

Abstract
Mutations in COMP (cartilage oligomeric matrix protein) cause severe long bone shortening in mice and humans. Previously, we showed that massive accumulation of misfolded COMP in the ER of growth plate chondrocytes in our MT-COMP mouse model of pseudoachondroplasia (PSACH) causes premature chondrocyte death and loss of linear growth. Premature chondrocyte death results from activation of oxidative stress and inflammation through the CHOP-ER pathway and is reduced by removing CHOP or by anti-inflammatory or antioxidant therapies. Although the mutant COMP chondrocyte pathologic mechanism is now recognized, the effect of mutant COMP on bone quality and joint health (laxity) is largely unknown. Applying multiple analytic approaches, we describe a novel mechanism by which the deleterious consequences of mutant COMP retention results in upregulation of miR-223 disturbing the adipogenesis – osteogenesis balance. This results in reduction in bone mineral density, bone quality, mechanical strength and subchondral bone thickness. These, in addition to abnormal patterns of ossification at the ends of the femoral bones likely contribute to precocious osteoarthritis (OA) of the hips and knees in the MT-COMP mouse and PSACH. Moreover, joint laxity is compromised by abnormally thin ligaments. Altogether, these novel findings align with the PSACH phenotype of delayed ossification and bone age, extreme joint laxity and joint erosion, and extend our understanding of the underlying processes that affect bone in PSACH. These results introduce a novel finding that miR-223 is involved in the ossification defect in MT-COMP mice making it a therapeutic target.
Keywords: Pseudoachondroplasia, cartilage oligomeric matrix protein, cartilage, miR-223, osteogenesis, adipogenesis, bone architecture
Introduction
Cartilage oligomeric matrix protein (COMP) is an extracellular matrix (ECM) protein expressed by chondrocytes, ligament, tendon, synovium, smooth muscle and in fibrotic disease states [1–3]. COMP plays a critical role in the cartilage matrix organization by interacting with other matrix proteins including collagen types II and IX, matrilin3 and fibronectin and to promote collagen fibrillogenesis [4–9]. The role of COMP in early cartilage and bone morphogenesis is unknown as COMP null mice are normal suggesting that COMP is not required for these processes [10]. In contrast, mutations in COMP dramatically affect postnatal development of cartilage and bone causing skeletal dysplasias, multiple epiphyseal dysplasia to pseudoachondroplasia (PSACH), which have mild to severe epiphyseal and spinal involvement and short stature, respectively [11–17]. Precocious OA occurs in both conditions [13]. The lack of cartilage and bony tissues from PSACH patients has severely restricted the ability to do longitudinal studies, thereby limiting a mechanistic understanding of the abnormal processes causing short stature and the development of therapeutics. To circumvent these limitations, a mouse model expressing the most common D469del-COMP mutation (MT-COMP) was generated and recapitulates the clinical PSACH phenotype.
Using the MT-COMP mouse, we defined the molecular mechanisms driving the short stature. We have previously shown that the premature death of growth plate chondrocytes contributes to limb shortening in the MT-COMP mice, confirming the increased chondrocyte death observed in PSACH growth plate and cultured PSACH chondrocytes [18, 19]. Cellular trafficking defect of COMP was observed in MT-COMP (and PSACH) chondrocytes causing enlarged rER cisternae [20–22]. COMP localized to these large rER cisternae, as did other matrix proteins including types II and IX collagens and MATN3, prematurely forming an extensive intracellular matrix network [22–25]. Accumulation of misfolded COMP proteins in the ER triggers the unfolded protein response (UPR), but the folding and degradation pathways were not effectively activated [26, 27]. MT-COMP expression induced persistent ER stress leading to PERK and C/ebp-homologous protein (CHOP) activation, oxidative stress, inflammation, decreased mitochondrial metabolism and DNA damage, thereby priming chondrocytes for necroptotic cell death [26, 27]. These important studies defined the MT-COMP driven molecular mechanisms involved in the loss of growth plate chondrocytes and loss of linear bone growth. However, little is known about the bone quality, and the causes of joint erosion and extreme joint laxity in PSACH [13–15].
In these studies, longitudinal developmental data on the long bone architecture and joint function from the MT-COMP mouse was examined from P14 through 16 weeks to identify the effects of MT-COMP expression on bone structure, properties and joint function. We define a novel mechanism involving miR regulation of bone development not previously been described in skeletal dysplasias.
Results
MT-COMP femurs are shorter, have a reduced bone mineral density and altered structural bone parameters
μCT examination at 3, 4, 6, 8, 12 and 16 weeks was performed to define the effects of MT-COMP expression on bone architecture and quality. An overall decrease in femoral length in MT-COMP mice (Fig. 1A and B) was found with the greatest reduction (16%) observed in 3 week old mice. Total BMD was reduced by 15% at P21, by 9% at 12 weeks (Fig. 1C), 17% in the epiphysis and 38% in the metaphysis at 16 weeks (Fig. 2C). BMD in the distal and proximal ends of the femur (which is defined as the terminal 20% of the total length) (Fig. 1A), showed a 14% reduction at P21, P28 and 12 weeks (Fig. 1F and G). Moreover, there was abnormal ossification at the end of the femurs (Fig. 1D and E). The MT-COMP femoral micro-architecture and structural indexes were significantly altered at P21, P28 and 16 weeks (Fig. 2). The MT-COMP femurs showed a decrease in BV/TV (42% at P21 and P28; 18% in the epiphysis and 46% in the metaphysis at 16 weeks) (Fig. 2). They also had reduced number of trabeculae (34% at P21;39% at P28; 38% in the metaphysis at 16 weeks), decreased trabecular thickness (20% at P21; 15% at P28; 18% in the epiphysis and 16% in the metaphysis at 16 weeks) and increased trabecular spacing (66% at P21; 81% at P28; 16% in the epiphysis and 22% in the metaphysis at 16 weeks) resulting in a lower connectivity density (70% at P21; 47% at P28; 37% in the metaphysis at 16 weeks) (Fig. 2). Unexpectedly, an increase in connectivity density of 135% was found in the MT-COMP epiphysis at 16 weeks (Fig. 2C).
Figure 1. MT-COMP femurs are shorter and have a reduced bone mineral density.
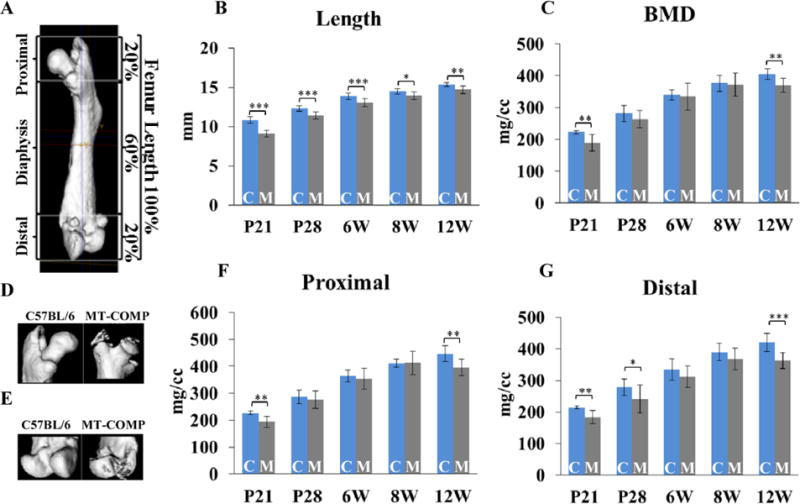
A. Schematic showing measurements performed on the femurs at P21, P28, 6, 8 and 12 weeks. The proximal and distal ends represent 20% of the total femoral length at all ages. B. Total femoral lengths at P21, P28, 6, 8 and 12 weeks. C. Bone mineral density (BMD) from femurs from P21 to 12 weeks. D. Proximal and E. distal ends of C57BL\6 control and MT-COMP femurs reconstructed from μCT. F. BMD of proximal and G. distal ends of C57BL\6 control and MT-COMP femurs. Blue bars = C57BL\6 control and gray bars = MT-COMP; *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 2. MT-COMP femurs have impaired trabecular bone at all ages.
