

Summary
Class 2 CRISPR-Cas systems endow microbes with diverse mechanisms for adaptive immunity. Here, we analyzed prokaryotic genome and metagenome sequences to identify an uncharacterized family of RNA-guided, RNA-targeting CRISPR systems which we classify as Type VI-D. Biochemical characterization and protein engineering of seven distinct orthologs generated a ribonuclease effector derived from Ruminococcus flavefaciens XPD3002 (CasRx) with robust activity in human cells. CasRx-mediated knockdown exhibits high efficiency and specificity relative to RNA interference across diverse endogenous transcripts. As one of the most compact single effector Cas enzymes, CasRx can also be flexibly packaged into adeno-associated virus. We target virally encoded, catalytically inactive CasRx to cis-elements of pre-mRNA to manipulate alternative splicing, alleviating dysregulated tau isoform ratios in a neuronal model of frontotemporal dementia. Our results present CasRx as a programmable RNA-binding module for efficient targeting of cellular RNA, enabling a general platform for transcriptome engineering and future therapeutic development.
Graphical abstract
Introduction
Mapping of transcriptome changes in cellular function and disease has been transformed by technological advances over the last two decades, from microarrays (Schena et al., 1995) to next-generation sequencing and single cell studies (Shendure et al., 2017). However, interrogating the function of individual transcript dynamics and establishing causal linkages between observed transcriptional changes and cellular phenotype requires the ability to actively control or modulate desired transcripts.
DNA engineering technologies such as CRISPR-Cas9 (Doudna and Charpentier, 2014; Hsu et al., 2014) enable researchers to dissect the function of specific genetic elements or correct disease-causing mutations. However, simple and scalable tools to study and manipulate RNA lag significantly behind their DNA counterparts. Existing RNA interference technologies, which enable cleavage or inhibition of desired transcripts, have significant off-target effects and remain challenging engineering targets due to their key role in endogenous processes (Birmingham et al., 2006; Jackson et al., 2003). As a result, methods for studying the functional role of RNAs directly have remained limited.
One of the key restrictions in RNA engineering has been the lack of RNA-binding domains that can be easily retargeted and introduced into target cells. The MS2 RNA-binding domain, for example, recognizes an invariant 21-nucleotide (nt) RNA sequence (Peabody, 1993), therefore requiring genomic modification to tag a desired transcript. Pumilio homology domains possess modular repeats with each protein module recognizing a separate RNA base, but they can only be targeted to short 8 nt RNA sequences (Cheong and Hall, 2006). While previously characterized type II (Batra et al., 2017; O’Connell et al., 2014) and VI (Abudayyeh et al., 2016; East-Seletsky et al., 2016) CRISPR-Cas systems can be reprogrammed to recognize 20–30 nt RNAs, their large size (~1200 amino acids, aa) makes it difficult to package into AAV for primary cell and in vivo delivery.
Reasoning that diverse RNA-targeting CRISPR systems and their associated defense nucleases remain largely unexplored and may harbor advantageous properties, we conducted bioinformatic analysis of prokaryotic genomes to identify sequence signatures of CRISPR-Cas repeat arrays and mine previously uncharacterized, compact Cas ribonucleases that could be developed into RNA targeting tools. We demonstrate that engineered Type VI-D CRISPR effectors can be used to efficiently knockdown endogenous RNAs in human cells and manipulate alternative splicing, paving the way for RNA targeting applications and further effector domain fusions as part of a transcriptome engineering toolbox.
Results
Computational identification of a Type VI-like Cas ribonuclease family
We first sought to identify previously undetected or uncharacterized RNA-targeting CRISPR-Cas systems by developing a computational pipeline for class 2 CRISPR-Cas loci, which require only a single nuclease for CRISPR interference such as Cas9, Cas12a (formerly Cpf1), or Cas13a (formerly C2c2) (Makarova et al., 2015; Shmakov et al., 2015). To improve upon previous strategies for bioinformatic mining of CRISPR systems, which focus on discovering sets of conserved Cas genes involved in spacer acquisition (Shmakov et al., 2015), we defined the minimal requirements for a CRISPR locus to be the presence of a CRISPR repeat array and a nearby effector nuclease. Using the CRISPR array as a search anchor, we first obtained all prokaryotic genome assemblies and scaffolds from the NCBI WGS database and adapted algorithms for de novo CRISPR array detection (Bland et al., 2007; Edgar, 2007; Grissa et al., 2007) to identify 21,175 putative CRISPR repeat arrays (Figure S1A).
