

Summary
Monitoring enzymatic activities at the cell surface is challenging due to the poor efficiency of transport and membrane integration of FRET-based biosensors. Therefore, we developed a hybrid biosensor with separate donor and acceptor that assemble in situ. The directed evolution and sequence-function analysis technologies were integrated to engineer a monobody variant (PEbody) that binds to R-Phycoerythrin (R-PE) dye. PEbody was used for visualizing the dynamic formation/separation of intercellular junctions. We further fused PEbody with the enhanced cyan fluorescent protein and an enzyme-specific peptide at the extracellular surface to create a hybrid FRET biosensor upon R-PE capture for monitoring membrane-type-1 matrix metalloproteinase (MT1-MMP) activities. This biosensor revealed asymmetric distribution of MT1-MMP activities, which were high and low at loose and stable cell-cell contacts, respectively. Therefore, directed evolution and rational design are promising tools to engineer molecular binders and hybrid FRET biosensors for monitoring molecular regulations at the surface of living cells.
Keywords: FRET biosensor, Monobody, R-Phycoerythrin, MT1-MMP, Cell-cell contacts
In Brief
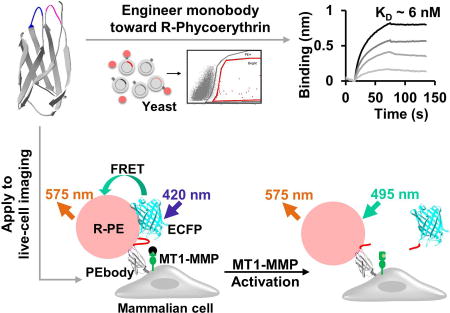
Limsakul et al. demonstrate that directed evolution and sequence-function analysis are promising tools for engineering molecular binders and hybrid FRET biosensors, which reveal new distinct subcellular features of MT1-MMP molecular regulations at the extracellular surface of live cells.
Introduction
Numerous genetically encoded intramolecular FRET biosensors have been developed to visualize various molecular events in live cells with high spatiotemporal resolution (Wang et al., 2005; Wang et al., 2008). However, improper protein folding frequently causes biosensor entrapment within cellular compartments (Shaner et al., 2007), particularly when these biosensors are targeted through the secretory pathways to extracellular membrane to monitor local molecular events. The entrapped biosensors typically do not respond to stimuli as designed and often contribute significantly to unwanted signal noise during analysis. To overcome this deficiency, we sought to develop a hybrid FRET biosensor consisting of an enhanced CFP (ECFP; donor) and an R-Phycoerythrin (R-PE; acceptor) to be assembled in situ at the extracellular surface of plasma membrane. Since R-PE is a cell-impermeable fluorescent dye with a high extinction coefficient and large Stokes shift (Glazer, 1985), the ECFP/R-PE pair is expected to provide strong FRET signals specifically at the plasma membrane with minimal intracellular background noise. However, R-PE cannot be genetically encoded (Isailovic et al., 2006). Therefore, a protein scaffold fused to ECFP is needed to capture R-PE for FRET functionality.
Directed evolution technology is a powerful tool used to engineer protein domains and scaffolds, particularly when rational design alone is insufficient (Arnold, 1998). This technology has been used to develop numerous fluorescent proteins with improved properties including enhanced brightness, modified spectra, and increased photo-stability (Shaner et al., 2004; Shaner et al., 2013; Shaner et al., 2008). Directed evolution and rational design based on sequence and structure information have also been applied to optimize the sensing components or linker lengths for genetically encoded FRET biosensors (Hires et al., 2008; Ibraheem et al., 2011; Komatsu et al., 2011).
Several protein scaffolds have been successfully optimized by directed evolution for different applications, including diagnostics (Binz et al., 2005), therapeutics (Wittrup et al., 2012), and imaging (Gulyani et al., 2011). Among these, a short 94-residue monobody (Figure 1A), derived from the tenth type III domain of human fibronectin, is a versatile non-antibody protein scaffold with a structure similar to the immunoglobulin heavy chain domain (Koide et al., 1998). The seven β-strands of the monobody can be randomized to create libraries of variants for protein binding sites (Batori et al., 2002; Koide et al., 1998), with the BC and FG loops proximally positioned to form a binding interface for target biomolecules with high flexibility and affinity (Carr et al., 1997; Koide et al., 1998).
Figure 1. The development of PEbody.

(A) The structure of the G9 monobody (modified from PDB ID: 1TTG). (B) The schematic diagram of the yeast display monobody library and the selection of the R-PE-binding monobody clones via FACS. (C) The R-PE binding capability of different monobody mutants as indicated: G9, a mutant with the FG loop of S4 (G9BC/S4FG), a mutant with the BC loop of S4 (S4BC/G9FG), and S4. The R-PE binding capability is defined as the ratio of the % of R-PE-positive yeast to the % of V5-positive yeast. The V5 epitope tag fused at C-terminus of PEbody was used as the indicator of protein expression on the yeast surface, see Figure S1C. (D) The improvement of R-PE-binding monobodies after further rounds of mutagenesis and sequence-function analysis. Eight mutants with different amino acid sequences in the FG loop were predicted and their R-PE binding capabilities were analyzed through flow cytometry. (E) Testing the specificity of R-PE-binding monobody. The binding capability of different dyes, including PerCP-Cy5.5, FITC, Alexa488, streptavidin-PE (SA-PE), and R-PE, to PEbodies displayed on the yeast surface was measured by flow cytometry. (F) The determination of binding affinity between R-PE and PEbody by bio-layer interferometry. Different concentrations of R-PE were used to determine kon and koff parameters which were used to calculate KD values. Data in (C-E) are represented as mean ± SD. The asterisk indicates a significant difference (*P < 0.05, **P < 0.01, and ***P < 0.001 with the two-tailed Student’s t test). See also Figure S1.
Utilizing directed evolution and sequence-function analysis, we developed a monobody variant, PEbody, which serves as a specific binding partner for R-PE. The multivalent interaction between PEbody and R-PE significantly enhances signals at the cell-cell contact, allowing the precise monitoring of the dynamic formation and dissociation of cell-cell contacts. We have further applied PEbody for the assembly of a new ECFP/R-PE hybrid FRET biosensor at the extracellular surface of cancer cells to monitor the proteolytic activity of MT1-MMP, which is a key molecule regulating pericellular matrix degradation during cancer metastasis (Covington et al., 2006; Gálvez et al., 2002; Hotary et al., 2003; Nawrocki-Raby et al., 2003; Rozanov et al., 2004; Woskowicz et al., 2013). The results revealed that MT1-MMP is differentially regulated depending on the maturity of cell-cell contacts, with high and low proteolytic activities at loose and stable cell-cell contacts, respectively. Thus, our hybrid biosensors can serve as potent tools to study the dynamics of molecular activities at the cell surface with subcellular resolutions.
