

Summary
Superoxide radical anion (O2•−) and other Reactive Oxygen Species (ROS) are constantly produced during respiration. In mitochondria, the dismutation of O2•− is accelerated by the mitochondrial superoxide dismutase 2 (SOD2), an enzyme that has been traditionally associated with antioxidant protection. However, increases in SOD2 expression promote oxidative stress, indicating that there may be a prooxidant role for SOD2. Here we show that SOD2, which normally binds manganese, can incorporate iron and generate an alternative isoform with peroxidase activity. The switch from manganese to iron allows FeSOD2 to utilize H2O2 to promote oxidative stress. We found that FeSOD2 is formed in cultured cells and in vivo. FeSOD2 causes mitochondrial dysfunction and higher levels of oxidative stress in cultured cells and in vivo. We show that formation of FeSOD2 converts an antioxidant defense into a prooxidant peroxidase that leads to cellular changes seen in multiple human diseases.
Keywords: peroxidase, mitochondrial superoxide dismutase (SOD2), metals, mitochondrial metabolism, free radicals, oxidative stress
eTOC blurb
Ganini et al demonstrates that incorporation of iron over manganese by the mitochondrial superoxide dismutase (SOD2) generates a prooxidant peroxidase in mitochondria of human cells and mice. They show that FeSOD2 formation leads to mitochondrial dysfunction and oxidative stress.
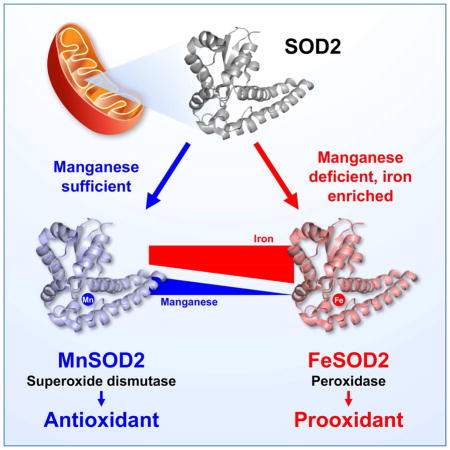
Introduction
Most reactive oxygen species, including the superoxide anion free radical (O2•−), are unavoidable products of aerobic respiration formed in mitochondria (Boveris and Cadenas, 1975). These species are major determinants of health, since variations in their levels widely impact cellular metabolism, signaling and phenotype (Fridovich, 1995). Manganese superoxide dismutase (SOD2) is a mitochondria-resident enzyme widely recognized for its role in accelerating the dismutation of O2•− into H2O2 and O2 (Weisiger and Fridovich, 1973). This activity, which has been widely associated with protective roles, prevents oxidative damage to sensitive iron-sulfur cluster enzymes that are critical for the energetic metabolism taking place in mitochondria (Flint et al., 1993; Gardner et al., 1995; Li et al., 1995). However, it is becoming clear that under various conditions, an increase in SOD2 expression not only fails to provide further protection but promotes oxidative stress to mitochondria. This suggests the occurrence of a possible damaging activity associated with dysregulated SOD2 expression (Ansenberger-Fricano et al., 2013; Connor et al., 2005; Menon et al., 2007; Tao et al., 2014).
Studies have indicated that the antioxidant or prooxidant fate of SOD2 depends on whether there is a parallel upregulation of H2O2-detoxifying enzymes, such as peroxiredoxins and glutathione peroxidases (Cox et al., 2006; Miar et al., 2015). This concept is supported by the finding that SOD2 expression becomes an accurate predictor of prostate and breast cancer risk and outcomes when normalized against the expression of glutathione peroxidase-1 and/or catalase, both of which eliminate H2O2 (Cox et al., 2006; Miar et al., 2015). For that reason, an optimal balance between SOD2 levels and that of downstream H2O2 scavengers is critical for antioxidant protection.
Indeed, most enzymes with prooxidant roles in vivo use H2O2 and iron, as an essential cofactor. It is well-established that the metabolism of iron occurs predominantly in mitochondria. Except in the case of certain diseases, iron trafficking and delivery to specific proteins are tightly regulated processes (Hentze et al., 2004). Under numerous conditions associated with disease states, iron handling in mitochondria is dysregulated along with an increase in SOD2. We hypothesized that incorporation of iron instead of manganese into SOD2 could lead to a gain of function, converting SOD2 into a prooxidant peroxidase that enables it to utilize H2O2 for the oxidation of other molecules. In testing this hypothesis we found that, indeed, the biochemical activity of SOD2 depends on the metal it incorporates. While MnSOD2 has negligible peroxidase activity in vitro, FeSOD2 behaves like a typical metal peroxidase. We also found that FeSOD2 loses its antioxidant superoxide dismutase function. We show that iron incorporation into SOD2 occurs in mitochondria under physiologic and pathogenic conditions both in cell culture and in mice. Furthermore, the switch to a prooxidant peroxidase imparts changes to the cellular metabolism and gene expression profiles of cells that mimic those seen in human disease states. Hence, our results indicate that a switch in metal cofactor may be mechanistically associated with the evolution of disease states characterized by deficits in mitochondrial energetic/respiratory functions associated with iron accumulation and SOD2 overexpression, such as often seen in cancers.
Results
It is becoming clear that increases in SOD2 expression do not necessarily afford further antioxidant protection to mitochondria and can, under certain conditions, promote oxidative damage (Ansenberger-Fricano et al., 2013; Connor et al., 2005; Menon et al., 2007; Tao et al., 2014). This led us to surmise that SOD2 may display an alternative function as a prooxidant. Because most prooxidant enzymes are iron-dependent, and both SOD2 and iron accumulate in mitochondria during many disease states, we tested the hypothesis that iron incorporation into SOD2 occurs and leads to a switch in the activity of SOD2 from an antioxidant superoxide dismutase to a prooxidant peroxidase.
To examine this idea, human SOD2 was prepared incorporated with manganese, its canonical metal cofactor, or with iron (Fig. S1). Incorporation with manganese was found to be indispensable for the antioxidant superoxide dismutase activity of this enzyme (Beyer and Fridovich, 1991; Vance and Miller, 1998, 2001; Whittaker, 2003). As expected, human SOD2 incorporated with iron (Fig. S1C) had negligible superoxide dismutase activity (Fig. S1D) but showed prominent peroxidase activity towards a classical peroxidase substrate, Amplex Red (Fig. 1A and Fig. S1E). Amplex Red is a colorless molecule that can be oxidized by H2O2 in the presence of peroxidases to the red, fluorescent resorufin (Summers et al., 2013). Complementarily, we also tested the ability of FeSOD2 and MnSOD2 to form protein radicals in the absence of substrates, a feature characteristic of iron-dependent peroxidases. SOD2 protein radical formation was highly H2O2-dependent with FeSOD2 and negligible with MnSOD2 (Fig. 1B). By using a single endpoint measurement for the oxidation of Amplex Red to resorufin (Summers et al., 2013; Zhao et al., 2012), we further demonstrated that FeSOD2 oxidizes Amplex Red and that this activity is dependent on H2O2 (Fig. 1C). These results indicate that FeSOD2 can use H2O2 to oxidize a substrate and that, in its absence, self-peroxidation occurs. Both of these characteristics are hallmark features of iron-dependent peroxidases.
Figure 1. Peroxidase activity of FeSOD2.

(A) Amplex Red oxidation was determined in samples containing 0.2 mg/mL SOD2 (8.6 μM), 100 μM Amplex Red, 860 μM H2O2 (100 equivalents to SOD2, μM:μM) prepared in 100 mM phosphate buffer, pH 7.4, with 25 μM DTPA. (B) Effect of H2O2 in the SOD2 free radical formation was studied using ELISA. Samples contained 0.2 mg/mL SOD2 (8.56 μM) and different concentrations of H2O2 (expressed as μM:μM equivalents to SOD2) in the presence of 100 mM DMPO prepared in 100 mM phosphate buffer, pH 7.4, with 25 μM DTPA for 1h at 37oC. Reaction was stopped by the addition of 200 U/mL catalase. (C) After being incubated in the dark for 1h, samples containing 10 μM FeSOD2, 100 μM Amplex Red and H2O2 (expressed as μM:μM equivalents to FeSOD2) were analyzed for their final resorufin yield. Samples were prepared in 100 mM phosphate buffer, pH 7.4, with 25 μM DTPA at 25°C. (D) Protein radical formation was determined by ELISA in samples containing 10 μM FeSOD2, 100 mM DMPO and H2O2 (expressed as μM:μM equivalents to FeSOD2); where indicated, samples also contained 100 μM Amplex Red. (E) Samples contained 10 μM FeSOD2, 100 mM DMPO, 500 μM H2O2 and, where indicated, also contained 100 μM Amplex Red or deferoxamine (DFO). Samples were prepared in 100 mM phosphate buffer, pH 7.4, with 25 μM DTPA. Reaction was stopped by the addition of 200 U/mL catalase. SOD2 and CAT bands are indicated. (F) Oxidation of Amplex Red was studied in samples containing 10 μM FeSOD2, MnCl2 or Fe(NH4)2(SO4)2, 100 μM Amplex Red, 1 mM H2O2, and 25 μM DTPA or 25 μM EDTA prepared in 100 mM phosphate buffer, pH 7.4, incubated for 1h in the dark at 25°C. Data represented as mean ± SD. See also Supplementary Fig. S1. * indicates p-value < 0.05 versus control and & indicates p-value < 0.05 as indicated in the graph.
In fact, protein radical formation and the oxidation of Amplex Red are competing reactions. As shown in Fig. 1D, FeSOD2 samples treated with different concentrations of H2O2 showed 5 times lower yields of protein radicals when in the presence of Amplex Red (Fig. S1E). Western blot-based assays for protein radicals showed that Amplex Red dramatically reduces the yield of protein radicals (Fig. 1E). The high-affinity chelator deferoxamine (DFO) had negligible effects on the formation of protein radicals by FeSOD2 and H2O2. Analyzing the formation of protein radicals in monomers of FeSOD2, only a peroxidase substrate, such as Amplex Red, was able to prevent its formation, which further supports a primary peroxidase activity of native FeSOD2. Chelators such as EDTA and DTPA had minor effects or no effect at all on the yield of Amplex Red oxidation by FeSOD2 in the presence of H2O2 (Fig. 1F). DTPA at 25 μM was present in the buffer of all in vitro experiments shown in this manuscript. Our buffers were Chelex-treated to decrease contaminating trace metals. As shown in Fig. 1F, unchelated iron or manganese in the presence of H2O2 failed to oxidize Amplex Red. Taken together, our results indicate that incorporation of iron changes human SOD2 to a prooxidant peroxidase.