Up to 20 kilobases (kb) of genomic DNA sequence flanking each CRISPR array was extracted to identify predicted protein-coding genes in the immediate vicinity. Candidate loci containing signature genes of known class 1 and class 2 CRISPR-Cas systems such as Cas3 or Cas9 were excluded from further analysis, except for Cas12a and Cas13a to judge the ability of our pipeline to detect and cluster these known class 2 effector families. To identify new class 2 Cas effectors, we required candidate proteins to be >750 residues in length and within 5 protein-coding genes of the repeat array, as large proteins closely associated with CRISPR repeats are key characteristics of known single effectors. The resulting proteins were classified into 408 putative protein families using single-linkage hierarchical clustering based on homology.
To discard protein clusters that reside in close proximity to CRISPR arrays due to chance or overall abundance in the genome, we next identified additional homologous proteins to each cluster from the NCBI non-redundant protein database and determined their proximity to a CRISPR array. Reasoning that true Cas genes would have a high co-occurrence rate with CRISPR repeats, >70% of the proteins for each expanded cluster were required to exist within 20 kb of a CRISPR repeat. These remaining protein families were analyzed for nuclease domains and motifs.
Among the candidates, which include the recently described Cas13b system (Smargon et al., 2017), we identified a family of uncharacterized putative class 2 CRISPR-Cas systems encoding a candidate CRISPR-associated ribonuclease containing 2 predicted HEPN ribonuclease motifs (Anantharaman et al., 2013) (Figure 1A). Importantly, they are among the smallest class 2 CRISPR effectors described to date (~930 aa). The Type VI CRISPR-Cas13 superfamily is exemplified by sequence-divergent, single-effector signature nucleases and the presence of two HEPN domains. Other than these two RxxxxH HEPN motifs (Figure S2A), our candidate effectors have no significant sequence similarity to previously described Cas13 enzymes, so we designated this family of putative CRISPR ribonucleases as Type VI Cas13d, or Type VI-D (Figure S2B).
Figure 1. Type VI CRISPR-Cas13d is a family of single effector CRISPR ribonucleases.
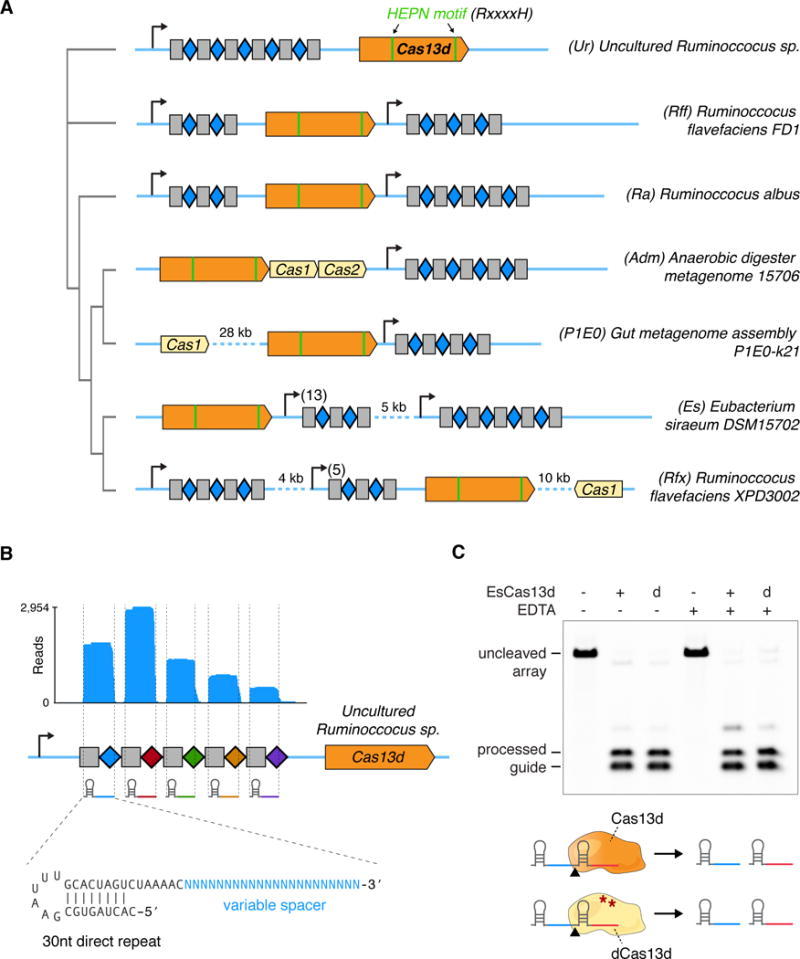
(A) Maximum-likelihood phylogenetic tree of Cas13d effectors used in this study, with the full Cas13d CRISPR locus depicted along with conserved HEPN RNase domains. Grey rectangles denote CRISPR direct repeats (DRs) and blue diamonds indicate spacer sequences. (B) RNA sequencing of a heterologously expressed Cas13d locus from an uncultured Ruminococcus sp. sample. Mature gRNAs mapping to the CRISPR array indicate a processed 30 nt DR and a variable spacer length from 14–26 nt. Co-fold analysis of direct repeat truncation indicates a strong hairpin structure. (C) Purified E. siraeum Cas13d and catalytically dead Cas13d (dCas13d) protein are each sufficient to process a guide array into its two component gRNAs. Addition of EDTA does not impair gRNA processing. ‘d’, dCas13d(R295A, H300A, R849A, H854A).
