CC, Hu Y. Identification and characterization of functional homologs of nitrogenase cofactor biosynthesis protein NifB from methanogens. Proc Natl Acad Sci USA. 2015;112:14829–14833. doi: 10.1073/pnas.1510409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilcoxen J, et al. Electron paramagnetic resonance characterization of three iron-sulfur clusters present in the nitrogenase cofactor maturase NifB from Methanocaldococcus infernus. J Am Chem Soc. 2016;138:7468–7471. doi: 10.1021/jacs.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Averill BA, Herskovitz T, Holm RH, Ibers JA. Synthetic analogs of the active sites of iron-sulfur proteins. II Synthesis and structure of the tetra(mercapto-μ3-sulfido-iron) clusters, (Fe4S4(SR)4)2−. J Am Chem Soc. 1973;95:3523–3534. doi: 10.1021/ja00792a013. [DOI] [PubMed] [Google Scholar]
- 16.Barclay JE, Davies SC, Evans DJ, Hughes DL. Lattice effects in the Mössbauer spectra of salts of [Fe4S4{S(CH2)nOH}4]2−. Crystal structures of [PPh4]2[Fe4S4{S(CH2)nOH}4] (n=2, 3 and 4) Inorg Chim Acta. 1999;291:101–108. [Google Scholar]
- 17.de Carvalho LM, Schwedt G. Sulfur speciation by capillary zone electrophoresis. determination of dithionite and its decomposition products sulfite, sulfate and thiosulfate in commercial bleaching agents. J Chromatogr A. 2005;1099:185–190. doi: 10.1016/j.chroma.2005.08.084. [DOI] [PubMed] [Google Scholar]
- 18.Münchow V, Steudel R. The Decomposition of aqueous dithionite and its reactions with polythionates SnO2−6 (n=3–5) studied by ion-pair chromatography. Z anorg allg Chem. 1994;620:121–126. [Google Scholar]
- 19.Vincent KA, et al. Instantaneous, stoichiometric generation of powerfully reducing states of protein active sites using Eu(II) and polyaminocarboxylate ligands. Chem Commun. 2003;2003:2590–2591. doi: 10.1039/b308188e. [DOI] [PubMed] [Google Scholar]
- 20.Spatzal T, Perez KA, Howard JB, Rees DC. Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. Elife. 2015;4:e11620. doi: 10.7554/eLife.11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC. Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science. 2014;345:1620–1623. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brychkova G, Grishkevich V, Fluhr R, Sagi M. An essential role for tomato sulfite oxidase and enzymes of the sulfite network in maintaining leaf sulfite homeostasis. Plant Physiol. 2013;161:148–164. doi: 10.1104/pp.112.208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol. 2012;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kertesz MA. Riding the sulfur cycle - metabolism of sulfonates and sulfate esters in gram-negative bacteria. FEMS Microbiol Rev. 2000;24:135–175. doi: 10.1016/S0168-6445(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 25.Available at the open Joint Genome Institute (JGI) database (https://img.jgi.doe.gov/cgi-bin/m/main.cgi)
- 26.Mishanina TV, Libiad M, Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol. 2015;11:457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 28.Mühlenhoff U, et al. Compartmentalization of iron between mitochondria and the cytosol and its regulation. Eur J Cell Biol. 2015;94:292–308. doi: 10.1016/j.ejcb.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Mettert EL, Kiley PJ. Fe–S proteins that regulate gene expression. Biochim Biophys Acta. 2015;1853:1284–1293. doi: 10.1016/j.bbamcr.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Brien E, et al. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science. 2017;355 doi: 10.1126/science.aag1789. pii: eaag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berggren G, et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature. 2013;499:66–69. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esselborn J, et al. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat Chem Biol. 2013;9:607–609. doi: 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shima S, et al. Reconstitution of [Fe] hydrogenase using model complexes. Nat Chem. 2015;7:995–1002. doi: 10.1038/nchem.2382. [DOI] [PubMed] [Google Scholar]
- 34.Tanifuji K, et al. Combining a nitrogenase scaffold and a synthetic compound into an artificial enzyme. Angew Chem Int Ed. 2015;54:14022–14025. doi: 10.1002/anie.201507646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinisch T, Ward TR. Artificial metalloenzymes based on the biotin-streptavidin technology: challenges and opportunities. Acc Chem Res. 2016;49:1711–1721. doi: 10.1021/acs.accounts.6b00235. [DOI] [PubMed] [Google Scholar]
- 36.Lin YW. Rational design of metalloenzymes: from single to multiple active sites. Coord Chem Rev. 2017;336:1–27. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files and from the corresponding authors upon reasonable request.