

Summary
Rapid and efficient synaptic vesicle fusion requires a pool of primed vesicles, the nearby tethering of Ca2+ channels, and the presence of the phospholipid PIP2 in the target membrane. While the presynaptic active zone mediates the first two requirements, it is unclear how fusion is targeted to membranes with high PIP2 content. Here, we find that the C2B domain of the active zone scaffold RIM is critical for action potential-triggered fusion. Remarkably, the known RIM functions in vesicle priming and Ca2+-influx do not require RIM C2B domains. Instead, biophysical experiments reveal that RIM C2 domains, which lack Ca2+ binding, specifically bind to PIP2. Mutational analyses establish that PIP2 binding to RIM C2B and its tethering to the other RIM domains are crucial for efficient exocytosis. We propose that RIM C2B domains are constitutive PIP2-binding modules that couple mechanisms for vesicle priming and Ca2+ channel tethering to PIP2-containing target membranes.
eTOC Blurb (in Brief)
De Jong et. al. demonstrate that the RIM C2B domain is important for neurotransmitter release. RIM C2B binds to the phospholipid PIP2, and this interaction directs synaptic vesicle priming and Ca2+ influx to the PIP2-containing plasma membrane for efficient exocytosis.
Introduction
Information transfer between neurons is initiated by rapid fusion of synaptic vesicles in presynaptic nerve terminals. Synaptic vesicle exocytosis is restricted to the active zone, a protein complex that is tightly attached to the presynaptic membrane and that controls the fusion machinery (Südhof, 2012). The membrane fusion reaction itself is driven by a conserved molecular machinery, composed of SNARE proteins and Munc18, among others (Jahn and Fasshauer, 2012). This machinery is highly homologous to those of secretory pathways in non-neuronal cells and unicellular organisms (Wickner, 2010) that operate without active zone-like scaffolds, and fusion itself is thought to occur independent of active zone proteins (Südhof, 2013).
To ensure the spatiotemporal precision of synaptic vesicle fusion, three fundamental requirements must be met. First, vesicles must undergo maturation steps to become highly fusogenic, broadly referred to as docking and priming (Kaeser and Regehr, 2017; Südhof, 2012). Second, voltage gated Ca2+ channels must be anchored near fusion ready vesicles such that local Ca2+ levels rise and fall quickly in response to a presynaptic action potential for rapid, synchronous fusion triggering (Eggermann et al., 2012). Third, efficient fusion relies on specific lipids, in particular the presence of phosphatidylinositol 4,5-bisphosphate (PIP2) in the target membrane (Hay et al., 1995; Milosevic et al., 2005; Di Paolo et al., 2004). The importance for PIP2 in fusion arises from its ability to boost presynaptic Ca2+ entry and the Ca2+-affinity of the fusion machinery (Van den Bogaart et al., 2012; Suh et al., 2010). In many secretory cells, PIP2 is not evenly distributed across membranes, but is restricted to the plasma membrane, where it forms small clusters (Aoyagi et al., 2005; van den Bogaart et al., 2011; Laux et al., 2000). While the exact distribution of PIP2 within a nerve terminal is not known, PIP2 defines plasma membrane identity (Di Paolo and De Camilli, 2006) and is essential for synaptic vesicle exocytosis (Di Paolo et al., 2004). It is further often hypothesized that PIP2 is clustered at the active zone (van den Bogaart et al., 2011; Lauwers et al., 2016). Hence, mechanisms that target the fusion machinery to PIP2-containing membranes are necessary. While a growing body of knowledge strongly supports the notion that the active zone provides molecular mechanisms for the first two requirements, it is unclear how these active zone functions are targeted to PIP2-containing membranes.
Active zones, which are composed of RIM, RIM-BP, ELKS, Munc13, Piccolo/Bassoon and Liprin-α, control the docking and priming of synaptic vesicles and the nearby anchoring of Ca2+ channels (Südhof, 2012). RIMs recruit Ca2+-channels in a tripartite complex with RIM-BPs (Han et al., 2011; Hibino et al., 2002; Kaeser et al., 2011; Liu et al., 2011). This recruitment is mediated by the central RIM PDZ domain, which binds to Ca2+ channels, and a proline-rich PxxP motif between two C-terminal C2 domains that binds to RIM-BPs, which in turn bind to Ca2+-channels as well (Figure 1A). Via its N-terminal zinc finger domain, RIM activates and anchors Munc13 to control the number of docked and primed vesicles (Andrews-Zwilling et al., 2006; Augustin et al., 1999; Camacho et al., 2017; Deng et al., 2011; Han et al., 2011; Imig et al., 2014). Thus, RIM proteins have evolved as essential scaffolds that participate in these key active zone functions (Figure 1A).
Figure 1. The RIM1 C2B domain is crucial for synaptic vesicle release.

(A) Domain structure of RIM1α (Zn: zinc finger domain with surrounding α-helical regions, PxxP: proline-rich region) and key RIM functions that are assigned to specific domains.
(B) Overview of rescue proteins.
(C, D) Example traces (C) and quantification of average amplitudes (D) of single evoked IPSCs in cultured hippocampal neurons. cDKO neurons are neurons from RIM1/2 conditional knockout mice infected with a lentivirus expressing Cre recombinase, and rescue proteins were expressed from a second, independent lentivirus. Control neurons are neurons from the same culture infected with a virus expressing truncated, inactive cre. Control n = 6 independent cultures/30 cells, cDKO 6/30, cDKO + RIM WT 6/31, cDKO + RIM ΔC2B 6/32.
(E, F) Example traces (E) and average IPSC paired pulse ratios (F) at various interstimulus intervals. Control n = 4 independent cultures/21 cells, cDKO 4/18, cDKO + RIM WT 4/21, cDKO + RIM ΔC2B 4/21. *** p < 0.001 genotype vs cDKO by two-way ANOVA.
(G–J) Example images (G) and quantification (H–J) of synapses using STED microscopy. Average RIM intensity within Bassoon objects (H), average Bassoon intensity within Bassoon objects (I), and average surface area of Bassoon objects (J) are shown. Control n = 3 independent cultures/10 fields of view (23.5 μm x 23.5 μm, typically containing 100 – 300 Bassoon objects per field of view), cDKO 3/7, cDKO + RIM WT 3/8, cDKO + RIM ΔC2B 3/11.
(K–M) Schematic representation (K) and intensity profile analysis (L, M) of side view synapses. Shaded area represents the ROI, the dotted line indicates distance = 0 nm centered to the peak of Bassoon labeling. Average intensity profiles of RIM (L) and Bassoon (M) are shown. Data are shown as mean (solid line) ± SEM (shaded area). Control n = 3 independent cultures/67 ROIs, cDKO 3/72, cDKO + RIM WT 3/83, cDKO + RIM ΔC2B 3/87.
All data are mean ± SEM, * p < 0.05, ** p < 0.01, *** p < 0.001 analyzed by one-way ANOVA (Kruskal Wallis; D, H–J) unless otherwise noted, all comparisons to cDKO. For assessment of synaptic transmission upon expression of RIM WT and RIM ΔC2B in wild type neurons and analysis of RIM ΔC2A rescue, see Figure S1, for STED overview images see Figure S2A.
The two RIM C2 domains, termed C2A and C2B, remain poorly understood despite being highly conserved sequences across RIM proteins (Wang and Südhof, 2003). C2 domains are widely known as Ca2+ sensors (Südhof, 2013), but RIM C2 domains cannot bind Ca2+ (Dai et al., 2005; Guan et al., 2007). Several interactors for RIM C2A or C2B have been identified in vitro. These include Liprin-α, SNAREs, Ca2+ channel β-subunits, and synaptotagmins (Coppola et al., 2001; Kaeser et al., 2012; Kiyonaka et al., 2007; Schoch et al., 2002), and RIM C2B domains also dimerize (Guan et al., 2007), but the physiological relevance of these interactions at synapses remains unclear. In C. elegans, removal of the C2B domain leads to a behavioral phenotype similar to RIM null mutants (Koushika et al., 2001). Together with the high conservation, this finding suggests important roles for RIM C2B, but it is unknown if this is due to defects in expression and localization of RIM, or due to a more direct role in synaptic transmission. Notably, RIM C2B contains a lysine-rich polybasic stretch with high homology to several other C2 domains (Corbalan-Garcia and Gómez-Fernández, 2014). In such C2 domains, this motif binds to PIP2 (Bai et al., 2004; Coudevylle et al., 2008; Guerrero-Valero et al., 2009), and mutations in this motif lead to biochemical defects and impaired synaptic transmission (Van den Bogaart et al., 2012; Groffen et al., 2010; Li et al., 2006).
Given the central scaffolding role for RIM at the active zone, constitutive binding of its C2 domains to PIP2 could provide an attractive mechanism to target synaptic vesicle docking, priming and Ca2+ channel tethering to PIP2-containing membranes. Here, we tested this hypothesis and found that both RIM C2 domains bind PIP2. Deleting the RIM C2B domain or disrupting PIP2 binding to the RIM C2B domain strongly impaired synaptic transmission, whereas similar manipulations of the RIM C2A domain did not have strong functional effects. Remarkably, while binding of the C2B domain to PIP2 was required for rescue of action potential triggered vesicle fusion, removing C2B did not impair the known RIM functions in vesicle priming and Ca2+-influx. Finally, RIM C2B domains were inactive unless they were attached to the N-terminal RIM domains. We propose that RIM C2B domains tether essential active zone functions to PIP2-containing membranes for rapid and efficient exocytosis.
Results
The RIM C2B domain is crucial for synaptic vesicle release
Based on the high conservation of the RIM C2B domain (Wang and Südhof, 2003), the strong behavioral defects upon RIM C2B deletion in C. elegans (Koushika et al., 2001), and the general importance of C2 domains in vesicle fusion (Südhof, 2013), we hypothesized that RIM C2B is critical for synaptic vesicle release. To test this hypothesis, we expressed full length RIM1α (RIM WT) and a RIM1 mutant lacking the C2B domain (RIM ΔC2B) in cultured hippocampal neurons obtained from RIM1/2 conditional knockout mice (Figure 1B). In this preparation, the Rims1 and Rims2 genes contain essential exons flanked by loxp sites, and we expressed Cre recombinase from a lentivirus to remove all RIM1 and RIM2 isoforms (cDKO) (Kaeser et al., 2008, 2011). Control neurons are neurons from the same litter infected with a lentivirus that expressed an inactivated variant of Cre recombinase (Kaeser et al., 2008). We delivered RIM using separate lentiviruses because bicistronic expression of Cre and rescue proteins, as described before (Kaeser et al., 2011), is impractical for full length RIM1α due to the packaging size limit of lentiviruses. This approach yielded reliable removal of endogenous RIM, and exogenous RIM expression reached levels above wild type control neurons (Figure S1B). RIM WT expression fully rescued the amplitude of action potential evoked γ-aminobutyric acid (GABA)-ergic inhibitory postsynaptic currents (IPSCs) and nearly completely restored paired pulse ratios (PPRs, Figures 1C–1F), which are inversely correlated with vesicular release probability (p) (Zucker and Regehr, 2002). RIM ΔC2B was expressed at levels comparable to RIM WT (Figure S1B), but showed no significant increase in the IPSC amplitude compared to cDKO neurons (Figures 1C and 1D). Furthermore, the PPR of RIM ΔC2B was indistinguishable from cDKO at all intervals tested (Figures 1E and 1F). Thus, the RIM C2B domain is necessary to enhance p, and removing C2B abolishes rescue activity. Importantly, expression of RIM WT or RIM ΔC2B in control neurons did not enhance or suppress release (Figures S1C–S1H), indicating that the effects in the rescue conditions are not due to overexpression or dominant negative roles. Furthermore, using the same experimental design, we found that deletion of RIM C2A has no strong effect on synaptic transmission (Figures S1I–S1P). We conclude that the C2B domain of RIM1 is crucial to set p at inhibitory synapses.