To determine if the availability of iron and manganese to cells are determinants in the formation of human FeSOD2 or MnSOD2, we performed experiments with mammalian cells overexpressing SOD2 (Mn11) compared to parental breast epithelial cells (MCF7/neo) (Zhang et al., 1999) cultivated under different iron-to-manganese ratios. The commonly used culture medium RPMI 1640 contains very low levels of both iron and manganese (Fig. 2A); however, by adding other components and supplements, the complete medium had nearly 3.8 μM of iron while still displaying only trace levels of manganese. Fetal bovine serum (FBS) was the component which contributed most of the iron available in the complete medium (Fig. 2A). These cell lines were cultivated in RPMI 1640-based medium with or without 25 μM manganese chloride supplementation for at least 21 days. Manganese supplementation had no effect on SOD2 protein levels (Fig. 2B) nor on the cytosolic Cu,Zn-SOD1 superoxide dismutase activity in either cell line (Fig. 2C, gray columns). Even though manganese supplementation in neo cells led to a ca. 5 fold increase in the level of intracellular manganese (Fig. S2A), the activity of MnSOD2 was not increased after manganese supplementation (Fig. 2C). In contrast, the SOD2-overexpressing cell line Mn11 accumulated more intracellular total manganese (13.5 times higher) when supplemented (Fig. S2A), and more importantly, showed an increased MnSOD2 superoxide dismutase activity (Fig. 2C, black columns). Mn11 cells in regular medium had twofold higher MnSOD2 superoxide dismutase activity than neo cells but, when cultivated in manganese supplemented medium, Mn11 displayed fourfold higher MnSOD2 superoxide dismutase activity. In summary, Mn11 cells grown in manganese-supplemented medium showed four times higher MnSOD2 superoxide dismutase activity and protein, whereas the same cell line cultivated in regular medium accumulated half of the SOD2 protein with no superoxide dismutase activity (Fig. 2C). Based on this finding, we surmised that the superoxide dismutase-inactive SOD2 accumulating in Mn11 was bound to iron.
Figure 2. Formation of the peroxidase FeSOD2 in SOD2-overexpressing cells cultivated in regular medium.

(A) Iron and manganese content of RPMI 1640 medium (high glucose with glutamine, Gibco®, Life Technologies) with or without 10% fetal bovine serum (ATCC®). The complete medium additionally contains 1 mM pyruvic acid, 0.4 mM uridine, 50 μg/mL geneticin, 100 μg/mL streptomycin and 100 U/mL penicillin. Metals were quantified by ICP-OES. (B) SOD2 protein abundance and (C) SODs specific activities in MCF-7-derived control (neo) and SOD2-overexpressing cells (Mn11) cultivated for 21 days in regular medium or manganese-supplemented medium (25 μM MnCl2). (D) Metal analysis by ICP-OES of the purified SOD2 from the neo and Mn11 cells. Peroxidase activity of SOD2 purified from neo and Mn11 cells was determined by Amplex Red oxidation (E) and by protein radical formation (F). The Amplex Red oxidation was measured in samples containing 20 μg/mL of SOD2, 100 μM H2O2 and 100 μM Amplex Red prepared in 100 mM phosphate buffer, pH 7.4, with 25 μM DTPA. Oxidation of Amplex Red of samples incubated in the dark did not differ from those shown in the figure, in which samples were exposed to instrumental light every 5 min for 60 min. In (F) samples were prepared as described in (E), except that Amplex Red was omitted and DMPO (100 mM) was added. Data represented as mean ± SD. See also Supplementary Fig. S2. * indicates p-value < 0.05 versus control; & indicates p-value < 0.05 as indicated in the graph; and NS indicates no statistically significant difference, p-value > 0.05 as indicated in the graph.
As suspected, SOD2 purified from Mn11 cells grown in unsupplemented RPMI medium (Fig. S2) was predominantly bound to iron, whereas in Mn11 cells cultivated in manganese-supplemented medium, SOD2 was largely in the manganese-containing form, MnSOD2 (Fig. 2D). Oxidation of Amplex Red in the presence of H2O2 was catalyzed only by SOD2 purified from Mn11 cultivated in unsupplemented medium (Fig. 2E). SOD2 purified from Mn11 cultivated in regular medium also produced higher levels of protein radicals when incubated with H2O2 (Fig. 2F), consistent with the formation of the peroxidase FeSOD2. Supplementation of the culture media with manganese suppressed the formation of protein radicals on SOD2 purified from Mn11 cells due to the formation of MnSOD2 (Fig. 2F, compare the sixth and eighth columns). With regard to H2O2-dependent protein radical formation, it is noteworthy that our sensitive ELISA assay for protein radicals showed no statistically significant difference in protein radical formation between SOD2 purified from neo and Mn11 cells grown in manganese-supplemented medium (Fig. 2F, fourth and eighth columns); however, the direct measurement of metal incorporation into SOD2 showed that SOD2 purified from Mn11 cells supplemented with MnCl2 had some FeSOD2. Ultimately, the most sensitive and reliable assay for the detection of FeSOD2 was the direct measurement of metal bound to the purified SOD2. Collectively, these results indicate that inadequate manganese led to the formation of the peroxidase FeSOD2 particularly in cells with elevated SOD2 expression, such as Mn11 cells.
It was also investigated whether FeSOD2 formation in cells had any functional effect on mitochondrial metabolism and respiration. Lactate production, a surrogate indicator of higher glycolytic metabolism, was highest in Mn11 cells cultivated in unsupplemented medium (Fig. 3A). Higher lactate was observed in tandem with lower steady-state levels of intracellular ATP (Fig. 3B). Cultivation of the cells in manganese-supplemented medium reversed these effects and was sufficient to simultaneously reduce lactate and restore ATP levels comparable to those observed in neo cells. Manganese supplementation did not have any measurable effect on neo cells with regard to lactate or ATP, suggesting that manganese supplementation is particularly important to prevent FeSOD2 formation when SOD2 expression is upregulated. Steady-state levels of ATP in Mn11 cells cultivated in unsupplemented medium were reduced by 75% (Fig. 3B, black columns) in comparison to levels achieved with manganese supplementation. A brief incubation with the glycolysis inhibitor 2-deoxy-glucose (Barban and Schulze, 1961) lowered ATP levels in all cell lines tested, whether grown in manganese-supplemented or regular medium. These experiments strongly suggested that glycolysis indeed contributes to the ATP generation in neo and Mn11 cells independent of manganese-availability (Fig. 3B, gray columns). In addition to measuring ATP, we also quantified different components of the mitochondrial electron transport chain (ETC) to define whether SOD2 bound to either iron or manganese affected the functioning of the ETC. Mn11 cells cultivated in manganese-deficient medium had decreased levels of Complex II and IV, which could explain the lower levels of ATP measured in this cell line (Fig. 3C). Manganese supplementation increased the levels of those components close to that found in neo cells (quantification can be found in Fig. S3A–B), implicating the peroxidase activity of FeSOD2 as a possible contributor to the degradation of Complexes II and IV. Assay for bioenergetics of cells using extracellular flux analyzes showed that manganese supplementation had only a minor effect on the mitochondrial function of neo cells (Fig. 3D, black and red lines). Quantification of basal respiration, mitochondrial respiration and mitochondrial spare capacity showed decreases of 10% in neo cells supplemented with manganese (Fig. S3C–E). In contrast, manganese supplementation increased the oxygen consumption of Mn11 cells (Fig. 3D, green and blue lines). Basal oxygen consumption and mitochondrial respiration were significantly 50% higher (Fig. S3C–D) and spare mitochondrial respiration was nearly 3 times higher (Fig. S3E).
Figure 3. Dysfunctional mitochondrial metabolism in cells with FeSOD2.

(A) Glycolysis was assessed by the quantification of lactate excreted to the culture medium for 24 hours. (B) Intracellular ATP was determined in cells under basal conditions or in the presence of the glucose uptake inhibitor 2-deoxy-glucose (50 mM for 30 min). In (C), representative Western blots for the different complexes of mitochondria of cells are depicted. Samples were prepared from total cell homogenates (30 μg proteins per lane), and laminin B1 staining was used as loading control. See Supplementary Figs. S3A and B for densitometry of Complex IV and II stainings. Data represented as mean ± SD. * indicates p-value < 0.05 versus control (basal neo cells); # indicates p-value < 0.05 versus basal; & indicates p-value < 0.05 as indicated in the graph; and NS indicates no statistically significant difference, p-value > 0.05 as indicated in the graph.
In order to obtain a more complete picture of the impact of FeSOD2 on cellular metabolism, microarray analyses using gene set enrichment (GSEA) and gene ontology were performed (Fig. 4 and in Fig. S4A). These analyses showed that the gene sets related to mitochondrial components were upregulated in MnSOD2-containing cells (Fig. 4B and F). Other upregulated gene sets were related to mitosis (Fig. S4B), organelle localization (Fig. 4C), organization of the organelle envelope (Fig. 4D), and microtubule organization (Fig. 4D). Downregulated gene sets were associated with amino acid transport (Fig. 4H), antigen binding (Fig. S4C), lipoprotein binding (Fig. S4D), and pigment metabolism and biosynthesis (Fig. 4G). Even though we detected an increase in genes associated with mitosis, we failed to observe any changes in the growth of the cells with or without manganese (Fig. S5A). Microarray analyses did not show significant differences in genes related to iron metabolism in neo and Mn11 cells cultivated with or without manganese, which we further confirmed by measuring the abundance of two major iron transporters in cells, transferrin receptor and DMT1 (Fig. S5B). Collectively, our gene expression results show that formation of MnSOD2 in cells leads to a robust increase in genes required to support mitochondrial respiration, energetic function and amino acid uptake/metabolism, as well as general cellular organization.
Figure 4. Alterations in gene sets based on gene ontology in Mn11 cells with the antioxidant MnSOD2 instead of the peroxidase FeSOD2.

Microarray and GSEA analyses were performed to show the effects of accumulation of the antioxidant MnSOD2 instead of peroxide FeSOD2 in cells. The effect of manganese supplementation in Mn11, which specifically promotes MnSOD2 formation instead of FeSOD2, was compared to the non-MnSOD2 effects of the manganese supplementation detected in the isogenic parental cell line, neo. The final contrast was (MnMn11-Mn11) versus (Mnneo-neo). An infographic for this contrast can be found in the Supplementary Fig. S3C. Gene sets were ranked by Normalized Enrichment Score (NES). Heat maps for the genes that contributed to the enrichment of the top gene sets are shown in B–H and Supplementary Fig. S3D–F.