Results
Directed evolution of an R-PE binder
We first applied directed evolution technology to engineer a monobody variant for the capture of R-PE and subsequent in situ assembly of a FRET biosensor at the extracellular surface. The G9 monobody, initially engineered to bind to the Src Homology 3 (SH3) of Fyn (Huang et al., 2012), was used as a template to generate mutant libraries by applying site-saturation mutagenesis (Miyazaki and Arnold, 1999) in the BC (residues 26–30) and FG (residues 77–81) loops of G9 (Figure S1A). These libraries were displayed on the yeast surface and used to screen for R-PE binders (Figure 1B and Figure S1B). After three rounds of selection by fluorescence-activated cell sorting (FACS), a mutant named Single4 (S4) containing the FINFK and WRWWY sequences in the BC and FG loops, respectively, was identified as an efficient R-PE binder (Figures 1C and S1F). The R-PE binding capability of S4, which is defined as the ratio between the % of R-PE binding yeast (gated by R-PE) and the % of monobody expressing yeast (gated by a labeling tag V5 on the monobody, Figure S1C), was 500-fold greater than the original G9 (Figure 1C), but was not further improved by additional rounds of cell sorting. It is therefore possible that the S4 sequence represents a locally optimal solution around the neighboring mutation space. Replacing a single BC or FG loop of G9 with that of S4, however, was not sufficient to increase the binding capability, suggesting that mutations on both loops are necessary for the overall enhanced R-PE-binding of S4 (Figure 1C).
Based on the structure and sequence of S4, we found that the FG variants consistently showed larger improvement on target binding capability than the BC variants (Figures S1D and S1E) (Batori et al., 2002). Consequently, it is possible to improve R-PE binding capability of S4 by further optimizing sequences in the FG loop of S4 while holding the BC loop constant. Additional mutagenesis was focused on the FG loop of S4 to create a new library (Figure S1F). However, no variant showed significant improvement of R-PE binding capability comparing to S4 after two more rounds of cell sorting (Figure S1G). Therefore, a rational approach was explored to further engineer and improve S4 for R-PE binding.
We surveyed the mutation space around the S4 monobody sequence by choosing 10 variants for sequence-function analysis, including 5 clones each with the highest and lowest binding affinities toward R-PE (Figure S1G). The high-affinity sequences (along with S4) were used to identify the residues contributing positively to R-PE-binding, while the low-affinity sequences allowed us to identify and exclude residues with negative contribution. Amino acids (AAs) at each position were ranked by their contribution scores. For example, the contribution score of amino acid A at the position j was calculated by averaging the R-PE binding capability among all mutants containing A at position j (see Method, Figures S1H and S1I). We then selected the highest ranked AAs for each position. This selection resulted in eight mutant sequences, including S4 (Figure 1D). The flow cytometry results showed that two mutants, M4 and M5, have significantly improved binding capability toward R-PE compared to S4 (Figure 1D). The mutant M4 was named as PEbody, having an R-PE-binding capability of 96%, and was used for all of the following experiments. The mutant M5 had similar R-PE-binding capability but less efficiency in protein production and hence was not further tested. Comparing the amino acid sequences of PEbody and S4 revealed that replacing arginine with proline at position 78 in S4 caused the increase in R-PE binding for PEbody (Figures 1D).
To further characterize the specificity and binding affinity of PEbody in recognizing R-PE, we stained the PEbody-expressing yeast cells with various fluorescent dyes, such as PerCP-Cy5.5, FITC, Alexa Fluor 488, and R-PE with streptavidin, that have been extensively used in flow cytometry applications. Only R-PE-based dyes could bind to PEbody, indicating that it is specific to R-PE (Figure 1E). The binding affinity between PEbody and R-PE was further characterized by using biolayer interferometry (BLI), with the KD determined to be 5.7 ± 3.5 nM (mean±SEM) (Figure 1F) which is consistent with the KD calculated using the yeast surface display (Feldhaus et al., 2003). The mean fluorescence intensity (MFI) of the R-PE bound to the yeast cell surface was plotted against increasing R-PE concentrations. We estimated the KD to be 9.6 ± 3.0 nM (mean±SEM) using nonlinear least-squares regression of the relationship between the MFI and the R-PE concentration (Figure S1J). These results suggest that PEbody suffices as an efficient and specific R-PE binder.
Characterization of PEbody in mammalian cells
PEbody was then displayed and characterized for R-PE-binding at the surface of human embryonic kidney cells (HEK293, Figure 2A). R-PE staining was found to be specific in cells expressing PEbody at the surface, especially efficient at the cell-cell contacts, but not in those expressing the non-specific G9 monobody or lacking the construct altogether (Figures 2B and S2A). These results further demonstrate that PEbody can be expressed in mammalian cells to serve as a specific binder of R-PE.
Figure 2. Characterization of PEbody on the surface of live mammalian cells.

(A) A schematic diagram of R-PE binding to PEbody displayed on the surface of a live mammalian cell. The green arrow represents the excitation wavelength (495 nm) and the orange arrow represents the emission wavelength (575 nm) of R-PE. (B) The differential interference contrast (DIC) and the R-PE intensity images of HEK293 cells expressing the G9 monobody or PEbody. (C) The R-PE staining of HEK293 cells expressing ECFP-G9 monobody or ECFP-PEbody. The respective DIC and overlay images of HEK293 cells expressing ECFP-PEbody are shown in the bottom panel. (D) The average R-PE or ECFP intensity at free end and cell-cell contact regions. Bar graphs represent mean ± SD (n = 33 and 69, where n is the number of analyzed cell free ends and cell-cell contacts, respectively). (E) The comparison of FRET/ECFP ratio between HEK293 cells expressing (i) ECFP-PEbody and (ii) PEbody-ECFP. Bar graphs are represented as mean ± SD (n = 36 and 42, where n is the number of analyzed cell contacts). The asterisk indicates a significant difference (***P < 0.001 with Wilcoxon rank sum test). Scale bar: 20 µm. See also Figure S2.
Because R-PE effectively binds to PEbody expressed on the cell surface, the R-PE/PEbody system can be utilized as a new acceptor for FRET biosensor applications. ECFP was hence fused with PEbody to create a FRET-based biosensor and allow energy transfer from ECFP to R-PE upon its capture by PEbody (Figures S2B and S2C), with the length of the linker between ECFP and PEbody affecting the FRET efficiency (Figure S2D). Indeed, upon the addition of R-PE to the cell culture medium, the R-PE signal dramatically increased at cell surfaces particularly at the cell-cell contacts, while exhibiting a corresponding ECFP signal decreased due to its energy transfer to R-PE (Figures S2E and S2F). The fluorescence images of HEK293 cells also revealed the expression of ECFP/PEbody and clear R-PE staining at the cell surface, with relatively strong signals at the cell-cell contacts compared to cell edges without contacts (Figure 2C, middle panel). We noticed that the R-PE signal at the cell-cell contact was 3.6-fold brighter than that at the free end, while the ECFP signal only increased 1.4-fold, which reflects the accumulated copies of local PEbody (Figure 2D). These results reveal high enrichment of R-PE at local sites when multiple PEbodies are present. We hypothesize that this phenomenon is due to the multivalent interaction between R-PE and PEbody. In contrast to the efficient binding between PEbody and R-PE, which resulted in a strong FRET between R-PE and ECFP, we observed no binding of R-PE in the cells expressing the control ECFP-G9 construct (Figure 2C, top panel). Consistently, cells without PEbody did not gain R-PE signals at the cell surface upon R-PE addition in the cell culture medium (Figure S2F, red broken line), verifying the specificity of PEbody in R-PE binding.
Interestingly, fusing PEbody at the N-terminus of ECFP showed weaker R-PE staining, indicating that the position and orientation of ECFP is important for the accessibility of PEbody to R-PE (Figure 2E). Taken together, these results indicate that the fusion of PEbody at the C-terminus of ECFP provides a platform for the development of biosensors based on FRET between ECFP and R-PE to visualize active molecular events at extracellular surface.