RIM C2B is not necessary for localizing RIM to the active zone
We hypothesized that the severe phenotype of RIM ΔC2B rescue might be due to perturbed recruitment of RIM ΔC2B to the active zone. To test this hypothesis, we performed stimulated emission depletion (STED) microscopy on cultured neurons stained for the inhibitory synapse marker vGAT, the active zone marker Bassoon, and RIM (Figures 1G–1M and S2A). RIM signals in control synapses co-localized well with Bassoon. To measure RIM levels at active zones, we detected Bassoon objects within vGAT positive synapses, and measured RIM staining intensities within Bassoon objects. While RIM rescue levels appeared slightly higher than control, we observed no difference between RIM WT and RIM ΔC2B (Figure 1H). In addition, no differences were found in the average Bassoon intensity, or the size of Bassoon objects (Figures 1I, 1J). To evaluate whether RIM ΔC2B localized normally within the presynaptic terminal, we selected synapses in side view, defined as a cluster of vGAT positive vesicles that contains an elongated band of Bassoon at the edge of the cluster (Wong et al., 2018). We then measured the staining intensity profile along a 250 nm wide rectangle perpendicular to the Bassoon band (Figure 1K), and aligned all profiles to the peak Bassoon intensity. Peak intensity of RIM co-localized well with Bassoon in control neurons, and peak intensity of RIM WT and RIM ΔC2B rescue proteins, while higher in intensity than endogenous RIM, did not shift relative to Bassoon (Figure 1L). The Bassoon signal did not change across conditions (Figure 1M). Despite complete absence of RIM in Western blotting (Figure S1B, and (Kaeser et al., 2011)), a small peak remained in cDKO in STED, similar to the 25% background with the same antibody in confocal analyses (Wang et al., 2016). These data establish that the RIM C2B domain is not required for RIM active zone localization.
The RIM C2B domain mediates a new function of RIM
We next assessed whether the RIM C2B domain participates in the two established RIM functions, which are synaptic vesicle priming via recruitment of Munc13 (Andrews-Zwilling et al., 2006; Camacho et al., 2017; Deng et al., 2011), and enhancing Ca2+ influx by tethering of presynaptic Ca2+ channels (Han et al., 2011; Kaeser et al., 2011; Muller et al., 2012). To test if RIM C2B is required to recruit Munc13 to the active zone, we quantified Munc13 levels within Bassoon objects using STED microscopy (Figures 2A–2D and S2B–S2D). RIM cDKO led to a reduction in Munc13 at the active zone, consistent with the reduction of Munc13 in RIM cDKO neurons (Deng et al., 2011). Strikingly, RIM ΔC2B and RIM WT rescue led to Munc13 levels slightly above control (Figure 2B), and the Munc13 peak intensity localized to the active zone in control as well as rescue conditions (Figure 2C). We next measured the size of the primed vesicle pool (readily releasable pool, RRP) by the application of hypertonic sucrose (Rosenmund and Stevens, 1996) and observed that RIM WT and RIM ΔC2B rescued RRP size to similar levels (Figures 2E and 2F). Thus, despite the failure to rescue action potential induced release, RIM ΔC2B is able to recruit Munc13 to active zones and to restore the RRP.
Figure 2. RIM1 C2B domains are dispensable for the functions of RIM in Munc13 recruitment, vesicle priming and Ca2+ influx.
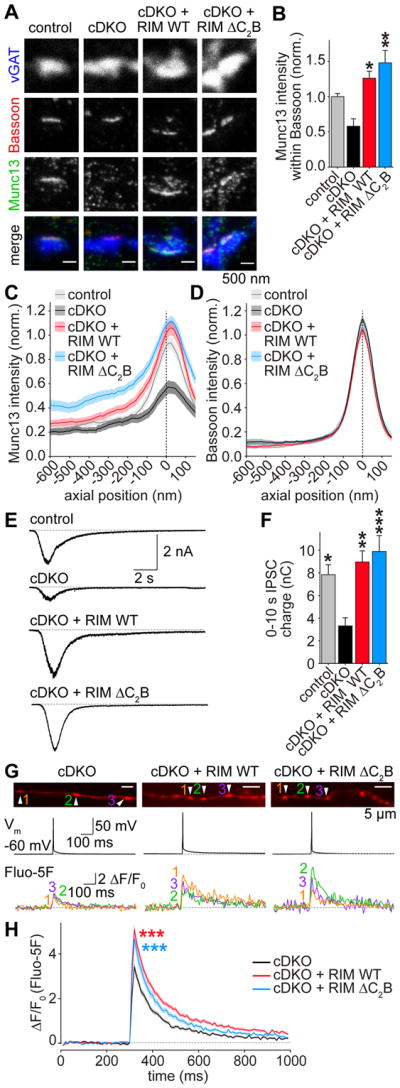
(A, B) Example images (A) and average Munc13 intensity (B) of synapses in STED microscopy. Control n = 3 independent cultures/7 fields of view, cDKO 3/6, cDKO + RIM WT 3/6, cDKO + RIM ΔC2B 3/6.
(C, D) Intensity profile for Munc13 (C) and Bassoon (D) in side view synapses. Control n = 3 independent cultures/60 ROIs, cDKO 3/53, cDKO + RIM WT 3/64, cDKO + RIM ΔC2B 3/66.
(E, F) Example traces (E) and quantification of IPSC charge (F) integrated over 10 s, induced by a focal 10 s puff of hyperosmolar sucrose (500 mM) as a measurement of the RRP. Control n = 3 independent cultures/13 cells, cDKO 3/15, cDKO + RIM WT 3/13, cDKO + RIM ΔC2B 3/13.
(G, H) Example boutons (G) and quantification (H) of presynaptic Ca2+ imaging. Individual cells were filled with Alexa 647 and the Ca2+ indicator Fluo-5F through a patch pipette. In G, images of Alexa 647 fluorescence in axons (top), the somatic membrane potentials (middle) and Fluo-5F fluorescence traces of individual boutons during a single action potential (bottom) in response to a brief somatic current injection are shown. Arrowheads in Alexa 647 images indicate boutons from which Fluo-5F signals at the bottom were obtained. H shows the average Fluo-5F ΔF/F0 during a single action potential. cDKO n = 3 independent cultures/8 cells/153 boutons, cDKO + RIM WT 3/8/160, cDKO + RIM ΔC2B 3/8/93. *** p < 0.001 genotype vs cDKO by 2-way ANOVA, for first 6 frames after stimulation.
All data are mean ± SEM, *p < 0.05, ** p < 0.01, *** p < 0.001 vs cDKO analyzed by one-way ANOVA (Kruskal Wallis; B,F) unless otherwise noted, all comparisons to cDKO. For overview STED images and quantification of Bassoon signals, see Figures S2B–S2D.
To test whether the RIM C2B domain contributes to presynaptic Ca2+ influx, we loaded individual cells with the low affinity Ca2+ indicator Fluo-5F through a patch pipette and measured Ca2+ influx in individual presynaptic boutons in response to a single action potential induced by a brief somatic current injection (Figures 2G and 2H). Both RIM WT and RIM ΔC2B significantly increased presynaptic Ca2+ influx compared to cDKO, and no difference was observed between the two rescue conditions. Thus, the C2B domain is not required to boost presynaptic Ca2+ influx. Taken together, these data establish that the RIM C2B domain is critical to enhance p, but it does so independent of the known roles of RIM in vesicle priming or Ca2+ influx.
RIM C2 domains bind to PIP2
What is the function of RIM C2B domains in synaptic transmission? The RIM C2B domain contains a lysine-rich polybasic sequence that is highly conserved among C2 domains, and this sequence mediates binding to the phospholipid PIP2 in several other C2 domains (reviewed in (Corbalan-Garcia and Gómez-Fernández, 2014)). To test if RIM1 C2B binds to PIP2, we performed liposome binding assays in which we measured co-sedimentation of purified C2B with heavy liposomes (Figures 3A and 3B). While we detected only weak binding of C2B to liposomes composed of phosphatidylcholine (PC) and phosphatidylserine (PS), addition of PIP2 enhanced co-sedimentation of the C2B domain with liposomes, suggesting direct binding of C2B to PIP2. Changing the surface charge of the liposomes by adding extra PS did not alter the amount of binding, suggesting that the interaction is specific for PIP2 and not simply mediated by changes in membrane charge (Figures 3A and 3B). We then tested how different lipid composition of the liposomes influenced binding of the RIM C2B domain using co-floatation assays. We found that the binding was almost entirely specific for PIP2 (Figure 3C), and other lipids, including the phosphoinositides PI and PIP, only showed weak binding.
Figure 3. RIM1 C2B domains bind specifically to the phospholipid PIP2.

(A, B) Example Coomassie stained SDS-PAGE gel (A) and average C2B domain content in the pellet (B) of a lipid co-sedimentation experiment using heavy liposomes of various compositions and purified His-C2B domains. PC: phosphatidylcholine, PS: phosphatidylserine. Averages ± SEM are obtained from 3 independent experiments. Statistical significance was assessed by Students t-test, with * p < 0.05 vs the 75:25:0 condition.
(C) Image of a Coomassie stained SDS-PAGE of a lipid co-floatation assay to test for PIP2 specificity of lipid-C2B binding. The assay was performed with purified RIM1 C2B and various liposome compositions, as indicated.
(D) Analysis of PIP2 binding to RIM1 C2B using NMR spectroscopy. Superposition of 1H-15N HSQC spectra of 15N-labeled RIM1 C2B alone (black contours) and in the presence of 100 μM diC4-PIP2 (orange contours), a water soluble PIP2 analogue.
(E) Superposition of 1H-15 N HSQC spectra of RIM1 C2B acquired in the presence of different diC4-PIP2 concentrations as indicated by the color code. Symbols indicate cross-peak shifts for which the dissociation constants (Kd) in panel F were calculated.
(F) Analysis of the binding affinity of PIP2 to 15N-labeled RIM1 C2B using NMR spectroscopy. Plots of 15N chemical shift perturbations (corresponding to labels in panel E) as a function of diC4-PIP2 concentration for selected cross-peaks. The data for each cross-peak were fitted with a standard single-site binding model, yielding the Kd per cross-peak.
(G) Analytical ultracentrifugation of RIM1 C2B at various concentrations. The c(s) distributions shown were normalized to the total signal in each distribution. The Kd of the dimer is indicated in the figure.
(H) Expansions showing the well-resolved methyl resonances of 1D 1H-NMR spectra of RIM1 C2B alone (left) and in the presence of 200 μM diC4-PIP2 (right). The line widths (in Hz) are indicated next to each resonance and are comparable in the two spectra.
