

Abstract
During extracellular matrix (ECM) assembly, fibronectin (FN) fibrils are irreversibly converted into a detergent-insoluble form which, through FN’s multi-domain structure, can interact with collagens, matricellular proteins, and growth factors to build a definitive matrix. FN also has heparin/heparan sulfate (HS) binding sites. Using HS-deficient CHO cells, we show that the addition of soluble heparin significantly increased the amount of FN matrix that these cells assemble. Sulfated HS glycosaminoglycan (GAG) mimetics similarly increased FN assembly and demonstrated a dependence on GAG sulfation. The length of the heparin chains also plays a role in assembly. Chains of sufficient length to bind to two FN molecules gave maximal stimulation of assembly whereas shorter heparin had less of an effect. Using a decellularized fibroblast matrix for proteolysis, detergent fractionation, and mass spectrometry, we found that the predominant domain within insoluble fibril fragments is FN’s major heparin-binding domain HepII (modules III12–14). Multiple HepII domains bind simultaneously to a single heparin chain in size exclusion chromatography analyses. We propose a model in which heparin/HS binding to the HepII domain connects multiple FNs together to facilitate the formation of protein interactions for insoluble fibril assembly.
Keywords: Fibronectin, heparin, extracellular matrix, glycosaminoglycans, matrix assembly
1. Introduction
The biophysical properties of the extracellular matrix (ECM) are essential for generating and maintaining the shape, rigidity, stability, and structure of all tissues. The proportions of fibrous glycoproteins, collagens, and polysaccharides that are cell-assembled into an ECM framework determine tissue properties and support tissue-specific cell activities [1, 2]. Fibronectin (FN) is a ubiquitous ECM protein and a main component of the ECM in most tissues. It is assembled into a fibrillar matrix in a cell-mediated process that requires cell contractility and FN-FN interactions. Initially fibrils are soluble in deoxycholate (DOC) detergent but over time nascent fibrils are irreversibly converted into a form that is insoluble in DOC [3–5]. This insoluble FN matrix serves an important role as a foundational matrix for other ECM proteins to build upon [6–8]. As such, FN’s multi-domain structure provides binding sites for deposition of other ECM proteins including collagens, matricellular proteins, proteoglycans/glycosaminoglycans (GAGs), and FN itself [3]. Its N-terminal FN-binding domain is essential for fibril assembly [9], and its primary interaction site is in the first type III modules III1–2 [10, 11]. The N-terminal domain also interacts with other FN-binding sites located throughout the molecule [10–14]. Two of these sites are in heparin binding domains, HepII (spanning modules III12–14) and HepIII (III4–6) which differ in their heparin binding properties [15]. Heparan sulfate (HS) on proteoglycans is abundant in the ECM and on the cell surface. In fact, the HepII domain has been implicated in modulating cell responses to FN through interactions with cell surface syndecan proteoglycans [16–19].
There is evidence that GAGs are involved in matrix assembly. Mutant Chinese hamster ovary (CHO) cells that lack HS GAGs do not assemble a FN matrix [20] and show a deficiency in cell contractility [21]. In contrast, FN assembly by BHK or HT1080 cells that produce HS can be reduced by adding excess heparin or GAGs to the cell culture [17, 20] suggesting that the level of HS determines whether it promotes or inhibits matrix assembly. Interactions involving the cytoplasmic domain of transmembrane syndecan-2 inside and its GAG chains outside have roles in matrix assembly [17, 18, 20]. Given the prevalence of HS binding sites on ECM proteins and evidence for syndecan-ECM interactions in assembly, it is surprising that we do not have a better understanding of the role of GAGs in fibrillogenesis.
To test the requirement for HS in FN assembly, we examined the matrix formed by CHO-677 cells, which do not synthesize HS [22]. CHO cells have been used extensively in matrix assembly studies to identify requirements for ECM proteins and domains, ECM receptors, and intracellular signaling [18, 20, 23–26]. Here we show that HS-deficient CHO cells require exogenous heparin or HS to stimulate the initiation of assembly of DOC-soluble fibrils and to promote the conversion to the DOC-insoluble form. GAG sulfation levels and GAG length are key parameters in promoting fibril assembly. We propose that heparin/HS acts early in the assembly process to bring together FN molecules to promote interactions required for fibrillogenesis.
2. Results
2.1 Heparin Addition is Required for FN Fibril Assembly by CHO-677 Cells
Mutant CHO-677 cells do not synthesize HS [22] due to significantly reduced expression of EXT1, a glycosyltransferase required for HS elongation [27]. Hamster FN was not detected in whole cell lysates or conditioned medium of CHO-677 or wild type CHO-K1 cells (data not shown). Matrix assembly was analyzed for confluent cell monolayers (Fig. S1) and neither cell line produced a significant amount of FN matrix in the absence of an exogenous supply of FN (Fig. 1A). Therefore, all subsequent experiments were performed in the presence of exogenous FN. While CHO-K1 cells assembled a dense FN matrix when the culture medium was supplemented with rat plasma FN, assembly by CHO-677 cells was significantly lower with a 0.7 ± 0.04 mean fluorescence intensity of the FN matrix compared to the wild type cells (p = 0.001) (Fig. 1A). To investigate the effect of heparin on matrix assembly, increasing concentrations of heparin were added along with FN. Heparin significantly increased the amount of FN matrix fibrils assembled by CHO-677 cells, with a 1.3 ± 0.1-fold increase at 50 μg/ml heparin and 1.5 ± 0.1-fold increase with 100 μg/ml heparin compared to the no heparin condition for these cells (p < 0.05) (Fig. 1B). Higher magnification shows that the FN matrix assembled in response to heparin stimulation has the typical fibrillar organization of a fibroblast matrix (Fig. 1C). The matrix assembled by CHO-K1 cells was more robust than that of CHO-677 cells and therefore, heparin did not have a significant effect on fibril formation or organization (Fig. 1B, C).
Figure 1. Heparin increases FN matrix in HS-deficient CHO cells.

Wild-type CHO-K1 and HS-deficient CHO-677 cells were grown for 3 days and, except for the “No FN” samples, exogenous FN was added at 25 μg/ml for the last 24 h. Samples were fixed and immunostained for FN, matrix was visualized by fluorescence microscopy, and fluorescence intensities were quantified. (A) Cells were grown with (+FN) or without FN (No FN). The fold change in mean fluorescence intensities of +FN samples for CHO-677 compared to CHO-K1 cells is shown in yellow (mean ± SE) (n=3, *p = 0.001). Scale bar = 50 μm. (B) CHO-K1 and CHO-677 cells were supplemented with FN alone (left), or together with 50 μg/ml (middle) or 100 μg/ml (right) heparin for 24 h. The fold change from the no heparin condition (No Hep) is noted for the CHO-677 cells (mean ± SE, n=3, *p < 0.05). Scale bar = 50 μm. (C) Higher magnification images of CHO-K1 and CHO-677 cells in (B) with FN and with or without heparin addition. Scale bar = 20 μm.