See also Figure S1, S2 and S3
CRISPR-Cas13d systems are derived from gut-resident microbes, so we sought to expand the Cas13d family via alignment to metagenomic contigs from recent large-scale microbiome sequencing efforts. Comparison of Cas13d proteins against public metagenome sequences without predicted open reading frames (ORFs) identified additional full-length systems as well as multiple effector and array fragments that cluster in several distinct branches (Figure S1B). To generate full-length Cas13d ortholog proteins and loci from the different branches of the Cas13d protein family, we obtained genomic DNA samples from associated assemblies and performed targeted Sanger sequencing to fill in gaps due to incomplete sequencing coverage, such as for the metagenomic ortholog ‘Anaerobic digester metagenome’ (Adm) (Treu et al., 2016).
Cas13d CRISPR loci are largely clustered within benign, Gram-positive gut bacteria of the genus Ruminococcus, and exhibit a surprising diversity of CRISPR locus architectures (Figure 1A). With the exception of the metagenomic AdmCas13d system, Cas13d systems lack the key spacer acquisition protein Cas1 (Yosef et al., 2012) within their CRISPR locus, highlighting the utility of a class 2 CRISPR discovery pipeline without Cas1 or Cas2 gene requirements. Cas13d direct repeats (DRs) are highly conserved in length and predicted secondary structure (Figure S2C), with a 36 nt length, an 8–10 nt stem with A/U-rich loop, and a 5′-AAAAC motif at the 3′ end of the direct repeat (Figure S2D). This conserved 5′-AAAAC motif has been previously shown to be specifically recognized by a type II Cas1/2 spacer acquisition complex (Wright and Doudna, 2016). In fact, Cas1 can be found in relative proximity to some Cas13d systems (within 10–30 kb for P1E0 and Rfx) while the remaining Cas13d-containing bacteria contain Cas1 elsewhere in their genomes, likely as part of another CRISPR locus.
CRISPR-Cas13d possesses dual RNase activities
To assess if the Cas13d repeat array is transcribed and processed into CRISPR guide RNAs (gRNA) as predicted (Deltcheva et al., 2011), we cloned the Cas13d CRISPR locus from an uncultured Ruminococcus sp. sample (Ur) into a bacterial expression plasmid. CRISPR systems tend to form self-contained operons with the necessary regulatory sequences for independent expression, facilitating heterologous expression in E. coli (Gasiunas et al., 2012). RNA sequencing (Heidrich et al., 2015) revealed processing of the array into ~52nt mature gRNAs, with a 30 nt 5′ direct repeat followed by a variable 3′ spacer that ranged from 14–26 nt in length (Figure 1B).