For experiments assessing PIP2 binding of RIM1 C2A, see Figure S3.
We next used nuclear magnetic resonance (NMR) spectroscopy and a water soluble PIP2 analogue (diC4-PIP2) to characterize its interaction with the RIM1 C2B domain. 1H-15N heteronuclear single quantum coherence (HSQC) spectra of C2B in the absence and presence of 100 μM diC4-PIP2 revealed specific cross-peak shifts (Figure 3D), demonstrating a direct interaction. To measure the binding affinity, we repeated the experiment at increasing concentrations of diC4-PIP2 (0–250 μM), which resulted in progressive shifts of the same cross-peaks (Figure 3E). We fitted the perturbations of individual chemical shifts from the cross-peaks that were most affected by diC4-PIP2 binding as a function of diC4-PIP2 concentration, and obtained Kds that ranged from 32 to 79 μM (Figure 3F). The Kd values are within the range of those found for other C2 domains (Van den Bogaart et al., 2012; Montaville et al., 2008), and the variability is expected considering that some shifts are relatively small. These results establish RIM1 C2B as a PIP2 binding module.
It was previously suggested that C2B forms a weak dimer via an interface that partly overlaps with the polybasic sequence (Guan et al., 2007), which thus might interfere with PIP2 binding. Using analytical ultracentrifugation, we found that the C2B homodimer affinity was very weak (Kd ~ 3 mM, Figure 3G), which makes it very unlikely to be physiologically relevant. Additionally, we found that the addition of PIP2 did not change the line widths of methyl resonances of 1D 1H-NMR spectra of RIM1 C2B, indicating that PIP2 does not change the very weak dimerization of C2B (Figure 3H).
We finally characterized the PIP2 binding of RIM1 by analyzing its C2A domain with the same assays (Figures S3A–S3F). We observed a specific interaction of C2A with PIP2-containing liposomes, and titrations monitored by 1H-15N HSQC spectra yielded a Kd value that was comparable to those measured for C2B. Thus, RIM1 can bind to PIP2-containing membranes via its C2A and C2B domains.
Distinct PIP2 binding modes of the RIM C2 domains
We next designed mutations to interfere with PIP2 binding. Since RIM1 C2B contains a polybasic sequence that is conserved among PIP2-binding C2 domains (Figure S3G), we based our mutations on homologous PIP2-binding sequences (Corbalan-Garcia and Gómez-Fernández, 2014). Using lipid co-sedimentation, we found that mutating single or multiple lysine (K) residues to glutamates (E) in the conserved polybasic sequence strongly impaired PIP2 binding (Figures 4A and 4B). From these mutants, we selected a double mutant in which K1513 and 1515 were mutated to E (K1513/1515E, named C2B 2E from hereon) for further analysis, because these lysines are conserved and have been well characterized in homologous C2 domains in biochemical and functional assays (Figures 4C and 4D) (Van den Bogaart et al., 2012; Groffen et al., 2010; Guerrero-Valero et al., 2009; Li et al., 2006). The 1H-15N HSQC spectrum of C2B 2E exhibited only small perturbations compared to that of WT C2B (Figure 4E), showing that the mutation did not perturb the folding of the domain. Importantly, no cross peak shifts were observed after the addition of diC4-PIP2 in RIM1 C2B 2E (Figure 4F), demonstrating that the 2E mutation abolished PIP2 binding.
Figure 4. Mutations in the polybasic sequence of RIM1 C2B abolish PIP2 binding.

(A, B) Example gels (A) and quantification (B) of liposome co-sedimentation assays using His-C2B domains. Binding was quantified as percent of total input, and the signal in the no lipids sample was set to 0% for normalization. Data shown as mean ± SEM from 3 independent experiments. Liposome composition (PC:PS:PIP2): PC/PS: 65:35:0, PC/PS/PIP2 65:30:5.
(C) Sequence alignment (also see Figure S3G) of the polybasic sequence of the C2B domains of rat RIM1 (Uniprot ID Q9JIR4), RIM2 (Q9JIS1), RIM3 (Q9JIR3), RIM4 (Q8CIX1), synaptotagmin-1 (SYT1, P21707) and human extended synaptotagmin 2 (ESYT2, A0FGR8). The lysine residues mutated in RIM1 C2B 2E are highlighted in orange. Conservation of mutated lysines is shown in bold. Numbers on top indicate residue positions in RIM1.
(D) Ribbon diagram of the RIM1 C2B domain showing the locations of the mutated residues (PDB: 2Q3X, (Guan et al., 2007)).
(E) Superposition of 1H-15N HSQC spectra of 15N-labeled RIM1 C2B WT (black contours) and RIM1 C2B 2E (orange contours) domains.
(F) Analysis of PIP2 binding to 15N-labeled RIM1 C2B 2E using NMR spectroscopy. Superposition of 1H-15N HSQC spectra of RIM1 C2B 2E alone (orange contours) and RIM1 C2B 2E in the presence of 100 μM diC4-PIP2 (blue contours).
For mutational analyses of C2A-PIP2 binding and of binding of RIM C2 domains to PIP3 and various PIP2 isomers, see Figure S4.
The polybasic sequence is only partially conserved in RIM1 C2A (Figure S3G). When we tested whether the lysine and arginine (K809 and R811) residues in C2A, at corresponding positions to K1513 and K1515 in C2B, were necessary for PIP2 binding of C2A, we found that mutating them to glutamates (KR mutant) did not completely abolish diC4-PIP2 binding (Figures S4A and S4B). Instead, RIM1 and RIM2 C2A contain two unique arginines at the bottom of the β-sandwich (R834 and R835, Figures S3G and S4A) that could potentially bind to PIP2, and mutation of one of these arginines has been associated with autosomal cone-rod dystrophy in human genetic studies (Johnson et al., 2003). 1H-15N HSQC spectra showed that mutation of both arginines to glutamate (C2A 2E mutant) abolished binding of RIM1 C2A to diC4-PIP2 without perturbing its folding (Figures S4C and S4D). Hence, RIM1 C2 domains, and likely other C2 domains, can bind to PIP2 through different motifs located at distinct regions of the β-sandwich.
Finally, we tested if the binding of the RIM C2 domains to phosphoinositides is restricted to PIP2 phosphorylated at the 4 and 5 positions at the inositol ring (PI(4,5)P2). We found that isomers other than PI(4,5)P2, as well as PIP3, induced only weak or no binding of C2B to liposomes in lipid co-sedimentation assays (Figures S4E – S4H). Hence, phosphoinositide binding of RIM C2B is mostly specific for PI(4,5)P2. Importantly, the 2E mutation of either C2A or C2B inhibited binding to all PIP2 isomers.
The polybasic region of RIM C2B is critical for synaptic vesicle release
To investigate the functional importance of the interactions of RIM C2 domains with PIP2, we generated a RIM1 mutant that harbors the 2E mutations of both C2A and C2B (RIM 4E, Figure 5A). RIM 4E was expressed at levels comparable to RIM WT, was efficiently recruited to synapses, and restored the priming deficit of RIM cDKO (Figures S5A–S5F). The amplitude of evoked IPSCs, however, was severely reduced compared to control and RIM WT rescued cells, and was statistically indistinguishable from that of cDKO neurons (Figures 5A–5C). In addition, RIM 4E showed no rescue of PPRs (Figures 5D and S5G) or release induced by brief action potential trains (10 action potentials at 10Hz, Figures S5H and S5I). Thus, the RIM 4E mutant did not rescue p and phenocopied the RIM ΔC2B rescue experiments (Figures 1, 2).
Figure 5. The PIP2-binding interface of C2B controls synaptic vesicle release.

(A) Overview of rescue proteins.
(B, C) Example traces (B) and quantification of average amplitudes (C) of single evoked IPSCs. Control n = 8 independent cultures/34 cells, cDKO 8/32, cDKO + RIM WT 8/39, cDKO + RIM 4E 8/38.
(D) IPSC paired pulse ratios at various interstimulus intervals. Control n = 3 independent cultures/11 cells, cDKO 3/12, cDKO + RIM WT 3/14, cDKO + RIM 4E 3/14.
(E–H) Identical to A–D, but for mutations in only the C2A (A2E) or C2B (B2E) domains. Control n = 4 independent cultures/15 cells, cDKO 4/16, cDKO + RIM WT 4/17, cDKO + RIM A2E 4/19, cDKO + RIM B2E 4/19.
All data shown as mean ± SEM, *p < 0.05, ** p < 0.01, *** p < 0.001 for IPSC amplitudes analyzed by one-way ANOVA (Kruskal Wallis; C, G), for PPRs analyzed by two-way ANOVA (D, H), all comparisons to cDKO. For expression analysis of rescue proteins, RRP measurements, example traces for PPRs and analyses of stimulation trains, see Figure S5.
To test if the phenotype of RIM 4E is due to mutations in C2A, C2B, or both, we generated variants of RIM in which only C2A (RIM A2E) or C2B (RIM B2E) was mutated (Figure 5E). As observed with RIM 4E, RIM A2E or B2E mutants expressed normally (Figure S5J). While RIM A2E led to significant rescue of the IPSC amplitude, RIM B2E did not show any rescue (Figures 5F and 5G). Similarly, RIM A2E rescued PPRs as efficiently as RIM WT, but RIM B2E did not display any rescue at all (Figure 5H and Figure S5K). Thus, we could not detect a statistically significant defect in synaptic transmission upon abolishing PIP2 binding to C2A, consistent with the finding that removing the C2A domain does not impair rescue (Figures S1H–S1P). However, mutating the polybasic region in RIM C2B alone leads to absence of rescue. This establishes a central role for this motif in synaptic vesicle fusion.
Known protein interactions of RIM C2B do not mediate its role in exocytosis
Is the effect of the C2B 2E mutation solely due to a loss of PIP2 binding, or does the mutation affect other molecular interactions as well? Previous studies have proposed that multiple proteins interact with RIM1 C2B, including Liprin-α (Schoch et al., 2002), synaptotagmin-1 (Coppola et al., 2001; Schoch et al., 2002), the SNARE proteins syntaxin-1 and SNAP-25 (Coppola et al., 2001) and β subunits of Ca2+ channels (Cavβ, Kiyonaka et al., 2007), although several of these interactions could not be reproduced by NMR spectroscopy (Guan et al., 2007). To test if C2B 2E mutations interfere with these interactions, we performed GST affinity purifications from mouse brain, and detected potential interactors by western blotting. We could not detect any binding to SNAREs and CaVβ4 (Figure 6A). To further characterize potential binding to SNAREs, we acquired 1H-15N HSQC NMR spectra of 15N-labeled C2B WT without or with unlabeled syntaxin-1 or SNAP-25 (Figures S6A and S6B). No binding between C2B and these SNARE proteins was observed. We did observe weak Ca2+-dependent binding of C2B to synaptotagmin-1 in GST affinity purification, which was not affected in C2B 2E (Figure S6C) and hence is unlikely to explain the strong impairment in exocytosis of RIM1 C2B 2E. Furthermore, we suspect that this binding is largely artifactual due to polyacidic impurities upon purification of GST-RIM1 C2B (Figures S6D–S6E), which was previously observed for synaptotagmin C2 domains (Ubach et al., 2001). In line with this conclusion, this interaction could not be observed using purified protein preparations and NMR spectroscopy (Guan et al., 2007).