Visualization of cell-cell contact dissociation and formation
Due to the multivalent interaction between R-PE and PEbody at cell-cell contacts, our hybridized assembly of ECFP/PEbody and R-PE could be ideally suited for monitoring cell-cell contact dissociation and formation. We performed an experiment in which cell-cell contacts between HEK293 cells were first disrupted with EGTA for 1 hour while the change of fluorescence signals was monitored. As shown in Figures 3A and 3B, cells started to contract upon the addition of EGTA, causing the local morphological thickening of cell-cell contacts and a slight, transient increase in R-PE signal, followed by sharp decrease when cells started to dissociate physically. Once EGTA was washed away, cells started to reform the junctions. During this phase, R-PE signal significantly increased while ECFP intensity was less changed (Figures 3C and 3D). Overall, the results showed a rapid increase in the junctional R-PE signal when cells form cell-cell contacts, and a fast decrease of the signal when cells dissociate. As such, the multivalent binding property between R-PE and PEbody at the cell-cell contacts can be used as a sensitive marker for monitoring the maturity of cell-cell junctions during the dynamic formation and dissociation processes, which is difficult to be monitored using ECFP or other genetically encoded fluorescent proteins alone.
Figure 3. The change of the R-PE and ECFP signals upon cell-cell contact dissociation and formation.

(A and C) The ECFP, R-PE, overlay, and R-PE/ECFP ratio images in (A) the EGTA-induced cell-cell contact dissociation and (C) the reformation of cell-cell contacts after washing out EGTA. The color bar indicates the R-PE/ECFP intensity ratio, with hot and cold colors representing the high and low ratios, respectively. The white boxes and arrows indicate the regions of interest where an R-PE/ECFP intensity ratio was quantified for (B) and (D). (B and D) Upper panel: The normalized time courses of the average R-PE (left y-axis) and ECFP intensity (right y-axis); Middle panel: The average R-PE/ECFP ratio during the periods of cell-cell contact dissociation (B) and reformation (D). The removal of EGTA was performed at time = 60 min after the addition of EGTA in (B), and time = 0 min in (D). The black arrow with broken line represents the time point where multiple cells start to separate (B) or form the contact (D). Bottom panel: The bar graphs represent the normalized average intensities of R-PE and ECFP. The level of R-PE dye was maintained in the medium at all times during imaging. The normalized intensity value for each cell was established by dividing the fluorescent signals of each individual cell with that when cell contact was completely dissociated (dimmest R-PE intensity). Data in (B) and (D) are represented as mean ± SEM (n = 8 and 9 where n is the number of analyzed cell contacts in (B) and (D), respectively; ***P < 0.001 with Wilcoxon rank sum test). Scale bar: 10 µm.
Visualization of the surface MT1-MMP activity
To demonstrate the functionality of the ECFP-PEbody construct in engineering hybrid FRET biosensors, we inserted an MT1-MMP substrate peptide (CRPAHLRDSG) flanked by linkers in between ECFP and PEbody. R-PE can then be applied to produce an ECFP/PEbody/R-PE hybrid FRET biosensor to visualize the in situ substrate cleavage by MT1-MMP at the extracellular surface (Figure 4A) (Lu et al., 2013; Ouyang et al., 2010). The functionality of the FRET biosensor was verified in MT1-MMP deficient HeLa cells transfected with full-length MT1-MMP. The cells were pretreated with MMP inhibitor, GM6001, and monitored before and after inhibitor washout (Lu et al., 2013). After inhibitor washout, MT1-MMP proteolysis of the substrate in the biosensor allows ECFP to diffuse away from R-PE, which leads to a reduction in energy transfer. We observed a decrease in FRET/R-PE ratio of approximately 50%, while R-PE intensity remained relatively constant (Figures 4B, 4C, S3A and Movie S1). In contrast, the FRET/R-PE ratio was not affected by inhibitor washout in the control HeLa cells deficient in MT1-MMP (Figures 4B(ii) and 4C (black line)), confirming the specificity of this hybrid FRET biosensor. We have also compared this hybrid ECFP/PEbody/R-PE biosensor with a previously developed ECFP/YPet MT1-MMP FRET biosensor (Ouyang et al., 2010). After we quantified the biosensor signals at the plasma membrane where MT1-MMP localizes, it is clear that the overall kinetics of MT1-MMP activities monitored by both biosensors had similar characteristics, while our hybrid biosensor produced significantly cleaner images (Figures S3B–D). These results indicate that our new ECFP/PEbody/R-PE hybrid biosensor provides high sensitivity and clean membrane signals with low intracellular noise.
Figure 4. The ECFP/PEbody hybrid FRET biosensor for visualizing MT1-MMP activity on the surface of live cancer cells.

(A) The design strategy and the sensing mechanism of the biosensor. The biosensor was anchored on the extracellular surface using the transmembrane domain of PDGFR. The N-terminus of the transmembrane domain was linked to PEbody to allow for the R-PE binding. A MT1-MMP substrate sequence flanked by flexible linkers was inserted between ECFP and PEbody. Left: R-PE-staining of the intact biosensor allowed the energy transfer from ECFP to R-PE; Right: Following activation, MT1-MMP cleaved the biosensor substrate sequence, disrupted FRET, and reduced the FRET/R-PE ratio. (B) The DIC and FRET/R-PE ratio images before and after GM6001 inhibitor washout of the representative HeLa cells expressing the MT1-MMP ECFP/PEbody biosensor (i) with or (ii) without the full-length MT1-MMP gene. The color bar indicates the FRET/R-PE ratio, with hot and cold colors representing the high and low ratios, respectively. The white boxes indicate the region where the ratio was quantified for (C). Scale bar: 20 µm. (C) The average quantified time course of the normalized FRET/R-PE ratio from multiple HeLa cells in the GM6001 washout assay was compared in cells with and without the full-length MT1-MMP gene. Data are normalized with an individual basal FRET/R-PE ratio before GM6001 washout and represented as mean ± SD (n = 64 and 29; n is the number of analyzed cell contacts with and without the full-length MT1-MMP, respectively). See also Figure S3.
Asymmetric dynamics of the cell-surface MT1-MMP activity
FRET/R-PE ratio images clearly revealed distinct kinetics of MT1-MMP activation at different subcellular locations upon inhibitor washout (Figure 5A). The quantified time courses of FRET/R-PE ratio showed similar basal values before GM6001 inhibitor washout (Figure 5B). After washout, however, the ratio values decreased at different rates in different cell-cell contacts (Figures 5C and Movie S2). To explored these differences, actin-GFP was used as an indicator to investigate different cell-cell contact types (Drees et al., 2005; Zhang et al., 2005). We found that the rapid decrease of FRET/R-PE ratio, representing fast MT1-MMP activation, was predominantly observed at loose cell-cell contacts where actin fibers formed wide and diffusive branches (Figures S4A–C) (Burridge and Wittchen, 2013; Zhang et al., 2005). Conversely, stable cell-cell contacts where actin filaments accumulate in packed bundles at the cell borders (Zhang et al., 2005) showed a slow decrease of FRET/R-PE ratio, representing slow MT1-MMP activation (Figures S4A–C). Interestingly, images of cells with mCherry-tagged MT1-MMP showed that the MT1-MMP protein localized at cell-cell contacts without particular spatial preferences (Figures S4D and S4E) (Remacle et al., 2013). Taken together, these results indicate that although the total amount of MT1-MMP is similar at both stable and loose contacts, the proteolytic activity of MT1-MMP may be inhibited at the stable cell-cell contacts by the local microenvironment.