Mitochondrial metabolism is central to the overall energetic balance of the cell and is a major target of oxidative stress. Therefore, we decided to study the consequences of FeSOD2 formation in mitochondria of cells under oxidative stress using three approaches: 1) acute toxicity induced by bolus H2O2 treatment (Fig. 5A–B); 2) low sublethal, continuous, external generation of H2O2 by a glucose oxidase/glucose system (Fig. S5C, and Fig. 5C–D); and 3) sublethal intoxication with the O2•−/H2O2-producing agent paraquat (1,1′-dimethyl-4,4′-bipyridinium, Fig. 5E). Addition of 100 μM H2O2 to neo cells, grown either in the presence or absence of manganese supplementation, led to an approximately 30% reduction in cellular viability (Fig. 5A, first and second columns). In contrast, the SOD2-overexpressing cell line Mn11 was more sensitive to bolus H2O2 treatment when cultivated in regular medium (50% viability loss), but more resistant (20% viability loss) when cultivated in manganese-supplemented medium (Fig. 5A, third and fourth columns). When the experiment was repeated with bolus 75 μM H2O2 instead, the higher loss of viability of Mn11 cells with FeSOD2 compared to neo cells (Fig. 5B, third black column) was completely mitigated by the mitochondrial antioxidant mitoTEMPO (Fig. 5B, third gray column). Pretreatment with N-acetyl cysteine, a general antioxidant, also mitigated the higher loss of cell viability in Mn11 with FeSOD2 challenged with bolus 100 μM H2O2 (Fig. S5B).
Figure 5. Cells with FeSOD2 show increased susceptibility to loose cell viability and accumulate damage in mitochondria.

The parental cell line, neo, and the SOD2-overexpressing cell line, Mn11, were exposed to (A) 100 μM H2O2 for 1 h, (B–C) 50 mU/mL glucose oxidase (GOX) for 4 h, or (D) 750 μM paraquat for 10 h. (A) Cell viability was determined in cells treated with H2O2 for 1 h using the MTT assay. (B) MtDNA damage or (C) mitochondrial protein radical formation was determined in cells treated with 50 mU/mL GOX for 4 h. In (C), medium also contained 40 mM DMPO. (D) Cells were incubated with 40 mM DMPO and then treated with paraquat (750 μM) for 10 h. Fluorescence immunohistochemistry was prepared for the detection of protein-centered radicals (anti-DMPO in red). Slides were covered with mounting media with DAPI (nuclear counterstaining in blue) just before confocal microscope imaging. Representative images are depicted. Data are represented as mean ± SD. * indicates p-value < 0.05 versus control; & indicates p-value < 0.05 as indicated in the graph; and NS indicates no statistically significant difference, p-value > 0.05 as indicated in the graph.
We next studied the effect of low levels of H2O2 production in cells. A low level of glucose oxidase/glucose in medium was used to generate an external, continuous production of H2O2. Under the conditions used for these experiments, cellular viability was not affected (Fig. S5D); however, when we analyzed the mtDNA for lesions, Mn11 cultivated in regular medium had higher levels of mtDNA lesions, which were completely prevented when this same cell line was cultivated in manganese-supplemented medium (Fig. 5C). Similarly, this sublethal treatment of cells with glucose oxidase/glucose induced higher mitochondrial protein radical formation in Mn11 cells cultivated in regular medium (Fig. 5D, third gray column), whereas Mn11 cells cultivated in manganese-supplemented medium did not show statistically significant formation of mitochondrial protein radicals (Fig. 5D, fourth gray column). Neo cells with or without manganese supplementation showed the same low level of mitochondrial protein radical formation (Fig. 5D, first and second gray columns).
We also used paraquat, an agent used to generate intracellular O2•− which disproportionates to form H2O2. When exposed to this toxic environmental agent, Mn11 cells cultivated in regular medium accumulated more protein radicals than neo cells (red staining for anti-DMPO in Fig. 5E, upper confocal images). Even more striking was the fact that Mn11 cells cultivated in manganese-supplemented medium did not show differences in protein radical formation compared to neo cells (Fig. 5E, lower confocal images). Under the conditions used for this experiment, cells were intact, as can be seen by the nuclear staining by DAPI in blue. In contrast to cells with MnSOD2, cells with FeSOD2 showed higher susceptibility to oxidative stress, free radical damage and consequent loss in cellular viability.
Since iron incorporation into SOD2 occurred only in cells cultivated in a medium with low levels of manganese-to-iron, we explored in vivo the role of low levels of manganese-to-iron in metal incorporation into SOD2 in mice. The NIH-31 diet, a standard natural-ingredient diet for mice, contains 0.1836 g/kg manganous oxide 60%. Without this additive, the background level of manganese is approximately 40 ppm. We then used three diets: 1) control diet, with the regular 150 ppm manganese and regular 275 ppm iron; 2) manganese-deprived diet, with no added manganese additive, 40 ppm manganese and the regular 275 ppm iron; and 3) iron-enriched diet, with ten times more iron than control diet, at 2700 ppm, and the regular 150 ppm manganese. Five-week-old mice were fed one of the three diets for 4 weeks (Fig. S6A). Weight gains and total blood hemoglobin were comparable among the groups (n = 9 per group), and the serum parameters alanine aminotransferase (ALT), creatinine, glucose, sodium and iron were also not different; however, total iron binding capacity (TIBC) and total cholesterol were lower in animals fed the iron-enriched diet (Fig. S6B). Since those parameters were lower, we chose to focus our studies on the liver because this organ is central for whole-body metal homeostasis and cholesterol metabolism.
Levels of iron in whole liver were increased only in animals fed the iron-enriched diet (Fig. 6A, black bars); however, in isolated liver mitochondria, iron was increased not only in animals fed an iron-enriched diet, but also in animals fed the manganese-deficient diet (Fig. 6B, black bars). In contrast, lower levels of manganese were found in whole tissue and isolated mitochondria of animals fed either the manganese-deprived diet or the iron-enriched diet (Fig. 6A–B, gray bars). In fact, the lower levels of manganese seen in liver correlated with the low level of manganese detected in blood of animals fed either the manganese-deprived diet or the iron-enriched diet (Fig. 6C).
Figure 6. Animals fed with a manganese-deficient diet or an iron-enriched diet show iron accumulation and manganese deprivation in liver.
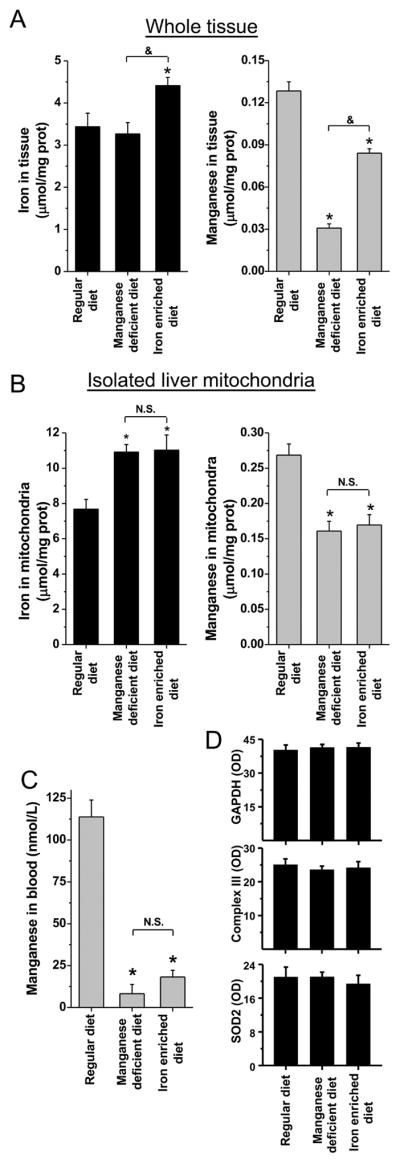
Animals were fed with control, iron- or manganese-manipulated diets for 4 weeks. PBS-perfused livers, freshly isolated mouse-liver mitochondria, and blood were collected and stored. Concentrations of iron (black bars) and manganese (gray bars) in whole liver homogenates (A) and isolated mitochondria (B) were determined by ICP-OES. In (C), manganese concentrations in total blood were determined by ICP-MS. In parallel, samples of whole liver homogenates were probed for housekeeping protein and glycolytic enzyme GAPDH, mitochondrial Complex III, and SOD2 (Fig. S6C). In (D) the densitometries (OD, A.U.) are shown. An infographic with additional information about the animal protocol can be found in the Supplementary Fig. S5A. Data are represented as mean ± SD. * indicates p-value < 0.05 versus control; & indicates p-value < 0.05 as indicated in the graph; and NS indicates no statistically significant difference, p-value > 0.05 as indicated in the graph.
Even though animals showed alterations in the levels of iron and manganese in the mitochondria, there were no differences for the abundance of GAPDH, a key enzyme of the glycolytic pathway, total mass of mitochondria, judged by the abundance of the Complex III component UQCRC2, or SOD2 (Fig. 6D and Fig. S6C). Even though total protein levels of SOD2 were not altered, superoxide dismutase activities of MnSOD2 were indeed decreased in animals fed either the manganese-deprived diet or the iron-enriched diet (Fig. 7A).
Figure 7. Animals fed with a manganese-deficient diet or an iron-enriched diet show mitochondrial damage with accumulation of the peroxidase FeSOD2.

Animals were fed with control, iron- or manganese-manipulated diets for 4 weeks. (A) Activities of SODs in whole liver were determined. For experiments shown in (B–D), animals were injected with DMPO (1g/kg) 1h before sacrifice, and liver and freshly-isolated mitochondria were obtained. Protein radicals were determined in isolated mitochondria by ELISA in (B) and by Western blotting in (C). In (D), mitochondrial Complex I was isolated from samples of liver mitochondria, and anti-DMPO Western blotting (protein radicals) was performed. In (E), activities of Complex I+III, II+III and citrate synthase were determined in isolated liver mitochondria samples. Native SOD2 was isolated from whole liver homogenates as described earlier for cells. In (F), SOD2s isolated from the mouse livers were used for the detection of peroxidase activity by the formation of SOD2-protein radicals. Samples contained 35 μM SOD2 (0.87 mg/mL), 100 mM DMPO, 250 μM H2O2 and, when present, 100 μM Amplex Red. In (G), concentrations of iron (black bars) and manganese (gray bars) in the SOD2s isolated from the mouse livers were determined by ICP-MS. Data are represented as mean ± SD. * indicates p-value < 0.05 versus Regular diet.