Figure 6. PIP2- but not Liprin-α-binding of RIM C2B is required for rescue.

(A) Example images of western blots of GST affinity purifications to test for binding of RIM C2 domains to syntaxin-1, SNAP-25, synaptobrevin/VAMP-2 (syb-2), Cavβ4 subunits and Liprin-α from mouse brain lysate. Each experiment was performed in at least 3 independent repeats.
(B, C) Representative gels (B) and quantification (C) of affinity binding assays with GST-Liprin-α3-LH1 -LH2 on beads and His-tagged RIM C2B variants in solution. Arrows in B indicate GST- Liprin-α3-LH1 and -LH2 bands; arrowheads indicate His-RIM C2B band. Average molar His-RIM C2B/GST-Liprin-α3-LH ratio was obtained from 3 independent replicates.
(D, E) Representative gels (D) and quantification (E, average of 3 independent repeats) of lipid co-sedimentation of RIM1, RIM3 and RIM4 His-C2B domains with heavy liposomes with increasing content of PIP2. Liposome composition (PC:PS:PIP2): 0% PIP2 (65:35:0), up to 5% PIP2 (65:30:5). Dotted line in E represents a Hill-fit through the data points.
(F) Overview of rescue proteins.
(G, H) Example traces (G) and quantification of average amplitudes (H) of single evoked IPSCs. Control n = 4 independent cultures/21 cells, cDKO 4/21, cDKO + RIM WT 4/22, cDKO + RIM 1–3 3/16, cDKO + RIM1-4 4/21.
(I) IPSC paired pulse ratios at various interstimulus intervals. Number of observations as in H.
All data shown as mean ± SEM, * p < 0.05, ** p < 0.01, *** p < 0.001 for protein interactions by Students t-test (C), for IPSC amplitudes analyzed by one-way ANOVA (Kruskal Wallis; H), for PPRs analyzed by two-way ANOVA (I); all comparisons to cDKO. For additional analyses of RIM C2B-SNARE interactions and synaptotagmin-1 binding, see Figure S6, for assessment of rescue RIM expression, example traces of PPRs, and a rescue experiment in which the RIM C2B domain was replaced by the Extended synaptotagmin 2 C2C domain, see Figure S7A–S7J.
As reported previously (Schoch et al., 2002), RIM1 C2B bound to Liprin-α in GST affinity purifications, and we found that the 2E mutation in C2B did not impair binding (Figure 6A). While testing binding of the C2B domains of RIM3 and RIM4, two short RIM homologues that have no known function in synaptic transmission, we found that neither bound to Liprin-α (Figure 6A). To further characterize the RIM C2B - Liprin-α interaction, we performed affinity binding assays using the Liprin-α3 LH1 or LH2 domain fused to GST (GST-Liprin-α3-LH1 and -LH2) on beads, and His-tagged C2B variants in solution, which do not show the ribonucleic acid contaminants that lead to artifacts using GST-C2 domains (Figures S6D and S6E). We found that RIM1 C2B, RIM1 C2B 2E and RIM2 C2B bound specifically to LH2 (Figures 6B and 6C). In contrast, neither RIM3 C2B nor RIM4 C2B showed any appreciable binding. Thus, the interaction of RIM1 C2B with Liprin-α is not affected by the 2E mutation, and RIM3 and RIM4 C2B do not bind to Liprin-α. In contrast, the polybasic sequence is fully conserved between RIM1, -3 and -4 C2B (Figure S3G), and we found that RIM1, -3 and -4 C2B bind to PIP2 to a similar extent in lipid co-sedimentation experiments (Figures 6D and 6E). Thus, RIM3 and RIM4 C2B bind to PIP2 but not to Liprin-α.
One possible scenario for the function of RIM C2B is that it recruits Liprin-α to PIP2-enriched membranes, and that synergistic binding to Liprin-α and PIP2 is required to control synaptic transmission. Another scenario is that the C2B domain does not work autonomously in synaptic transmission, but tethers the biochemical activities of other RIM domains to PIP2 containing membranes. A previous study supports the latter possibility, because expression of the RIM2 C2B domain alone did not confer rescue of synaptic transmission (Kaeser et al., 2012). To directly test whether Liprin-α binding is involved in rescue, we made use of the finding that RIM3 and RIM4 C2B do not bind to Liprin-α, but bind to PIP2. We created hybrid constructs of RIM1 where the C2B domain is replaced with that of RIM3 (RIM 1–3) or RIM4 (RIM 1–4), and tested their effect on synaptic transmission (Figures 6F–6I). RIM 1–3 and RIM 1–4 were expressed at levels comparable to RIM WT (Figures S7A). RIM 1–4 fully rescued evoked release as well as PPRs, indistinguishable from RIM WT rescue (Figures 6G–6I and S7B). These findings directly demonstrate that Liprin-α binding to C2B is not required for rescue. Somewhat surprisingly, RIM 1–3, while having biochemical properties identical to RIM 1–4 in terms of PIP2 and Liprin-α binding, showed only partial rescue of evoked release, and strongly diminished rescue of PPRs (Figures 6G–6I). These observations suggest that C2B may have additional unidentified roles that are not conserved in RIM3 C2B. To further test whether C2B might have a second activity besides PIP2 binding, we exchanged C2B with another PIP2-binding C2 domain. The C2C domain of extended synaptotagmin-2 has a polybasic sequence nearly identical to RIM C2B (Figures 4C and S3G), and binds to PIP2 but not Ca2+ (Giordano et al., 2013). The resulting hybrid construct (RIM ΔC2B–C2C, Figure S7C) was efficiently expressed in cultured neurons and rescued vesicle priming (Figures S7D–S7F), but failed to rescue evoked release as well as PPRs (Figures S7G–S7J). We conclude that RIM1 C2B has likely an additional function in synaptic transmission, or, more trivially, that artificially fusing RIM3 C2B or extended synaptotagmin-2 C2C with RIM1 affects the packing of the C-terminal C2 domain against the rest of the protein, which in turn reduces rescue activity.
C2B needs to be attached to RIM to mediate rescue
Our results thus far suggest that C2B tethers RIMs known roles in release to membranes that contain PIP2. However, it is also possible that C2B has an autonomous function at the active zone, and does not need to be attached to the N-terminal RIM domains to increase synaptic vesicle fusion. To distinguish between these possibilities, we expressed RIM C2B alone or together with RIM ΔC2B (RIM ΔC2B + C2B, Figures 7A and S7K), and measured the effect on synaptic transmission. Strikingly, we observed no rescue of IPSC amplitudes or PPRs by RIM C2B alone or by RIM ΔC2B + C2B, but only RIM WT was sufficient for rescue (Figures 7B–7D and S7L). These results indicate that C2B needs to be attached to the N-terminal domains of RIM to execute its role in synaptic vesicle release, directly establishing a tethering function.
Figure 7. Tethering of RIM C2B to N-terminal RIM domains is essential for rescue.

(A) Overview of rescue proteins.
(B, C) Example traces (B) and quantification of average amplitudes (C) of single evoked IPSCs. Control n = 3 independent cultures/16 cells, cDKO 3/15, cDKO + RIM WT 3/16, cDKO + RIM Δ C2B 3/15, cDKO + RIM C2B 3/15, cDKO + RIM ΔC2B + RIM C2B 3/15.
(D) Quantification of IPSC paired pulse ratios at various interstimulus intervals. Number of observations as in C.
(E, F) Example traces (E) and quantification of average amplitudes (F) of single evoked NMDA receptor EPSCs in cultured hippocampal neurons. Control n = 4 independent cultures/18 cells, cDKO 4/19, cDKO + RIM WT 4/20, cDKO + RIM ΔC2B 4/19.
(G) Quantification of EPSC paired pulse ratios at various interstimulus intervals. Number of observations as in F.
(H) Working model for the roles of RIM C2B at the active zone. C2B targets RIM to PIP2-containing membranes, recruiting its direct interaction partners and additional important fusion machinery to colocalize vesicle priming, Ca2+ influx and fusion with PIP2. Many proteins that are important for release are activated by PIP2 (highlighted in blue, with effect of PIP2 indicated in italic). C2B has an additional activity in synaptic vesicle release that may depend on PIP2 at the active zone.
All data shown as mean ± SEM, *p < 0.05, ** p < 0.01, *** p < 0.001 for IPSC and EPSC amplitude analyzed by one-way ANOVA (Kruskal Wallis; C,F), for PPRs analyzed by two-way ANOVA (D, G); all comparisons to cDKO. For analyses of rescue protein expression and example traces of PPRs, see Figure S7K–S7M.
RIM C2B has a universal role in synaptic transmission
The results discussed so far demonstrate that RIM1 C2B has a crucial function in regulating p at inhibitory synapses. Is this function unique to GABAergic synaptic transmission, or shared among many synapses? To address this, we recorded N-methyl-D-aspartate (NMDA) receptor mediated excitatory postsynaptic currents (EPSCs) in cultured hippocampal neurons in control neurons, cDKO neurons, and cDKO neurons rescued with full length RIM or RIM ΔC2B. When measuring single evoked EPSC amplitudes, we found that RIM ΔC2B did not show any rescue (Figures 7E and 7F). Additionally, RIM ΔC2B failed to rescue PPRs (Figures 7G and S7M), indicating a defect in p at excitatory synapses. These results establish that the role of C2B is not restricted to inhibitory synapses, but instead may be important at diverse synapses throughout the brain.
Discussion
The presynaptic active zone controls vesicle docking, priming and the anchoring of Ca2+ channels. The fusion reaction itself is mediated by SNARE proteins (Jahn and Fasshauer, 2012; Südhof, 2013) and requires PIP2 in the target membrane (Di Paolo et al., 2004). We found here that both C2 domains of RIM bind to PIP2 and that the interaction between the C2B domain and PIP2 is essential for RIM’s role in synaptic vesicle fusion. Importantly, the C2B domain cannot act autonomously to mediate this role, but needs to be attached to the other RIM domains. In contrast to interactions of other presynaptic C2 domain proteins with PIP2, PIP2 interactions of RIM are not controlled by Ca2+-binding to RIM C2B, and may thus fulfill constitutive, activity-independent functions. We propose that the C2B domains cluster the vesicle priming and Ca2+ channel tethering functions of RIM to PIP2 in the target membrane to enable fast and efficient exocytosis (Figure 7H).
PIP2 binding of RIM C2 domains
We found that RIM1 C2B binds specifically to PIP2-containing membranes via a binding region that is similar to those of homologous C2 domains (Figures 3 and 4, and (Corbalan-Garcia and Gómez-Fernández, 2014)). Indeed, as observed previously for other PIP2 binding C2 domains (Van den Bogaart et al., 2012; Guerrero-Valero et al., 2009; Li et al., 2006), mutations in the polybasic sequence of RIM1 C2B (K1513E, K1515E) abolish binding to PIP2. We further found that RIM C2A binds to PIP2. Surprisingly, this interaction was largely mediated by R834 and R835 located at the bottom of the β-sandwich, an area that has not been implicated in PIP2 binding of other C2 domains. This finding suggests a new mode of PIP2 binding, which may also be employed by other C2 domains. While we identified essential roles for PIP2 binding to C2B in synaptic transmission, we could not detect a strong effect of PIP2 binding to the C2A domain in synaptic transmission at hippocampal synapses. It is interesting, however, that a point mutation in R834 is associated with a form of cone-rod dystrophy (CORD7), a form of late onset blindness (Johnson et al., 2003). Thus, the C2A-PIP2 interaction may play important roles that are either too mild to detect in our experimental paradigm, act on processes not tested here, or may be more pronounced in other synapses.