Figure 5. A distinct distribution of MT1-MMP activity at different subcellular cell-cell contacts.

(A) The representative FRET/R-PE ratio images of HeLa cells co-expressing the ECFP/PEbody MT1-MMP FRET biosensor and the full-length MT1-MMP. After GM6001 inhibitor washout, the discrete regions of the membrane showed distinctive FRET/R-PE ratios. Hot and cold colors indicate high and low ratios, respectively. The position 1 and 2 with white boxes are used to quantify the FRET/R-PE ratio in (B). Scale bar: 20 µm. (B) The quantified time course of the FRET/R-PE ratio was compared between positions 1 and 2 in (A). (C) The quantified time course of the normalized FRET/R-PE ratio from multiple cells. The normalized FRET/R-PE ratios illustrated in green or red represent results predominantly from the loose or stable cell-cell contacts, respectively. (D) The average time course of the normalized FRET/R-PE ratio quantified from the loose or stable cell-cell contacts with or without Cytochalasin D or nocodazole treatment. (E) The bar graphs represent the average half-time of signal reduction in (D) with n = 34, 30, 18, and 38, respectively, where n is the number of analyzed cell contacts. (F) The comparison of the MT1-MMP diffusion speed between free edge regions (denoted as loose cell-cell contact) and cell-cell contacts (denoted as stable cell-cell contact) (n = 7 for each group, where n is the number of cells from three independent experiments. Data in (C-D) are normalized with an individual basal FRET/R-PE ratio before GM6001 washout and data in (D-F) are represented as mean ± SD. The asterisk indicates a significant difference (**P < 0.01 and ***P < 0.001 with Wilcoxon rank sum test). See also Figures S4 and S5.
The cytoskeleton plays a crucial role in controlling cell-cell adhesions and MT1-MMP dynamics (Gálvez et al., 2002; Nawrocki-Raby et al., 2003; Woskowicz et al., 2013). Actin filaments and microtubules regulate the localization and trafficking of MT1-MMP (Ouyang et al., 2008; Remacle et al., 2003; Zucker et al., 2002), so we reasoned that they may also mediate the observed asymmetric distribution of MT1-MMP activity. To investigate the relationship between the MT1-MMP activity and the cytoskeletal components, cytochalasin D and nocodazole were applied to cells to disrupt actin filaments and microtubules, respectively. The application of either inhibitor resulted in a uniformly rapid decrease of the FRET/R-PE ratio at different cell-cell contacts, similar to the loose contacts in MT1-MMP-expressing HeLa cells after inhibitor washout (Figures 5D and 5E). These results indicate that actin filaments and microtubules play crucial roles in the regulation of MT1-MMP activity at different cell-cell contacts.
To further explore the spatially asymmetric regulation of MT1-MMP activity, we measured the apparent diffusion rate of MT1-MMP using the fluorescence recovery after photobleaching (FRAP) assay. The full-length MT1-MMP-mCherry fusion protein was monitored before and after photobleaching (Figure S5). A mathematical model was used to quantify the diffusion rate of MT1-MMP-mCherry based on the recovery images (Lu et al., 2008). The analysis showed that the average diffusion rate of MT1-MMP-mCherry at the membrane regions lacking cell-cell contacts was 2.2-fold greater than at regions with stable cell-cell contacts (4.40±2.17 vs. 1.98±0.82 µm2/sec) (Figures 5F and S5). These results indicate that the MT1-MMP molecules are less mobile at stable contacts, which is consistent with the results from our biosensor that showed less proteolytic activity of MT1-MMP at stable contacts. This possibly reflects its inhibited forms in the presence of inhibitory interacting molecules at these local microenvironments.
Discussion
FRET biosensors are valuable molecular imaging tools that allow for real-time visualization of specific molecular activities during important cellular processes. Through an integration of iterative directed evolution and rational design (Figure 1), we developed a hybrid FRET biosensor composed of ECFP and an engineered PEbody that functions as a selective and effective binder of the R-PE fluorescent dye. Because R-PE is cell-impermeable, this hybrid biosensor can be assembled in situ at the live cell surface to monitor extracellular molecular activities with high spatiotemporal resolution. Indeed, the binding between R-PE and PEbody can be clearly detected on the cell surface with minimal intracellular background noise, particularly at cell-cell contacts (Figure 2). Our design provides an alternative approach to the SNAP-tag and Halo-tag technologies (Brun et al., 2009; Maurel et al., 2008), with the advantage of allowing for the direct and reversible assembly of biosensors without additional chemical modifications. Integrating PEbody into a FRET biosensor, we successfully applied our hybrid biosensor to monitor cell-cell junction maturity during the dynamic formation and dissociation of cell-cell contacts (Figure 3), utilizing the multivalent binding property of R-PE toward PEbody. Therefore, the integration of the directed evolution technology and rational sequence-function analysis can be used as powerful tools to engineer protein binding scaffolds for the assembly of efficient hybrid FRET biosensors capable of monitoring subcellular molecular events at live cell surfaces (Figures 4 and 5).
Monobody is widely used as a protein scaffold because of its small size, flexible loops, and proper folding within living cells (Batard et al., 2002; Koide et al., 1998). Accordingly, with the G9 monobody serving as a starting template for directed evolution (Huang et al., 2012), FACS screenings directly allowed the identification of the S4 variant with improved, specific R-PE binding capability. However, additional mutagenesis and screenings failed to identify a better binder to R-PE than S4. This may be due to the nonlinear relation between the library sequence and the functional readout, as well as our limited library size (Arnold, 1998; Benatuil et al., 2010; Lutz, 2010). Further improvement was only achieved by the integration of screening and rational design. Optimizing amino acid residues in both BC and FG loops resulted in the higher affinity of PEbody toward R-PE. As such, the integration of directed evolution and rational analysis provided powerful tools for the molecular engineering of novel proteins.
PEbody was integrated into a FRET-based biosensor for monitoring the proteolytic activity of MT1-MMP. We found that although the amount of MT1-MMP protein is relatively uniform at cell-cell contacts (Gálvez et al., 2002; Remacle et al., 2013), the proteolytic activity of MT1-MMP is surprisingly spatially heterogeneous. Perhaps the E-cadherin complexes of stable cell-cell contacts can recruit antagonistic molecules to create an inhibitory microenvironment, suppressing MT1-MMP-mediated cadherin cleavage and thereby maintaining stable junctions (Liu et al., 2010). Consistent with this hypothesis, FRAP results indicated that the MT1-MMP molecules are less mobile at stable cell-cell contacts which could account for the lower activity of MT1-MMP. This supports the notion that MT1-MMP molecules may be more restricted by inhibitory partners at these local regions. The results from FRAP experiment also agree well with the results of cytoskeletal disruption. Cytochalasin D or nocodazole treatment eliminated this differential distribution of MT1-MMP activity at different cell-cell contacts. It is hence possible that the distinct characteristics of the cytoskeleton at different cell-cell contacts play a role in determining the local microenvironment and regulating MT1-MMP distinctively at different cell-cell contacts.