The amounts of DOC-insoluble FN matrix assembled by wild type and mutant cells without and with heparin were quantified. When increasing concentrations of heparin were added to the mutant cells, the amounts of DOC-insoluble FN matrix were not statistically different after 24 h with 50, 100, or 200 μg/ml heparin, but insoluble FN was decreased when 25 μg/ml heparin or less was added (Fig. 2A). The 50 μg/ml heparin concentration was thus chosen for subsequent experiments. Heparin addition did not have an effect on insoluble FN levels in wild type CHO-K1 cells at either 12 or 24 h. Levels of DOC-insoluble material compared to 12h-no heparin samples differed by 0.9 ± 0.3 and 1.4 ± 0.4 for 12 h and 24 h heparin treatments, respectively (Fig. 2B left). Addition of heparin to CHO-677 cells had a significant effect on insoluble FN levels. Compared to 12 h-no heparin samples, heparin induced an average fold increase in DOC-insoluble FN of 2.7 ± 0.4 at 12 h and 3.8 ± 0.95 at 24 h (Fig. 2B right). Heparin treatment for 24 h is able to rescue the assembly defect of CHO-677 cells since the addition of heparin yielded DOC-insoluble matrix levels equivalent to CHO-K1 cells at the same time. With time, there was a modest increase in insoluble FN without heparin (2.1 ± 0.6-fold at 24 h compared to 12 h) suggesting that the HS-deficiency slows rather than completely stops assembly. The effects of heparin addition were not due to changes in cell numbers or morphology since heparin had no effect on the cell growth rate and no overt differences in the cell shape of CHO-677 cells were detected (data not shown).
Figure 2. Increased DOC-insoluble FN matrix with heparin treatment.

(A) DOC-insoluble material was isolated from CHO-677 cells 24 h after addition of 25 μg/ml FN alone or together with increasing concentrations of heparin from 12.5 to 200 μg/ml. Samples were separated on a 6% polyacrylamide-SDS gel followed by FN immunoblotting with R457 anti-70 kDa antiserum. The fold increase for each sample was calculated relative to no heparin. (B) DOC-insoluble material was isolated from CHO-K1 and CHO-677 cells at 12 or 24 h after addition of 25 μg/ml FN alone or together with 50 μg/ml heparin. The average fold change for each sample was calculated relative to no heparin at 12 h. The difference in DOC-insoluble FN between CHO-K1 cells and CHO-677 cells at 12h with no heparin was 2.8 ± 0.9. The bar graphs below the blots represent the averages ± SE for each sample from 3 independent experiments and for each a representative blot is shown (n = 3, *p < 0.05).
To determine how early in the assembly process heparin begins to act, we examined FN fibrils formed by CHO-677 cells starting at 1 h after FN was added alone or together with heparin. We compared fibril fluorescence intensities at each time point and found that significantly more fibrils were detected in the presence of heparin than in cultures without added heparin. As early as 1 h there was already a 1.5 ± 0.1 mean fold increase in fluorescence intensity (p < 0.05) (Fig. 3A). The difference between plus and minus heparin conditions was significant throughout the time course and reached 1.9 ± 0.3-fold at 24 h (p < 0.05) (Fig. 3A). There was also a gradual increase in FN fluorescence between 1 h and 24 h in the absence of heparin (1.6 ± 0.2-fold) to a level equivalent to 1–2 h with heparin addition. A comparison of mean fluorescence intensities for CHO-677 cells shows that heparin stimulates a continuous increase over 24 h while fluorescence intensities without heparin appear to plateau at 4 h (Fig. 3B). Thus, heparin has a significant stimulatory effect on FN fibril formation by cells that are deficient in HS production.
Figure 3. Heparin increases matrix at early stages of assembly.
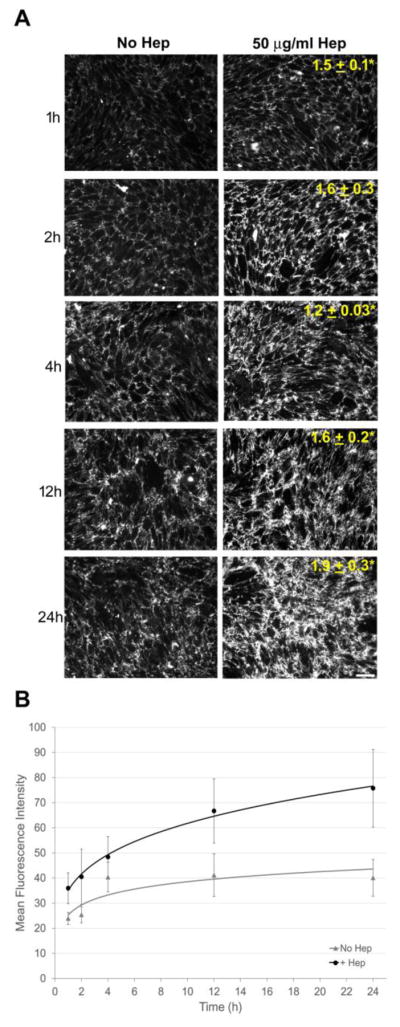
(A) CHO-677 cells were treated as in Figure 1 and analyzed at 1, 2, 4, 12, and 24 h after addition of FN and heparin followed by immunostaining for FN and fluorescence microscopy to visualize the matrix. Mean fluorescence fold change ± SE at each time point relative to No Hep at the same time is noted on the images. Scale bar indicates 50 μm. (n = 3, *p < 0.05). (B) Plot indicates the mean fluorescence intensity ± SE as a function of time for the No Hep and + Hep conditions (n=3).
Wild type CHO-K1 cells are quite efficient at assembling FN fibrils and both DOC soluble and insoluble material and matrix fibrils are readily detectable 4 h after addition of FN (Fig. S2). This is not the case for CHO-677 cells whose deficiency limits the formation of DOC-insoluble matrix such that FN is primarily DOC-soluble at 4 h (Fig. 4A). Heparin stimulated assembly of DOC-soluble FN fibrils detectable as early as 0.5 h, the earliest time tested. FN levels in the matrix were increased on average at least 3.1 ± 0.8-fold at 1 h and 3.9 ± 1.1-fold at 2 h (Fig. 4B). The significant increases in DOC-soluble FN fibrils within 1 h of heparin addition demonstrate a key role for heparin in the initial stages of assembly when DOC-soluble fibrils are being formed.
Figure 4. Increased DOC-soluble FN at early times of treatment.

(A) Cells were treated as in Figure 1 legend. The DOC-soluble and insoluble matrix fractions of CHO-677 cells were immunoblotted after treatment for 4 h. An immunoblot of DOC-soluble material with anti-GAPDH antibodies was used as a loading control. (B) The DOC-soluble FN matrix fractions of CHO-677 cells were immunoblotted after 1 h or 2 h with FN plus or minus heparin. The mean fold change ± SE relative to no heparin at each time point, normalized to GAPDH, was calculated from 3 independent experiments. A representative blot is shown.
2.2 Disaccharide Sulfation is Important for FN Matrix Insolubility
To determine the GAG specificity for stimulating matrix assembly, we used synthetic GAG-mimetics that represent naturally occurring HS sulfation motifs [28, 29]. These GAG-mimetics are synthesized by attaching disaccharides with varying degrees of sulfation to ligation sites along a soluble linear polymer of fixed length. The three GAG-mimetics used in the following experiments were: D2S6, D2A6, and D0A0. In the heparin mimetic D2S6, disaccharides were triple-sulfated whereas D2A6 resembled HS in having two sulfates per disaccharide, and D0A0 was unsulfated (Fig. S3A) [28, 29]. The main advantage of using synthetic GAG-mimetics is that, unlike natural HS or heparin, all of the GAGs in a mimetic have identical numbers of sulfates and sulfation patterns. The ligation efficiencies of unsulfated and double sulfated GAGs to the polymer were equivalent (~ 80%), but the higher negative charge of the triple sulfated GAGs resulted in a lower ligation efficiency (~ 40%). To confirm the ability of the HS-mimetic D2A6 to bind to FN, a glutathione S-transferase-III12–15+V (GST-HepII) fusion protein was incubated with the HS GAG-mimetics attached to Streptavidin agarose beads and beads were immunostained with anti-III12–15+V (anti-HepII) antiserum to visualize the binding. Quantification of the fluorescent signal revealed that the HepII domain bound to D2A6 significantly more than to D0A0 (Fig. S3B). To confirm that synthetic molecules function similarly to heparin, the effect of heparin-mimetic D2S6 and HS-mimetic D2A6 on assembly by CHO-677 cells was compared to heparin. D2S6 and D2A6, but not D0A0, promoted FN assembly at least as well as heparin as determined by quantitative imaging (Fig. 5A). Higher magnification images revealed a similar fibrillar organization to the matrix with either heparin or the GAG-mimetics (Fig. 5A). Direct comparisons of DOC-insoluble FN levels showed a significant increase with heparin or D2S6 compared to no heparin addition (Fig. 5B). In addition, both D2S6 and D2A6 had significantly more insoluble FN compared to the unsulfated D0A0, with an average fold increase of 2.1 ± 0.3 for D2S6 and 4.2 ± 0.9 for D2A6 (Fig. 5B). The heparin-mimetic increased insoluble FN matrix similarly to heparin while the HS-mimetic had an even greater effect than heparin. It is possible that the HS-mimetic D2A6 was more effective than the heparin-mimetic D2S6 because it had a higher density of GAGs coupled to the polymer. Matrix levels with D0A0 were similar to no heparin conditions. These results show that GAG sulfation makes a significant contribution to the assembly process.