RIM C2B is critical for action potential mediated release
RIM ΔC2B did not rescue action potential evoked vesicle release in RIM cDKO, demonstrating that C2B is essential for the function of RIM in synaptic transmission (Figures 1 and 2). This is in line with a study in C. elegans, where ΔC2B phenocopies behavioral impairments caused by deletion of RIM (Koushika et al., 2001). The strong defect in PPR, contrasted by rescue of vesicle priming and Munc13 levels and localization (Figure 2), establishes a defect in vesicular release probability p. RIM boosts Ca2+ influx (Han et al., 2011; Kaeser et al., 2011; Muller et al., 2012), which may explain a reduction in p. Previously, RIM C2B was shown to inhibit voltage dependent Ca2+ channel inactivation to enhance Ca2+ currents in transfected, non-neuronal cells (Kaeser et al., 2012; Kiyonaka et al., 2007; Uriu et al., 2010), suggesting a direct role for C2B in presynaptic Ca2+ influx. However, our experiments establish that RIM C2B domains do not boost p through enhancing Ca2 -influx. First, we found that RIM ΔC2B rescues Ca2+ influx (Figure 2). Second, train stimulation, which leads to high residual Ca2+ in nerve terminals, fails to boost vesicle fusion in RIM 4E expressing neurons, indicating that the role of C2B is downstream of Ca2+ influx (Figure S5). Third, RIM C2B domains alone do not increase action potential triggered release or its Ca2+ dependence in RIM cDKO neurons (Figure 7 and (Kaeser et al., 2011, 2012)). Finally, the effect in heterologous cells was observed for all RIMs, including RIM3 (Uriu et al., 2010), but we found that substitution of RIM1 C2B with RIM3 C2B did not fully rescue p (Figure 6). Thus, RIM C2B does not boost Ca2+ entry to enhance p. Importantly, RIM cDKO at the Calyx of Held leads to reduced Ca2+ sensitivity of fusion when Ca2+ entry is bypassed with Ca2+ uncaging (Han et al., 2011), supporting that roles for RIM in p independent of Ca2+ entry exist. We conclude that C2B mediates a new function of RIM, independent of its activities in vesicle docking, priming and Ca2+ entry.
Roles for RIM C2B binding to PIP2 in action potential triggered release
In rescue experiments using mutations in the polybasic sequence of C2B, we identified a critical role for PIP2 binding to the RIM C2B domain for action potential induced release (Figure 5). Could other interactions of the RIM C2B domain contribute to the function of C2B in vesicle fusion? As outlined above, it is unlikely that the previously identified interaction with β subunits of Ca2+ channels contributes via boosting Ca2+ influx. The RIM1 C2B domain was also identified to bind to Liprin-α proteins (Schoch et al., 2002), and this interaction was not affected by the C2B 2E mutation (Figure 6). Remarkably, Liprin-α did not bind to RIM4 C2B, but replacing RIM1 C2B with the C2B domain of RIM4 fully rescued fusion. Hence, binding of Liprin-α to RIM is either not important for neurotransmitter release, or functional redundancy with other RIM interactions masks important roles for Liprin-α RIM C2B domain interactions.
Surprisingly, substitution with the RIM3 C2B domain, which, like RIM4 C2B, binds PIP2 but not Liprin-α, led to incomplete rescue of synaptic transmission (Figure 6). In addition, exchanging C2B with the C2C domain of extended synaptotagmin-2, which, like C2B, binds PIP2 but not Ca2+ (Giordano et al., 2013), failed to rescue p or IPSC amplitudes (Figure S7). Thus, substituting C2B with another PIP2-binding C2 domain is insufficient, indicating that RIM1 C2B has additional roles in vesicle fusion that are also present in RIM4 C2B. Interestingly, our data show that C2B alone is insufficient to boost fusion in cDKO neurons (Figure 7 and (Kaeser et al., 2011, 2012)), and that C2B must be connected to the N-terminal RIM domains to rescue exocytosis (Figure 7). This indicates that its role at the active zone requires close proximity to the other RIM domains and their binding partners, and it is possible that C2B engages in intramolecular interactions with other RIM domains.
Does RIM C2B localize synaptic vesicle priming and Ca2+ influx to PIP2-containing membranes?
PIP2 is required at the target membrane for efficient vesicle fusion (Hay et al., 1995; Milosevic et al., 2005; Di Paolo et al., 2004). While the exact distribution of PIP2 at the active zone is unknown, studies in non-neuronal cells showed that PIP2 is clustered in small domains within the plasma membrane (van den Bogaart et al., 2011; Honigmann et al., 2013; Laux et al., 2000). Functions of many core components of the synaptic vesicle fusion machinery depend on interactions with PIP2, for example those of synaptotagmin, syntaxin, Munc13 and Ca2+ channels (Bai et al., 2004; van den Bogaart et al., 2011; Li et al., 2006; Shin et al., 2010; Suh et al., 2010). Multiple additional proteins that regulate synaptic exocytosis bind to PIP2, including CAPS, Doc2, PKC, and Rabphilin, and endocytosis is also dependent on PIP2 (Koch and Holt, 2012; Lauwers et al., 2016). Hence, optimal fusion requires co-localization of PIP2 with these proteins, and mis-targeting of release machinery away from PIP2-containing membranes would lead to impaired release with multiple proteins operating at sub-optimal capacity.
We propose that RIM C2B domains target release machinery to PIP2-containing membranes (Figure 7H). This targeting mechanism could have several important implications for synaptic vesicle exocytosis. If PIP2 is present at the active zone in small clusters, as has been observed in non-neuronal cells (Aoyagi et al., 2005; van den Bogaart et al., 2011; Honigmann et al., 2013), this could provide for nanoscale scaffolding at PIP2-rich fusion sites to integrate the functions of RIM, and potentially its PIP2-dependent interaction partners, for efficient docking, priming and Ca2+ triggering. Recent findings provide further support for this model. Within an active zone, synaptic vesicle exocytosis is restricted to a few sites (Maschi and Klyachko, 2017; Tang et al., 2016). These sites align in a nano-columnar structure with postsynaptic receptor clusters and contain RIM as a central component. Remarkably, the diameter of these RIM clusters is ~80 nm (Tang et al., 2016), very similar to the diameter of syntaxin-associated PIP2 clusters, which was determined to be 73 nm in non-neuronal secretory cells (van den Bogaart et al., 2011). The exact distribution of PIP2 within an active zone and the underlying clustering mechanisms are currently unknown because the available tools have not allowed measuring PIP2 clustering at synapses. In the model in which RIM targets release to PIP2-containing fusion sites, we favor that PIP2 clusters RIM and its activities in exocytosis because there are likely many more PIP2 molecules than RIM molecules in a nerve terminal. But it is possible that RIM has an active role in generating PIP2 clusters, or that PIP2 activates other RIM activities. Ultimately, the functional effects of these alternative possibilities are similar: they all lead to fusion sites where essential activities of RIM and PIP2 are tethered to one another for efficient exocytosis.
Because the extent of nanoscale clustering of PIP2 at the active zone is not well known, other potential roles of PIP2-RIM interactions need to be considered. It is well established that PIP2 defines the plasma membrane identity (Di Paolo and De Camilli, 2006) independent of how it is clustered. Hence, it is possible that the targeting function of RIM-PIP2 interactions is necessary to assemble fusion machinery in the correct orientation for efficient release, or that this interaction provides a proofreading mechanism to ensure that exocytosis occurs at the correct target membrane. The apparent specificity of C2B for the PI(4,5)P2 isomer (Figure S4) would be in line with the latter model, as this isomer is almost exclusively present at the plasma membrane (Di Paolo and De Camilli, 2006). Finally, because RIM C2B has additional roles in fusion (Figures 6 and S7), it is possible that these roles are activated by PIP2. In a similar fashion, Ca2+-binding to synaptotagmin-1 C2B is likely activated by PIP2 (Bai et al., 2004; Van den Bogaart et al., 2012).
We have identified an unexpected, important role for RIM C2B in the control of synaptic vesicle exocytosis. Our data suggest that the function of RIM at the active zone reaches beyond docking, priming and tethering of Ca2+ channels, but directly regulates the efficiency of fusion by interacting with essential membrane lipids. RIM and PIP2 have important roles in fusion outside the nervous system (Hay et al., 1995; Milosevic et al., 2005; Yasuda et al., 2010), and many additional C2 domain proteins are Ca2+ independent, but have polybasic sequences in their C2 domains similar to RIM. Hence, the constitutive tethering of C2 domain scaffolds to PIP2-rich membranes may provide for a mechanism across diverse secretory pathways to target exocytosis to specific membrane domains.
STAR Methods
Contact for reagent and resource sharing
Requests for reagents and resource sharing should be directed to the Lead Contact, Pascal S. Kaeser, at kaeser@hms.harvard.edu
Experimental models and subject details
Mice
RIM1/2 conditional knockout mice were previously described (Kaeser et al., 2011) and were maintained as homozygote lines in which the Rims1 gene (RRID:IMSR_JAX:015832 (Kaeser et al., 2008)) and the Rims2 gene (RRID:IMSR_JAX:015833 (Kaeser et al., 2011)) contained essential exons flanked by loxp sites. All animal experiments were approved by the Harvard University Animal Care and Use Committee.
Neuronal cell cultures and lentiviral infection
Dissociated high-density hippocampal cultures were prepared from newborn RIM1/2 conditional double knockout mice of either sex as described (Kaeser et al., 2008; Maximov et al., 2007). Lentiviruses were produced in HEK293T cells. HEK cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and were split every 2 days to maintain optimal growth rate. For virus production, HEK cells in neuronal culture media were transfected using Fugene (Promega) with 3rd generation lentiviral packaging plasmids (REV, RRE and VSV-G) and a separate lentiviral plasmid (FSW or FUGW) encoding the recombinant gene of interest. After 48 h, the culture medium was harvested and centrifuged for 5 min at 700 × g, and the supernatant was used immediately for infection. Neuronal cultures were infected at 4 days in vitro (DIV) with lentiviruses expressing GFP-Cre with enhanced nuclear localization driven by a ubiquitin (for electrophysiology) or synapsin promoter (for Ca2+-imaging) to generate cDKO neurons or an inactivate, truncated variant of GFP-Cre to generate control neurons (Kaeser et al., 2011). For rescue expression of full length RIM or mutant RIM, cultures were infected with a separate rescue virus containing ubiquitin-promotor driven RIM open reading frame with an HA tag immediately following the PxxP motif (plasmid pAJ14063, (Kaeser et al., 2011)).