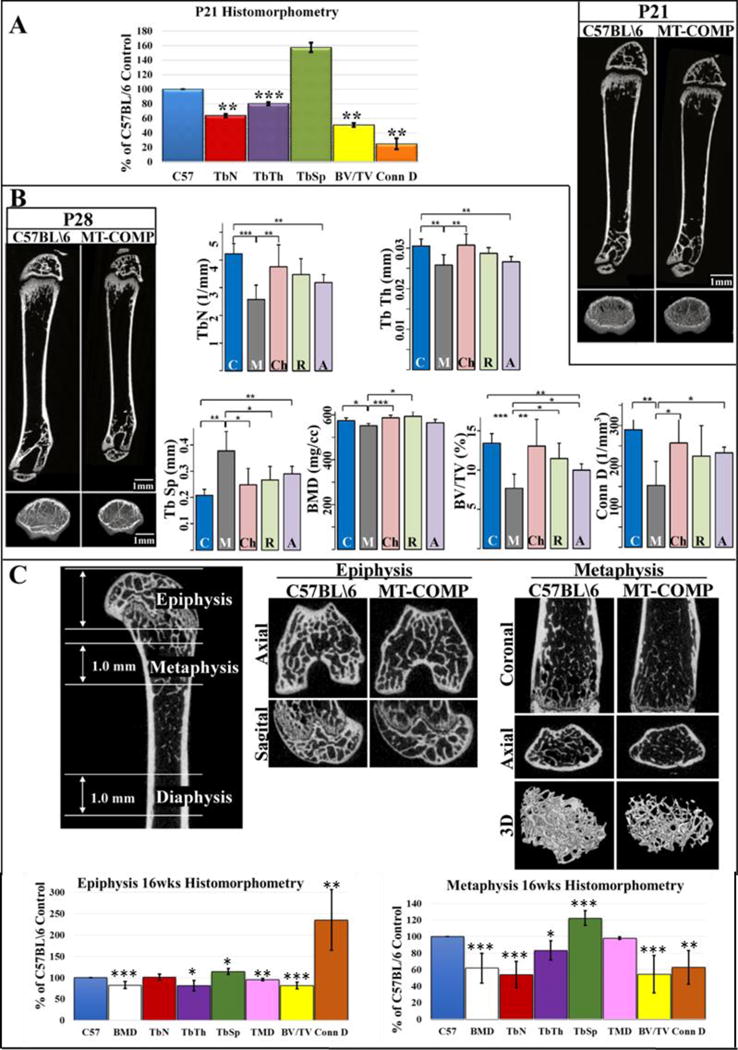
A. Comparison of microarchitecture and structural indices of metaphyses from C57BL\6 control and MT-COMP of P21 mice shown as a percentage of C57BL/6 control and μCT images of femurs. B. μCT images of femurs from C57BL\6 control and MT-COMP mice at P28 with comparison of metaphyseal microarchitecture and structural indices with and without resveratrol and aspirin treatments or Chop ablation (Chop−/−). Blue bars = C57BL\6 control (C); dark gray bars = MT-COMP (M); pink bars = Chop−/−/MT-COMP (Ch); green bars = MT-COMP treated with resveratrol (R); purple bars = MT-COMP treated with aspirin (A). C. μCT images of femurs from C57BL\6 control and MT-COMP mice with comparison of microarchitecture and structural indices from epiphyses and metaphyses at 16 weeks shown as a percentage of C57BL/6 control. Abbreviations: BMD, bone mineral density; Tb N, trabecular number; Tb Th, trabecular thickness; Tb Sp., trabecular spacing; TMD, tissue mineral density; BV/TV, bone volume/total volume, Conn D, connectivity density; Ct Th., cortical thickness; Ct. Po., cortical porosity. *P < 0.05; **P < 0.01; ***P < 0.001.
MT-COMP femurs have reduced cortical thickness, increased cortical porosity and an inferior extrinsic and intrinsic mechanical bone quality
MT-COMP cortical bone was thinner (17% at P21 and P28; 10% at 16 weeks) and porosity was increased (75% at P21; 20% at P28; 23% at 16 weeks) (Fig. 3A–C). Consistent with these findings, both vascular and lacunar porosity was increased at 16 weeks. Interestingly, the bone mineral density distribution was slightly increased (3.4%; P<0.05) and heterogeneity (full-width at half maximum) was equivalent to C57BL/6 controls (Fig. 3D).
Figure 3. MT-COMP femurs have reduced cortical bone thickness, increased porosity and inferior extrinsic and intrinsic bone mechanical quality.
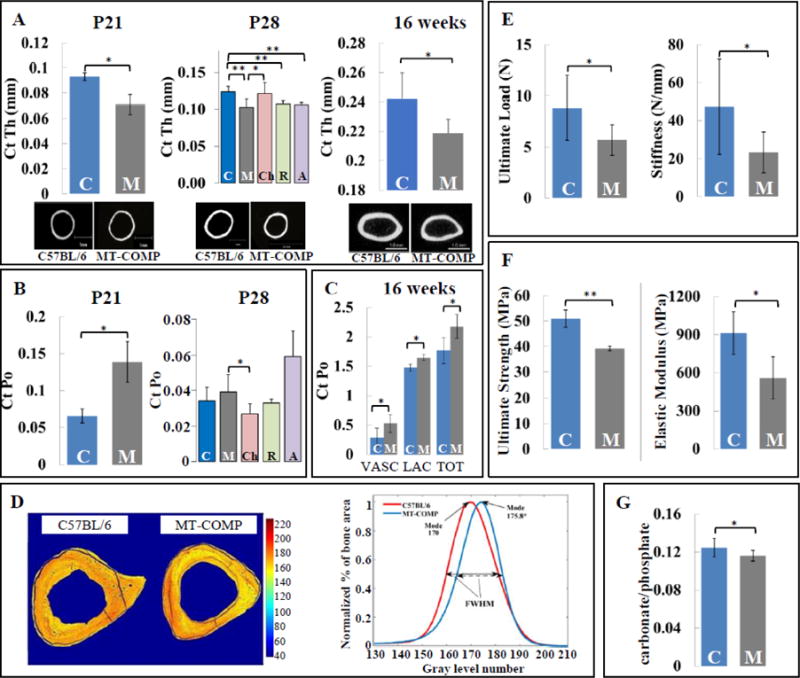
A. Femur cortical thickness of C57BL\6 control and MT-COMP at P21, P28 and 16 weeks. B. Femur cortical porosity for C57BL\6 control and MT-COMP at P21 and P28. C. Tibial cortical porosity for C57BL\6 control and MT-COMP at 16 weeks. D. Gray level representation of the bone mineral density shown on cross-sectional images of tibiae from C57BL\6 control and MT-COMP mice at 16 weeks and the corresponding bone mineral density histograms. E. Extrinsic and F. intrinsic mechanical properties of C57BL\6 control and MT-COMP femurs at P28. G. Carbonation content of C57BL\6 control and MT-COMP femurs at P28. Blue bars = C57BL\6 control (C); dark gray bars = MT-COMP (M); pink bars = Chop−/−/MT-COMP (Ch); green bars = MT-COMP treated with resveratrol (R); purple bars = MT-COMP treated with aspirin (A). *P < 0.05; **P < 0.01.
To determine if these changes in microarchitecture and structural indexes affected mechanical strength, femurs were subjected to 3-point bending testing and Raman spectroscopy at P28. As shown in Figure 3E, the ultimate load was 35% lower and stiffness was reduced by 51% in the MT-COMP mice. As shown in Figure 3F, the MT-COMP femurs have lower ultimate strength (23% reduction) and elastic modulus (39% decrease), indicating that the decrease in strength was not just a result of the decreased size of the mutant femurs, but due to the intrinsic properties of the bone material. Carbonation content was significantly lower in MT-COMP mice indicating superior crystallite qualities (Fig. 3G).