Interestingly, animals fed either the manganese-deprived diet or the iron-enriched diet showed significantly higher levels of mitochondrial protein free radicals in vivo (Fig. 7B), and those levels were inversely proportional to the activity of the antioxidant MnSOD2 (Fig. 7A, gray bars, and Fig. 7B). In an attempt to determine whether protein radicals were formed in Complex I of the mitochondrial OXPHOS, a known sensitive target for free radicals, we isolated the proteins from Complex I by immuno-precipitation. After performing anti-DMPO Western blotting, higher levels of free radicals on proteins of Complex I were detected in the samples from animals fed either the manganese-deprived diet or the iron-enriched diet (Fig. 7C). Furthermore, by determining the activity of Complex I+II and Complex II+III, we detected a specific decrease in the activity of Complex I only in the mitochondria of the animals fed the iron-enriched diet (Fig. 7D). Citrate synthase activity was also performed to show the use of comparable amounts of total mitochondrial proteins in those assays.
Intriguingly, mitochondrial iron accumulation alone could not explain the higher formation of mitochondrial protein free radicals and Complex I loss in activity seen in animals fed the iron-enriched diet because the actual iron levels in mitochondria of animals fed the manganese-deprived diet or the iron-enriched diet were comparable (Fig. 6B). If damage were caused by the iron overload exclusively, the mitochondrial damage would have been comparable in animals fed the manganese-deprived diet or the iron-enriched diet.
Because of our discovery that low levels of manganese-to-iron predispose the formation of the peroxidase FeSOD2, we hypothesized that the difference between the two dietary treatments was a difference in the formation of FeSOD2 in the mitochondria of animals fed the manganese-deprived or iron-enriched diets. Hence, we isolated native SOD2 from the whole livers and performed an assay for the peroxidase prooxidant activity of FeSOD2 with the isolated proteins. SOD2 from animals fed the iron-enriched diet showed a remarkably higher level of peroxidase activity (Fig. 7E, first lane). Protein self-oxidation, as a result of its peroxidase activity, was prevented by the addition of the peroxidase substrate Amplex Red (Fig. 7E, second lane). Even though at a much lower yield, SOD2 isolated from animals fed the manganese-deprived diet also showed some peroxidase activity which was prevented by the peroxidase substrate Amplex Red (Fig. 7E, last two lanes). Indeed, the determination of iron and manganese incorporated into the SOD2s showed that animals fed the iron-enriched diet had the highest level of the peroxidase FeSOD2, with nearly 80% of the total SOD2 pool being incorporated with iron (Fig. 7F, third column). Animals fed the manganese-deprived diet showed nearly 30% of the total pool of SOD2 incorporated with iron (Fig. 7F, second column). Animals fed the control diet had negligible levels of the peroxidase FeSOD2 (Fig. 7F, first column).
Discussion
Optimal mitochondrial metabolism is a central component of numerous biosynthetic processes and maintains energetic homeostasis by supplying large amounts of ATP (Darley-Usmar et al., 1987; Melov et al., 1999). In eukaryotes, SOD2 is an essential mitochondrial enzyme as indicated by previous studies showing that SOD2 knockout mice have extensive cardiac remodeling, neurodegeneration, and perinatal death (Fridovich, 1995; Lebovitz et al., 1996; Melov et al., 1999; Melov et al., 1998). SOD2 is broadly accepted as a key component of the mitochondrial antioxidant defense system; however, there are also reports showing that overexpression of SOD2 can negatively impact mitochondria both structurally and functionally (Ansenberger-Fricano et al., 2013; Connor et al., 2007; Kim et al., 2005; Larosche et al., 2009; Rodriguez et al., 2000). Such studies have shown increased tissue oxidative damage in animals and cells overexpressing SOD2, particularly when they are chronically exposed to toxicants (Connor et al., 2007; Kim et al., 2005; Larosche et al., 2009; Rodriguez et al., 2000). In the present work, we investigated the molecular basis of the prooxidant function of SOD2 in mitochondria of cells and animals and focused our efforts on: 1) characterizing the consequences of SOD2 overexpression on cellular energetic metabolism; and 2) investigating the prooxidant role of FeSOD2 in vivo.
We have previously shown that cells overexpressing SOD2 have a shift in metabolism from oxidative phosphorylation to glycolysis (Hart et al., 2015). In fact, here we show that this shift in energy metabolism is associated with the formation of FeSOD2 in the mitochondria of cells overexpressing SOD2. Even though there was no change in mitochondrial mass, cells with FeSOD2 showed low levels of ATP production and higher glycolytic rate. After being supplemented with manganese, MnSOD2 was formed and cells showed higher levels of ATP production, lower glycolytic rates, increased levels of expression of the mitochondrial components and higher mitochondrial function. Formation of FeSOD2 in unsupplemented Mn11 leads to blunted capacity to produce ATP through oxidative phosphorylation. Since those cells with FeSOD2 still had nearly twice the mitochondrial superoxide dismutase activity of MnSOD2 compared to neo cells, we show that the mitochondrial dysfunction was most probably due to the accumulation of the peroxidase FeSOD2. We attribute this effect to the higher level of oxidative damage to components of the electron transport chain in these cells (Fig. 3C).
Organisms have developed many mechanisms to control the uptake and availability of intracellular iron (Hentze et al., 2004), and many key processes occur exclusively within mitochondria (Toyokuni et al., 2017). It is also well established that under conditions of oxidative stress, iron can be released from iron-sulfur clusters (Cantu et al., 2009; Gardner et al., 1995). Interestingly, recent reports have shown that many human disease states, such as cancers, neurodegenerative diseases and exposure to environmental poisons, are associated with increases in the levels of iron within mitochondria due to oxidative stress (Cantu et al., 2009; Chiang et al., 2017; Toyokuni et al., 2017). In contrast, mechanisms that regulate homeostatic levels of cellular manganese are less clear (Horning et al., 2015). In mammalian cells, excessive manganese is sequestered into the endoplasmic reticulum and Golgi apparatus (Horning et al., 2015). And, while accumulation of manganese in those organelles is cytotoxic (Seo et al., 2013), this toxicity is observed only in experiments carried out with supraphysiological millimolar concentrations added to the medium. In our work, we detected increases in intracellular total manganese in parental and the SOD2-overexpressing MCF-7-derived cell lines supplemented with 25 μM MnCl2 for 21 days. We also identified a small 10% decrease in mitochondrial respiration in the parental cell line cultivated with MnCl2, but we also detected an overall 50% increase in mitochondrial function in Mn11 cells. More importantly, the growth curves of cells supplemented with manganese were no different from those of the age-matched cells cultured in regular medium. Overall this indicates that at 25 μM manganese, manganese was essentially non-toxic to the cells used in this work.
Manganese metabolism is known to overlap with that of iron, since both metals compete for absorption, metabolism and excretion at the cellular level (Fitsanakis et al., 2010; Park et al., 2013; Seo et al., 2013; Sigel and Sigel, 2000). In this work, we show that the low level of manganese in regular media is sufficient for the MCF-7-derived parental cell line, neo, to have SOD2 mostly incorporated with manganese. However, overexpression of SOD2 in the Mn11 cell line, to levels often observed in some cancers (Hart et al., 2015), leads to the incorporation of iron into SOD2 due to limited manganese availability. Even though iron was being incorporated into SOD2, Mn11 cells were not starved for iron since microarray data analyses and the abundance of the iron transporters DMT1 and transferrin receptor, known to be upregulated when cells are under iron starvation (Gunshin et al., 2001; Hentze et al., 2004), did not show any perturbation in the Mn11 cell line compared to its isogenic parental cell line, neo.
Though it is well established that SOD in bacteria (Beyer and Fridovich, 1991; Ganini et al., 2015) and yeast (Culotta et al., 2006; Luk et al., 2003; Luk et al., 2005) can incorporate iron in lieu of manganese, this is the first study reporting that misincorporation of SOD2 with iron occurs in mammalian cells, that FeSOD2 has drastically different biochemical activity and that this metal switch may be important for the functioning of mitochondria and cellular metabolism known to occur in most cancers. As a cautionary note, our results suggest that the iron-to-manganese balance may also contribute to experimental variability as well as confound the results from studies examining redox homeostasis and signaling. The formation of manganese-dependent SOD2 incorporated with iron is believed to be deleterious to organisms due to the loss of antioxidant superoxide dismutase activity (Aguirre and Culotta, 2012; Culotta et al., 2006; Lee et al., 2013; Luk et al., 2003; Luk et al., 2005; Whittaker, 2003). In our experiments with cells, we show that under different oxidative-stress conditions, Mn11 cells with FeSOD2 lose viability even though those cells had approximately twofold higher SOD2-antioxidant activity compared to the control cell line, neo (Fig. 2C). Even though authors have claimed that the loss of MnSOD2 superoxide dismutase activity conferred cytotoxicity 38,40,42,43, our findings show that the formation of FeSOD2 is the most important cause for oxidative stress. The iron incorporation into SOD2 promotes the switch of the canonical antioxidant SOD2 into a deleterious, prooxidant peroxidase, which leads to oxidative damage in the presence of H2O2 and, as we have shown, an increase in the susceptibility to oxidative stress.
Using mice, we showed that by decreasing the ratio of manganese-to-iron in the diet of mice, liver SOD2 incorporated iron (Fig. 7F). In turn, the peroxidase FeSOD2 in the liver of animals induced higher levels of mitochondrial free radicals and mitochondrial dysfunction (Fig. 7B–D). Due to the nature of the natural-ingredient diet NIH-31, experiments with a customized diet completely devoid of manganese were not feasible; however, the removal of the additional supplementation of manganese in the standard NIH-31 diet, without altering its iron content, was enough to elicit the formation of FeSOD2 and cause higher levels of free radical formation in the liver mitochondria of mice. It is important to emphasize that even though animals fed the manganese-deprived diet appeared physiologically normal, they showed significantly higher levels of the peroxidase FeSOD2 (Fig. 7F) and higher levels of mitochondrial protein free radicals (Fig. 7B). In the animals fed an iron-enriched diet, FeSOD2 was the main form of SOD2 found in the liver of mice and, as expected, induced high levels of mitochondrial protein free radicals (Fig. 7B–C), mitochondrial dysfunction (Fig. 7D) and physiological alterations (Fig. S5B). Our customized iron-enriched diet had ten times more iron than the regular mouse diet NIH-31. Anemic humans often are given high doses of iron, but since those patients are defective in iron absorption/retention/metabolism, they are unlikely to suffer from iron accumulation and poisoning; however, an extrapolation for the equivalent daily dose as tablets of over-the-counter iron supplement (usually 65 mg elemental iron/tablet) for regular individuals is 1.1 tablet or 69 ppm iron for 1-to 3-year-old infants, 1.23 tablets or 80 ppm for men older than 19 years, and 2.8 tablets or 180 ppm for women from 19–50 years (Trumbo et al., 2001). The dose of iron used in our experiments in mice (ten times higher iron corresponds to 10.8 ppm of daily iron intake) is actually lower than the supplementation to anemic humans.