Significance
By integrating iterative directed evolution and rational analysis, we succeeded in systematically engineering and optimizing a genetically encoded binder of a fluorescent dye. We further introduced a novel design for FRET biosensors which combines genetically encoded fluorescent protein and PEbody as the binding scaffold for a cell-impermeable fluorescent dye R-PE to visualize molecular events on the extracellular surface in live cells. A hybrid FRET biosensor based on this design allowed us to monitor cell-cell junction maturity with high sensitivity and precision during the dynamic formation and dissociation of cell-cell contacts. This biosensor further revealed a distinctive distribution of proteolytic activity of MT1-MMP (a key enzyme for the tumor cells invasion) at different cell-cell contacts (Hotary et al., 2003). The readout from this biosensor could be used to detect the invasiveness of single cells from clinical samples, and to screen for anti-cancer drugs. Our design for this biosensor can also be readily adapted to develop other FRET biosensors for the monitoring of molecular activities at the extracellular surface as well as the endocytosis of surface molecules, due to the low-pH stability of both R-PE and ECFP (Fredj et al., 2012; Zhai et al., 2005). As such, our method can be employed to systematically design FRET biosensors capable of monitoring important molecular dynamics at the surface and other subcellular regions of live cells.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yingxiao Wang (yiw015@eng.ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains and Growth Conditions
Saccharomyces cerevisiae EBY100 (a GAL1-AGA1::URA3 ura3–52 trp1 leu2Δ1 his3Δ200 pep4::HIS2 prb1Δ1.6R can1 GAL) was used throughout the work for yeast surface display library (Boder and Wittrup, 1997). EBY100 was routinely cultured at 30 °C in rich media (YPD). Once transformed with the yeast display plasmid pYD1 (Thermo Fisher Scientific, Cat. No. V835-01), cells were grown in synthetic complete medium minus tryptophan (SC-Trp with 2% (w/v) glucose), and monobody library expression was induced in galactose media (SC-Trp with 2% (w/v) galactose). The pYD1 plasmid also carries ampicillin resistance for amplification in E. coli (DH5α; Thermo Fisher Scientific, Cat. No. 18258012), which was cultured in Luria Bertani (LB; Fisher Scientific, Cat. No. BP14262) medium at 37 °C.
Cell Lines
HEK293 cells (ATCC, Cat. No. CRL-1573, Gender: N/A) and HeLa cells (ATCC, Cat. No. CCL-2, Gender: female) were grown in DMEM (Thermo Fisher Scientific, Cat. No. 11995073) supplemented with 10% FBS (Thermo Fisher Scientific, Cat. No. 10438026) plus 100U/ml penicillin, 100 µg/ml streptomycin (Thermo Fisher Scientific, Cat. No. 15140122) at 37°C with 5% CO2.
METHOD DETAILS
Library Construction
Two generations of R-PE-binding monobody libraries were created by site-saturation mutagenesis. For the first-generation libraries, five codons in the BC (residues 26–30) and FG loops (residues 77–81) of the G9 monobody underwent site-saturation mutagenesis by using NNK degenerate primers (IDT) with Q5 DNA polymerase (NEB, Cat. No. M0491), where N represents an equimolar distribution of A, T, G, and C; K denotes an equimolar distribution of T and G, to yield the BC and FG libraries. After generating the library by PCR, the fragments containing library were then extracted from agarose gel, inserted into pYD1 vector between the KpnI and XhoI restriction sites, transformed into E. coli (DH10B), and purified plasmid library with Qiagen HiSpeed Plasmid Maxi kit. These plasmids were then transformed into Saccharomyces cerevisiae (EBY100). The libraries were screened twice by FACS (BD FACSAria) using R-PE (Sigma-Aldrich, Cat. No. 52412-1MG-F) as a ligand (Figures S1B and S1C). The clones with the best binding capability and fluorescence intensity were combined to generate the R-PE-binding monobody library.
To construct the combined library, the selected DNA libraries of BC and FG loops were amplified by the primer P1 (forward) and P2 (reverse) to produce the BC library, and the primer P3 (forward) and P4 (reverse) to generate the FG library. P1 anneals to a region in the G9 sequence upstream of the randomized BC loop; whereas P2 anneals to a region between the BC and FG loop. P3 was complementary to the 20 nucleotides at the 5’ end of P2; P4 anneals to a region downstream of the FG loop. These two DNA fragments were assembled through the overlap extension PCR with a 20-nucleotide sequence complementary. The combined library was then transformed into S. cerevisiae jointly with the original pYD1 vector which has a complementary sequence of G9 upstream of the BC loop and downstream of the FG loop. The size of the resulting R-PE-binding monobody library was approximately 1×106 individual clones.
Selection of the R-PE-Binding Monobody Using FACS
The surface expression of the variant proteins was induced by culturing yeast in 2% galactose-containing medium (synthetic complete medium minus tryptophan, SC-Trp) at 20°C for 24–48 hours during which the OD600 increased 2–4-fold. In order to identify and select the yeast cells that displayed the G9 monobody variants with the high R-PE-binding capability, the yeast cells were incubated with 10 µg/mL R-PE in wash buffer containing the phosphate buffered saline (PBS, Sigma-Aldrich) with 0.5% bovine serum albumin (BSA) (Sigma-Aldrich) for 1 hour at room temperature. The level of the expressed proteins on the yeast surface was also measured using the anti-V5 epitope antibody (Thermo Fisher Scientific, Cat. No. R960-25). In each step of staining, the unbound reagents were washed away with wash buffer.
Each of the BC and FG libraries were sorted twice by FACS using R-PE as a ligand. In the first round of sorting, the top 10% of the R-PE positive cells were collected for further expansion. In the second round of sorting, 0.1% of the R-PE positive cells were selected (Figure S1B). Later, the R-PE-binding monobody library was constructed by combining both selected BC and FG libraries. For the combined library, the top 10%, 0.5%, and 0.1% of the R-PE binders were consecutively screened in three rounds of FACS. After the third round of sorting, the cells, which were most efficient in binding to R-PE, were selected and seeded into wells of 96-well plates for DNA sequencing (Genewiz).
Improvement of the R-PE-Binding Monobody
After three rounds of FACS, the S4 monobody variant, which efficiently binds to R-PE, was identified. To further improve the R-PE-binding monobody, the second generation of the S4-based monobody library was generated by site-saturation mutagenesis. The sequence in the BC loop of S4 was fixed; while the sequence of the FG loop was subjected to site-saturation mutagenesis using the NNK degenerate primers.
To increase the selection stringency of the R-PE binders, only the top 0.1% of the brightest R-PE positive cells were selected in the first and second round of FACS. In the second round of FACS, cells were further seeded into wells of 96-well plates. 20 single colonies were then randomly selected to measure their R-PE-binding capability by flow cytometer (BD Accuri C6). Later, ten individual clones, including five clones of the highest and lowest R-PE-binding capability, were subjected to the DNA sequence analysis. The R-PE binding capability was defined as the ratio between the % of R-PE binding yeasts (gated by R-PE) and the % of monobody expressing yeast (gated by a labeling tag V5 on the monobody).
The DNA sequences of S4 and those 10 representative clones, were used to predict the mutants with the improved R-PE-binding capability (Figure S1G). The amino acids in the FG loop of each clone were evaluated based on the results from flow-cytometry analysis. The contribution score of the amino acid residue A at position j in the FG loop was calculated by averaging the relative R-PE binding capability among all mutants of A at j. If the contribution score of A at position j was higher than 10% of the maximum contribution score at that position, amino acid residues with the highest ranking for this position were then selected for further examinations (Figures S1H and S1I). As a result, amino acids W, R/P, W/F, W, and Y/N for positions 77–81 of the FG loop sequence were selected for experimental examination. A combination of these residues led us to the eight individual mutant sequences (Figure 1D). The R-PE binding capability of these mutants were later examined by flow cytometry (BD Accuri C6). Data from flow cytometry were collected from about 100,000 events per sample with three independent experiments. Our approach greatly reduced the searching space and provided an efficient path to reach an optimized R-PE binder.