MT-COMP femurs showed delayed ossification and increased adipocytes
To determine the cause of reduced BMD, cell populations in femurs were evaluated in MT-COMP mice at P14, P21 and P28 (Fig. 4A). The secondary ossification center (SOC) was minimally developed in the MT-COMP femur at P14 compared to the fully formed control SOC indicating delay of ossification (Fig. 4A). Interestingly, a 74% increase in the number of adipocytes was observed in the SOC of the P21 MT-COMP mice (Fig. 4A and B). Consistent with the immunostaining findings, microarray analysis from the MT-COMP murine knee showed increased adipogenic C/ebp mRNA levels P21 and P28 (data not shown). Specifically, C/ebp ε and C/ebp γ levels were higher at P21 (59% C/ebp ε and 46% C/ebp γ) and C/ebps β, δ, ε and γ at P28 (21% C/ebp β, 23%, C/ebp δ, 38% C/ebp ε and 26% C/ebp γ). RT-PCR confirmed increases in C/ebp δ and C/ebp ε at P21 (Fig. 4D).
Figure 4. Increased miR-223 alters the balance of adipogenesis and osteogenesis in MT-COMP femurs.
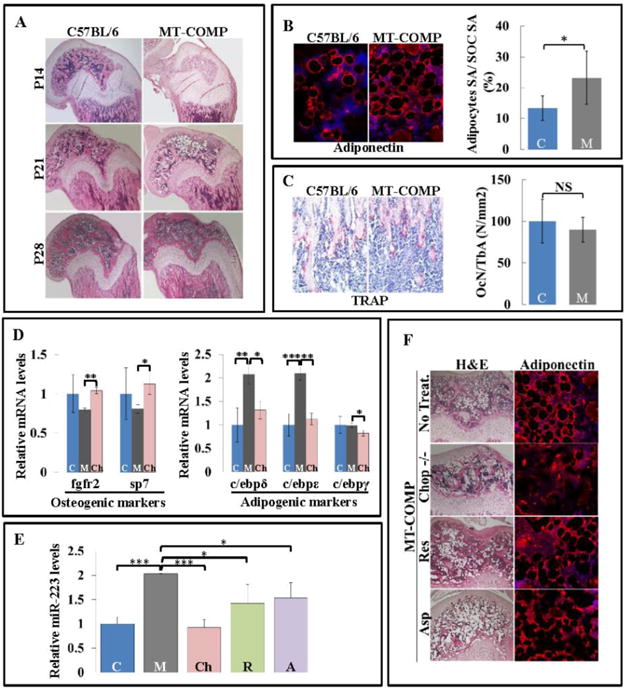
A. Distal end of femurs from C57BL/6 and MT-COMP stained with H&E at P14, P21 and P28. B. Adiponectin immunostaining and quantification of adipocytes surface area relative to the SOC surface area at P21. C. TRAP staining (red) counterstained with hematoxylin (blue) to detect osteoclasts in the secondary spongiosa and quantification in C57BL\6 control and MT-COMP mice at P21. D. C57BL/6 control, MT-COMP and Chop−/−/MT-COMP mice mRNA level of Fgfr2 and Sp7, both osteogenic markers, at P28 and C/ebp δ, C/ebp ε, and C/ebp γ, adipogenesis markers at P21. E. miR-223 levels in P21 C57BL/6 control, MT-COMP, Chop−/−/MT-COMP and MT-COMP mice treated with resveratrol or aspirin. F. H&E and adiponectin immunostaining of C57BL\6 control, MT-COMP, Chop−/−/MT-COMP and MT-COMP treated with resveratrol or aspirin SOCs at P21. Blue bars = C57BL\6 control (C); dark gray bars = MT-COMP (M); pink bars = Chop−/−/MT-COMP (Ch); green bars = MT-COMP treated with resveratrol (R); purple bars = MT-COMP treated with aspirin (A); *P < 0.05; **P < 0.01; ***P < 0.001
Based on the lower BMD, delayed ossification and increased adipocytes, osteoclast and osteoblast populations were evaluated. There was no difference in the number of osteoclasts in the secondary spongiosa relative to the trabecular bone area by TRAP staining (Fig. 4C). However, mRNA levels of osteoblasts markers, Fgfr2 and Sp7, were reduced in MT-COMP femurs at P28 by 20% and 36%, respectively, which was confirmed by RT-PCR (Fig. 4D). This indicates impaired bone formation. The balance between adipogenesis and osteogenesis is regulated by miR-223 by increasing expression of C/ebps and inhibiting Fgfr2 dependent osteogenesis [28]. Indeed, miR-223 was elevated two fold in the MT-COMP femur at P21 consistent with the finding that mRNA levels of adipogenic and osteogenic markers were perturbed, (Fig. 4E).
Bone parameters improve with dampening of MT-COMP chondrocyte stress
In previous work, we showed that CHOP/DDIT3 plays a critical role in the MT-COMP retention in the ER and loss of CHOP reduces inflammation and growth plate chondrocyte death [26, 27]. To determine whether loss of Chop normalized bony abnormalities, femurs from Chop−/−/MT-COMP mice were examined. At P21, miR-223 was restored to C57BL/6 control levels; at P28 the expression of osteogenic markers Fgfr2 and Sp7 and at P21 adipogenic markers (C/ebp δ, C/ebp ε, and C/ebp γ) were also at levels comparable to C57BL/6 controls (Fig. 4D and E). Importantly, fewer adipocytes were observed in the SOC in Chop−/−/MT-COMP mice consistent with an improvement in the balance between osteogenesis and adipogenesis (Fig. 4F). Moreover, the micro-architecture and structural indexes of Chop−/−/MT-COMP femurs at P28 were normalized with increased BMD, trabeculae number, trabecular thickness, connectivity density, BV/TV, cortical thickness and with decreased trabecular spacing, cortical porosity compared to MT-COMP femur (Fig. 2B; 3A and B).
Based on our previous work showing that aspirin and resveratrol treatment lessens the negative impact of MT-COMP expression on growth plate chondrocytes [29], we next determined whether these therapies could also improve MT-COMP bone quality. Resveratrol (antioxidant) or aspirin (anti-inflammatory) treatments from birth to P21 reduced the levels of miR-223 by 32% and 27%, respectively (Fig. 4E). However, these treatments had no effect on the number of adipocytes in the SOC (Fig. 4F). BV/TV was improved by resveratrol (49%) and aspirin (30%) treatments but only resveratrol increased BMD by 7% and decreased the trabecular spacing by 29% (Fig. 2B). Microarchitecture and structural indexes improved but to a lesser degree than with ablation of Chop (Fig. 2B).
MT-COMP mice have thinner articular cartilage, subchondral bone and ligaments together with lax joints
Since early onset joint erosion is a significant complication in PSACH, we examined articular cartilage morphology and subchondral bone in knee joints of adult 16 week old MT-COMP mice. Thickness map of the articular cartilage shows that MT-COMP articular cartilage was 17% thinner than controls particularly in articular cartilage where the patella glides in the trochlear groove of the femur (Fig. 5A). The subchondral bone thickness of both the medial and lateral femoral condyles was reduced by 5% in MT-COMP mice (Fig. 5B). PSACH individuals have extreme joint laxity and it is unclear whether this is because of ligament abnormalities. To determine whether MT-COMP joints are hypermobile, anterior-posterior and varus-valgus knee laxity was measured at 16 weeks (Fig. 5C and D). While anterior-posterior total displacement (i.e. laxity) was increased and anterior stiffness was decreased in MT-COMP mice, there was no difference in posterior stiffness. Furthermore, varus stiffness and valgus stiffness were both decreased and total varus-valgus displacement was increased in MT-COMP mice (Fig. 5C and D). Interestingly, MT-COMP patellar ligaments had a 70% reduction in surface area compared to controls at P28 (Fig. 5E and F).