Even though it is known that, in humans, serum levels of manganese are dependent on both iron and manganese concentrations in the diet (Fitsanakis et al., 2010; Rahman et al., 2013) and endogenous iron status (Park et al., 2013; Rahman et al., 2013), there are no studies with humans exploring metal incorporation into SOD2. Studies are needed to precisely determine the importance of iron incorporation into SOD2 in different human diseases at different ages. One interesting place to start would be the Friedreich’s ataxia disease, a devastating human genetic disorder which main characteristic is iron overload in the mitochondria (Puccio and Koenig, 2000). From the results presented here, it is plausible to assume that the formation of the peroxidase FeSOD2 may predispose humans to oxidative stress as well as to produce changes to the mitochondrial and cellular metabolism that are characteristic of many human diseases.
STAR ★ Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Material requests should be directed to and will be fulfilled by the Lead Contact Dr. Ronald P. Mason, Free Radical Biology Group, NIEHS, NIH, Research Triangle Park, NC 27709, USA (mason4@niehs.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The female cell lines neo and Mn11 (Zhang et al., 1999) were derived from the human breast cancer cell line MCF-7. They were cultivated in RPMI 1640 high glucose (2 g/L) with glutamine (300 mg/L) from Gibco® (Thermo Scientific, Grand Island, NY), and added streptomycin (100 mg/L)/penicillin (100 U/L), 1 mM pyruvate, 0.4 mM uridine, 50 μg/mL Geneticin® (Invitrogen™, Thermo Scientific, Grand Island, NY) and 10% FBS (ATCC, Manassas, VA). When indicated, cells were cultivated for at least 21 days with manganese-supplemented medium (regular medium with 25 μM MnCl2). Since the half-life of SOD2 in mouse liver, a high metabolic-rate tissue, is approximately 6 days (Kim et al., 2012), we adopted a minimum of 21 days of supplementation in order to guarantee that most of the SOD2 was endogenously de novo replenished. Cultures cultivated with regular medium or manganese-supplemented medium were age-matched.
In vivo Animal Studies
Male mice C57BL/6J were purchased from Jackson Laboratories (Bar Harbor, ME). Three animals were housed in a cage for one week before the start of the feeding experiment. The animals were housed under controlled and standard conditions of temperature and humidity with a 12 h light/dark cycle. After the first week eating the regular pelleted mouse chow, NIH-31, animals were fed with the customized diets ad libitum. Customized diets were obtained irradiated from Harlan Laboratories Inc. (Madison, WI). Customized diets were based on the composition of the natural-ingredient diet for mice, NIH-31. Control diet, TD.150479, had the standard 150 ppm manganese and 275 ppm iron. Manganese-deprived diet, TD.150480, had 40 ppm manganese and the standard 275 ppm iron. Iron-enriched diet, TD.150481, had 2700 ppm iron and the standard 150 ppm manganese. All animals were treated in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and the animal study proposal was revised and approved by the NIEHS Animal Study Proposal review board.
METHODS DETAILS
Buffers, preparation of cellular homogenates and determination of protein concentrations
In vitro experiments with SOD2 were prepared in Chelex-treated 100 mM phosphate buffer, pH 7.4, with the chelator diethylene triamine pentaacetic acid (DTPA) at 25 μM. Cellular homogenates were prepared with a non-denaturing extraction buffer containing TBS, 0.1% Triton X-100 and complete protease inhibitor cocktail (Roche, Indianapolis, IN). Extraction buffer was added to the cell pellets and lysed by sonication followed by centrifugation at 12,000 g for 10 min at 4°C. The lysates were determined for protein concentration using a BCA protein assay kit from Pierce™ Biotechnology (Thermo Scientific, Rockford, IL).
Expression and purification of human SOD2
Human SOD2 gene was recombined from pDONR 221 into a pDEST 527 expression plasmid (destination vector kindly supplied by Dr. Dominic Esposito, SAIC, Frederick, MD). This vector was transformed into Rosetta 2 DE3 pLacI competent cells by heat shock. Colonies resistant to ampicillin and chloramphenicol were grown in Luria-Bertani broth with the selecting agents. Cultures of bacteria were prepared at OD 0.05 and incubated under agitation (220 rpm) at 37°C until they reached OD 0.6–0.8 (approximately 2h). SOD2 expression was induced by adding 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the cultures and incubating for 4h with agitation (220 rpm) at RT.
SOD2 was purified from native bacterial lysates using HisPur Cobalt Purification kits from Pierce™ Biotechnology (Thermo Scientific, Rockford, IL) following the supplier’s instructions (Supplementary Fig. S1). Purified SOD2 was extensively buffer-exchanged with Chelex-treated 10 mM phosphate buffer, pH 7.4, using ultrafiltration devices (Amicon®, Merk Millipore, Billerica, MA), followed by desalting against the same buffer using Zeba Spin Desalting Columns (Pierce™ Biotechnology, Thermo Scientific, Rockford, IL).
Incorporation of manganese and iron into human SOD2
In vitro manganese and iron incorporation into SOD2 was done as described by Beyer and Fridovich, except the buffer compositions in the dialysis steps were as described by Yamakura and collaborators (Beyer and Fridovich, 1991; Yamakura et al., 1998). After the dialyses, artificially-metallated SOD2 was concentrated (10-kDa cut-off filter devices from Amicon®, Merck Millipore, Billerica, MA) and desalted/buffer exchanged for Chelex-treated 10 mM phosphate buffer, pH 7.4 (Zeba™ spin columns from Pierce™ Biotechnology, Thermo Scientific, Rockford, IL).
Superoxide dismutase activity of MnSOD2
Superoxide dismutase activity of MnSOD2 was measured by the inhibition of cytochrome c reduction by a constant flux of superoxide radicals generated by xanthine oxidase in the presence of xanthine (McCord and Fridovich, 1969). In cellular homogenates, cyanide was used to selectively inhibit the superoxide dismutase activity of Cu,Zn-SOD1.
Metal analyses
Samples were analyzed for metals using inductively coupled plasma with optical emission spectrometry (ICP-OES) or inductively coupled plasma with mass spectrometry (ICP-MS) by the Chemical Analysis Laboratory (CAIS) in the Center for Applied Isotope Studies at the University of Georgia (Athens, GA). Samples were extensively digested using nitric acid before analysis.
Peroxidase activity of FeSOD2
The peroxidase activity of FeSOD2 was assessed by resorufin formation (ε571nm = 54.0 mM−1cm−1 and calculated ε560nm = 85.16 mM−1cm−1) from 100 μM Amplex Red (Molecular Probes®, Thermo Scientific, Grand Island, NY) in the presence of excess H2O2 (ε240nm = 0.0436 mM−1cm−1). Due to possible light-dependent artifacts (Summers et al., 2013; Zhao et al., 2012), samples without H2O2, without SOD2, and complete samples read by a single final measurement (Fig. 1C) were also prepared to determine the possible contribution of an instrumental light-dependent artifact on the final resorufin yield.
SOD2 protein radical formation
The spin trap 5,5′-dimethyl-1-pyrroline N-oxide (DMPO) and immuno-spin trapping were used to tag and detect protein radicals (Gomez-Mejiba et al., 2009; Mason, 2004, 2016) formed by the peroxidase activity of SOD2. ELISAs and Western blots were prepared using 7.5 μg/mL of mouse monoclonal anti-DMPO. ELISA had anti-mouse IgG conjugated to HRP as a secondary antibody (1:500, Thermo Scientific, Grand Island, NY). ELISA was revealed using LumiGlo substrate (KPL, Gaithersburg, MD), and the luminescence was detected on a GenIos plate reader (Tecan, Morrisville, NC). For Western blot, the secondary antibody was anti-mouse IgG conjugated to IRDye 800CW (1:15,000, Li-Cor Biosciences, Lincoln, NE), and the images were obtained on an Odyssey scanner (Li-Cor Biosciences, Lincoln, NE).
Purification of SOD2 from cells
Frozen cell pellets equivalent to 1–4 × 107 cells were resuspended in native extraction buffer [TBS, pH 7.4, 0.1% Triton X-100 and complete protease inhibitor cocktail with EDTA (Roche, Indianapolis, IN)]. Samples were then sonicated for 1 min on ice, and cleared by centrifugation at 10,000 g for 10 min at 4°C. Homogenates were adjusted to 4 mg of protein/mL with native extraction buffer. Ammonium sulfate salt was added to the samples up to 60% of saturation. Samples were then incubated at RT for 30 min before being submitted to centrifugation at 10,000 g for 10 min at 25°C. See Supplementary Fig. S4 for additional details. The supernatant, enriched with SOD2, was then dialyzed using 10kDa Slide-A-lyzer® dialysis cassettes (Pierce™ Biotechnology, Thermo Scientific, Rockford, IL) against a more than 1,000-fold excess of 10 mM Tris buffer, pH 7.2 (3 buffer changes over 24 h). Samples were concentrated using 10-kDa cut-off ultrafiltration devices (Amicon®, Merk Millipore, Billerica, MA). With an ÄKTA FPLC system (GE Healthcare Bio-Sciences, Pittsburgh, PA) set at 1 mL/min and operating with the same buffer used for the dialysis, samples were injected into a MonoQ 10/100 column (anion exchange column from GE Healthcare Bio-Sciences, Pittsburgh, PA). Proteins were eluted from the column using a gradient of NaCl up to 1 M over 20 min, and 0.5 mL fractions were collected for further analyses of SOD2 abundance and purity. Positive fractions for SOD2 (dot blot assay anti-SOD2, Supplementary Fig. S4) were pooled and the buffer exchanged for a 10 mM Chelex-treated phosphate buffer, pH 7.4, using 10-kDa cut-off ultrafiltration devices (Amicon®, Merk Millipore, Billerica, MA). The final SOD2 was 85% pure as judged by SDS-PAGE and Coomassie blue staining.