Measuring KD by Flow Cytometry
The protein-protein dissociation constant KD of different monobody variants was measured using the yeast surface display as described (Feldhaus et al., 2003). Antigen concentration ranged from 10-fold lower to 10-fold higher than the expected KD; concentrations from 0.5 nM to 50 nM were used to label 2×106 induced yeast cells which were incubated at room temperature for 1 hour. The unbound R-PE was then washed away and the samples were analyzed by flow cytometry. The mean fluorescence intensity (MFI) of the R-PE bound to the yeast cell surface was plotted against the increasing R-PE concentration. The KD was calculated from nonlinear least-squares regression of the relationship between the MFI and the R-PE concentration (Figure S1J).
The dissociation constant (KD) of the PEbody and R-PE using a yeast display method by flow-cytometry analysis (Feldhaus et al., 2003) was calculated to be 9.6 ± 3.0 nM (mean±SEM). Similarly, the KD of anti-V5 antibody toward V5 epitope tag, which is located at C-terminus of PEbody as an indicator of protein expression on the yeast surface (Figure S1C), was measured to be about 2 nM (Figure S1J), which is consistent with previously reported values (Kastritis et al., 2011).
Measuring KD by Bio-Layer Interferometry
PEbody proteins were expressed with N-terminal 6× His tag in Escherichia coli and purified by nickel chelation chromatography as previously described (Wang et al., 2005). Binding kinetics of PEbody and R-PE were measured by bio-layer interferometry (ForteBio BLItz system). PEbody with His tag was loaded onto an anti-HIS biosensor for 15 sec to establish baselines. A variety of concentrations of R-PE (40, 80, 160, and 320 nM) were introduced and their association to immobilized PEbody was monitored for 60 sec, after which it was allowed to dissociate into PBS with 0.5% BSA (pH 7.4) for 60 s. KD values were calculated from the 1:1 binding model to be 5.7 ± 3.5 nM (mean ± SEM) by determined kon and koff parameters.
Construction of the MT1-MMP FRET Biosensor
The MT1-MMP substrate peptide sequence (CRPAHLRDSG, the scissile bond is underlined) flanked by the GGSGGT linker peptides was inserted between the C-terminus of ECFP and the N-terminus of PEbody (GGSGGTCRPAHLRDSGGGTGGS) (Ouyang et al., 2010). The flexible linker (GGSGGT) was added to extend the cleavable peptide and to allow its access by MT1-MMP. The biosensor sequence was then inserted to the pDisplay vector (Thermo Fisher Scientific, Cat. No. V66020) between the BglII and SalI restriction sites. The pDisplay vector contains an N-terminal murine Ig κ-chain leader sequence, which directs the protein cargo to the secretory pathway, and a C-terminal transmembrane (TM) domain of the platelet-derived growth factor receptor β (PDGFR-β), which directs target proteins to the extracellular surface of the plasma membrane (Ouyang et al., 2008). The mCherry-conjugated MT1-MMP was constructed by fusing mCherry sequence at the C-terminal of MT1-MMP with GGS and inserted to pcDNA3.1 between HindIII and XhoI restriction sites (Ouyang et al., 2008).
Cell Culture and Transfection
Cell culture reagents were purchased from Thermo Fisher Scientific. The human embryonic kidney cells (HEK293) and the cervical cancer cells (HeLa) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 2 mM of L-glutamine, 100 units/mL of penicillin, 100 µg/mL of streptomycin, and 1mM of sodium pyruvate at 37°C with 5% CO2. The DNA plasmids were transfected into cells with Lipofectamine 3000 (Thermo Fisher Scientific, Cat. No. L3000015). To visualize MT1-MMP activity in HeLa cells, the ECFP/PEbody MT1-MMP FRET biosensor was co-transfected with the full-length MT1-MMP gene. 36–48 hours after transfection, R-PE (10 µg/mL) was added into the culture medium and incubated with the cells for 30 minutes. The unbound R-PE was then washed out and the samples were analyzed by fluorescence microscopy.
Microscopy, Image Acquisition, and Analysis
Cells expressing the exogenous biosensor proteins were cultured in a glass bottom dish (Cell E&G) coated with 20 µg/mL fibronectin for 16–20 hours before imaging. Cells were then incubated with R-PE in serum-free DMEM without phenol red at 37°C for 30 min, washed twice using PBS, and maintained in the starvation medium (DMEM with 0.5% FBS). The images were taken with a Nikon Eclipse Ti inverted microscope with a cooled charge-coupled device camera and analyzed using MetaFluor 7.8 or MetaMorph 7.8 software (Molecular Devices). The FRET ratio images were obtained by dividing the fluorescence image pixel-by-pixel of FRET wavelength (420/40 nm excitation, 575/20 emission, with 560 LP dichroic mirror) by the R-PE wavelength (495/10 nm excitation, 575/20 emission, with 560 LP dichroic mirror) after the background subtraction. The images were analyzed and displayed in the intensity modified display mode by using MetaFluor and Fluocell software (Lu et al., 2011; Lu et al., 2008).
For data presentation, the normalized values were shown to clearly compare the differences among the experimental groups and to minimize the cell-cell heterogeneity. The pre-stimulation baseline for each cell was established by averaging the FRET or other fluorescent signals of each individual cell before stimulation. This provides an internal normalization reference for establishing the stimulation-dependent FRET changes of an individual cell which reduces the cell-cell heterogeneity and noise. This normalization procedure had been used for Figures 4, 5, S3, and S5. In Figure 3, the normalization was calculated by dividing the intensity at each time point of an individual cell with the intensity when that cell contact was completely dissociated (minimum R-PE intensity was observed during time = 50–60 minutes after addition of EGTA).
Dissociation and Formation of Cell-Cell Contacts
For R-PE-label experiment, R-PE (10 µg/mL) was directly added onto HEK293 cells expressing ECFP-PEbody under microscope. For cell-cell contact dissociation experiments, HEK293 cells expressing ECFP-PEbody were stained with R-PE (10 µg/mL) and cell-cell junctions were disrupted by the addition of 2 mM EGTA (Sigma-Aldrich) to monitor the change of R-PE and ECFP signal at cell-cell contacts. Once EGTA was washed away, cells started to reform the junctions. During this phase, R-PE signal was monitored together with ECFP signal at reforming cell-cell contacts.
Inhibitor Washout FRAP Assay
For inhibitor washout assays, HeLa cells were pre-incubated with 20 µM GM6001 (EMD Millipore, Cat. No. 364205), a broad-spectrum matrix metalloproteinase inhibitor, in the starvation medium for 16–20 hours. The inhibitor was then washed out with the Hank's balanced salt solution (HBSS, with Ca2+ and Mg2+, Thermo Fisher Scientific). For FRAP experiments, cells expressing the MT1-MMP-mCherry fusion protein were imaged using a 100× objective as a reference for setting the pinhole position which allows strong light to pass through. Photobleaching was performed by exciting mCherry in the chosen pinhole region with the full power of excitation light for 1 minute. Cells were then monitored for the recovery of the fluorescence intensity. To estimate the diffusion coefficients, our previous published finite element mathematical model and algorithm were used (Lu et al., 2008; Vandenberg and Dedecker, 2017). This method is flexible to handle different imaging and photobleach protocols, as well as variable cell geometry. Briefly, two images from the recovery time course were used to estimate a diffusion coefficient, based on a linear correction between the temporal and spatial variations of fluorescence signal. The estimated diffusion coefficient was considered to be sufficiently accurate when the correlation calculated from a pair of images is larger than 0.7 (Lu et al., 2008).