Figure 5. MT-COMP mice have thinner articular cartilage, subchondral bone and ligaments together with lax joints.
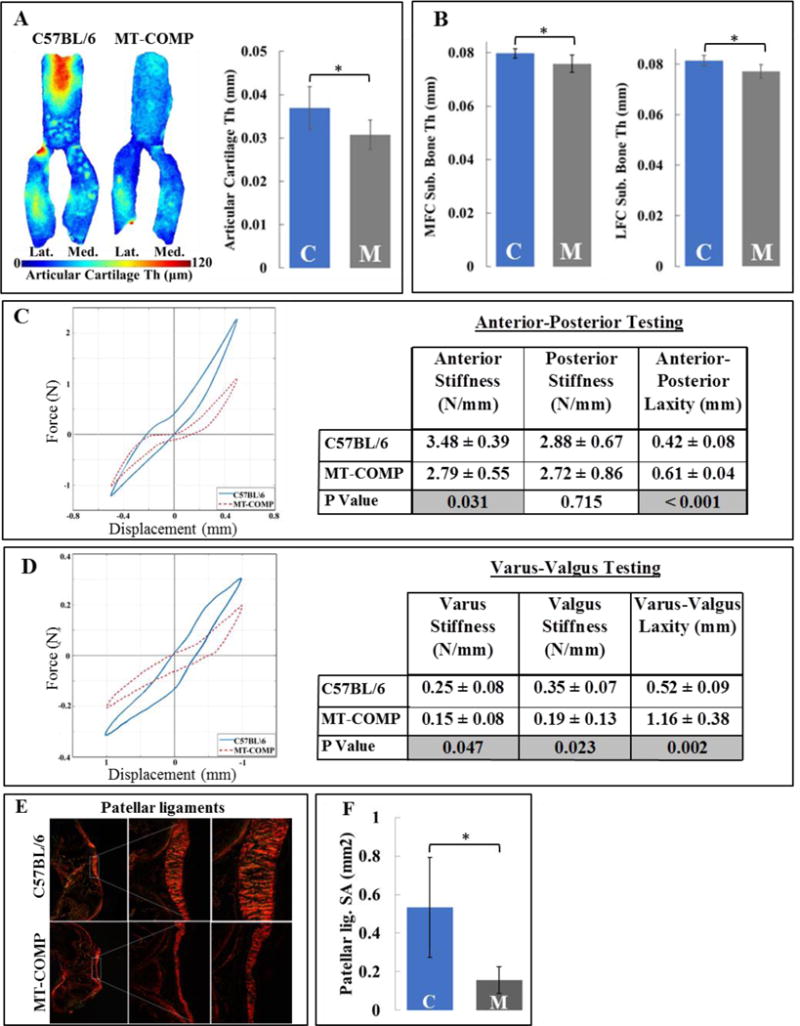
A. Thickness map and quantification of articular cartilage of C57BL\6 control and MT-COMP mice. B. Comparison of medial (MFC Sub. Bone thickness) and lateral (LFC Sub. Bone thickness) femoral subchondral bone thickness. Blue bars = C57BL\6 control and gray bars = MT-COMP; *P < 0.05. C. Anterior-posterior knee laxity and comparison of laxity parameters D. Varus-valgus knee laxity and comparison of laxity parameters. Laxity is plotted for C57BL\6 control (blue) and MT-COMP (red dotted line) mice. E. Picro-Sirius red staining of patellar ligaments from C57BL\6 control and MT-COMP mice at P28. F. Quantification of surface area from C57BL\6 control and MT-COMP mice patellar ligaments at P28. Blue bars = C57BL\6 control and gray bars = MT-COMP. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
The findings of this study for first time link the effects of intracellular mutant protein accumulation in chondrocytes to abnormal ossification through miR-223. While it is known that miR-223 regulates adipogenesis-osteogenesis balance [30], this process has not previously been associated with mutant COMP ER accumulation and the downstream pathological consequences. This enlarges the understanding of the abnormal bone pathology in the MT-COMP mouse that recapitulates PSACH. Moreover, the results begin to define the effects of mutant COMP on ligament structure and joint laxity towards the goal of defining the mechanism(s) of abnormal PSACH joint function.
miR-223 drives abnormal ossification
Total BMD was reduced significantly along with connectivity density, a measure of number of redundant connections between trabecular structures per unit volume, which is consistent with the observed increase in trabecular spacing and decrease in trabecular number and thickness (Figs. 1 and 2). In MT-COMP mice, irregular ossification was observed in the subchondral bone at P28 and the subchondral bone was thinner at 16 weeks likely due to the ossification delay in SOC or early OA (Fig. 1 and 5). Subchondral bone supports the articular cartilage, and based on known OA pathology, the subchondral bone first thins and then later becomes hyperossified [31]. The abnormalities in subchondral bone and the ends of the MT-COMP femur may contribute to the thinning of the articular cartilage (discussed below) and correlate with early joint erosion in PSACH.
Previously, we described the mechanism by which the growth plate chondrocytes expressing MT-COMP die prematurely resulting in a decrease of linear growth and shorter bones [26, 27]. In this work, we define the molecular mechanisms affecting MT-COMP bone quality. After determining that ossification was abnormal, a histological evaluation of bone development revealed a delay of ossification in the femoral SOC at P14. By P21 there were more adipocytes compared to the control mice, in which the SOC is well developed at P14, some adipocytes are evident at P21 and very few remain at P28 (Fig. 4). Importantly, this is remarkably similar to delayed and abnormal ossification reported in PSACH [13, 32].
Bone marrow mesenchymal stem cells (MSCs) give rise to both osteoblasts and adipocytes [33, 34]. MSCs exhibit an epigenetic plasticity enabling them to trans-differentiate from adipocytes to osteoblasts and vice-versa [35]. Bone marrow adipocytes are distinct from other adipocytes as they express both adipocyte markers and Sp7, an osteoblast specific marker [36]. Differentiation of MSCs is regulated by chemical, physical and biological factors including microRNAs [37–40]. To define this process in the MT-COMP mouse, we evaluated genes associated with bone development and the microRNAs that regulate this process. We found that C/ebps, adipogenic markers, were increased and two osteogenic genes Fgfr2 and Sp7 were expressed at lower levels in P21 and P28 hind limb joints of MT-COMP mice. miR-223, which regulates the balance between osteogenesis and adipogenesis, was increased at P21 in the MT-COMP mice (Fig. 4). Others have shown that growth plate chondrocytes produce extracellular matrix vesicles containing miRNAs that are specifically enriched for miR-223 [41, 42]. miR-223 has been shown to repress osteogenesis by targeting Fgfr2 for degradation, lower Fgfr2 levels increases expression of C/ebps and enhances adipogenesis [28] consistent with our findings the MT-COMP mice. Dysregulation of the adipogenic/osteogenic balance, resulting in an increase in adipose tissue and low BMD in the MT-COMP mice is similar to osteoporosis and other conditions [30, 43].
Next, we asked what mechanisms upregulate miR-223. IL-1β and TNF-α, inflammatory molecules, are known to upregulated miR-223 in fibroblasts and monocytes [44, 45] and both were increased in MT-COMP growth plate chondrocytes. This MT-COMP inflammatory environment may be responsible for the increases in miR-223 (Fig. 4E) [26, 29]. Additionally, miR-223 expression is upregulated by C/ebps through binding to the promoter regions of miR-223 leading to a constitutive upregulation of miR-223 and repression of osteogenesis [28]. Both upregulation of C/ebps and inflammation were present in MT-COMP mice and together likely contributed to increased miR-223 levels (Fig. 6).