Figure 5. Soluble heparin/HS GAG-mimetics increase matrix assembly by CHO-677 cells.

(A) Cells were supplemented with 25 μg/ml FN alone, or together with 2.8 μM (50 μg/ml) heparin, or with 5 μM of D2S6, D2A6, or D0A0 for 24 h followed by immunostaining for FN and fluorescence microscopy to visualize the matrix. Mean FN fluorescence fold increase ± SE relative to the FN alone control is noted in yellow (n=2). Scale bar indicates 50 μm. Higher magnification images are shown below for each. Scale bar indicates 20 μm. (B) DOC-insoluble matrix fractions from CHO-677 cells 24 h in the presence of 25 μg/ml FN without heparin, or in the presence of 2.8 μM heparin, 5 μM D2S6, D2A6, or D0A0 were immunoblotted for FN. The average fold changes ± SE relative to 0 μM heparin or to D0A0 are noted below. Values are 3 ± 0.5 and 3.2 ± 0.9 for heparin and D2S6, respectively, and 2.1 ± 0.3 and 4.2 ± 0.9 for D2S6 and D2A6, respectively. All averages are n = 3, p < 0.05. A representative blot is shown with intervening lanes within the same gel cropped.
2.3 Characterization of Insoluble FN Matrix Fragments
To gain a better understanding of the FN domain interactions that form insoluble fibrils, we isolated FN matrices assembled by mouse NIH 3T3 fibroblasts since these cells assemble a dense, FN-rich matrix with a large amount of detergent-insoluble material. Cell cultures were decellularized to remove intracellular and transmembrane components according to our established procedure [4]. This decellularization does not interfere with the organization of the matrix fibrils [30]. Following decellularization, proteolysis with chymotrypsin in situ cleaved matrix fibrils into various sized fragments. These matrix fibril fragments were then subjected to a series of detergent solubilization and centrifugation steps to isolate three matrix fractions, the cleavage buffer soluble (CBS), the DOC-soluble (DS), and the DOC-insoluble (DI) fractions. The progress of proteolysis over time was followed by immunoblotting with an antiserum raised against FN modules III1–6 (anti-III1–6) (Fig. 6A, left). At a short digestion time of 2h, fibril fragments ranged in size from 100 kDa down to less than 25 kDa (Fig. 6A, left). After 6h of digestion, the higher molecular weight fragments were lost and some of the lower molecular weight fragments, 50 kDa and smaller, increased in amount. The range of fragment sizes at each time point was similar between CBS, DS, and DI fractions, but relative levels of individual fragments varied among these fractions. In particular, there was a ~ 32 kDa band present in DS and DI but not in CBS fractions and that was also apparent in the DI fraction with an antiserum against full-length FN (R39) (Fig. 6A, middle *). Additional regions of FN present in fibril fragments were identified by immunoblotting samples with other FN domain specific antisera. Bands similar in size to anti-III1–6 bands (52 and 32 kDa) were detected with antiserum against HepII at 4 h (Fig. 6A, right). There was an additional band detected with anti-HepII at ~28 kDa (between 25 and 32 kDa, marked by =) that was not apparent with anti-III1–6, but appeared to be detected with R39, suggesting that it contains HepII domain epitopes. No fragments were detected with the anti-N-terminal 70 kDa antiserum (Fig. 6A, right). While bands in DI samples were always less intense than in DS samples, the band patterns were similar indicating that the interactions in DOC-soluble fibrils are converted into an insoluble form.
Figure 6. Identification of FN domains in DOC-insoluble matrix fibril fragments.

(A) Decellularized matrices from NIH3T3 fibroblasts were subjected to 2 h or 6 h proteolysis with chymotrypsin followed by detergent solubilization. Cleavage buffer soluble (CBS), DOC-soluble (DS), and DOC-insoluble (DI) matrix fractions were separated by SDS-PAGE on a 10 % polyacrylamide gel. FN fragments were detected by immunoblotting 2 h and 6 h digests with anti-III1–6 antiserum (R184). The sample in the FN DI lane was digested for 9 h and was immunoblotted with anti-full length rat FN antiserum (R39). DI samples after 4 h of digestion were immunoblotted with anti-(HepII) antiserum or anti-70 kDa antiserum (R457, 1:250 dilution) to visualize FN specific fragments. Anti-HepII antibodies detected band(s) between 25 and 32 kDa, which are not apparent in the anti-III1–6 blot. Asterisks denote the band at ~ 32 kDa and = marks the ~ 28 kDa band referred to in the text. (B) Fragments of about 28 kDa (between 25 and 32 kDa) from 6 h and 24 h digestions of decellularized matrix were submitted for mass spectrometry. Schematics of FN modules are colored-coded to indicate the total number of spectra from all the FN peptides identified in each of the modules. See color legend for ranges of total spectral counts. The majority of peptides were localized in the HepII domain (III12–14) but, at 6 h, fragments containing III4–5 and III8–10 were also detected.
To determine the specific region(s) of FN that are within the 28–32 kDa fragments unique to DOC fractions, gel slices containing the DOC-insoluble fragments from 6 h and 24 h digestions were submitted for mass spectrometric analyses. All domains of FN can be detected by mass spectrometry as indicated by the overall sequence coverage of identified peptides (data not shown; [31]). For each of the analyzed bands, the number of spectral counts (MS2 spectra from identified peptides) was tabulated (Fig. 6B). A greater number of spectral counts for a certain peptide is in part because there is more of that peptide present during elution allowing for it to be sampled multiple times. For the submitted gel band, the modules in which peptides were identified are shown in Fig. 6B, color-coded to show the total number of spectra that were identified from all the peptides found in each of the modules. Very few spectral counts were identified in modules other than type IIIs. The majority of the spectra were derived from the III12–14 region (HepII), the main heparin-binding domain in FN. Two other regions III4–5 and III8–10 were detected in the 6 h samples but dramatically less at 24 h (Fig. 6B). For the 6 h digested ~ 28 kDa band, 93% of all spectral counts came from the modules colored blue (Fig. 6B). For the 24 h digestion, 99% of the spectral counts in the ~ 28 kDa band came from the three domains. More specifically, 55% and 88% of all spectral counts came from the HepII domain in the 6 h and 24 h digested samples, respectively, with the greatest number of spectral counts coming from module III13 containing the “cationic cradle” heparin-binding site [32] (Fig. 6B). These results show that the HepII domain of FN is more prevalent than other domains in insoluble matrix fragments.