Method details
Lentiviral rescue constructs
To obtain a RIM ΔC2B construct, full length RIM1α was truncated at the residue immediately preceding C2B, following the definition of (Guan et al., 2007), the last residues being …FLDGL-stop (pAJ15020). In all constructs where C2B is substituted with another domain, the substituting domain was fused directly to RIM ΔC2B without additional linker sequences. RIM3 C2B (residues GPAQI…SPSCS, pAJ15019) and RIM4-C2B (residues GPAQF…CGERS, pAJ15032) were obtained from rat, Extended Synaptotagmin-2 C2C (residues RLRQL…PQAMT, Addgene #66831, (Giordano et al., 2013), pAJ17016) from human. For co-expression of RIM ΔC2B and C2B in Figure 8, RIM1 C2B (residues GPAQL…PCIRS-stop) was fused to Cre with a 2A sequence (pAJ17015). Successful expression of both open reading frames was confirmed by western blotting. To obtain RIM1 ΔC2A, the sequence between residues DAPQV…HDESS in RIM1α was substituted with a short linker sequence (GSGA, pAJ17013)). Point mutations in C2A and C2B were generated by site-directed mutagenesis (pAJ14064, pAJ14065, pAJ14066). Expression of rescue constructs was confirmed by western blotting for every culture.
Electrophysiology
Whole-cell patch-clamp recordings were performed in cultured hippocampal neurons at DIV14-17 as described in (Maximov et al., 2007; Wang et al., 2016) at room temperature. Extracellular solution contained (in mM) 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.4) and 10 Glucose. Borosilicate glass pipettes (2.5 – 4.5 MΩ) were filled with intracellular solution containing (in mM) 40 CsCl, 90 K-gluconate, 1.8 NaCl, 1.7 MgCl2, 3.5 KCl, 0.05 EGTA, 10 HEPES (pH 7.4), 2 ATP-Mg, 0.4 GTP-Na2, 10 phosphocreatine, 4 QX314-Cl. Cells were clamped at −70 mV, with Rseries < 15 MΩ, and Rseries was compensated online until Raccess = 0.6 – 1.1 MΩ (typically 50–70%). Action potential-evoked IPSCs were elicited by local electric stimulation and recorded in extracellular solution supplemented with 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μM (2R)-amino-5-phosphonovaleric acid (D-AP5). Evoked EPSCs were recorded in extracellular solution supplemented with 20 μM CNQX and 20 μM picrotoxin (PTX), and cells were clamped at +40 mV. For RRP measurements, 500 mM hypertonic sucrose was applied for 10 seconds by local perfusion (Rosenmund and Stevens, 1996), in the presence of 20 μM CNQX, 50 μM D-AP5 and 1 μM tedrodotoxin (TTX). The integral of the first 10 s of the response was used as measure of the RRP. Data were acquired using a Multiclamp 700B amplifier and a Digidata 1550 digitizer, recorded at 10 kHz, and filtered at 2 kHz. Data were analyzed using pClamp and custom written programs in MATLAB. For all electrophysiological experiments and analyses, the experimenter was blind to the experimental condition.
Ca2+ imaging
Imaging of presynaptic Ca2+-transients was performed as described in (Wang et al., 2016) with minor modifications. Neurons were recorded at DIV14-16 with whole-cell patch clamp electrophysiology at room temperature in extracellular solution supplemented with 20 μM CNQX, 50 μM D-AP5 and 20 μM PTX. Intracellular solution contained (in mM) 140 K-Gluconate, 0.1 EGTA, 2 MgCl2, 4 Na2ATP, 1 NaGTP, 0.3 Fluo5-F, 0.03 AlexaFluor-647 10 HEPES (pH 7.4). Because of the limited time window during which this experiment needs to be done and its laborious nature, we could only analyze three conditions at a time, and hence we compared RIM cDKO neurons to cDKO neurons rescued with RIM WT or RIM ΔC2B. After filling for 7 minutes in current clamp (Vm < −50 mV at I = 0 pA), boutons were identified in the Alexa channel, based on their typical bead-like morphology. After 10 minutes of filling, cells were held at Vm = −60 mV, and action potentials were triggered using a somatic current injection (5 ms, 800–1500 pA). Fluo5-F was excited with a pE-4000 LED at 470 nm (CoolLED) at 75% intensity. Images were acquired using a Slicescope upright microscope (Scientifica), with 60×1.0 numerical aperture (NA) objective, a multiple bandpass excitation/emission filter set (LED-DA/FI/TR/Cy5A, Semrock) and ORCA-Flash4.0 CMOS camera (Hamamatsu) at 100 Hz and 2×2 pixel binning. Images were analyzed in ImageJ (NIH). For all Ca2+ imaging experiments and analyses, the experimenter was blind to the experimental condition.
Immunofluorescence of cultured neurons
Neuronal cultures at DIV14-16 were washed once with PBS and fixed with 4% paraformaldehyde in PBS for 10 minutes at RT. Cells were permeabilized in blocking solution containing 0.1% Triton X-100, 3% BSA in PBS, and incubated with primary antibodies against RIM (BD-Biosciences 610907, at 1:500 dilution for confocal microscopy, SySy 140 003, 1:500 for STED microscopy), synaptophysin (SySy 101 004, 1:500), vGAT (SySy 131 004, 1:500), Munc13-1 (SySy 126 103, 1:200), Bassoon (Enzo life sciences SAP7F407, 1:1000) and MAP-2 (Sysy 188 002, 1:1000) in blocking solution overnight at 4°C. Staining with AlexaFluor- or Oregon Green-coupled secondary antibodies (Life Technologies, 1:1000 for confocal microscopy, 1:200 for STED microscopy) was performed for 1 hr at room temperature. Coverslips were air dried at room temperature and mounted on to glass slides in mounting medium and stored at 4 °C until imaging.
STED imaging and analysis
STED microscopy was performed as described in (Wong et al., 2018). Images were acquired with a Leica SP8 Confocal/STED 3X microscope with an oil immersion 100X 1.44 NA objective. 23.3 × 23.3 μm2 synapse-rich areas were scanned at a sampling frequency of ~10 nm/pixel. Triple-color sequential confocal scans were followed by a dual-color sequential STED scans. Alexa 633, Alexa Fluor 555 and Oregon green 488 were excited with 633 nm, 555 nm and 488 nm white light lasers respectively at 2–5% of 1.5 mW laser power in this order of sequence. During STED scanning, Oregon green 488 and Alexa Fluor 555 signals were depleted with 592 nm (75% of max power) and 660 nm (25% of max power) time-gated depletion lasers. 4-times line accumulation and 3-times frame averaging were applied during STED scanning. Identical settings were applied to all samples within an experiment. In all STED experiments, vGAT was acquired in the confocal channel. To measure protein levels at active zones, masks that contained Bassoon signal within vGAT signals were created using MATLAB. These masks were used to measure fluorescence intensity of proteins in the second STED channel. For intensity profiles, side view vGAT positive synapses (Wong et al., 2018) were selected manually, and a rectangular 0.25 μm × 1.0 μm region of interest (ROI) was placed perpendicular to the Bassoon signals in ImageJ. Profiles were aligned on peak intensity of the Bassoon signal and in each channel normalized to the peak fluorescence in control. Note that for RIM and Munc13, peak localization relative to Bassoon is not always identical, and hence the average normalized control peak intensity is slightly below 1. All quantitative analyses were performed on original images without adjustments and were done identically for all experimental conditions. For all image acquisition the experimenter was blind to the experimental condition.
Confocal microscopy
Images were acquired at an Olympus FV1000 confocal microscope using a 60×1.4 NA oil-immersion objective, and identical settings were applied to all samples within an experiment. Images were analyzed in ImageJ and MATLAB as described in (Wang et al., 2016). Synaptic levels of RIM were obtained by measuring RIM signal within synaptophysin puncta. 10 fields of view were quantified per culture per genotype. The experimenter was blind to the experimental condition of the culture.
Protein expression and purification
GST fusion proteins were expressed and purified according to standard procedures. The following proteins were produced: rat RIM1α C2A (residues QVLPG…. WYKLQ plasmids pAJ13026, pAJ14016, pGEX-KT Rim1 C2A WT, pGEX-KT Rim1 C2A WT R834, R835E and pGEX-KT Rim1 C2A WT K809,R811E), RIM2 C2A (residues QFLSG…WYKLQ, pAJ13027) RIM1α C2B domain (residues GPAQL…PCIRS, (Guan et al., 2007), pAJ13005, pAJ13014, pGEX-KT Rim1 C2B WT and pGEX-KT Rim1 C2B K1513,K1515E), RIM2 C2B domain (residues GPAQL…SYSRS, pAJ13028), RIM3-C2B (residues GPAQI…SPSCS, pAJ15025), RIM4-C2B (residues GPAQF…CGERS, pAJ15026), rat Liprin-α3-LH1 (residues CEVMP…LEEEL, pMYW12035), rat Liprin-α3-LH2 (residues ELSNQ…ELDGS, pMYW12036), and rat Syntaxin 1a (residues 2–253 KDRTQ…SDTKK, pGEX-KT Syntaxin1). In brief, fusion proteins were expressed at 25°C in E. coli BL21 (DE3) for 20 h with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), except GST-Liprin-α3-LH1 and -LH2, which were expressed at 18°C for 20 h with 50 μM IPTG. For GST-pulldown experiments, cells were resuspended in PBS, and lysed using lysozyme and brief sonication. Proteins were purified using glutathione-sepharose resin in PBS and used within 5 days of purification.
For co-floatation assays, analytical ultracentrifugation and NMR spectroscopy, cells were resuspended in PBS buffer containing a protease inhibitor cocktail and lysed using an Avestin EmulsiFlex-C5 homogenizer. The RIM1α C2A and C2B domains were purified by the same procedure except that the RIM1α C2B lysate was first treated with protamine sulfate (1% (w/v)) for 1 h at 4°C followed by centrifugation. The proteins were then isolated by affinity chromatography on glutathione-sepharose. Resin was washed with PBS and PBS containing 1M NaCl. Remaining nucleic acid contaminants bound to the protein were then cleared with nuclease treatment (~600 units per liter of cell culture) in 50 mM Tris (pH 8.0), 2 mM MgCl2 for 2 h at room temperature with gentle rotation of the beads. The GST tag was cleaved with thrombin on the resin at room temperature for 3 h. The eluted protein was further purified by size-exclusion chromatography on a Superdex 75 16/60 column using standard buffer (20 mM MES (pH 6.0), 150 mM NaCl, 1 mM EDTA, and 0.5 mM TCEP). Uniform 15N-labeling was achieved by growing the bacteria in 15NH4Cl as the sole nitrogen sources.
Rat Syntaxin1A (residues 2–253) in pGEX-KT was expressed by induction with 0.4 mM IPTG at an O.D600 of 1 at 25°C for 18 hours. Cells were resuspended in resuspension buffer (RB; PBS, 1mM EDTA, 5mM DTT and protease inhibitors). Cleared lysates were applied to glutathione sepharose resin, washed with 100 ml RB, 200 ml RB + 1% Triton X-100, 200 ml RB + 1 M NaCl and 100 ml RB. The protein was then treated with nuclease for 1 hour at room temperature. The GST tag was removed by thrombin cleavage at 4°C overnight. The protein was further purified by ion exchange chromatography on a Source Q column using buffer A: 25 mM Tris pH 7.4, 1 mM TCEP and B: 25 mM Tris pH 7.4, 1M NaCl, 1mM TCEP. Source Q fractions (~38% NaCl) were frozen in liquid N2 and stored at −80°C.