To characterize Cas13d properties in vitro, we next purified Eubacterium siraeum Cas13d protein (EsCas13d) based on its robust recombinant expression in E. coli (Figure S3) and found that EsCas13d was solely sufficient to process its matching CRISPR array into constituent guides without additional helper ribonucleases (Figure 1C, Table S1), a property shared by some class 2 CRISPR-Cas systems (East-Seletsky et al., 2016; Fonfara et al., 2016; Smargon et al., 2017). Furthermore, inactivating the positively charged catalytic residues of the HEPN motifs (Anantharaman et al., 2013) (dCas13d: R295A, H300A, R849A, H854A) did not affect array processing, indicating a distinct RNase activity dictating gRNA biogenesis analogous to Cas13a (East-Seletsky et al., 2016; Liu et al., 2017).
Cas effector proteins typically form a binary complex with mature gRNA to generate an RNA-guided surveillance ribonucleoprotein capable of cleaving foreign nucleic acids for immune defense (van der Oost et al., 2014). To assess if Cas13d has programmable RNA targeting activity as predicted by the presence of two HEPN motifs, EsCas13d protein was paired with an array or a mature gRNA along with a cognate in vitro-transcribed target. Based on the RNA sequencing results, we selected a mature gRNA containing a 30 nt direct repeat and an intermediate spacer length of 22 nt.
Cas13d was able to efficiently cleave the complementary target ssRNA with both the unprocessed array and mature gRNA in a guide-sequence dependent manner, while non-matching spacer sequences abolished Cas13d activity (Figure 2A). Substitution with dCas13d or the addition of EDTA to the cleavage reaction also abolished guide-dependent RNA targeting, indicating that Cas13d targeting is HEPN- and Mg2+ -dependent (Figure 2B). To determine the minimal spacer length for efficient Cas13d targeting, we next generated a series of spacer truncations ranging from the unprocessed 30 nt length down to 10 nt (Figure S4A). Cleavage activity dropped significantly below a 21 nt spacer length, confirming the choice of a 22 nt spacer (Figure S4B).
Figure 2. Programmable RNA targeting by Cas13d in vitro.
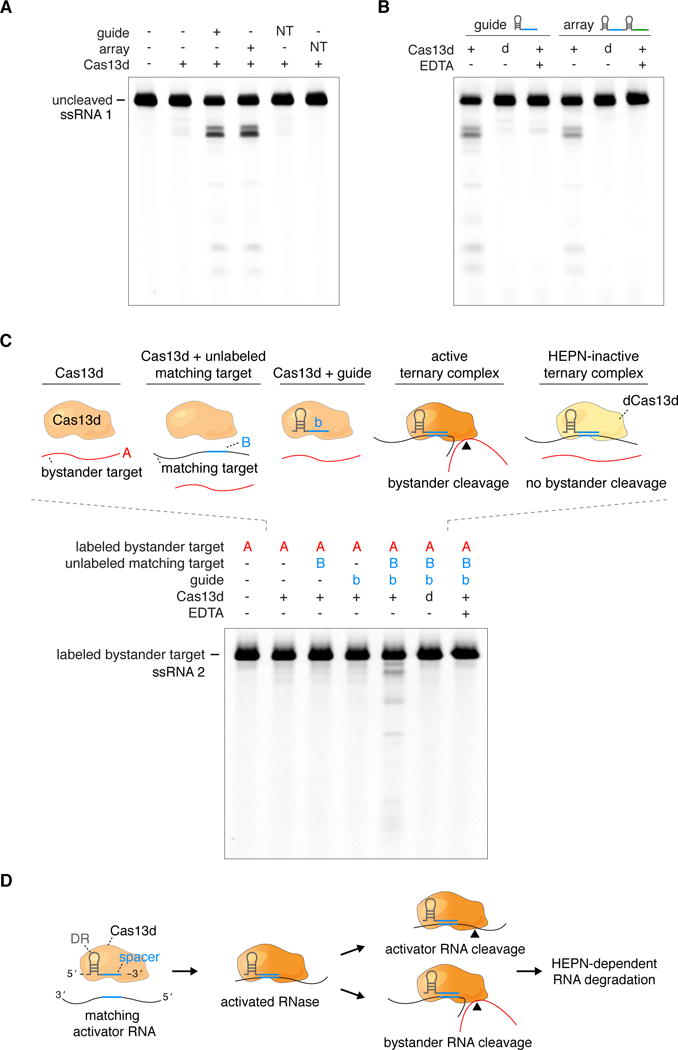
(A) E. siraeum Cas13d requires a matching guide array or mature gRNA to efficiently cleave complementary ssRNA targets. Denaturing gel depicts cleavage reactions incubated at 37°C for 1 hour. NT, non-targeting. (B) Substitution with dCas13d or addition of EDTA abrogate Cas13d-mediated RNA targeting with both the guide and array. ‘d’, dead Cas13d. (C) Denaturing gel depicting guide-target match dependent activation of Cas13d cleavage activity. Scrambled target RNA (‘A’) is fluorescently labeled, while guide-complementary activator target RNA (‘B’) is unlabeled. RNA cleavage activity is abolished by the individual removal of guide RNA or complementary target RNA, as well as the addition of EDTA or the catalytic inactivation of Cas13d (indicated as ‘d’). (D) A model for guide and target-dependent activation of Cas13d RNase activity. The ternary Cas13d:gRNA:target RNA complex is capable of cleaving the complementary target RNA or bystander RNAs.