2.4 Heparin Chain Length Affects Matrix Insolubility
The HepII domain was a prominent component of DOC-insoluble FN fibril fragments suggesting that this domain plays a central role in fibril assembly. Heparin could promote fibril formation by binding multiple FN molecules, bringing them into close proximity and thus facilitating the FN-FN interactions that link these molecules together. HS chains of 7–8 disaccharides in length bound to the HepII domain of FN with a similar affinity as unfractionated heparin, whereas heparin chains smaller than 7 disaccharides bound with a 5–10 fold lower affinity than unfractionated heparin [33, 34]. The unfractionated heparin used in our experiments has an average molecular weight of 18 kDa, which is 30–35 disaccharides. Since this is 4–5 times longer than the minimum length for full binding to FN, each heparin could potentially bind 4–5 FN molecules on average.
To determine if the stimulatory effect of heparin requires a chain length that can bind to more than one FN molecule at the same time, we compared unfractionated heparin with heparins of defined length. We used heparin chains that were either 8 disaccharides (16 saccharides; H016) or 15 disaccharides (30 saccharides; H030), which can bind to one or two FN HepII domains, respectively. We confirmed that H016 and H030 can bind to FN using a competition experiment. Heparin-agarose beads were incubated with a maltose binding protein (MBP)-HepII fusion protein [10] alone or together with excess unfractionated heparin, H016, or H030. All three heparins inhibited MBP-HepII binding to heparin-agarose, although 2–4-fold higher concentrations of H030 and H016 were needed to achieve the same inhibition as unfractionated heparin (Fig. S4), similar to the reported relative binding activities of shorter heparins.
To test the effect of heparin chain lengths on matrix insolubility, increasing concentrations of H016 or H030 were added to cells along with exogenous FN and the levels of insoluble FN were compared to the effects of unfractionated heparin. Addition of H030 increased insoluble FN levels to the same extent as 2.8 μM (50 μg/ml) heparin (Fig. 7B). Increasing the concentration of H030 did not further increase the formation of DOC-insoluble matrix. This was seen by immunofluorescent staining of the matrix fibrils as well, with a comparable matrix assembled by the cells when the highest H030 dose was added as when the unfractionated heparin was added (Fig. 7A). For H016, insoluble FN matrix reached 40–60% of the amount induced by heparin or H030, and FN fibril intensity was less than that of unfractionated heparin or H030 (Fig. 7A, B). However, H016 did have an effect on assembly yielding on average a 2-fold increase at its highest dose over the control with no heparin added. Therefore, a heparin chain length able to bind at least two FNs is needed for maximum effect on matrix insolubility.
Figure 7. Stimulation of matrix assembly varies with heparin chain length.

(A) CHO-677 cells were supplemented with FN alone (No Hep), or together with 2.8 μM unfractionated heparin (Hep), or with 11.2 μM of either H016 or H030 for the last 24 h of growth followed by immunostaining for FN and fluorescence microscopy to visualize the matrix. Scale bar indicates 50 μm. (B) DOC-insoluble matrix fractions of CHO-677 cells were isolated 24 h after addition of 25 μg/ml FN alone (0), or with the indicated μM concentrations of unfractionated heparin (Hep), H016, or H030. 2.8 μM heparin is equivalent to 50 μg/ml heparin. The mean fold change relative to unfractionated heparin is noted below. For three independent experiments, the average fold changes for H016 relative to Hep are 0.4 ± 0.1 for 2.8 μM, 0.4 ± 0.2 for 5.6 μM, and 0.6 ± 0.1 for 11.2 μM H016 (p < 0.05). For H030, average fold changes are 1.2 ± 0.2 for 2.8 μM, and 1.1 ± 0.4 for 5.6 and 11.2 μM H030. Representative blots are shown. For the right-hand blot, intervening lanes within the same gel were cropped.
2.5 Two or More HepII Domains Bind Simultaneously to a Single Heparin Chain
To test whether a single heparin chain can indeed bind to multiple FN molecules at the same time, we performed size exclusion chromatography with a recombinant MBP-HepII fusion protein plus or minus heparin. MBP-HepII alone eluted in fractions 15 – 18 with the peak at fraction 16 (Fig. 8A, yellow curve). With the addition of unfractionated heparin to MBP-HepII, the monomer peak was dramatically reduced and the protein eluted significantly earlier, beginning near the void volume in fraction 9 and extending to fraction 13 with the peak in fractions 11–12 (Fig. 8A, gray curve). The average size of the unfractionated heparin is 18 kDa, insufficient to explain the large shift in the MBP-HepII peak. SDS-PAGE analyses confirmed the shift of MBP-HepII with heparin to an earlier elution time and also showed that MBP, which contaminates the MBP-HepII preparation, did not shift from fractions 20–22 in the presence of heparin (Fig. 8B). The shift of the MBP-HepII elution profile with heparin demonstrates that MBP-HepII and heparin formed a complex with two or more HepII domains linked together by heparin.
Figure 8. Heparin binds two or more HepII domains.

(A) 1 μM MBP-HepII alone (yellow) or preincubated with 10 μM unfractionated heparin (Hep) (gray) for 1 h was subjected to size exclusion chromatography. The elution profiles show A280 (milli-absorbance units, mAU) for fractions from the void volume (fraction 9, which contains protein aggregates) to ~ 20 kDa (fraction 22). MW standards eluted in fractions 9 (670 kDa), 14 (158 kDa), 21 (44 kDa), and 23 (17 kDa). (B) Fractions from (A) were analyzed by SDS-PAGE using 10% polyacrylamide SDS gels and silver staining. Molecular mass standards are indicated on the left. * indicates MBP-HepII, ~ indicates MBP at 42 kDa, which contaminates the MBP-HepII preparation. (C) Size exclusion chromatographs for 1 μM MBP-HepII alone (yellow) or incubated for 1 hr with 10 μM H030 (blue), H016 (orange), or unfractionated heparin (dotted gray). mAU is plotted versus fraction number. Elution profiles were confirmed by SDS-PAGE and silver staining (data not shown).
To determine the effect of heparin chain length on complex formation, size exclusion chromatography was performed in the presence of the shorter heparin chains, H030 and H016. MBP-HepII plus H030 heparin was largely shifted to an earlier elution time in fractions 11–14 with a small shoulder that overlaps with the monomeric MBP-HepII in fraction 15 (Fig. 8C, blue curve). This profile indicates that H030 heparin also forms a complex with more than one HepII but, because H030-HepII complexes do not elute as early as with unfractionated heparin, it appears that fewer proteins can bind per H030 chain.
On the other hand, MBP-HepII preincubated with H016 heparin showed only a slight shift in the elution profile to fractions 14–18 with the peak fraction at 15 (Fig. 8C, orange curve). The overlap between MBP-HepII plus and minus H016 suggests that binding of this shorter heparin does not induce the formation of protein complexes. Instead it seems likely that binding of ~4 kDa H016 induces a change in HepII conformation that is detected by a shift in the elution profile. The observed shifts in elution times are indicative of protein complexes linked by heparin and the differing elution times with different length heparins indicate that longer heparin chains bind two or more HepII domains.