Mitochondria isolation from cultured mammalian cells
Pelleted frozen cells were used for isolation of mitochondria using a classical differential centrifugation procedure (Darley-Usmar et al., 1987). Frozen cells were resuspended in ice-cold mitochondria isolation buffer (10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid [HEPES] pH 7.4, 250 mM sucrose, 10 mM 2-[2-[2-[2-[bis(carboxymethyl)amino]ethoxy]ethoxy]ethyl-(carboxymethyl)amino]acetic acid [EGTA] pH 7.4, and 5 mg/mL BSA). The solution was transferred into a Dounce tissue homogenizer and after 12 strokes at 600 rpm, cellular debris and unbroken cells were pelleted from the solution by centrifugation at 1,000g for 10 min at 4°C. The supernatants, containing isolated mitochondria, were transferred into a new tube and centrifuged for 10 min at 10,000g and 4°C. The pellets containing the isolated mitochondria were washed once with ice-cold mitochondria isolation buffer without BSA. The final mitochondrial pellets obtained were then resuspended in the same isolation buffer without BSA and kept on ice.
Determination of lactate production and intracellular ATP
Lactate production was determined in cells seeded at 1×105 in 24-well plates. After 24 h, the medium was changed. After an additional 24 h, medium was collected for lactate quantification. Media samples were ultrafiltrated in 10-kDa cut-off filter devices (Amicon®, Merck Millipore, Cambridge, MA), and lactate was quantified using the Lactate Assay Kit (MAK064 from Sigma-Aldrich, St. Louis, MO). ATP was determined in cells seeded in 96-well plates at 2.5 × 103 cells per well. After cells were maintained in a humidified CO2 incubator overnight, the medium was aspirated, and 100 μL of regular or manganese-supplemented RPMI 1640 with pyruvate and uridine was added to each well. Control wells also had 20 mM 2-deoxy-glucose in the RPMI 1640. Plates were incubated for 1h. ATP was quantified using an ATP Determination Kit (Molecular Probes™, Thermo Scientific, Grand Island, NY).
Microarray and gene set enrichment analysis (GSEA)
Total RNA was isolated from cells (independent triplicates) using the RNeasy Midi® kit (QIAGEN, Valencia, CA) with in-column DNAse treatment. Gene expression analysis was conducted using Agilent Whole Human Genome 4×44 arrays (014850, Agilent Technologies, Palo Alto, CA). Total RNA samples were labeled with Cy3 according to the manufacturer’s protocol and hybridized for 17 h in a rotating hybridization oven. Slides were washed and then scanned with an Agilent Scanner. Data were obtained using the Agilent Feature Extraction software (v. 12), using defaults for all parameters.
Cell viability and cell growth determination
Cells seeded in 96- or 24-well plates, 2.5 × 102 and 2.5 × 103 cells per well, respectively, were let to adhere overnight in the humidified CO2 incubator. After treatment (cell viability assays) or at a given time-point (growth curve determination), the vital dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the media at 0.5 mg/mL, and plates were kept in the humidified CO2 incubator for 1h. Media were aspirated and intracellular oxidized MTT, formazan, was solubilized by DMSO for absorbance measurements at 560 nm using 96-well plates. Pretreatment with mitoTEMPO (100 μM) was performed for 1 h before treatment with H2O2 and cell viability assays with MTT. Pretreatment with N-acetyl cysteine (10 mM at pH 7.4) was performed for 3 h. These plates were washed twice with PBS and added of fresh medium before cells were challenged with H2O2 and the cell viability was assessed by the MTT assay.
Determination of mtDNA lesions
Cells were seeded in 6-well plates at 1.5 × 106 cells per well. After being treated, cells were trypsinized and washed once with PBS. Cell pellets were frozen at −80°C. Mitochondrial DNA was isolated using the Genomic-tip 20/G and the Genomic DNA Buffer Set Kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. Quantification of mtDNA lesions was carried out using mitochondrial-specific primers and the QPCR technique described by Santos et al. (Santos et al., 2006).
Clinical chemistry of mice serum and hemoglobin determination in total blood
Serum alanine transaminase (ALT), creatinine, glucose, sodium and total cholesterol were determined in an automated clinical chemistry analyzer Olympus AU400e, from Beckman Coulter Inc. (Irving, TX). All reagents were from Beckman Coulter Inc. (Melville, NY). Serum iron and total iron binding capacity (TIBC) were determined in the same clinical chemistry analyzer using reagents from Sekisui Diagnostics (Framingham, MA). Hemoglobin in total blood was determined using the classical Drabkin’s method (reagent from Sigma-Aldrich, St. Louis, MO) using a standard curve of bovine hemoglobin (Sigma-Aldrich, St. Louis, MO).
Mitochondrial isolation of mouse liver
Mitochondria from liver were isolated from freshly harvested livers using the classical differential centrifugation procedure previously described for mitochondria isolation from cells in culture (Darley-Usmar et al., 1987). Instead, using a pair of surgical scissors, freshly harvested livers were finely chopped into the ice-cold mitochondria isolation buffer before subjected to the Dounce homogenizer.
Activity of OXPHOS complexes and immunoprecipitation of Complex I
Activities of isolated OXPHOS complexes were determined using classical spectrophotometric assays using homogenates of isolated liver mitochondria (Trounce et al., 1996). Activity of Complex I+III was determined by the rotenone-sensitive rate of cytochrome c reduction (ε at 550 nm = 18.7 mM−1.cm−1). Mitochondrial homogenates (50 μg/mL in 50 mM Tris buffer, pH 8.0) in the presence of BSA (0.25 mg/mL), NADH (0.2 mM) and cyanide (0.3 μM), were added of cytochrome c at 50 μM. After 5 min of baseline recording for cytochrome c reduction at 550 nm, rotenone at 2.5 μM was added to the samples and absorbance was followed for an additional 5 min. The difference in rate for the sample after and before rotenone was used to calculate the specific activity of Complex I+III. Activity of Complex II+III was determined by following for 5 min the cytochrome c reduction (ε at 550 nm = 18.7 mM−1.cm−1) of samples containing mitochondrial homogenate (50 μg/mL in 25 mM potassium phosphate buffer, pH 7.2 with 5 mM MgCl2) added of rotenone (2.5 μM), succinate (20 mM), cyanide (2 mM), and finally cytochrome c (35 μM). Citrate synthase activity was determined following for 3 min the formation of thio-nitrobenzoate at 412 nm (ε at 412 nm = 13.6 mM−1.cm−1) of samples containing mitochondrial homogenate (10 μg/mL in 200 mM Tris buffer, pH 8.0), acetyl coenzyme A (62 μg/mL), 5,5′-dithiobis(2-nitrobenzoic acid) (0.1 mM), Triton X-100 (0.1 %, V/V), and finally oxaloacetate (0.25 mM).
For the isolation of Complex I, we used a Complex I Immunocapture Kit from Abcam (Cambridge, MA) with lauryl maltoside from Abcam (Cambridge, MA) following the manufacturer’s instructions.
Bioenergetics of cells in culture
Mitochondrial respiration of cells in culture was determined using the SeaHorse Metabolic Analyzer (Agilent Technologies, Santa Clara, CA). Cells were seeded at 5×104 per well onto 24-well SeaHorse plates using regular RMPI 1640 medium with or without manganese. Plates were incubated in CO2-incubators at 37°C overnight. In the following day, media was changed to the SeaHorse media containing 25 mM glucose and 1 mM pyruvate, pH 7.4. Cell plates were incubated in a non-CO2 incubator at 37°C for 40 minutes before transferred into the SeaHorse Analyzer. A plate containing probes is used to measure media acidification and oxygen consumption in real-time. After recording media acidification (glycolysis) and initial oxygen consumption (basal respiration) three times for 3 minutes, cells were sequentially exposed to oligomycin (10 μM), CCCP (10 μM) and a mixture of rotenone and antimycin (10 μM each). Oligomycin inhibits oxygen consumption related to ATP synthesis, the uncoupler CCCP induces maximal mitochondrial respiration, and the mixture of rotenone and antimycin ultimately inhibits mitochondrial respiration. Total mitochondrial respiration was calculated by subtracting the levels seen after addition of rotenone and antimycin from the basal oxygen consumption levels. Spare mitochondrial capacity was calculated by subtracting the basal oxygen consumption rate from the high levels of maximal mitochondrial oxygen respiration seen after the CCCP addition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Origin 6.0. Data obtained with SOD2 protein, cell cultures and in vivo animals are represented as means ± standard deviation and were analyzed using Student’s t-test. Assessment of differences between more than two samples, were calculated using ANOVA statistics with post-hoc tests based on Student’s t-test. Values of p-value lower than 0.05 were considered significant. All in vitro and cell culture experiments were performed at least with three independent replicates and assays were performed with three pseudoreplicates (n=3 from 3 preparations or cell cultures). Experiments were independently repeated at least three independent days. Any additional details are included in the figure legends. For the experiments using animals, tissues of three animals of each group were processed on each day. Assays with isolated mitochondria or whole tissue homogenates were performed in three pseudoreplicates each day (n=3 from 3 animals). Experiments were repeated at two independent days. For microarray analysis, the scanner reads were background subtracted and normalized in R using limma (Ritchie et al., 2015). Results of Gene Set Enrichment Analysis (GSEA) of the full transcriptome of cells are reported for the gene sets with p-value lower than 0.001 and False Discover Rate (FDR) lower than 0.25. For additional details about the microarray data processing and GSEA analysis, please refer to the Data and Software Availability session.
DATA AND SOFTWARE AVAILABILITY
Microarray data processing
Data was processed using R (version 3.1.3) and RStudio (version 0.98.1103). Background correction, using normexp and the saddle method, and data normalization, using the quantile method, were done using the limma package (version 3.22.6) (Ritchie et al., 2015). Annotation was made using the hgug4112a database. Heat maps were prepared in R using the gplot package (version 2.16.0).