See also Figure S4.
RNA-targeting class 2 CRISPR systems have been proposed to act as sensors of foreign RNAs (Abudayyeh et al., 2016; East-Seletsky et al., 2016), where general RNase activity of the effector nuclease is triggered by a guide-matching target. To assay for a similar property in Cas13d, RNase activity of the binary EsCas13d:gRNA complex was monitored in the presence of a matching RNA target. We observed that EsCas13d can be activated by target RNA to cleave bystander RNA targets (Figure 2C), albeit inefficiently relative to its activity on the complementary ssRNA target. Bystander cleavage is guide sequence- and HEPN-dependent, as the presence of non-matching bystander target alone was insufficient to induce cleavage while substitution of dCas13d or addition of EDTA abolished activity. These results suggest that bystander RNase activity may be a general property of RNA-targeting class 2 systems in CRISPR adaptive bacterial immunity (Figure 2D).
To assess the generalizability of Cas13d reprogramming, we first generated twelve guides tiling a complementary RNA target and observed efficient cleavage in all cases (Figure 3A). Cas13d was unable to cleave a ssDNA (Figure S4C) or dsDNA (Figure S4D) version of the ssRNA target, indicating that Cas13d is an RNA-specific nuclease. Further, RNA target cleavage did not appear to depend on the protospacer flanking sequence (PFS) (Figure 3A) in contrast to other RNA-targeting class 2 systems, which require a 3′-H (Abudayyeh et al., 2016) or a double-sided, DR-proximal 5′-D and 3′-NAN or NNA (Smargon et al., 2017). Although we initially observed a slight bias against an adenine PFS (Figure S4E), varying the target PFS base with a constant guide sequence resulted in no significant differences (P=0.768) in targeting efficiency (Figure S4F).
Figure 3. Characterization of Cas13d target substrate preference.
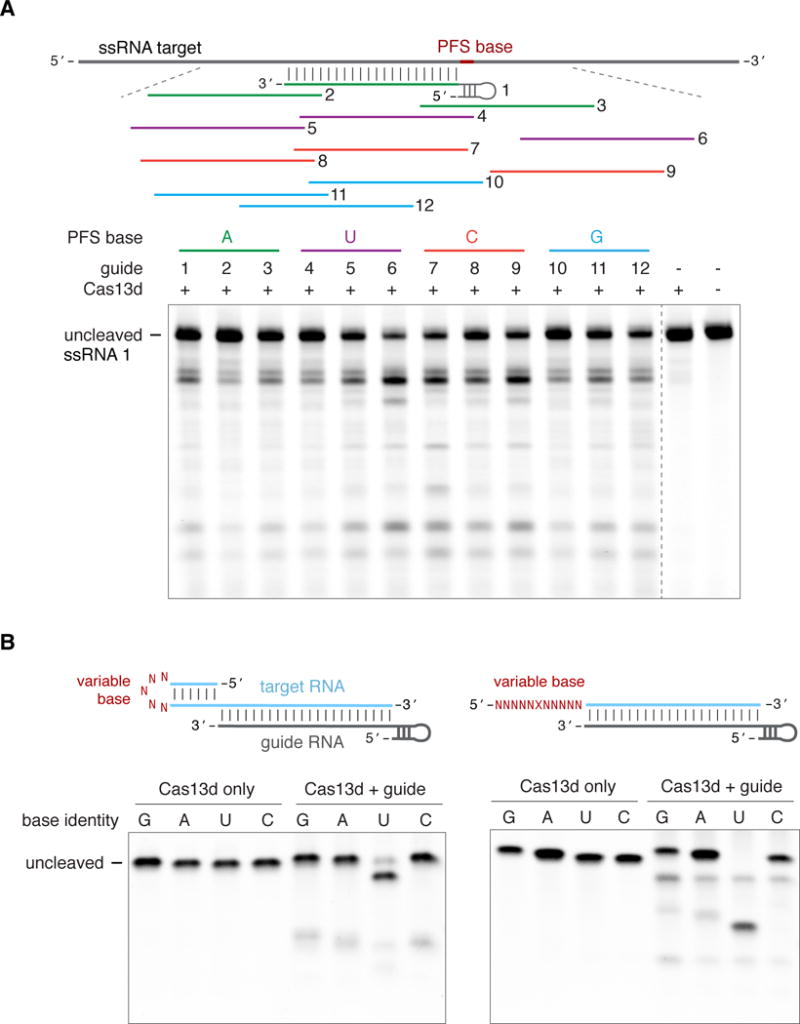
(A) Cas13d can be generalizably reprogrammed with multiple guides and does not exhibit a protospacer flanking sequence (PFS) requirement. RNA cleavage by EsCas13d and 12 guides tiling the target RNA is shown. Control lanes are from a separate gel run in parallel. (B) Cas13d preferentially cleaves uracil bases in the loop of a hairpin or a linear homopolymer repeat, which is interrupted every 5 nt by a transition mutation (X) to enable synthesis.
See also Figure S4.