Figure 6. Model showing the mechanism by which MT-COMP alters adipogenesis and osteogenesis thereby affecting murine bone.
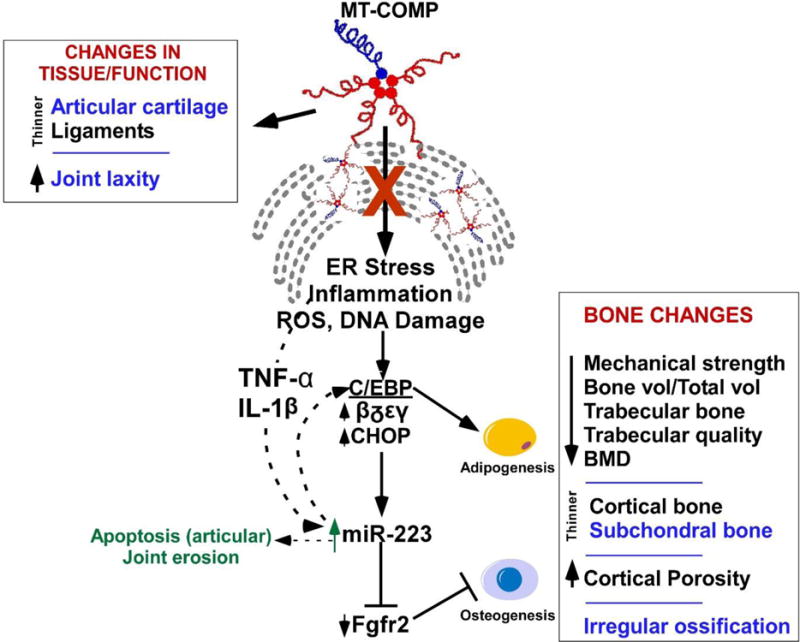
The alteration in adipogenesis negatively impacts mechanical strength of bone, bone volume, trabecular bone, trabecular bone quality, articular cartilage, bone mineral density, and cortical thickness and porosity. MT-COMP expression also results in thinner ligaments, articular cartilage and increased joint laxity. Thinner articular cartilage, subchondral bone, increased joint laxity and irregular ossification of the ends of the bone (shown in blue) may contribute to early joint erosion.
Abnormal articular cartilage
In addition to altering the osteogenesis-adipogenesis balance, miR-223 has been shown to stimulate apoptosis through mitochondrial dysfunction [46]. Mitochondrial dysfunction is present in rat chondrosarcoma cells expressing MT-COMP in vitro [47]. Chondrocyte death plays a critical role in PSACH pathology decreasing long bone growth and reducing the number of articular chondrocytes available for cartilage maintenance. Since PSACH is associated with precocious and severe OA, we next evaluated the articular cartilage of MT-COMP mice. The articular cartilage was relatively normal at P21 (data not shown) and was thinner in the knee joint in 16 week old MT-COMP mice compared to control mice (Fig. 5) indicating that the articular cartilage prematurely thins in MT-COMP mice. Others have shown that increases in miR-223 are associated with the early stages of joint erosion [48] which suggests that MT-COMP stimulated increase in miR-223 contributes to articular chondrocyte death.
Rescue of bone pathology
We have previously shown that MT-COMP induced ER stress through C/ebp-homologous protein (CHOP) in chondrocytes results in ROS production, DNA damage, inflammation [26, 27] and the chondrocyte pathology is dampened by the loss of Chop or treatment with either resveratrol (antioxidant) or aspirin (anti-inflammatory). Based on this information, we asked if these therapies would improve the long bone quality in the MT-COMP mice. Chop deletion restored miR-223 and osteogenic marker levels to normal. Additionally, the loss of Chop in MT-COMP mice lowered both adipocyte markers and the number of adipocytes in SOC showing that ER stress stimulated by MT-COMP plays a role in ossification processes in mice (Fig. 4). The microarchitectural and structural parameters of the Chop−/−/MT-COMP femurs were improved compared to those in the MT-COMP mice and similar to those of control mice (Fig. 2). Aspirin and resveratrol treatments resulted in some improvements in bone quality and reduced miR-223 although to a lesser degree than Chop ablation (Figs. 2 and 4). Taken together, this data suggests that treatments or loss of Chop dampens the negative impact on long bones by suppressing chondrocyte stress [26, 27] and the down-regulation of miR-223, thereby restoring the balance between osteogenesis and adipogenesis.
Mechanical strength
Next, we assessed the effect of lower bone quality on the mechanical strength of the MT-COMP femurs. Both BMD and bone quality, which determine bone strength [49, 50], were significantly compromised in the MT-COMP mice. Three-point bending analysis showed that the strength of the MT-COMP femur was decreased at P28 (Fig. 3E and F). The decrease in strength was caused by intrinsic material property differences in BMD and trabecular quality (Fig. 3E and F) and not because of the smaller size of the MT-COMP femurs (Fig. 1).
Although, the MT-COMP femurs had inferior mechanical strength, individuals with PSACH are not known to have a higher risk of fracture [51]. Similarly, MT-COMP mice do not spontaneously fracture. Resistance to fracture is related not only to bone strength, but also to fracture toughness and fatigue strength [52]. At P28, the post yield deflection was higher (data not shown) and heterogeneity was not compromised at 16 weeks (Fig. 3D) indicating that MT-COMP femurs were less brittle and could sustain more damage before fracture. This is supported by Raman measurements in MT-COMP femurs at P28 which showed no change in collagen matrix (data not shown), a crucial measure of fracture toughness [53]. Lower carbonate content in the bone mineral phase was observed in MT-COMP femurs at P28 (Fig. 3G) and is associated with superior crystallite qualities [54]. Bone turnover was not altered in MT-COMP mice (Fig. 4C) and therefore a decrease in carbonation is not attributable to higher rate of bone turnover. Together, these findings suggest that MT-COMP mice have better crystalline mineral quality compared to control (C57) due to slower bone maturation (Fig. 3G). The degree of cortical bone mineralization impacts fracture susceptibility [55]. Although uCT suggested that BMD was reduced in MT-COMP mice, cortical bone had higher local mineralization as assessed by gray level measurement (Fig. 3D). When measuring the mineralization qbSEM and SEM excludes all the pores in bones, which may explain this discrepancy. Overall superior mineral crystal quality, higher degree of cortical mineralization, increased ductility and normal collagen matrix may compensate for higher porosity, lower BMD and negative changes in the microarchitecture of the MT-COMP bones and may explain the absence of spontaneous fracture in MT-COMP mice. This may also be true in PSACH and needs to be assessed [51].
Lax joints
Next, we evaluated joint function in the MT-COMP mice because extreme joint laxity is associated with PSACH. An increase in both anterior-posterior and varus-valgus laxity was observed (Fig. 5). Loss of articular cartilage is associated with increased varus-valgus laxity and both are altered in MT-COMP mice and may increase OA susceptibility in these mice [56, 57]. Joint capsules and ligaments together regulate joint mobility/laxity [58, 59]. Patellar knee ligaments were thinner in MT-COMP mice at P28 which may underlie the increase in joint laxity. However, MT-COMP expression was restricted to cells expressing type II collagen in the MT-COMP mouse [18]. Since the major collagen in mature ligaments is type I collagen, it is unclear how MT-COMP expression in cartilage is related to thinner ligaments. Ligament precursor cells have the potential to differentiate into either chondrocytes or fibroblastic ligament cells and inflammation induces abnormal differentiation of ligament cells [60]. Perhaps early expression of MT-COMP and the resulting inflammation in precursor cells may account for decreased ligament width.