GSEA analyses
Differences in log2-transformed intensities between the cells grown with and without manganese supplementation were calculated in R and RStudio. GSEA (Subramanian et al., 2005) was performed with the full transcriptome data set for the two contrasts, Mn11 differences versus neo differences, using the full GO annotation gene set database, c5.all.v5.0.symbols (Ashburner et al., 2000), 1,000 permutations, and defaults for other parameters. Top gene sets with False Discovery Rate (FDR) lower than 0.25 and p-value lower than 0.001 were ranked according to the Normalized Enrichment Score (NES). The original microarray data is available at the Gene Expression Omnibus under the GEO accession code GSE106206.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies and Immunoprecipitation kits | ||
| Anti-DMPO from mouse | This laboratory | N/A |
| HRP-conjugated anti-mouse | Thermo Scientific | Cat#32430, RRID:3AB_1185566 |
| IRDye® 800 CW-conjugated anti-mouse | LI-COR Biosciences | Cat#926-32212, RRID: AB_621847 |
| Anti-SOD2 from rabbit | Abcam | Cat#ab13533, RRID: AB_300434 |
| IRDye® 800 CW-conjugated anti-rabbit | LI-COR Biosciences | Cat#926-32213, RRID: AB_621848 |
| IRDye® 680 LT-conjugated anti-mouse | LI-COR Biosciences | Cat#926-68022, RRID: AB_10715072 |
| Complex I Immunocapture Kit | Abcam | Cat#ab109711 |
| Anti-GAPDH from mouse | Abcam | Cat#ab125247, RRID: AB_11129118 |
| Anti-murine Total OXPHOS Cocktail | Abcam | Cat#ab110413, RRID: AB_2629281 |
| Anti-Lamin B1 from mouse | Abcam | Cat#ab16048, RRID: AB_443298 |
| Alexa 488-conjugated anti-mouse | Invitrogen | Cat#A11001, RRID: AB_2534069 |
| Anti-Complex III (UQCRC2) from mouse | Abcam | Cat#ab14745, RRID: AB_2213640 |
| Anti-Transferrin receptor from rabbit | Abcam | Cat#ab84036, RRID: AB_10673794 |
| Anti-DMT1 from mouse | Abcam | Cat#ab55735, RRID: AB_2239227 |
| Chemicals and Recombinant Proteins | ||
| Human SOD2 | This study | N/A |
| Chelex® 100 Resin | Bio-Rad | Cat#142-2832 |
| Protease inhibitor cocktail, Complete Mini | Roche | Cat#10948000 |
| Manganese(II) Chloride hydrate | Aldrich | Cat#529680 |
| Ammonium iron(II) sulfate | Aldrich | Cat#203505 |
| Equine cytochrome c | Sigma-Aldrich | Cat#C2506 |
| Nitric acid 70%, purified by redistillation | Aldrich | Cat#225711 |
| Amplex® Ultra Red | Molecular Probes | Cat#A36006 |
| DMPO | Dojindo Laboratories | Cat#D048 |
| BSA | Roche | Cat#03 117 057 001 |
| RPMI 1640 with glutamine | Gibco, Thermo Scientific | Cat#11875-093 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat#P0781 |
| Sodium Pyruvate (100 mM) | Sigma-Aldrich | Cat#S8636 |
| Uridine | Sigma-Aldrich | Cat#U3003 |
| Geneticin | Gibco, Thermo Scientific | Cat#10131-035 |
| Fetal Bovine Serum | ATCC | Cat#30-2020 |
| Lauryl Maltoside | Abcam | Cat#ab109857 |
| MitoTEMPO | Sigma-Aldrich | Cat#SML0737 |
| N-acetyl cysteine | Sigma-Aldrich | Cat#A7250 |
| EGTA | Sigma-Aldrich | Cat#03777 |
| HEPES | Sigma-Aldrich | Cat#H7006 |
| Sucrose | Sigma-Aldrich | Cat#S9378 |
| Acetyl coenzyme A lithium salt | Sigma-Aldrich | Cat#A2181 |
| 5,5′-Dithiobis(2-nitrobenzoic acid) | Sigma-Aldrich | Cat#D218200 |
| Triton™ X-100 | Sigma-Aldrich | Cat#X100 |
| Oxaloacetate | Sigma-Aldrich | Cat#O7753 |
| KCN | Sigma-Aldrich | Cat#60178 |
| NADH | Sigma-Aldrich | Cat#N4505 |
| Succinate | Sigma-Aldrich | Cat#S2378 |
| Oligomycin A | Sigma-Aldrich | Cat#75351 |
| CCCP | Sigma-Aldrich | Cat#C2759 |
| Rotenone | Sigma-Aldrich | Cat#R8875 |
| Antimycin A | Sigma-Aldrich | Cat#A8674 |
| SeaHorse XF base medium | Agilent | Cat#102353-100 |
| Critical Commercial Assays | ||
| Amicon® Ultra, centrifugal filters | Thermo Scientific | Cat#UFC900324 and Cat#UFC801024 |
| Slide-A-Lyzer® Dialysis Devices | Thermo Scientific | Cat#66380 and Cat#66810 |
| Zeba spin columns | Thermo Scientific | Cat#89892 |
| BCA Protein Assay kit | Thermo Scientific | Cat#23225 |
| LumiGlo® Chemiluminescent Substrate Kit | KPL | Cat#54-61-01 |
| Lactate Assay kit | Sigma-Aldrich | Cat#MAK064 |
| ATP Determination Kit | Molecular Probes | Cat#A22066 |
| Rneasy Midi® Kit | QIAGEN | Cat#75144 |
| QIAshredder | QIAGEN | Cat#79654 |
| RNAse-free DNAse | QIAGEN | Cat#79254 |
| Whole Human Genome 4×44 Array Kits | Agilent | Cat#G4845A |
| Genomic-tip 20/G | QIAGEN | Cat#10223 |
| Genomic DNA Buffer Set Kit | QIAGEN | Cat#19060 |
| HisPur Cobalt Purification Kits | Thermo Scientific | Cat#89969 |
| ALT Blood Chemistry Kits | Beckman Coulter Inc. | Cat#OSR6607 |
| Creatinine Blood Chemistry Kits | Beckman Coulter Inc. | Cat#OSR6678 |
| Glucose Blood Chemistry Kits | Beckman Coulter Inc. | Cat#HE10900 |
| Sodium Blood Chemistry Kits | Beckman Coulter Inc. | Cat#A28937 |
| Total Cholesterol Blood Chemistry Kits | Beckman Coulter Inc. | Cat#OSR6116 |
| Iron Blood Chemistry Kits | Sekisui Diagnostics | Cat#157-30 |
| TIBC Blood Chemistry Kits | Sekisui Diagnostics | Cat#I7517-150 |
| Seahorse XF24 FluxPak mini | Agilent | Cat#100867-100 |
| Deposited data | ||
| Raw data for neo and Mn11 with and without manganese supplementation | This paper | GEO: GSE106206 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106206 |
| Experimental Models: Cell Lines | ||
| Human: neo and Mn11 female cell lines (MCF-7 derived) | Zhang et al., 1999 | N/A |
| Experimental Models: Organisms/Strains and Animal Chow | ||
| C57BL/6J | Jackson Laboratories | Cat#000664 |
| Control diet | Harlan Laboratories Inc. | Cat#TD.150479 |
| Manganese-deprived diet | Harlan Laboratories Inc. | Cat#TD.150480 |
| Iron-enriched diet | Harlan Laboratories Inc. | Cat#TD.150481 |
| Software and algorithms | ||
| Origin 6.0 Professional | Microcal Software, Inc. | https://www.originlab.com/ |
| Odyssey imaging system | LI-COR Biosciences | LI-COR Biosciences |
| R (version 3.1.3) | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Rstudio (version 0.98.1103) | RStudio, Inc. | https://www.rstudio.com/ |
| Limma package for R (version 3.22.6) | Ritchie et al., 2015 | http://bioconductor.org/packages/release/bioc/html/limma.html |
| Human reference database, hgug4112a (version 3.2.3) | Carlson M. Agilent ‘Human Genome, Whole’ annotation data. R package | https://bioconductor.org/packages/release/data/annotation/html/hgug4112a.db.html |
| Gplots package for R (version 2.16.0) | R package | https://cran.r-project.org/package=gplots |
| Gene Set Enrichment Analysis (GSEA), desktop application (version 3.0) | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Gene Ontology annotation, gene set for GSEA (database c5.all.v5.0.symbols) | Ashburner et al., 2000 | http://software.broadinstitute.org/gsea/downloads.jsp |
Supplementary Material
Significance.
Free radicals and oxidative stress are associated with many human diseases. Mitochondria are the primary source of free radicals in cells, as superoxide radical anion (O2•−) is constantly produced during respiration. SOD2 incorporated with manganese (MnSOD2) is the primary mitochondrial antioxidant that accelerates the removal of O2•−. However, it is well-known that increases in SOD2 expression promote oxidative stress, thus SOD2 also has an important prooxidant effect in cells and in vivo. We demonstrate here that the prooxidant effect of higher expression of SOD2 is due to the formation of SOD2 incorporated with iron. FeSOD2 is a peroxidase that utilizes H2O2 to promote damage and free radical generation. A switch in metal cofactor from manganese to iron changes SOD2 from an antioxidant to a prooxidant enzyme. Cells in culture overexpressing SOD2 showed FeSOD2 formation in mitochondria. Accumulation of FeSOD2 led to a shift in cellular metabolism from oxidative phosphorylation to glycolysis due to mitochondrial dysfunction. Cells with FeSOD2 were more susceptible to oxidative stress. Mice accumulated FeSOD2 in liver when fed a manganese-deficient diet or an iron-enriched diet. Their liver mitochondria had high levels of mitochondrial Complex I oxidation suggestive of mitochondrial dysfunction and oxidative damage. We show that the formation of FeSOD2 converts an antioxidant defense (MnSOD2) into a prooxidant enzyme (FeSOD2) that leads to cellular changes seen in multiple human diseases, such as cancers and iron overload diseases.
Highlights.
Mitochondrial SOD2 incorporated with iron (FeSOD2) is a prooxidant peroxidase.
Cells and animals accumulate FeSOD2 when manganese-to-iron levels are low.
FeSOD2 in cells and animals leads to oxidative stress and mitochondrial dysfunction.
Acknowledgments
This research was supported by the Intramural Research Program of the NIEHS/NIH.
The authors acknowledge Dr. Larry Oberley (in memoriam) and Dr. Frederick Domann from the University of Iowa for providing us the neo and Mn11 cell lines; Dr. Ann Motten, Dr. Marilyn Ehrenshaft and Ms. Mary Mason for helping in the revision of the manuscript; Ms. Jean Corbett, Dr. Robert Petrovich and Ms. Lori Edwards for valuable technical assistance; and Dr. Kevin Gerrish and Dr. Pierre R. Bushel for discussion and assistance regarding the microarray analysis. The authors declare no competing financial interests. This research was supported by the Intramural Research Program of the NIEHS/NIH.