3. Discussion
To determine whether heparin/HS makes a significant contribution to the ECM assembly process, we treated HS-deficient CHO-677 cells and found that heparin addition substantially stimulates FN fibril assembly. This stimulation began very soon after addition of heparin to the culture medium and higher levels of matrix were detected first as DOC-soluble FN fibrils, which were converted into DOC-insoluble matrix over time. Results with heparin- or HS-mimetics showed the enhancement depends on GAG sulfation. We developed a fractionation procedure using proteolysis of a decellularized fibroblast matrix and isolation of the detergent insoluble fibril fragments to identify FN domains that interact within insoluble matrix fibrils. This approach identified heparin-binding domains of FN, in particular the HepII domain, as the major components of the insoluble fraction. Heparin molecules and GAG-mimetics that were long enough to bind multiple FNs were the most effective in promoting assembly. Shorter heparin, able to bind a single FN, also stimulated assembly albeit not as effectively as heparins that can bind two or more FNs. Size exclusion chromatography data show that heparin chains of 30 or more saccharides form complexes with multiple HepII domains. We propose a model in which heparin/HS binding promotes FN-FN interactions and fibril conversion to insolubility by bringing multiple molecules into close proximity at the cell surface.
Our analyses of matrix fragments that are both resistant to prolonged proteolysis by chymotrypsin and insoluble in DOC detergent have shown a predominance of heparin-binding domains from FN. The majority of peptides identified after digestion of the matrix for 24 h were in the III12–14 HepII domain. This domain has the highest affinity for heparin among the FN domains [15]. Heparin addition significantly increased FN fibrils and insoluble FN levels assembled by HS-deficient CHO-677 cells. However, using biotinylated heparin, we were not able to detect heparin within the assembled FN fibrils (unpublished observations). Others have reported that heparin acts transiently and is not retained in the ECM [35, 36]. Results with heparin- and HS-mimetics show a requirement for sufficient GAG sulfation in FN assembly, and also showed that HS GAGs can increase insoluble matrix. The effect of the HS-mimetic D2A6 was in fact greater than that of heparin or D2S6. This could result from differences between heparin and HS sulfation levels. It is also possible that the presence of more GAG disaccharides attached to the polymer in D2A6 than in D2S6 has an effect on assembly. The higher ligation efficiency of double-sulfated compared to triple-sulfated GAG disaccharides is due to the high negative charge on the heparin-mimetic D2S6, which prevents close packing along the polymer, resulting in approximately half as many GAG disaccharides bound (~ 130 vs. ~ 60).
The effects of heparin, the mimetics, and sulfation on assembly may be mimicking FN interactions with syndecan transmembrane proteoglycans. GAG sulfation and syndecan-2 are required for FN assembly by fibrosarcoma cells [17]. Syndecan-4 binding to HepII enhances cell contractility and integrin signaling [16, 19, 21], both of which are involved in matrix assembly. In vivo evidence for an HS role in FN matrix organization is provided by conditional knockout in the mouse limb bud mesenchyme of EXT1, the HS synthesizing enzyme, which caused severe limb skeletal defects [37]. An expanded perichondrium layer with less densely packed FN matrix fibrils was observed suggesting that HS is needed for formation of a foundational FN matrix during normal tissue development. In our experiments in the absence of added heparin, CHO-677 cells gradually assembled a low level of exogenous FN that never reached the level of matrix observed in the presence of heparin. This suggests that the lack of HS in mutant cells may slow the assembly process and this would suggest that heparin/HS may act to increase the rate of FN polymerization. In this case, this GAG would not be absolutely necessary for assembly. We did not detect any effect of heparin on integrin-FN interactions using CHO-677 cells and the integrin domain of FN, III9–10, in suspension binding assays (IR, unpublished observations). CHO-677 cells lack HS but produce higher levels of chondroitin sulfate than wild type cells [22]. Chondroitin sulfate also binds to the HepII domain, albeit not as strongly as heparin [38]. This increase in chondroitin sulfate suggests an alternative explanation for the slow assembly without heparin. Perhaps chondroitin sulfate can take the place of heparin, although not as effectively, to induce FN-FN interactions and fibril formation.
The HepII domain contains the major heparin-binding site in FN, the cationic cradle formed by two β-strands in III13 [32]. This domain has binding activity for the N-terminal assembly domain I1–5 of FN and protein fragments spanning III12–14 inhibit incorporation of exogenous FN into insoluble fibrils [12]. III12–14 also binds to III2–3 to form the compact conformation of FN in solution; deletion of III12–14 in full length FN generated a more open conformation by velocity sedimentation [39]. Binding of heparin and HS can induce conformational changes in FN [15], which expose new binding sites for growth factors [36, 40] or cause matrix changes that affect cell differentiation [35, 41]. Conformational changes are also involved in FN matrix assembly [3, 42, 43] and our results suggest that cell-produced HS may be critical for inducing some of these changes.
Based on our results, we suggest a model in which heparin/HS binding primarily at the HepII domain increases the rate of formation of FN-FN interactions. GAGs could act as a bridge between individual FN dimers to facilitate their interactions, but would not necessarily be constitutively incorporated into matrix fibrils. In our experiments with soluble heparin addition, this bridging effect would be independent of intracellular signaling by transmembrane proteoglycans. Our results with heparin chains of different lengths support this idea. H016 heparin at 8 disaccharides is the minimum length for optimal binding to HepII [33, 34] and stimulated assembly compared to no heparin. A heparin disaccharide is about 0.86 nm in length [34, 44] whereas a typical type III module is 3.6 nm across in the N-to-C dimension [45]. Thus 8 disaccharides will cover ~ 6.9 nm, which is more than one but less than two type III modules. This length would be sufficient to span the ~ 6 nm extended heparin binding region described in the structure of the III13 cationic cradle plus putative heparin binding site (HBS)-2 in III14 [46]. H016 was not as effective as longer heparins. H030 heparin, which at ~ 13 nm is about twice the length of H016 and long enough to bind to two extended heparin binding regions simultaneously, increased insoluble FN to levels similar to that detected with unfractionated heparin. Size exclusion chromatography results further support this model. Both unfractionated heparin and H030 heparin formed complexes with HepII domains. Although the stoichiometry of HepII to heparin cannot be determined by this method, the relationship between the elution peaks shows that unfractionated heparin can bind more HepII domains than H030 heparin. H016 heparin induced only a small shift showing that it bound to the HepII domains but did not induce formation of protein complexes. Therefore, we suggest that heparin could be increasing fibril formation by binding at the HepII domains of two or more FNs simultaneously, bringing them together to increase the formation of FN-FN interactions, thus nucleating fibril formation by shortening the lag phase of polymerization. Independently of a bridging role, the stimulatory effect of heparin could include conformational changes induced by one-to-one heparin-FN binding. Such conformational changes would result in increased exposure of the various FN domains that are involved in the insoluble interactions, allowing these interactions to occur, which could explain the effect of the shorter H016 heparin on matrix assembly. Whatever the mechanism, it is clear that GAGs play an important role in matrix assembly raising the possibility that phenotypes caused by mutations in genes for GAG attachment or synthesis enzymes may arise from defects in FN matrix.
4. Materials and Methods
4.1 Cell Culture
NIH 3T3 mouse fibroblasts (ATCC, Manassas, VA) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Grand Island, NY) with 10% bovine calf serum (Hyclone, Logan, UT) and antibiotic/antimycotic cocktail (Corning Life Sciences, Oneonta, NY). Wild-type CHO-K1 (ATCC) and mutant CHO-677 cells (a gift from Dr. Jeffrey Esko, UCSD) [22] were maintained in DMEM containing 10% FetalClone Serum (Hyclone) and antibiotic/antimycotic cocktail. All cell lines used were tested and found to be free of mycoplasma contamination.
4.2 Reagents
Fibronectin was purified from fresh frozen rat plasma by gelatin-Sepharose affinity chromatography [47]. Heparin sodium salt from porcine intestinal mucosa (Grade I-A, ≥180 USP units/mg) (unfractionated heparin) was obtained from Sigma (St. Louis, MO). Different sized heparin GAG chains, H016 (average molecular weight 4650) and H030 (average molecular weight >9000), were obtained from Iduron (Galen Laboratory Supplies, Middletown, CT).