Figure 6 shows and summarizes the proposed novel mechanism by which MT-COMP affects bone health by disrupting bone ossification through miR-223 in our MT-COMP mice. In MT-COMP mice, the increased chondrocyte stress from intracellular retention of MT-COMP elevates C/EBPs and stimulates an inflammatory process through TNF-α and IL-1β up-regulating miR-223 synthesis. Elevated levels of miR-223 inhibit Fgfr2, creating an imbalance between osteogenesis and adipogenesis. While miR-223 is known to regulate the balance between adipogenesis and osteogenesis in bone [28], this is the first report of its involvement in the MT-COMP pathology. Consistent with our results, increases in miR-223 are known to decrease osteogenesis and induce mitochondrial dysfunction which increases apoptosis. Additionally, we observed abnormal ossification and thinning of articular cartilage in the MT-COMP mice. In MT-COMP mice, bone density and quality improved and levels of Fgfr2 and C/ebps were normalized by reducing miR-223 through loss of Chop. These findings open the door for RNA-based treatment [61] such as antisense-miR-223 and antisense delivery may be aided by the rich blood supply of the SOC. Indeed, anti-sense therapy successfully dampened the MT-COMP pathology in the growth plate chondrocytes in MT-COMP mice [62]. Others have shown that inhibiting miR-223 reduces articular cartilage erosion [46] suggesting that reducing miR-223 may also improve articular cartilage health. Increased joint laxity and premature OA results from abnormal joint tissue/function and are the most debilitating features of PSACH [13, 32]. Our novel joint ligament findings expand the understanding of the mechanisms and progression of PSACH joint erosion and set the stage for future joint function and treatment studies. Moreover, and importantly, based on these and our previous growth plate chondrocyte studies, some treatment approaches may improve the health of PSACH chondrocytes, bone and joints.
Experimental procedures
Generation of MT-COMP mice
Plasmids containing expression cassettes for mutant D469del‐COMP under control of a tetracycline‐inducible (TRE) promoter and recombinant tetracycline controlled trans‐activation factor (rtTA) under control of type II collagen promoter were generated as previously described [18]. Standard breeding was used to generate bigenic MT-COMP mice in C57BL/6 genetic background as well as Chop−/−/MT-COMP mice as previously described [26]. Male MT-COMP, Chop−/−/MT-COMP, and C57BL/6 used as control mice were administered DOX (500 ng/mL) through drinking water prenatally and postnatally. MT-COMP is expressed at a lower level (cycle threshold (Ct) 28) than the endogenous wild-type mouse COMP (Ct 24) in mice P21-P28 and the difference is approximately 16 fold lower. This gross approximation of the differences between mouse and human mutant COMP mRNAs is imprecise because two different primer sets were used to amplify human mutant and mouse COMP. These studies were approved by the Animal Welfare Committee at the McGovern Medical School at The University of Texas Health Science Center at Houston (UT Health).
μCT scanning
Femurs were obtained from MT-COMP and C57BL/6 control mice at ages P21, P28, 6, 8, and 12 weeks. At least 9 MT-COMP and C57BL/6 mice were examined at each age by low resolution μCT scanning to measure length and BMD. High resolution μCT scanning was performed on an additional 6 femurs from each group at ages P21, P28 and 16 weeks. Bone histomorphometry and detailed μ-CT analysis are described in supplementary method section.
Measurement of bone mineral density distribution (BMDD) and porosity
Five tibia samples from 16 week old MT-COMP and C57BL/6 control mice were prepared and processed for qbSEM analysis. Processing and analysis procedures are detailed in supplementary method section.
Femur mechanical testing
Femurs (n=8 MT-COMP and n=11 C57BB/6 control) were collected at P28 and frozen until testing in 3-point bending in the anterior to posterior direction using an Instron 5848 MicroTester for mechanical evaluation (Supplementary methods).
Articular cartilage morphology and subchondral bone morphometry
Articular cartilage morphology and subchondral bone morphometry from MT-COMP (n=6) and C57BL/6 control (n=6) were assessed using contrast-enhanced μCT imaging and details of analysis techniques are in supplementary method section.
Visualization of collagen I and III fibers by Picro-Sirius red staining
Hind limbs from P28 MT-COMP (n=6) and C57BL/6 control (n=6) mice were collected, fixed in 4% PFA overnight at 4°C, decalcified, embedded with paraffin and sectioned. Sections were stained with Picro-Sirius red (PSR) stain kit according to the manufacturer’s instructions (ab150681, Abcam, Cambridge, MA). The PSR stain was viewed using Zeiss Axio Scope A1 Polarized Light Microscope and Zen Pro software (Informer Technologies Inc.).
Surface area measurements of patellar ligaments
Surface areas of patellar ligaments in P28 mice were measured using Adobe Photoshop CS6.
Joint laxity assessment
Joint laxity was assessed in the anterior-posterior (A-P) and varus-valgus (Var-Valg) loading directions in hind limbs from MT-COMP (n=6) and C57BL/6 mice (n=6) from 16 week mice and testing methods are described in supplementary methods.
Histology
Hind limbs from P21 and P28 MT-COMP and C57BL/6 control mice were collected and femurs were analyzed. The femurs were fixed in 4% PFA overnight at 4°C, decalcified in 14% EDTA pH 7.4 for a week, embedded with paraffin and sectioned (5-μm). Immunostaining was performed as previously described [18]. Briefly sections were incubated overnight at 4°C with primary antibody for adiponectin used as a marker for adipocytes (ab701148, 1:300, Invitrogen, Carlsbad, CA). Primary antibodies were detected by using Alexa Fluor 594 for 1 hour at 37°C (A21207, 1:500, Invitrogen, Carlsbad, CA). Quantification of adipocytes in MT-COMP (n=8) and C57BL/6 control (n=8) mice was obtained by calculating the adipocytic cells surface area relative to the SOC surface area. Osteoclasts were detected in MT-COMP (n=7) and C57BL/6 control (n=6) mice using tartrate-resistant acid phosphatase (TRAP) histochemistry followed by counterstaining with Mayer’s hematoxylin. The acid phosphatase activity in the osteoclasts results in the development of a bright red staining. Osteoclast population density was determined by counting the number of osteoclasts (red stained cells displaying at least 3 nuclei) in the secondary spongiosa adjusted by the bone trabecular area.
Transcriptome analysis
Microarray analysis were performed on RNA extracted from knee joints (area encompassing epiphysis and epiphyseal growth plate) of three MT-COMP and three control mice at P21 and P28 as previously described [26].
miRNA microarray analysis
miRNAs were extracted from knee joints (area encompassing epiphysis and epiphyseal growth plate) of three MT-COMP and three C57BL/6 control mice at P21 using a miRNA Qiagen kit (Qiagen, CA, USA). Exiqon Services (Denmark) performed miRNA microarray analyses as described in supplementary method section.
RNA quantification
Total RNA was isolated from mouse knee joints using Trizol (Life Technologies Corp, Carlsbad, CA) and treated with RNAse-free DNAse1 (Life Technologies Corp, Carlsbad, CA). cDNA was made using iScript kit (Bio-Rad Laboratories, Hercules, CA). Levels of gene expression were measured by quantitative PCR using SYBR Green kit (Life Technologies Corp, Carlsbad, CA). All measurements were normalized to endogenous Hprt1 mRNA. miR-223 level was measured by quantitative PCR using Taqman probe (002295) and normalized to the endogenous snoRNA234 (001234 Life Technologies Corp, Carlsbad, CA). Relative changes in RNA levels were assessed using the comparative CT method.
Statistical analysis
Based on the Shapiro-Wilk test, the data was normally distributed. Comparisons were performed using an unpaired two-tailed T-test with the appropriate T-test given the variance F-test outcome. Significance is indicated as *p<0.05; **p<0.01; ***p<0.001.
Supplementary Material
Highlights.
Mutant COMP disturbs the balance between adipogenesis and osteogenesis through miRNA 223 which affects most measures of bone quality in MT-COMP mice.
Abnormal ossification at ends of long bone, thin articular cartilage and subchondral bone likely contributes to early joint erosion in pseudoachondroplasia.