His-tagged RIM C2A (pAJ14021 and pAJ14022) and C2B proteins (pAJ13023, 14006, 14046, 14047, 16001–16008) were expressed using the same protocol as GST-fusion proteins. Proteins were isolated in 300 mM NaCl, 10 mM imidazole, 50 mM NaH2PO4 (pH 8.0) with Ni-NTA agarose and eluted from the resin in the same buffer containing 100–300 mM imidazole. After overnight dialysis to 150 mM NaCl, 25 mM HEPES (pH 8.0), aliquots were stored at −80°C until use. UV spectra were obtained on a Nanodrop 2000c (Thermo Fisher).
Human SNAP-25A full length in pET28A was expressed in E.coli BL21 (DE3) cells by induction with 0.4 mM IPTG at an O. D600 of 0.8 at 23°C for 18 hours. Cells were resuspended in 50 mM Tris pH 8, 500 mM NaCl, 4 mM Imidazole, 1% Triton X-100, protease inhibitor. Cleared lysates were applied to Ni-NTA resin, washed with 100 ml resuspension buffer containing 20 mM Imidazole and treated with nuclease at 4°C overnight. The His-tag was removed by thrombin cleavage at room temperature for 1.5 hours. The protein was further purified by size exclusion chromatography on a Superdex S75 column (GE 16/60) equilibrated with 50 mM Tris pH 8, 150 mM NaCl. Fractions were concentrated to 50 μM, frozen in liquid N2 and stored at −80°C.
Liposome co-sedimentation
Liposome co-sedimentation was performed essentially as described in (Shin et al., 2010). Porcine brain-derived lipids were obtained from Avanti Polar lipids: L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′-bisphosphate) (PI(3,4)P2), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,5′-bisphosphate) (PI(3,5)P2), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′,5′-trisphosphate) (PI(3,4,5)P3), L-α-phosphatidylserine (PS) and L-α-phosphatidylcholine (PC). Unless stated otherwise, the term PIP2 refers to the PI(4,5)P2 isomer. Lipids were mixed in glass tubes and dried under N2, and resuspended in 100 mM NaCl, 25 mM HEPES (pH 8.0) and 500 mM sucrose by rigorous vortexing for 20 minutes and subsequent sonication to generate heavy liposomes. Buffer without sucrose was added to 12 ml, and liposomes were sedimented by ultracentrifugation at 150,000 × g for 30 min. The pellet was resuspended in buffer, and centrifuged again (20,800 × g 10 min). Liposome composition for each co-sedimentation experiment is indicated in the Figure legend. For binding assays, 200 μg liposomes were mixed with 10 μg protein in 1 ml buffer, and mixed for 10 minutes at 30°C while shaking at 10,00 0 RPM. Liposomes were pelleted for 10 min at 20,800 × g at 4°C, washed 3 times with buffer, and resuspended in 200 μl methanol:chloroform (2:1) and incubated for >30 minutes at −20°C to solubilize the lipids and precipitate the proteins. Proteins were pelleted for 15 min at 20,800 × g at room temperature and resuspended in 30 μL 1X SDS sample buffer. Total recovered protein was assessed by SDS-PAGE with Coomassie staining, and quantified with densitometry in ImageJ. Background protein binding to the no lipids control was subtracted from all other samples.
Liposome preparation for co-floatation
Mixtures of 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine] (PS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (PE), L-α-Phosphatidylinositol (PI), L-α-Phosphatidylinositol-4-phosphate (PIP) and L-α-Phosphatidylinositol-4,5-biphosphate (PIP2) (Avanti Polar Lipids) were prepared by mixing the lipids dissolved in chloroform in a glass test tube in the desired ratio, and chloroform was evaporated using a dry nitrogen stream. The lipids were placed in a vacuum chamber overnight to remove organic solvent. Lipid films were hydrated with reconstitution buffer (25 mM HEPES pH = 7.2, 150 mM NaCl) in an appropriate volume yielding 5 mM total lipid concentration. Lipids were vortexed >5 min then frozen and thawed five times. Large unilamellar vesicles were prepared by extruding the hydrated lipid solution through 0.08-μm polycarbonate membranes 23 times using an Avanti Mini-Extruder. The homogeneity of the vesicle size distribution was confirmed by dynamic light scattering on a Wyatt DynaPro instrument (Wyatt Technology, Santa Barbara, CA).
Liposome co-floatation assays
Liposomes containing PC:PE(99:1), PC:PS:PE(84:15:1), PC:PS:PE:PI (83:15:1:1), PC:PS:PE:PIP (83:15:1:1) and PC:PS:PE:PIP2 (83:15:1:1) were mixed with the proteins with a protein/lipid (P/L) ratio of 1:200 and incubated at room temperature for 1 hr. The liposomes and bound proteins were isolated by floatation on a Histodenz density gradient (40%:35%:30%) as described (Guan et al., 2008). Samples from the top of the gradient (35 μL) were taken and analyzed by SDS-PAGE and Coomassie blue staining.
NMR spectroscopy
All NMR data were acquired at 27 °C on Varian INOVA 600 spectrometers (Varian, Palo Alto, California, USA) with RIM1 C2 domain samples dissolved in standard buffer (20 mM MES (pH 6.0), 150 mM NaCl, 1 mM EDTA, 0.5 mM TCEP), using H2O/D2O 95:5 (v/v) as the solvent. All 2D1H-15N HSQC spectra testing phospholipid binding were acquired at 100 μM and 30 μM protein concentrations for C2A and C2B, respectively, and dibutanoyl phosphatidylinositol 4,5-bisphosphate (diC4-PIP2) concentrations ranging from 5 to 300 μM. Spectra testing for SNARE protein binding were performed using 20 μM 15N-labeled RIM1 C2B and 25 μM unlabeled Syntaxin or SNAP-25. The 1D 1H-NMR spectra were acquired with samples containing 100 μM RIM1α C2B domain and in the presence or absence of 200 μM diC4-PIP2. All 2D NMR spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed with NMRView (Johnson and Blevins, 1994). To derive Kd values from the diC4-PIP2 titrations, the 15N chemical shifts from selected cross-peaks, which were among those that had larger diC4-PIP2-induced chemical shift changes and were better resolved, were plotted as a function of diC4-PIP2 concentration. The data were then fitted to a standard single-site binding model using Sigma Plot.
Analytical ultracentrifugation
All centrifugation experiments were carried out in a Beckman-Coulter Optima XL-I (Beckman-Coulter, Brea, CA) at 20 °C; a standard sedimentati on velocity protocol was used (Zhao et al., 2013). The rotor speed was 50,000 rpm and the rotor was an An50-Ti (Beckman-Coulter, Brea, CA). A280 data were analyzed in SEDFIT using the c(s) distribution model (Schuck, 2000). The distributions were normalized, integrated, and rendered in GUSSI (Brautigam, 2015), and the resulting isotherm was examined in SEDPHAT (Schuck, 2003) to estimate the Kd, assuming and fixing the values of 1.85 S for the monomer and 3.1 S for the dimer.
GST pulldowns with His-tagged proteins
To test binding of C2B domains to Liprin sequences, 6 μg GST-Liprin-α3-LH1 or -LH2 was mixed with 15 μg His-tagged RIM C2B WT or mutant, in 100 mM NaCl, 25 mM HEPES (pH 7.4), 0.1% Triton X-100 and 1 mM DTT. After incubation for two hours at 4°C with gentle agitation, beads were washed 5 times with 500 μl buffer, and proteins were eluted from the beads with 1× SDS sample buffer. Binding was determined using Coomassie-stained 15% SDS-PAGE gels. Protein intensity was measured by densitometry in ImageJ, and molar His-RIM C2B to GST-Liprin-α3-LH was calculated by dividing both signals after correcting the raw signal for protein length.
GST pulldowns from mouse brain
To make brain lysate, brains from 6–10 week old wild type mice were homogenized in 10 ml ice cold buffer containing 100 mM NaCl, 4 mM EGTA, 25 mM HEPES (pH 7.4) and protease inhibitor cocktail (Sigma, containing AEBSP, Aprotinin, Bestatin, E-64, Leupeptin and Pepstatin A), and incubated with 1% Triton X-100 for 1 hr. The insoluble fraction was removed by ultracentrifugation at 118 K × g for 30 minutes. Supernatant was precleared from glutathione-binding proteins by incubation for 1 hr with 200 μl 50% glutathione-sepharose beads. Subsequently, 15 μg GST-fusion protein was mixed with 1 ml cleared brain lysate, and incubated for 1 hr at 4°C with gentle agitation. To the +Ca2+ pulldown conditions, 5 mM CaCl2 was added to obtain [Ca2+]free of 1 mM. Beads were washed 5 times with 1 ml buffer, and proteins were eluted from the beads with 100 μl 1X SDS sample buffer. Bound proteins were identified with SDS-PAGE and western blotting.
Western blotting
SDS-PAGE gels were transferred to nitrocellulose membranes (GE Healthcare) for 6.5 hr at 4°C in buffer containing (per l) 200 ml methanol, 14 g glycine and 6 g Tris. After blocking for 1 hr at RT in TBST with 10% non-fat milk powder and 5% normal goat serum (NGS), blots were stained for 2–3 hrs at RT with primary antibodies in TBST with 5% milk and 2.5% NGS. Primary antibodies used were R809 (RIM1/2 PDZ and C2A domains, (Schoch et al., 2002), 1:2000), HM1092 (RIM1/2 PDZ and C2A domains, this study, 1:1000), U1130 (RIM1/2 c-terminus, (Schoch et al., 2002), 1:2000), 4396 (Liprin-α LH2 domain, (Schoch et al., 2002), 1:5000), V216 (synaptotagmin-1 cytoplasmatic domain, Südhof laboratory, 1:1000), P913 (SNAP-25, Südhof laboratory 1:2000), P939 (Synaptobrevin-2, Südhof laboratory, 1:2000), 438B (Syntaxin-1, Südhof laboratory, 1:1000), HA (Covance MMS-101P, 1:1000) and β-actin (Sigma A1978, 1:5000). After washing 5 × 5 min with TBST, blots were stained for 1 hr with horseradish peroxidase-conjugated secondary antibodies in the same solution, and blots were washed 5 × 5 min. Protein bands were visualized using enhanced chemiluminescence.
Antibody production
HM1092 RIM antibodies were raised in rabbit against a GST-fusion protein containing the PDZ domain an part of the C2A domain of RIM1 (residues PGSAV…KVGHQ, identical to the immunogen of antibody R809, (Schoch et al., 2002), pAJ16009). The GST-fusion protein was purified as described above, and eluted from the beads with 10 mM glutathione for 3 hrs at 4°C. After overnight dialysis to PBS, the protein was snap-frozen in ethanol/dry ice and submitted to Cocalico Biologicals for immunization using standard procedures. Sera were screened using western blots on protein samples from neuronal RIM1/2 control and RIM1/2 cDKO cultures. The serum with highest specific immunoreactivity (HM1092) for RIM was selected for use.