QUANTIFICATION AND STATISTICAL ANALYSIS
All the experiments were replicated at least three times and represented biological replicates. Data are presented as mean values ± standard deviation (SD) or mean values ± standard error of the mean (SEM) as indicated in the figure legend. Statistical analyses were carried out using MATLAB (version R2015b) and were defined by P values calculated from the two-tailed Student’s t tests or Wilcoxon rank sum test by comparison with the relevant control as specified in the figures or figure legends. Levels of significance were P<0.05 (*), P<0.01 (**), P<0.001 (***). When the sample size n>=4, the Wilcoxon rank sum test was used since it is applicable to random samples of all distributions. For some flow cytometry experiments, when n=3, each data point is an average reading from more than 100,000 single cells. According to the central limit theorem, these data follow the Gaussian distribution. Therefore, we used the two-tailed Student’s t-tests.
Supplementary Material
Imaging of live HeLa cells co-expressing the MT1-MMP biosensor and the full-length MT1-MMP. Left panel shows movie of DIC images; Right panel shows time-lapse of ratio images (FRET/R-PE). Magnification, 100X. Movie was acquired with a 60-sec interval. Activation of MT1-MMP is observed at the cell periphery.
Imaging of live HeLa cells expressing the MT1-MMP biosensor and the full-length MT1-MMP gene. Left panel shows movie of DIC images; right panel shows time-lapse of ratio images (FRET/R-PE). Magnification, 100X. Movie was acquired with a 60-sec interval. The different levels of active MT1-MMP are recorded at the cell periphery.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-V5 antibody | Thermo Fisher Scientific | Cat#R960-25 |
| Bacterial and Virus Strains | ||
| MAX Efficiency™ DH5α™ Competent Cells | Thermo Fisher Scientific | Cat#18258012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GM6001 | EMD Millipore | Cat#364205 |
| R-Phycoerythrin | Sigma-Aldrich | Cat#52412-1MG-F |
| EGTA | Sigma-Aldrich | Cat#E3889 |
| Experimental Models: Cell Lines | ||
| HEK293 cells | ATCC | Cat#CRL-1573 |
| HeLa cells | ATCC | Cat#CCL-2 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae (EBY100) | N/A | N/A |
| Recombinant DNA | ||
| pYD1-PEbody | This paper | N/A |
| pDisplay-PEbody | This paper | N/A |
| pDisplay-ECFP-CRPAHLRDSG-PEbody | This paper | N/A |
| pCR3.1-MT1-MMP | Ouyang et al., 2008 | N/A |
| pCDNA3.1-MT1-MMP-mCherry | Ouyang et al., 2008 | N/A |
| Software and Algorithms | ||
| Fluocell | Lu S et al. 2008, Lu S et al. 2011 | https://code.google.com/archive/p/fluocell/ |
| MetaMorph | Molecular Devices | https://www.moleculardevices.com |
| MetaFluor | Molecular Devices | https://www.moleculardevices.com |
| PyMol | PyMol | https://www.pymol.org/ |
Highlights.
Directed evolution and rational design are used for biosensor development
PEbody can be applied to track cell-cell contact dynamics with high precision
Assembling a hybrid FRET biosensor at cell surface enhances signal-to-noise ratio
Biosensor reveals heterogeneous activity of MT1-MMP at cell-cell contacts
Acknowledgments
This work is supported by grants from NIH HL098472, HL109142, HL121365 (Y. Wang), NSF CBET1360341, DMS1361421 (Y. Wang and S.L.), UC San Diego, and Beckman Laser Institute, Inc. (Y. Wang) and NIH NS 085092 (X.L. Yang). This research was also supported by NSF China NSFC 11428207 (Y. Wang). P.L. acknowledges fellowship support from the Thai Ministry of Science and Technology. Q.P. acknowledges fellowship support from China Scholarship Council (CSC). J.L. acknowledges fellowship support from the Agency for Science, Technology and Research of Singapore. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
P.L., Q.P., and Y. Wang designed the research; Y. Wang supervised the research; P.L. and Q.P. performed experiments and analyzed data; P.L., Y.W., and S.L. carried out FRAP analysis; T.L., and X.G. carried out the KD analysis by bio-layer interferometry; P.L., S.L., and Y. Wang wrote the manuscript. Q.P., M.A., and A.Y.S. reviewed and edited the manuscript. All authors reviewed the manuscript.
Declaration of Interests
The authors declare no competing interests.
Supplemental Information includes five figures, and two movies.
References
- Arnold FH. Design by directed evolution. Accounts of chemical research. 1998;31:125–131. [Google Scholar]
- Batard P, Szollosi J, Luescher I, Cerottini JC, MacDonald R, Romero P. Use of phycoerythrin and allophycocyanin for fluorescence resonance energy transfer analyzed by flow cytometry: advantages and limitations. Cytometry. 2002;48:97–105. doi: 10.1002/cyto.10106. [DOI] [PubMed] [Google Scholar]
- Batori V, Koide A, Koide S. Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain. Protein engineering. 2002;15:1015–1020. doi: 10.1093/protein/15.12.1015. [DOI] [PubMed] [Google Scholar]
- Benatuil L, Perez JM, Belk J, Hsieh C-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Engineering Design and Selection, gzq002. 2010 doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nature biotechnology. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nature biotechnology. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Brun MA, Tan KT, Nakata E, Hinner MJ, Johnsson K. Semisynthetic fluorescent sensor proteins based on self-labeling protein tags. Journal of the American Chemical Society. 2009;131:5873–5884. doi: 10.1021/ja900149e. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol. 2013;200:9–19. doi: 10.1083/jcb.201210090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr PA, Erickson HP, Palmer AG. Backbone dynamics of homologous fibronectin type III cell adhesion domains from fibronectin and tenascin. Structure. 1997;5:949–959. doi: 10.1016/S0969-2126(97)00248-7. [DOI] [PubMed] [Google Scholar]
- Covington MD, Burghardt RC, Parrish AR. Ischemia-induced cleavage of cadherins in NRK cells requires MT1-MMP (MMP-14) Am J Physiol Renal Physiol. 2006;290:F43–51. doi: 10.1152/ajprenal.00179.2005. [DOI] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. α-catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nature biotechnology. 2003;21:163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- Fredj A, Pasquier H, Demachy I, Jonasson G, Levy B, Derrien V, Bousmah Y, Manoussaris G, Wien F, Ridard J. The single T65S mutation generates brighter cyan fluorescent proteins with increased photostability and pH insensitivity. 2012 doi: 10.1371/journal.pone.0049149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez BG, Matías-Román S, Yáñez-Mó M, Sánchez-Madrid F, Arroyo AG. ECM regulates MT1-MMP localization with β1 or αvβ3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. The Journal of cell biology. 2002;159:509–521. doi: 10.1083/jcb.200205026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer A. Light harvesting by phycobilisomes. Annual review of biophysics and biophysical chemistry. 1985;14:47–77. doi: 10.1146/annurev.bb.14.060185.000403. [DOI] [PubMed] [Google Scholar]