4.3 Preparation of Decellularized Matrices and Matrix Proteolysis
NIH 3T3 fibroblasts were plated at 6.3×106 cells/10-cm plate and grown for 10 days, changing media every 2–3 days. Plates were then decellularized to remove the cellular material and leave behind the fibrillar FN matrix as previously described [4]. For chymotryptic digestion, plates were washed with cleavage buffer (100 mM Tris-HCl, pH 7.8, 10 mM CaCl2), and then 2 ml of cleavage buffer with 50 μg/ml α-chymotrypsin were added and proteolysis was allowed to proceed at 37°C for the indicated amounts of time. Matrix fragments were then scraped into tubes, the reaction stopped with 2 mM phenylmethylsulfonyl fluoride (PMSF), centrifuged at 14,000 rpm for 15 minutes at 4°C, and the supernatant collected as the cleavage buffer soluble (CBS) fraction. The resulting pellet was washed once with cleavage buffer followed by addition of DOC lysis buffer (2% DOC, 20 mM Tris-HCl, pH 8, 2 mM EDTA) with 2 mM PMSF at 25 μl/10-cm plate pellet and incubated at room temperature for 15 minutes with vortexing every 5 minutes. Following centrifugation at 14,000 rpm for 15 minutes, the supernatant was collected as the DOC-soluble (DS) fraction. The remaining pellet was solubilized by the addition of SDS lysis buffer (4% SDS, 20 mM Tris-HCl, pH 8, 2 mM EDTA) with 2 mM PMSF at 25 μl/10-cm plate pellet and boiling for 5 minutes. This was followed by centrifugation, and the supernatant was transferred to a new tube as the DOC-insoluble (DI) fraction.
4.4 Mass Spectrometry
DOC-insoluble material from 6 h and 24 h digested matrices was separated by SDS-PAGE using 10 % polyacrylamide and stained with GelCode Blue (Pierce Chemical Co., Rockford, IL). In parallel, an aliquot of the same material was immunoblotted with antiserum against the III1–6 modules of FN (R184) and alignment of the blot with the stained gel was used to select bands for excision. These were submitted for mass spectrometry.
Proteins in polyacrylamide gel slices were digested using trypsin as in [48]. Dried extracted peptides were reconstituted in 20 μl of 0.1% formic acid pH 3. Five μl were injected per run using an Easy-nLC 1000 UPLC system. Samples were loaded directly onto a 45 cm long 75 μm inner diameter nano capillary column packed with 1.9 μm C18-AQ (Dr. Maisch, Germany) mated to metal emitter in-line with an Orbitrap Elite (Thermo Scientific, USA). The mass spectrometer was operated in data dependent mode with the 120,000 resolution MS1 scan (400–1800 m/z) in the Orbitrap followed by up to 20 MS/MS scans with CID fragmentation in the ion trap. Dynamic exclusion list was invoked to exclude previously sequenced peptides for 120 s if sequenced within the last 30 s.
Raw files were searched using MS-Amanda [49], Byonic [50], and Sequest HT algorithms [51] within the Proteome Discoverer 2.1 suite (Thermo Scientific, USA). 10 ppm MS1 and 0.4 Da MS2 mass tolerances were specified. Caramidomethylation of cysteine was used as fixed modification, oxidation of methionine, deamidation of asparagine and conversion of glutamine to pyro-glutamate (at peptide N-termini) were specified as dynamic modifications. Chymotrypsin and trypsin double digestion with maximum of 2 missed cleavages were allowed against the Uniprot Mouse database.
Scaffold (version Scaffold_4.7.5, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 99.5% probability by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [52]. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Spectral counts are the total number of identified MS2 spectra derived from and matching the peptide sequences of interest.
4.5 Immunofluorescence Microscopy
Cells were seeded on glass coverslips in 24-well plates and 25 μg/ml exogenous rat plasma FN was added either alone or together with heparin or the specified GAG-mimetic for the indicated amounts of time. Upon assay completion, cells were washed with PBS, fixed in 3.7% (w/v) formaldehyde (Sigma, St. Louis, MO) in PBS, and then stained with R457, an anti-FN antiserum against the N-terminal 70 kDa domain, diluted 1:100 as in [25]. Alexa-fluor 488 or Alexa-fluor 568 conjugated goat anti-rabbit IgG secondary antibody (Invitrogen, Eugene, OR) was diluted 1:600 and DAPI (Sigma, St. Louis, MO) was diluted 1:1000. Coverslips were mounted using ProLong Gold antifade reagent (Life Technologies, Grand Island, NY). In all experiments, cell densities were equivalent as determined by phase microscopy. Images were captured using a Nikon Eclipse Ti microscope equipped with a Hamamatsu C10600 ORCA-R2 digital camera. Total mean fluorescence measurements were performed using ImageJ by averaging 7 randomly selected fields of view from each coverslip in a given experiment. The fluorescence fold-change with heparin or a GAG-mimetic was calculated by normalizing to the no heparin condition, which was set to 1. The fold changes were averaged across 3 independent experiments, unless otherwise indicated, and are reported as the mean ± standard error.
4.6 Preparation of DOC-Soluble and DOC-Insoluble Fractions
CHO cells were seeded in a 24-well plate at 60,000 cells/well and grown for 2–3 days, at which point growth medium was replaced with fresh medium supplemented with exogenous rat plasma FN at 25 μg/ml and heparin or other reagents as needed and incubated for the indicated amounts of time. Cell lysates were separated into DOC-soluble and -insoluble fractions as previously described [53]. Total protein concentrations were measured in the DOC-soluble fraction using the BCA protein assay (Pierce Chemical Co., Rockford, IL). Protein concentrations in lysates of CHO-677 cells plus and minus heparin were equivalent within an experiment indicating that cells were of similar densities at collection. Equivalent amounts of the DOC-insoluble fractions were run proportionate to the soluble fraction concentrations.
4.7 Immunoblotting
Proteins were separated by SDS-PAGE on 6 or 10% polyacrylamide gels alongside Precision Plus Protein Standard (Bio-Rad, Hercules, CA) and then transferred to nitrocellulose membranes. Antibody incubations were performed as described [25]. Rabbit anti-FN polyclonal antisera were raised in house: R457 against the rat N-terminal 70 kDa domain [10]; R184 against an MBP fusion protein containing the rat FN III1–6 domain; and R39, against full-length rat plasma FN [25] (see Fig. 1C). Anti-III12–15+V (anti-HepII), which contains the HepII domain, was raised at the Pocono Rabbit Farm and Laboratory (Canadensis, PA) against a GST fusion protein containing this region of human FN and was validated by immunoblotting with the immunogen and with plasma FN and by immunofluorescence with mouse fibroblasts. Antiserum dilutions were: 1:2000 for R457, 1:100 for R184, 1:500 for R39, and 1:1000 for anti-HepII. DOC-soluble samples were probed with an antibody against GAPDH at 1:2000 to confirm equal loading (14C10, Cell Signaling Technology, Danvers, MA). Secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit IgG diluted 1:10,000 (Pierce Chemical Co., Rockford, IL). Blots were developed using SuperSignal West Pico ECL reagents (Pierce Chemical Co., Rockford, IL). Densitometry was performed on scanned films using ImageJ and exposures yielding signals within the linear range were quantified. Immunoblots of lysates from 3 independently performed experiments were analyzed and quantified signals were averaged across the 3 experiments.