Quantification and statistical analysis
For electrophysiological and STED experiments, statistical differences were assessed using a one-way Kruskal-Wallis ANOVA with tukey-kramer correction for multiple testing, comparing against the cDKO condition. For PPR, a two-way ANOVA with tukey-kramer correction for multiple testing was used, testing genotype against cDKO. P values reported for one- and two-way ANOVAs are derived from the post-hoc test as reported in each figure legend. For lipid co-sedimentation in Figures 3 and S3 and GST-pulldowns in Figure 6, a Student’s t-test was used. In all figures, * indicates p < 0.05, ** p < 0.01, and *** p < 0.001. No tests were used to estimate sample size. Except for NMR spectra and co-floatation assays, all experiments were performed at least 3 times to ensure reproducibility. The statistical tests used, the definition of n and the number of observations per experimental condition are specified in the figure legends. In all experiments, data is represented as mean ± standard error of the mean (SEM).
Supplementary Material
Highlights.
RIM C2B controls synaptic vesicle fusion downstream of priming and Ca2+ influx
C2B domains bind to PIP2 and PIP2 binding is required for RIM’s role in release
C2B domains need to be tethered to other RIM domains for mediating vesicle fusion
RIM C2B domains have additional functions for enhancing release probability
Acknowledgments
We thank Lydia Bickford and Jennifer Jiexin Wang for technical support, Hajnalka Nyitrai for help with antibody production, Dr. Thomas Südhof for antibodies, and Dr. Jonathan Cohen and members of the Kaeser laboratory for comments on the manuscript. This work was supported by grants from the NIH (R01NS083898 and R01MH113349 to PSK; R35 NS097333 to JR), the Lefler Foundation (to AdJ), the NWO (Rubicon Fellowship 825.12.028 to AdJ), the Harvard Brain Initiative (to PSK) and the Welch Foundation (I-1304 to JR). We also acknowledge the Neurobiology Imaging Facility (supported by a P30 Core Center Grant NS072030) and the Harvard Neurodiscovery Imaging Center.
Footnotes
Declaration of Interests
The authors have no conflicts of interest to declare.
Author Contributions
Conceptualization, A.d.J., J.R. and P.S.K.; Methodology, A.d.J., J.R. and P.S.K.; Formal Analysis, A.d.J., M.H., C.R., J.R. and P.S.K.; Investigation, A.d.J., M.H., C.R., M.Y.W. and C.B.; Writing-Original Draft, A.d.J. and P.S.K..; Supervision, J.R. and P.S.K.; Funding Acquisition A.d.J., J.R. and P.S.K.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Aoyagi K, Sugaya T, Umeda M, Yamamoto S, Terakawa S, Takahashi M. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem. 2005;280:17346–17352. doi: 10.1074/jbc.M413307200. [DOI] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, Dier M, Hell SW, Grubmüller H, Diederichsen U, et al. Membrane protein sequestering by ionic protein–lipid interactions. Nature. 2011;479:552–555. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015;562:109–133. doi: 10.1016/bs.mie.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Camacho M, Basu J, Trimbuch T, Chang S, Pulido-Lozano C, Chang SS, Duluvova I, Abo-Rady M, Rizo J, Rosenmund C. Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat Commun. 2017;8:15293. doi: 10.1038/ncomms15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola T, Magnin-Lüthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct Interaction of the Rab3 Effector RIM with Ca2+ Channels, SNAP-25, and Synaptotagmin. J Biol Chem. 2001;276:32756–32762. doi: 10.1074/jbc.M100929200. [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Gómez-Fernández JC. Signaling through C2 domains: More than one lipid target. Biochim Biophys Acta - Biomembr. 2014;1838:1536–1547. doi: 10.1016/j.bbamem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Coudevylle N, Montaville P, Leonov A, Zweckstetter M, Becker S. Structural determinants for Ca2+ and phosphatidylinositol 4,5-bisphosphate binding by the C2A domain of rabphilin-3A. J Biol Chem. 2008;283:35918–35928. doi: 10.1074/jbc.M804094200. [DOI] [PubMed] [Google Scholar]
- Dai H, Tomchick DR, García J, Südhof TC, Machius M, Rizo J. Crystal structure of the RIM2 C2A-domain at 1.4 Å resolution. Biochemistry. 2005;44:13533–13542. doi: 10.1021/bi0513608. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, Südhof TC. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of munc13. Neuron. 2011;69:317–331. doi: 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P. PI(4,5)P2-Dependent and Ca2+-Regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153:1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Arazola RD, Cornelisse LN, Lozovaya N, de Jong APH, Goriounova NA, Habets RLP, Takai Y, Borst JG, et al. Doc2b Is a High-Affinity Ca2+ Sensor for Spontaneous Neurotransmitter Release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Dai H, Tomchick DR, Dulubova I, Machius M, Sudhof TC, Rizo J. Crystal Structure of the RIM1a C2B Domain at 1.7 Å Resolution. Biochemistry. 2007;46:8988–8998. doi: 10.1021/bi700698a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN Domain to Membrane-Anchored SNARE complexes. Biochem. 2008;47:1474–1481. doi: 10.1021/bi702345m. [DOI] [PubMed] [Google Scholar]
- Guerrero-Valero M, Ferrer-Orta C, Querol-Audi J, Marin-Vicente C, Fita I, Gomez-Fernandez JC, Verdaguer N, Corbalan-Garcia S. Structural and mechanistic insights into the association of PKC-C2 domain to PtdIns(4,5)P2. Proc Natl Acad Sci. 2009;106:6603–6607. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF. ATP-dependent inositide phosphorylation required for Ca2+-activated secretion. Nature. 1995;374:173–177. doi: 10.1038/374173a0. [DOI] [PubMed] [Google Scholar]
- Hibino H, Pironkova R, Onwumere O, Vologodskaia M, Hudspeth aJ, Lesage F. RIM - binding proteins (RBPs) couple Rab3 - interacting molecules (RIMs) to voltage - gated Ca2+ channels. Neuron. 2002;34:411–423. doi: 10.1016/s0896-6273(02)00667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Müllar S, Diederichsen U, Fasshauer D, Grubmüller H, et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol. 2013;20:679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Südhof TC, Rhee JS, Brose N, Cooper BH. The Morphological and Molecular Nature of Synaptic Vesicle Priming at Presynaptic Active Zones. Neuron. 2014;84:416–431. doi: 10.1016/j.neuron.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMRVIEW: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Johnson S, Halford S, Morris AG, Patel RJ, Wilkie SE, Hardcastle AJ, Moore AT, Zhang K, Hunt DM. Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7) Genomics. 2003;81:304–314. doi: 10.1016/s0888-7543(03)00010-7. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG. The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol. 2017;43:63–70. doi: 10.1016/j.conb.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Chiu CQ, Deng L, Castillo PE, Sudhof TC. RIM1 and RIM1 Are Synthesized from Distinct Promoters of the RIM1 Gene to Mediate Differential But Overlapping Synaptic Functions. J Neurosci. 2008;28:13435–13447. doi: 10.1523/JNEUROSCI.3235-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Fan M, Südhof TC. RIM genes differentially contribute to organizing presynaptic release sites. Proc Natl Acad Sci U S A. 2012;109:11830–11835. doi: 10.1073/pnas.1209318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, et al. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Holt M. Coupling exo- and endocytosis: An essential role for PIP 2 at the synapse. Biochim Biophys Acta - Mol Cell Biol Lipids. 2012;1821:1114–1132. doi: 10.1016/j.bbalip.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E, Goodchild R, Verstreken P. Membrane Lipids in Presynaptic Function and Disease. Neuron. 2016;90:11–25. doi: 10.1016/j.neuron.2016.02.033. [DOI] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. Rafts, and Regulate Cell Cortex Actin Dynamics through a Common Mechanism. Cell. 2000;149:1455–1471. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shin OH, Rhee JS, Araç D, Rah JC, Rizo J, Südhof T, Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- Liu KS, Siebert M, Mertel S, Knoche E, Wegener S, Wichmann C, Matkovic T, Muhammad K, Depner H, Mettke C, et al. RIM-Binding Protein, a Central Part of the Active Zone, Is Essential for Neurotransmitter Release. Science. 2011;334:1565–1569. doi: 10.1126/science.1212991. [DOI] [PubMed] [Google Scholar]
- Maschi D, Klyachko VA. Spatiotemporal Regulation of Synaptic Vesicle Fusion Sites in Central Synapses. Neuron. 2017;94:65–73e3. doi: 10.1016/j.neuron.2017.03.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DGR, Südhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods. 2007;161:75–87. doi: 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Sorensen J, Lang T, Krauss M, Nagy G, Hauke V, Jahn R, Neher E. Plasmalemmal Phosphatidylinositol-4,5-Bisphosphate Level Regulates the Releasable Vesicle Pool Size in Chromaffin Cells. J Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaville P, Coudevylle N, Radhakrishnan A, Leonov A, Zweckstetter M, Becker S. The PIP2 binding mode of the C2 domains of rabphilin-3A. Protein Sci. 2008;17:1025–1034. doi: 10.1110/ps.073326608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Liu KSY, Sigrist SJ, Davis GW. RIM Controls Homeostatic Plasticity through Modulation of the Readily-Releasable Vesicle Pool. J Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan Ta, De Camilli P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J, et al. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol. 2010;17:280–288. doi: 10.1038/nsmb.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Leal K, Hille B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Chen H, Li TP, Metzbower SR, MacGillavry HD, Blanpied TA. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature. 2016;536:210–214. doi: 10.1038/nature19058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J, Lao Y, Fernandez I, Arac D, Südhof TC, Rizo J. The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry. 2001;40:5854–5860. doi: 10.1021/bi010340c. [DOI] [PubMed] [Google Scholar]
- Uriu Y, Kiyonaka S, Miki T, Yagi M, Akiyama S, Mori E, Nakao A, Beedle AM, Campbell KP, Wakamori M, et al. Rab3-interacting molecule γ isoforms lacking the rab3-binding domain induce long lasting currents but block neurotransmitter vesicle anchoring in voltage-dependent P/Q-type Ca2+ channels. J Biol Chem. 2010;285:21750–21767. doi: 10.1074/jbc.M110.101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Südhof TC. Genomic definition of RIM proteins: Evolutionary amplification of a family of synaptic regulatory proteins. Genomics. 2003;81:126–137. doi: 10.1016/s0888-7543(02)00024-1. [DOI] [PubMed] [Google Scholar]
- Wang SSH, Held RG, Wong MY, Liu C, Karakhanyan A, Kaeser PS. Fusion Competent Synaptic Vesicles Persist upon Active Zone Disruption and Loss of Vesicle Docking. Neuron. 2016;91:777–791. doi: 10.1016/j.neuron.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol. 2010;26:115–136. doi: 10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- Wong MY, Liu C, Wang SSH, Roquas ACF, Fowler S, Kaeser PS. Liprin-α 3 controls vesicle docking and exocytosis at the active zone of hippocampal synapses. Proc Natl Acad Sci. 2018 doi: 10.1073/pnas.1719012115. ePub ahead of print (2/8/2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Shibasaki T, Minami K, Takahashi H, Mizoguchi A, Uriu Y, Numata T, Mori Y, Miyazaki JI, Miki T, et al. Rim2a determines docking and priming states in insulin granule exocytosis. Cell Metab. 2010;12:117–129. doi: 10.1016/j.cmet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, Schuck P. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci. 2013:1–49. doi: 10.1002/0471140864.ps2012s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-Term Synaptic Plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.