Ligaments from MT-COMP mice are significantly smaller contributing to increased joint laxity in this mouse model which is similar to the extreme joint laxity associated with pseudoachondroplasia.
Acknowledgments
This work was supported by NIH #5R01 AR057117 grant to JTH and KLP, the Leah Lewis Family Foundation and the Rolanette and Berdon Lawrence Texas Bone Disease Program. We thank Brian Dawson at Baylor College of Medicine μCT core facility for technical assistance and advice in bone histomorphometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: All authors state that they have no conflicts of interest (employment, research funding, income (e.g., fees for consulting, expert testimony, or speaking), or ownership interests (e.g., stock, patents) in or from an organization that may gain or lose financially from the work being submitted for publication).
Author contributions
Design study: FC, KLP; Experimental conduct: FC, TM, AMA, CGA, SN, XB, MW, KG, LR, AV, MH, FC; Data Analysis: FC, KLP, TM, KB, AMA, CGA, SN, SJS, XB, MW, KG, LR; Draft manuscript: FC, KLP. Jacqueline T Hecht was the project director involved in all aspects and phases of the work.
Conflict of interest
The authors do not have conflict of interest.
References
- 1.Posey KL, Alcorn JL, Hecht JT. Pseudoachondroplasia/COMP – translating from the bench to the bedside. Matrix biology: journal of the International Society for Matrix Biology. 2014;37:167–73. doi: 10.1016/j.matbio.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Cesare PE, Carlson CS, Stollerman ES, Chen FS, Leslie M, Perris R. Expression of cartilage oligomeric matrix protein by human synovium. FEBS Lett. 1997;412(1):249–52. doi: 10.1016/s0014-5793(97)00789-8. [DOI] [PubMed] [Google Scholar]
- 3.Schulz JN, Nuchel J, Niehoff A, Bloch W, Schonborn K, Hayashi S, Kamper M, Brinckmann J, Plomann M, Paulsson M, Krieg T, Zaucke F, Eckes B. COMP-assisted collagen secretion–a novel intracellular function required for fibrosis. J Cell Sci. 2016;129(4):706–16. doi: 10.1242/jcs.180216. [DOI] [PubMed] [Google Scholar]
- 4.Acharya C, Yik JH, Kishore A, Van Dinh V, Di Cesare PE, Haudenschild DR. Cartilage oligomeric matrix protein and its binding partners in the cartilage extracellular matrix: interaction, regulation and role in chondrogenesis. Matrix biology: journal of the International Society for Matrix Biology. 2014;37:102–11. doi: 10.1016/j.matbio.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Adams JC. Thrombospondins: Multifunctional Regulators of Cell Interactions. Annual Review of Cell and Developmental Biology. 2001;17(1):25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 6.Sodersten F, Ekman S, Eloranta ML, Heinegard D, Dudhia J, Hultenby K. Ultrastructural immunolocalization of cartilage oligomeric matrix protein (COMP) in relation to collagen fibrils in the equine tendon. Matrix biology: journal of the International Society for Matrix Biology. 2005;24(5):376–85. doi: 10.1016/j.matbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Di Cesare PE, Chen FS, Moergelin M, Carlson CS, Leslie MP, Perris R, Fang C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix biology: journal of the International Society for Matrix Biology. 2002;21(5):461–70. doi: 10.1016/s0945-053x(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 8.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix biology: journal of the International Society for Matrix Biology. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halasz K, Kassner A, Morgelin M, Heinegard D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282(43):31166–73. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 10.Svensson L, Aszodi A, Heinegard D, Hunziker EB, Reinholt FP, Fassler R, Oldberg A. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol. 2002;22(12):4366–71. doi: 10.1128/MCB.22.12.4366-4371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs MD, Hoffman SMG, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nature Genetics. 1995;10(3):330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 12.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FFB, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nature Genetics. 1995;10(3):325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 13.Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. American Journal of Medical Genetics. 2001;106(4):244. [PubMed] [Google Scholar]
- 14.McKeand J, Rotta J, Hecht JT. Natural history study of pseudoachondroplasia. Am J Med Genet. 1996;63(2):406–10. doi: 10.1002/(SICI)1096-8628(19960517)63:2<406::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 15.Posey KL, Hecht JT. The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Current drug targets. 2008;9(10):869–77. doi: 10.2174/138945008785909293. [DOI] [PubMed] [Google Scholar]
- 16.Hecht JT, Deere M, Putnam E, Cole W, Vertel B, Chen H, Lawler J. Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudoachondroplasia musculoskeletal tissues. Matrix biology: journal of the International Society for Matrix Biology. 1998;17(4):269–78. doi: 10.1016/s0945-053x(98)90080-4. [DOI] [PubMed] [Google Scholar]
- 17.Hecht JT, Hayes E, Haynes R, Cole WG. COMP mutations, chondrocyte function and cartilage matrix. Matrix biology: journal of the International Society for Matrix Biology. 2005;23(8):525–33. doi: 10.1016/j.matbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Posey KL, Veerisetty AC, Liu P, Wang HR, Poindexter BJ, Bick R, Alcorn JL, Hecht JT. An inducible cartilage oligomeric matrix protein mouse model recapitulates human pseudoachondroplasia phenotype. Am J Pathol. 2009;175(4):1555–63. doi: 10.2353/ajpath.2009.090184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duke J, Montufar-Solis D, Underwood S, Lalani Z, Hecht JT. Apoptosis staining in cultured pseudoachondroplasia chondrocytes. Apoptosis. 2003;8(2):191–7. doi: 10.1023/a:1022926811397. [DOI] [PubMed] [Google Scholar]
- 20.Hecht JT, Hayes E, Snuggs M, Decker G, Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J, Duke PJ. Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biology. 2001;20(4):251–262. doi: 10.1016/s0945-053x(01)00136-6. [DOI] [PubMed] [Google Scholar]
- 21.Vranka J, Mokashi A, Keene DR, Tufa S, Corson G, Sussman M, Horton WA, Maddox K, Sakai L, Bächinger HP. Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biology. 2001;20(7):439–450. doi: 10.1016/s0945-053x(01)00148-2. [DOI] [PubMed] [Google Scholar]
- 22.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar-Solis D, Duke PJ, Cole WG. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22(4):759–67. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Merritt TM, Bick R, Poindexter BJ, Alcorn JL, Hecht JT. Unique matrix structure in the rough endoplasmic reticulum cisternae of pseudoachondroplasia chondrocytes. The American journal of pathology. 2007;170(1):293–300. doi: 10.2353/ajpath.2007.060530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix biology: journal of the International Society for Matrix Biology. 1998;17(8–9):625–33. doi: 10.1016/s0945-053x(98)90113-5. [DOI] [PubMed] [Google Scholar]
- 25.Merritt TM, Alcorn JL, Haynes R, Hecht JT. Expression of mutant cartilage oligomeric matrix protein in human chondrocytes induces the pseudoachondroplasia phenotype. J Orthop Res. 2006;24(4):700–7. doi: 10.1002/jor.20100. [DOI] [PubMed] [Google Scholar]
- 26.Posey KL, Coustry F, Veerisetty AC, Liu P, Alcorn JL, Hecht JT. Chop (Ddit3) is essential for D469del-COMP retention and cell death in chondrocytes in an inducible transgenic mouse model of pseudoachondroplasia. The American journal of pathology. 2012;180(2):727–37. doi: 10.1016/j.ajpath.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coustry F, Posey KL, Liu P, Alcorn JL, Hecht JT. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am J Pathol. 2012;180(2):738–48. doi: 10.1016/j.ajpath.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan X, Gao Y, Zhou J, Wang J, Zheng F, Guo F, Chang A, Li X, Wang B. miR-223 Regulates Adipogenic and Osteogenic Differentiation of Mesenchymal Stem Cells Through a C/EBPs/miR-223/FGFR2 Regulatory Feedback Loop. Stem Cells. 2015;33(5):1589–600. doi: 10.1002/stem.1947. [DOI] [PubMed] [Google Scholar]