4.8 Synthesis of Heparan Sulfate GAG-Mimetics
Unless otherwise stated, all chemicals and reagents were purchased from Sigma Aldrich, and used without further purification. GAG disaccharides were purchased from Dextra, UK. The synthesis and characterization of the GAG-mimetic glycopolymers has previously been described [29, 54]. Briefly, a Boc-protected N-methylaminooxy acrylamide precursor was subjected to RAFT polymerization using the chain initiator AIBN (2,2′-azobis(2-methylpropionitrile) in the presence of an azide-terminated chain transfer agent to form the azide-functionalized glycopolymer precursor backbone (Mn = 43 kDa, ð = 1.15, n = 167). Similarly, a biotin-containing chain transfer agent can be used to generate biotinylated polymers (vide infra, Mn = 51,600, ð = 1.21, n = 200). Following end-deprotection of the trithiocarbonate with butylamine to expose free thiol groups, the polymers were labeled with maleimide-functionalized AlexaFluor488 or TAMRA fluorophores. The removal of the side-chain protecting Boc groups was achieved by treatment with TMS chloride and phenol in DCM. The resulting polymer was reacted with the desired GAG disaccharide (1.1 equiv. per reactive aminooxy sidechain) at 50 °C for 72 hours. Following purification by size-exclusion chromatography on spin columns (Sephadex G-50, Roche cat # 11814427001), the final GAG-mimetic glycopolymers were characterized by 1H NMR and UV-VIS spectroscopy to determine the efficiency of disaccharide ligation and to establish polymer concentrations, respectively. Polymers with 170 or 212 ligation sites were used for the preparations reported here. Average ligation efficiencies for D2A6, D0A0, and D2S6 were approximately 80%, 81%, and 37%, respectively, which correspond to 136, 137, and 60 disaccharides per mimetic, respectively. The functionality of GAG-mimetics compared to heparin was determined using a growth factor signaling assay [29].
4.9 Size exclusion chromatography
A maltose binding protein-III12–15 fusion protein (MBP-HepII) was purified by amylose resin chromatography [10]. 1 μM MBP-HepII was preincubated for 1 h at room temperature with 10 μM of either unfractionated heparin, H030, or H016 heparin in 50 mM Tris-HCl, pH 7.8, 50 mM NaCl at a final volume of 500 μl per sample. Size exclusion chromatography was performed using a 24 ml Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with the above buffer at 4 °C with a flow rate of 0.7 ml/min on an AKTA pure FPLC (GE Healthcare). The elution profile of the column was characterized with a standard curve of elution volume versus log molecular weight of gel filtration standards ranging in size from 1,350 to 670,000 D (Bio-Rad). Absorbance at 280 nm of MBP-HepII in the eluate was measured continuously and recorded as milli-absorbance units (mAU) per elution volume. 500 μl fractions were collected and proteins in fractions across the relevant elution volumes were analyzed by electrophoresis on 10% polyacrylamide-SDS gels followed by silver staining.
4.10 Statistical analysis
Results are reported as the mean ± standard error for a minimum of 3 independent samples, unless specified otherwise. Computed means were compared using a two-tailed Student’s t-test, with p < 0.05 considered statistically significant.
Supplementary Material
Highlights.
We show a requirement for soluble heparin/heparan sulfate and glycosaminoglycan sulfation in fibronectin fibrillogenesis.
Mass spectrometry of matrix fibril fragments identified fibronectin’s HepII heparin-binding region as the predominant domain comprising insoluble regions of fibrils.
Heparin chains of sufficient length to bind to at least two fibronectin heparin binding domains gave maximal stimulation of assembly suggesting a new role for heparin/heparan sulfate as a linker that brings fibronectin molecules together to facilitate insoluble fibril assembly.
Acknowledgments
The authors are grateful to Dr. Tharan Srikumar, manager of the Molecular Biology Proteomics and Mass Spectrometry Core Facility, for scientific and technical assistance, to Yinan Wang, Christopher J. Fisher, and Ember Tota for assistance with polymer synthesis, and to Drs. Jaegeun Song and Sabine Petry for assistance with size exclusion chromatography. The authors would also like to thank members of the Schwarzbauer lab for helpful scientific discussions.
7. Funding
This research was supported by the National Institutes of Health (NIH) [R01 CA160611 to J.E.S.]. IR was supported by NIH pre-doctoral training grant T32 GM007388 to Princeton University Department of Molecular Biology. SAW was supported by NIH NRSA fellowship F32 GM077891. MLH and KG were supported in part by the NIH Pathway to Independence Award [NIBIB: 4 R00 EB013446], the NIH Director’s New Innovator Award [1DP2HS087954], and the Program of Excellence in Glycosciences [PEG, NHLBI: 4P01HL107150].
Abbreviations Used
- CBS
Cleavage buffer soluble
- DI
DOC-insoluble
- DS
DOC-soluble
- DOC
Deoxycholate
- HS
Heparan Sulfate
- MBP
Maltose binding protein
- PMSF
Phenylmethylsulfonyl fluoride
Footnotes
6. Competing Interests
No competing interests declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hynes RO, Yamada KM. Extracellular Matrix Biology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2012. p. 397. [Google Scholar]
- 2.Mecham RP. Biology of Extracellular Matrix. Springer; 2011. The Extracellular Matrix: an Overview; p. 425. [Google Scholar]
- 3.Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mao Y, Schwarzbauer JE. Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci. 2005;118(19):4427–4436. doi: 10.1242/jcs.02566. [DOI] [PubMed] [Google Scholar]
- 5.McKeown-Longo PJ, Mosher DF. Binding of plasma fibronectin to cell layers of human skin fibroblasts. J Cell Biol. 1983;97:466–472. doi: 10.1083/jcb.97.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20(5):495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP. Fibrillin assembly requires fibronectin. Mol Biol Cell. 2009;20(3):846–858. doi: 10.1091/mbc.E08-08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13(10):3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarzbauer JE. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J Cell Biol. 1991;113(6):1463–1473. doi: 10.1083/jcb.113.6.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguirre KM, McCormick RJ, Schwarzbauer JE. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J Biol Chem. 1994;269:27863–27868. [PubMed] [Google Scholar]
- 11.Hocking D, Sottile J, McKeown-Longo PJ. Fibronectin’s III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J Biol Chem. 1994;269(29):19183–19187. [PubMed] [Google Scholar]
- 12.Bultmann H, Santas AJ, Pesciotta Peters DM. Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J Biol Chem. 1998;273:2601–2609. doi: 10.1074/jbc.273.5.2601. [DOI] [PubMed] [Google Scholar]
- 13.Ingham KC, Brew SA, Huff S, Litvinovich SV. Cryptic self-association sites in type III modules of fibronectin. J Biol Chem. 1997;272(3):1718–1724. doi: 10.1074/jbc.272.3.1718. [DOI] [PubMed] [Google Scholar]
- 14.Maqueda A, Moyano JV, Hernández Del Cerro M, Peters DM, Garcia-Pardo A. The heparin III-binding domain of fibronectin (III4–5 repeats) binds to fibronectin and inhibits fibronectin matrix assembly. Matrix Biol. 2007;26(8):642–651. doi: 10.1016/j.matbio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Hynes RO. Fibronectins. Springer-Verlag; New York: 1990. [Google Scholar]
- 16.Bloom L, Ingham KC, Hynes RO. Fibronectin regulates assembly of actin filaments and focal contacts in cultured cells via the heparin-binding site in repeat III13. Mol Biol Cell. 1999;10:1521–1536. doi: 10.1091/mbc.10.5.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galante L, Schwarzbauer JE. Requirements for sulfate transport and the diastrophic dysplasia sulfate transporter in fibronectin matrix assembly. J Cell Biol. 2007;179(5):999–1009. doi: 10.1083/jcb.200707150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klass CM, Couchman JR, Woods A. Control of extracellular matrix assembly by syndecan-2 proteoglycan. J Cell Sci. 2000;113(3):493–506. doi: 10.1242/jcs.113.3.493. [DOI] [PubMed] [Google Scholar]