- Gulyani A, Vitriol E, Allen R, Wu J, Gremyachinskiy D, Lewis S, Dewar B, Graves LM, Kay BK, Kuhlman B. A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nature chemical biology. 2011;7:437–444. doi: 10.1038/nchembio.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires SA, Zhu Y, Tsien RY. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4411–4416. doi: 10.1073/pnas.0712008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three- dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- Huang R, Fang P, Kay BK. Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase. New biotechnology. 2012;29:526–533. doi: 10.1016/j.nbt.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraheem A, Yap H, Ding Y, Campbell RE. A bacteria colony-based screen for optimal linker combinations in genetically encoded biosensors. BMC Biotechnol. 2011;11:105. doi: 10.1186/1472-6750-11-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isailovic D, Sultana I, Phillips GJ, Yeung ES. Formation of fluorescent proteins by the attachment of phycoerythrobilin to R-phycoerythrin alpha and beta apo-subunits. Analytical biochemistry. 2006;358:38–50. doi: 10.1016/j.ab.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastritis PL, Moal IH, Hwang H, Weng Z, Bates PA, Bonvin AM, Janin J. A structure-based benchmark for protein–protein binding affinity. Protein Science. 2011;20:482–491. doi: 10.1002/pro.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. Journal of molecular biology. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Molecular biology of the cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Yang J, Pei J, Pei D, Wilson MJ. Regulation of MT1-MMP activity by beta-catenin in MDCK non-cancer and HT1080 cancer cells. J Cell Physiol. 2010;225:810–821. doi: 10.1002/jcp.22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Kim T-j, Chen C-E, Ouyang M, Seong J, Liao X, Wang Y. Computational analysis of the spatiotemporal coordination of polarized PI3K and Rac1 activities in micro-patterned live cells. PloS one. 2011;6:e21293–e21293. doi: 10.1371/journal.pone.0021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Ouyang M, Seong J, Zhang J, Chien S, Wang Y. The spatiotemporal pattern of Src activation at lipid rafts revealed by diffusion-corrected FRET imaging. PLoS Comput Biol. 2008;4:e1000127–e1000127. doi: 10.1371/journal.pcbi.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Wang Y, Huang H, Pan Y, Chaney EJ, Boppart SA, Ozer H, Strongin AY, Wang Y. Quantitative FRET imaging to visualize the invasiveness of live breast cancer cells. PloS one. 2013;8:e58569. doi: 10.1371/journal.pone.0058569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S. Beyond directed evolution--semi-rational protein engineering and design. Curr Opin Biotechnol. 2010;21:734–743. doi: 10.1016/j.copbio.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prezeau L, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–567. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Arnold FH. Exploring nonnatural evolutionary pathways by saturation mutagenesis: rapid improvement of protein function. Journal of molecular evolution. 1999;49:716–720. doi: 10.1007/pl00006593. [DOI] [PubMed] [Google Scholar]
- Nawrocki-Raby B, Gilles C, Polette M, Martinella-Catusse C, Bonnet N, Puchelle E, Foidart J-M, Van Roy F, Birembaut P. E-Cadherin mediates MMP down- regulation in highly invasive bronchial tumor cells. The American journal of pathology. 2003;163:653–661. doi: 10.1016/S0002-9440(10)63692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M, Huang H, Shaner NC, Remacle AG, Shiryaev SA, Strongin AY, Tsien RY, Wang Y. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer research. 2010;70:2204–2212. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M, Lu S, Li X-Y, Xu J, Seong J, Giepmans BN, Shyy JY-J, Weiss SJ, Wang Y. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. Journal of Biological Chemistry. 2008;283:17740–17748. doi: 10.1074/jbc.M709872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle A, Murphy G, Roghi C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J Cell Sci. 2003;116:3905–3916. doi: 10.1242/jcs.00710. [DOI] [PubMed] [Google Scholar]
- Remacle AG, Shiryaev SA, Golubkov VS, Freskos JN, Brown MA, Karwa AS, Naik AD, Howard CP, Sympson CJ, Strongin AY. Non-destructive and selective imaging of the functionally active, pro-invasive membrane type-1 matrix metalloproteinase (MT1-MMP) enzyme in cancer cells. Journal of Biological Chemistry. 2013;288:20568–20580. doi: 10.1074/jbc.M113.471508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanov DV, Deryugina EI, Monosov EZ, Marchenko ND, Strongin AY. Aberrant, persistent inclusion into lipid rafts limits the tumorigenic function of membrane type-1 matrix metalloproteinase in malignant cells. Exp Cell Res. 2004;293:81–95. doi: 10.1016/j.yexcr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature biotechnology. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nature methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. Journal of Cell Science. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- Vandenberg W, Dedecker P. Effect of probe diffusion on the SOFI imaging accuracy. Scientific Reports. 2017;7:44665. doi: 10.1038/srep44665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shyy JY-J, Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng. 2008;10:1–38. doi: 10.1146/annurev.bioeng.010308.161731. [DOI] [PubMed] [Google Scholar]
- Wittrup KD, Thurber GM, Schmidt MM, Rhoden JJ. Practical theoretic guidance for the design of tumor-targeting agents. Methods in enzymology. 2012;503:255. doi: 10.1016/B978-0-12-396962-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woskowicz AM, Weaver SA, Shitomi Y, Ito N, Itoh Y. MT-LOOP-dependent localization of membrane type I matrix metalloproteinase (MT1-MMP) to the cell adhesion complexes promotes cancer cell invasion. Journal of Biological Chemistry. 2013;288:35126–35137. doi: 10.1074/jbc.M113.496067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Hotary KB, Nan B, Bosch FX, Muñoz N, Weiss SJ, Cho KR. Expression of membrane type 1 matrix metalloproteinase is associated with cervical carcinoma progression and invasion. Cancer Research. 2005;65:6543–6550. doi: 10.1158/0008-5472.CAN-05-0231. [DOI] [PubMed] [Google Scholar]
- Zhang J, Betson M, Erasmus J, Zeikos K, Bailly M, Cramer LP, Braga VM. Actin at cell-cell junctions is composed of two dynamic and functional populations. Journal of cell science. 2005;118:5549–5562. doi: 10.1242/jcs.02639. [DOI] [PubMed] [Google Scholar]
- Zucker S, Hymowitz M, Conner CE, DiYanni EA, Cao J. Rapid trafficking of membrane type 1-matrix metalloproteinase to the cell surface regulates progelatinase a activation. Laboratory investigation. 2002;82:1673–1684. doi: 10.1097/01.lab.0000041713.74852.2a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Imaging of live HeLa cells co-expressing the MT1-MMP biosensor and the full-length MT1-MMP. Left panel shows movie of DIC images; Right panel shows time-lapse of ratio images (FRET/R-PE). Magnification, 100X. Movie was acquired with a 60-sec interval. Activation of MT1-MMP is observed at the cell periphery.
Imaging of live HeLa cells expressing the MT1-MMP biosensor and the full-length MT1-MMP gene. Left panel shows movie of DIC images; right panel shows time-lapse of ratio images (FRET/R-PE). Magnification, 100X. Movie was acquired with a 60-sec interval. The different levels of active MT1-MMP are recorded at the cell periphery.