- 29.Posey KL, Coustry F, Veerisetty AC, Hossain M, Alcorn JL, Hecht JT. Antioxidant and anti-inflammatory agents mitigate pathology in a mouse model of pseudoachondroplasia. Human molecular genetics. 2015;24(14):3918–28. doi: 10.1093/hmg/ddv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheller EL, Rosen CJ. What’s the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Ann N Y Acad Sci. 2014;1311:14–30. doi: 10.1111/nyas.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox LG, van Donkelaar CC, van Rietbergen B, Emans PJ, Ito K. Alterations to the subchondral bone architecture during osteoarthritis: bone adaptation vs endochondral bone formation. Osteoarthritis Cartilage. 2013;21(2):331–8. doi: 10.1016/j.joca.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Briggs MD, Wright MJ. Pseudoachondroplasia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 33.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9(5):641–50. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 34.Horwitz EM, Blanc KLe, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A, T. International Society for Cellular Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7(5):393–5. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 35.Meyer MB, Benkusky NA, Sen B, Rubin J, Pike JW. Epigenetic Plasticity Drives Adipogenic and Osteogenic Differentiation of Marrow-derived Mesenchymal Stem Cells. J Biol Chem. 2016;291(34):17829–47. doi: 10.1074/jbc.M116.736538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berry R, Rodeheffer MS, Rosen CJ, Horowitz MC. Adipose Tissue Residing Progenitors (Adipocyte Lineage Progenitors and Adipose Derived Stem Cells (ADSC) Curr Mol Biol Rep. 2015;1(3):101–109. doi: 10.1007/s40610-015-0018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, Zhang Y. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol. 2012;8(4):212–27. doi: 10.1038/nrendo.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang S, Deng Y, Gu P, Fan X. MicroRNAs regulate bone development and regeneration. International journal of molecular sciences. 2015;16(4):8227–53. doi: 10.3390/ijms16048227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamam D, Ali D, Kassem M, Aldahmash A, Alajez NM. microRNAs as regulators of adipogenic differentiation of mesenchymal stem cells. Stem Cells Dev. 2015;24(4):417–25. doi: 10.1089/scd.2014.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q, Cao J, Xie N, Velletri T, Zhang X, Xu C, Zhang L, Yang H, Hou J, Wang Y, Shi Y. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell death and differentiation. 2016;23(7):1128–39. doi: 10.1038/cdd.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Z, Rodriguez NE, Zhao J, Ramey AN, Hyzy SL, Boyan BD, Schwartz Z. Selective enrichment of microRNAs in extracellular matrix vesicles produced by growth plate chondrocytes. Bone. 2016;88:47–55. doi: 10.1016/j.bone.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rilla K, Mustonen AM, Arasu UT, Harkonen K, Matilainen J, Nieminen P. Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix biology: journal of the International Society for Matrix Biology. 2017 doi: 10.1016/j.matbio.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 43.Rendina-Ruedy E, Rosen CJ. Bone-Fat Interaction. Endocrinol Metab Clin North Am. 2017;46(1):41–50. doi: 10.1016/j.ecl.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsui S, Ogata Y. Effects of miR-223 on expression of IL-1beta and IL-6 in human gingival fibroblasts. J Oral Sci. 2016;58(1):101–8. doi: 10.2334/josnusd.58.101. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Wang R, Jiang J, Yang B, Cao Z, Cheng X. miR-223 is upregulated in monocytes from patients with tuberculosis and regulates function of monocyte-derived macrophages. Mol Immunol. 2015;67(2 Pt B):475–81. doi: 10.1016/j.molimm.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Kim D, Song J, Ahn C, Kang Y, Chun CH, Jin EJ. Peroxisomal dysfunction is associated with up-regulation of apoptotic cell death via miR-223 induction in knee osteoarthritis patients with type 2 diabetes mellitus. Bone. 2014;64:124–31. doi: 10.1016/j.bone.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 47.Coustry F, Posey KL, Liu P, Alcorn JL, Hecht JT. D469del COMP Retention in Chondrocytes Stimulates Caspase-Independent Necroptosis. American Journal of Pathology. 2011 doi: 10.1016/j.ajpath.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okuhara A, Nakasa T, Shibuya H, Niimoto T, Adachi N, Deie M, Ochi M. Changes in microRNA expression in peripheral mononuclear cells according to the progression of osteoarthritis. Mod Rheumatol. 2012;22(3):446–57. doi: 10.1007/s10165-011-0536-2. [DOI] [PubMed] [Google Scholar]
- 49.Bouxsein ML. Bone quality: where do we go from here? Osteoporos Int. 2003;14(Suppl 5):S118–27. doi: 10.1007/s00198-003-1489-x. [DOI] [PubMed] [Google Scholar]
- 50.Currey JD. Bone strength: what are we trying to measure? Calcif Tissue Int. 2001;68(4):205–10. doi: 10.1007/s002230020040. [DOI] [PubMed] [Google Scholar]
- 51.Gamble C, Nguyen J, Hashmi SS, Hecht JT. Pseudoachondroplasia and painful sequelae. American journal of medical genetics. 2015;167(11):2618–22. doi: 10.1002/ajmg.a.37253. Part A. [DOI] [PubMed] [Google Scholar]
- 52.Hernandez CJ, van der Meulen MC. Understanding Bone Strength Is Not Enough. J Bone Miner Res. 2017;32(6):1157–1162. doi: 10.1002/jbmr.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inzana JA, Maher JR, Takahata M, Schwarz EM, Berger AJ, Awad HA. Bone fragility beyond strength and mineral density: Raman spectroscopy predicts femoral fracture toughness in a murine model of rheumatoid arthritis. Journal of biomechanics. 2013;46(4):723–30. doi: 10.1016/j.jbiomech.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porter A, Patel N, Brooks R, Best S, Rushton N, Bonfield W. Effect of carbonate substitution on the ultrastructural characteristics of hydroxyapatite implants. J Mater Sci Mater Med. 2005;16(10):899–907. doi: 10.1007/s10856-005-4424-1. [DOI] [PubMed] [Google Scholar]
- 55.Follet H, Boivin G, Rumelhart C, Meunier PJ. The degree of mineralization is a determinant of bone strength: a study on human calcanei. Bone. 2004;34(5):783–9. doi: 10.1016/j.bone.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 56.Brandt KD, Radin EL, Dieppe PA, van de Putte L. Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis. 2006;65(10):1261–4. doi: 10.1136/ard.2006.058347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cross M. Clinical Terminology for Describing Knee Instability. Sports Medicine and Arthroscopy Reviews. 1996;4:313–318. [Google Scholar]
- 59.van der Esch M, Steultjens M, Knol DL, Dinant H, Dekker J. Joint laxity and the relationship between muscle strength and functional ability in patients with osteoarthritis of the knee. Arthritis Rheum. 2006;55(6):953–9. doi: 10.1002/art.22344. [DOI] [PubMed] [Google Scholar]
- 60.Asahara H, Inui M, Lotz MK. Tendons and Ligaments: Connecting Developmental Biology to Musculoskeletal Disease Pathogenesis. J Bone Miner Res. 2017 doi: 10.1002/jbmr.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Catuogno S, Rienzo A, Di Vito A, Esposito CL, de Franciscis V. Selective delivery of therapeutic single strand antimiRs by aptamer-based conjugates. J Control Release. 2015;210:147–59. doi: 10.1016/j.jconrel.2015.05.276. [DOI] [PubMed] [Google Scholar]
- 62.Posey KL, Coustry F, Veerisetty AC, Hossain M, Gattis D, Booten S, Alcorn JL, Seth PP, Hecht JT. Antisense Reduction of Mutant COMP Reduces Growth Plate Chondrocyte Pathology. Mol Ther. 2017;25(3):705–714. doi: 10.1016/j.ymthe.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.