- 19.Wilcox-Adelman SA, Denhez F, Goetinck PF. Syndecan-4 modulates focal adhesion kinase phosphorylation. J Biol Chem. 2002;277:32970–32977. doi: 10.1074/jbc.M201283200. [DOI] [PubMed] [Google Scholar]
- 20.Chung C, Erickson HP. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci. 1997;110(Pt 12):1413–1419. doi: 10.1242/jcs.110.12.1413. [DOI] [PubMed] [Google Scholar]
- 21.Midwood KS, Valenick LV, Hsia HC, Schwarzbauer JE. Coregulation of fibronectin signaling and matrix contraction by tenascin-C and syndecan-4. Mol Biol Cell. 2004;15:5670–5677. doi: 10.1091/mbc.E04-08-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lidholt K, Weinke JL, Kiser CS, Lugemwa FN, Bame KJ, Cheifetz S, Massagué J, Lindahl U, Esko JD. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci U S A. 1992;89(6):2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271(12):6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 24.Sechler JL, Rao H, Cumiskey AM, Vega-Colon I, Smith MS, Murata T, Schwarzbauer JE. A novel fibronectin binding site required for fibronectin fibril growth during matrix assembly. J Cell Biol. 2001;154(5):1081–1088. doi: 10.1083/jcb.200102034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sechler JL, Takada Y, Schwarzbauer JE. Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol. 1996;134(2):573–583. doi: 10.1083/jcb.134.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C, Keivens VM, O’Toole TE, McDonald JA, Ginsberg MH. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell. 1995;83(5):715–724. doi: 10.1016/0092-8674(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 27.Wei G, Bai X, Bame KJ, Koshy TI, Spear PG, Esko JD. Location of the glucuronosyltransferase domain in the heparan sulfate copolymerase (EXT1) by analysis of Chinese hamster ovary cell mutants. J Biol Chem. 2000;275:27733–27740. doi: 10.1074/jbc.M002990200. [DOI] [PubMed] [Google Scholar]
- 28.Huang ML, Smith RA, Trieger GW, Godula K. Glycocalyx remodeling with glycopolymer-based proteoglycan mimetics. Methods Mol Biol. 2016;1367:207–224. doi: 10.1007/978-1-4939-3130-9_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang ML, Smith RA, Trieger GW, Godula K. Glycocalyx remodeling with proteoglycan mimetics promotes neural specification in embryonic stem cells. J Am Chem Soc. 2014;136(30):10565–10568. doi: 10.1021/ja505012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh S, Bandini SB, Donnelly PE, Schwartz J, Schwarzbauer JE. A cell-assembled spatially aligned extracellular matrix to promote directed tissue development. J Mater Chem B Mater Biol Med. 2014;2(11):1449–1453. doi: 10.1039/C3TB21864C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastino AK, Greco TM, Mathias RA, Cristea IM, Schwarzbauer JE. Stimulatory effects of advanced glycation endproducts (AGEs) on fibronectin matrix assembly. Matrix Biol. 2017;59:39–53. doi: 10.1016/j.matbio.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busby TF, Argraves WS, Brew SA, Pechik I, Gilliland GL, Ingham KC. Heparin binding by fibronectin module III-13 involves six discontinuous basic residues brought together to form a cationic cradle. J Biol Chem. 1995;270(31):18558–18562. doi: 10.1074/jbc.270.31.18558. [DOI] [PubMed] [Google Scholar]
- 33.Ingham KC, Brew SA, Atha DH. Interaction of heparin with fibronectin and isolated fibronectin domains. Biochem J. 1990;272(3):605–611. doi: 10.1042/bj2720605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker A, Gallagher JT. Structural domains of heparan sulphate for specific recognition of the C-terminal heparin-binding domain of human plasma fibronectin (HEPII) Biochem J. 1996;317(3):871–877. doi: 10.1042/bj3170871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B, Lin Z, Mitsi M, Zhang Y, Vogel V. Heparin-induced conformational changes of fibronectin within the extracellular matrix promote hMSC osteogenic differentiation. Biomater Sci. 2015;3(1):73–84. doi: 10.1039/c3bm60326a. [DOI] [PubMed] [Google Scholar]
- 36.Mitsi M, Forsten-Williams K, Gopalakrishnan M, Nugent MA. A catalytic role of heparin within the extracellular matrix. J Biol Chem. 2008;283(50):34796–34807. doi: 10.1074/jbc.M806692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto Y, Matsumoto K, Irie F, Fukushi J, Stallcup WB, Yamaguchi Y. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J Biol Chem. 2010;285(25):19227–19234. doi: 10.1074/jbc.M110.105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barkalow FJ, Schwarzbauer JE. Interactions between fibronectin and chondroitin sulfate are modulated by molecular context. J Biol Chem. 1994;269(6):3957–3962. [PubMed] [Google Scholar]
- 39.Johnson KJ, Sage H, Briscoe G, Erickson HP. The compact conformation of fibronectin is determined by intramolecular ionic interactions. J Biol Chem. 1999;274:15473–15479. doi: 10.1074/jbc.274.22.15473. [DOI] [PubMed] [Google Scholar]
- 40.Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45(34):10319–10328. doi: 10.1021/bi060974p. [DOI] [PubMed] [Google Scholar]
- 41.Vogel S, Arnoldini S, Moller S, Schnabelrauch M, Hempel U. Sulfated hyaluronan alters fibronectin matrix assembly and promotes osteogenic differentiation of human bone marrow stromal cells. Sci Rep. 2016;6:36418. doi: 10.1038/srep36418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maurer LM, Ma W, Mosher DF. Dynamic structure of plasma fibronectin. Crit Rev Biochem Mol Biol. 2015;51(4):213–227. doi: 10.1080/10409238.2016.1184224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erickson HP. Protein unfolding under isometric tension-what force can integrins generate, and can it unfold FNIII domains? Curr Opin Struct Biol. 2017;42:98–105. doi: 10.1016/j.sbi.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulloy B, Forster MJ, Jones C, Davies DB. NMR and molecular-modelling studies of the solution conformation of heparin. Biochem J. 1993;293:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leahy DJ, Hendrickson WA, Aukhil I, Erickson HP. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 1992;258(5084):987–991. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- 46.Sharma A, Askari JA, Humphries MJ, Jones EY. SDI Crystal structure of a heparin- and integrin-binding segment of human fibronectin. EMBO J. 1999;18:1468–1479. doi: 10.1093/emboj/18.6.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engvall E, Ruoslahti E. Binding of soluble form of fibroblast surface protein, fibronectin, to collagen. Int J Cancer. 1977;20:1–5. doi: 10.1002/ijc.2910200102. [DOI] [PubMed] [Google Scholar]
- 48.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protoc. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 49.Dorfer V, Pichler P, Stranzl T, Stadlmann J, Taus T, Winkler S, Mechtler K, Amanda MS. a universal identification algorithm optimized for high accuracy tandem mass spectra. J Proteome Res. 2014;13(8):3679–3684. doi: 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics. 2012;Chapter 13(Unit 13.20) doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 52.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 53.Wierzbicka-Patynowski I, Mao Y, Schwarzbauer JE. Analysis of fibronectin matrix assembly. Curr Protoc Cell Biol. 2004;Chapter 10:10.12. doi: 10.1002/0471143030.cb1012s25. [DOI] [PubMed] [Google Scholar]
- 54.Huang ML, Cohen M, Fisher CJ, Schooley RT, Gagneux P, Godula K. Determination of receptor specificities for whole influenza viruses using multivalent glycan arrays. Chem Commun. 2015;51:5326–5329. doi: 10.1039/c4cc08613a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.