Abbreviations
- A.U
arbitrary units
- ATP5A
ATP synthase subunit alpha
- BCA
bicinchoninic acid
- DAPI
4,6-diamidino-2-phenylindole
- DFO
deferoxamine mesylate
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- DMT1
natural resistance-associated macrophage protein 2 or NRAM2
- DTPA
diethylenetriaminepentaacetic acid
- EDTA
ethylenediaminetetraacetic acid
- ETC
electron transport chain
- FDR
false discovery rate
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GO
gene ontology
- GOX
glucose oxidase
- GSEA
gene set enrichment analyses
- ICP-MS
inductively coupled plasma mass spectrometry
- ICP-OES
inductively coupled plasma optical emission spectrometry
- MCF-7
Michigan Cancer Foundation-7 cell line
- MTCO1
cytochrome c oxidase subunit 1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NDUFB8
NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 8
- NES
normalized enrichment score
- ES
enrichment score
- OD
optical density
- OXPHOS
oxidative phosphorylation
- PBS
phosphate-buffered saline
- RPMI 1640
Roswell Park Memorial Institute medium 1640
- SDHB
succinate dehydrogenase [ubiquinone] iron-sulfur subunit
- SOD2
mitochondrial superoxide dismutase [manganese dependent]
- TBS
Tris-buffered saline
- UQCRC2
cytochrome b-c1 complex subunit 2
Footnotes
Author contributions
D.G. and J.H.S. performed experiments and data analysis. R.P.M. supervised the project. D.G. and M.B. wrote the manuscript. D.G., J.H.S., M.B. and R.P.M. discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre JD, Culotta VC. Battles with iron: manganese in oxidative stress protection. J Biol Chem. 2012;287:13541–13548. doi: 10.1074/jbc.R111.312181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansenberger-Fricano K, Ganini D, Mao M, Chatterjee S, Dallas S, Mason RP, Stadler K, Santos JH, Bonini MG. The peroxidase activity of mitochondrial superoxide dismutase. Free Radic Biol Med. 2013;54:116–124. doi: 10.1016/j.freeradbiomed.2012.08.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barban S, Schulze HO. The effects of 2-deoxyglucose on the growth and metabolism of cultured human cells. J Biol Chem. 1961;236:1887–1890. [PubMed] [Google Scholar]
- Beyer WF, Fridovich I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J Biol Chem. 1991;266:303–308. [PubMed] [Google Scholar]
- Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975;54:311–314. doi: 10.1016/0014-5793(75)80928-8. [DOI] [PubMed] [Google Scholar]
- Cantu D, Schaack J, Patel M. Oxidative Inactivation of Mitochondrial Aconitase Results in Iron and H2O2-Mediated Neurotoxicity in Rat Primary Mesencephalic Cultures. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S, Kalinowski DS, Jansson PJ, Richardson DR, Huang ML. Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich’s ataxia. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Connor KM, Hempel N, Nelson KK, Dabiri G, Gamarra A, Belarmino J, Van De Water L, Mian BM, Melendez JA. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 2007;67:10260–10267. doi: 10.1158/0008-5472.CAN-07-1204. [DOI] [PubMed] [Google Scholar]
- Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280:16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- Cox DG, Tamimi RM, Hunter DJ. Gene x Gene interaction between MnSOD and GPX-1 and breast cancer risk: a nested case-control study. BMC cancer. 2006;6:217. doi: 10.1186/1471-2407-6-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dismutases: putting the metal to the pedal. Biochim Biophys Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darley-Usmar VM, Rickwood D, Wilson MT. Mitochondria, a practical approach. Oxford; Washington, DC: IRL Press; 1987. [Google Scholar]
- Fitsanakis VA, Zhang N, Garcia S, Aschner M. Manganese (Mn) and iron (Fe): interdependency of transport and regulation. Neurotox Res. 2010;18:124–131. doi: 10.1007/s12640-009-9130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint DH, Tuminello JF, Emptage MH. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Ganini D, Petrovich RM, Edwards LL, Mason RP. Iron incorporation into MnSOD A (bacterial Mn-dependent superoxide dismutase) leads to the formation of a peroxidase/catalase implicated in oxidative damage to bacteria. Biochim Biophys Acta. 2015;1850:1795–1805. doi: 10.1016/j.bbagen.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- Gomez-Mejiba SE, Zhai Z, Akram H, Deterding LJ, Hensley K, Smith N, Towner RA, Tomer KB, Mason RP, Ramirez DC. Immuno-spin trapping of protein and DNA radicals: “tagging” free radicals to locate and understand the redox process. Free Radic Biol Med. 2009;46:853–865. doi: 10.1016/j.freeradbiomed.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro ME, et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M. Manganese is essential for neuronal health. Annu Rev Nutr. 2015;35:71–108. doi: 10.1146/annurev-nutr-071714-034419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Oberley LW, Oberley TD. Induction of apoptosis by adenovirus-mediated manganese superoxide dismutase overexpression in SV-40-transformed human fibroblasts. Free Radic Biol Med. 2005;39:1128–1141. doi: 10.1016/j.freeradbiomed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics. 2012;11:1586–1594. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larosche I, Chournar A, Fromenty B, Letteron P, Abbey-Toby A, Van Remmen H, Epstein CJ, Richardson A, Feldmann G, Pessayre D, et al. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol. 2009;234:326–338. doi: 10.1016/j.taap.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Jouihan HA, Cooksey RC, Jones D, Kim HJ, Winge DR, McClain DA. Manganese supplementation protects against diet-induced diabetes in wild type mice by enhancing insulin secretion. Endocrinology. 2013;154:1029–1038. doi: 10.1210/en.2012-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Luk E, Carroll M, Baker M, Culotta VC. Manganese activation of superoxide dismutase 2 in Saccharomyces cerevisiae requires MTM1, a member of the mitochondrial carrier family. Proc Natl Acad Sci USA. 2003;100:10353–10357. doi: 10.1073/pnas.1632471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk E, Yang M, Jensen LT, Bourbonnais Y, Culotta VC. Manganese activation of superoxide dismutase 2 in the mitochondria of Saccharomyces cerevisiae. J Biol Chem. 2005;280:22715–22720. doi: 10.1074/jbc.M504257200. [DOI] [PubMed] [Google Scholar]
- Mason RP. Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radic Biol Med. 2004;36:1214–1223. doi: 10.1016/j.freeradbiomed.2004.02.077. [DOI] [PubMed] [Google Scholar]
- Mason RP. Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol. 2016;8:422–429. doi: 10.1016/j.redox.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Menon SG, Sarsour EH, Kalen AL, Venkataraman S, Hitchler MJ, Domann FE, Oberley LW, Goswami PC. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007;67:6392–6399. doi: 10.1158/0008-5472.CAN-07-0225. [DOI] [PubMed] [Google Scholar]
- Miar A, Hevia D, Munoz-Cimadevilla H, Astudillo A, Velasco J, Sainz RM, Mayo JC. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic Biol Med. 2015;85:45–55. doi: 10.1016/j.freeradbiomed.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Park S, Sim CS, Lee H, Kim Y. Blood manganese concentration is elevated in infants with iron deficiency. Biol Trace Elem Res. 2013;155:184–189. doi: 10.1007/s12011-013-9782-9. [DOI] [PubMed] [Google Scholar]
- Puccio H, Koenig M. Recent advances in the molecular pathogenesis of Friedreich ataxia. Hum Mol Genet. 2000;9:887–892. doi: 10.1093/hmg/9.6.887. [DOI] [PubMed] [Google Scholar]
- Rahman MA, Rahman B, Ahmed N. High blood manganese in iron-deficient children in Karachi. Public Health Nutr. 2013;16:1677–1683. doi: 10.1017/S1368980013000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AM, Carrico PM, Mazurkiewicz JE, Melendez JA. Mitochondrial or cytosolic catalase reverses the MnSOD-dependent inhibition of proliferation by enhancing respiratory chain activity, net ATP production, and decreasing the steady state levels of H2O2. Free Radic Biol Med. 2000;29:801–813. doi: 10.1016/s0891-5849(00)00362-2. [DOI] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- Seo YA, Li Y, Wessling-Resnick M. Iron depletion increases manganese uptake and potentiates apoptosis through ER stress. Neurotoxicology. 2013;38:67–73. doi: 10.1016/j.neuro.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel A, Sigel H. Manganese and its role in biological processes. New York: Marcel Dekker; 2000. [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers FA, Zhao B, Ganini D, Mason RP. Photooxidation of Amplex Red to resorufin: implications of exposing the Amplex Red assay to light. Methods Enzymol. 2013;526:1–17. doi: 10.1016/B978-0-12-405883-5.00001-6. [DOI] [PubMed] [Google Scholar]
- Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20:1646–1654. doi: 10.1089/ars.2013.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic Biol Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294–301. doi: 10.1016/S0002-8223(01)00078-5. [DOI] [PubMed] [Google Scholar]
- Vance CK, Miller AF. A simple proposal that can explain the inactivity of metal-substituted superoxide dismutases. J Am Chem Soc. 1998;120:461–467. [Google Scholar]
- Vance CK, Miller AF. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high Em. Biochemistry. 2001;40:13079–13087. doi: 10.1021/bi0113317. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- Whittaker JW. The irony of manganese superoxide dismutase. Biochem Soc Trans. 2003;31:1318–1321. doi: 10.1042/bst0311318. [DOI] [PubMed] [Google Scholar]
- Yamakura F, Rardin RL, Petsko GA, Ringe D, Hiraoka BY, Nakayama K, Fujimura T, Taka H, Murayama K. Inactivation and destruction of conserved Trp159 of Fe-superoxide dismutase from Porphyromonas gingivalis by hydrogen peroxide. Eur J Biochem. 1998;253:49–56. doi: 10.1046/j.1432-1327.1998.2530049.x. [DOI] [PubMed] [Google Scholar]
- Zhang HJ, Yan T, Oberley TD, Oberley LW. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res. 1999;59:6276–6283. [PubMed] [Google Scholar]
- Zhao B, Summers FA, Mason RP. Photooxidation of Amplex Red to resorufin: implications of exposing the Amplex Red assay to light. Free Radic Biol Med. 2012;53:1080–1087. doi: 10.1016/j.freeradbiomed.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.