While DNA-targeting class 2 CRISPR systems (Gasiunas et al., 2012; Jinek et al., 2012; Zetsche et al., 2015) and some RNA-targeting class 1 systems tend to cleave at defined positions relative to the target-guide duplex (Samai et al., 2015; Zhang et al., 2016), the Cas13d cleavage pattern varies for different targets (Figure 2A, 2C, S4H) and remains remarkably similar despite the guide sequence position (Figure 3A). This suggests that Cas13d may preferentially cleave specific sequences or structurally accessible regions in the target RNA. We tested Cas13d activity on targets containing variable homopolymer repeats in the loop region of a hairpin or as a linear single-stranded repeat. EsCas13d exhibited significant preference for uracil bases in both target structures, with lower but detectable activity at all other bases (Figure 3B).
Cas enzymes are found in nearly all archaea and about half of bacteria (Hsu et al., 2014; van der Oost et al., 2014), spanning a wide range of environmental temperatures. To determine the optimal temperature range for Cas13d activity, we next tested a spectrum of cleavage temperature conditions from 16–62°C and observed maximal activity in the 24–41°C range (Figure S4G, S4H). This temperature range is compatible with a wide range of prokaryotic and eukaryotic hosts, raising the possibility of adapting Cas13d for RNA targeting in different cells and organisms.
Cell-based activity screen of engineered orthologs
We next sought to develop the Cas13d nuclease into a flexible tool for programmable RNA targeting in mammalian cells. CRISPR orthologs from distinct bacterial species commonly exhibit variable activity (Abudayyeh et al., 2017; East-Seletsky et al., 2017), especially upon heterologous expression in human cells (Ran et al., 2015; Zetsche et al., 2017). We therefore sought to identify highly active Cas13d orthologs in a eukaryotic cell-based mCherry reporter screen.
By synthesizing human codon-optimized versions of 7 orthologs from distinct branches within the Cas13d family (Figure S1B), we generated mammalian expression plasmids carrying the catalytically active and HEPN-inactive proteins. Each protein was then optionally fused to N- and C-terminal nuclear localization signals (NLS). These Cas13d effector designs were HA-tagged and paired with two distinct guide RNA architectures, either with a 30 nt spacer flanked by two direct repeat sequences to mimic an unprocessed guide RNA (pre-gRNA) or a 30 nt direct repeat with 22 nt spacer (gRNA) predicted to mimic mature guide RNAs (Figure 4A). For each guide design, four distinct spacer sequences complementary to the mCherry transcript were then pooled to minimize potential spacer-dependent variability in targeting efficiency. We then assessed the ability of Cas13d to knockdown mCherry protein levels in a human embryonic kidney (HEK) 293FT cell-based reporter assay.
Figure 4. RNA knockdown activity screen of engineered Cas13d orthologs in human cells.
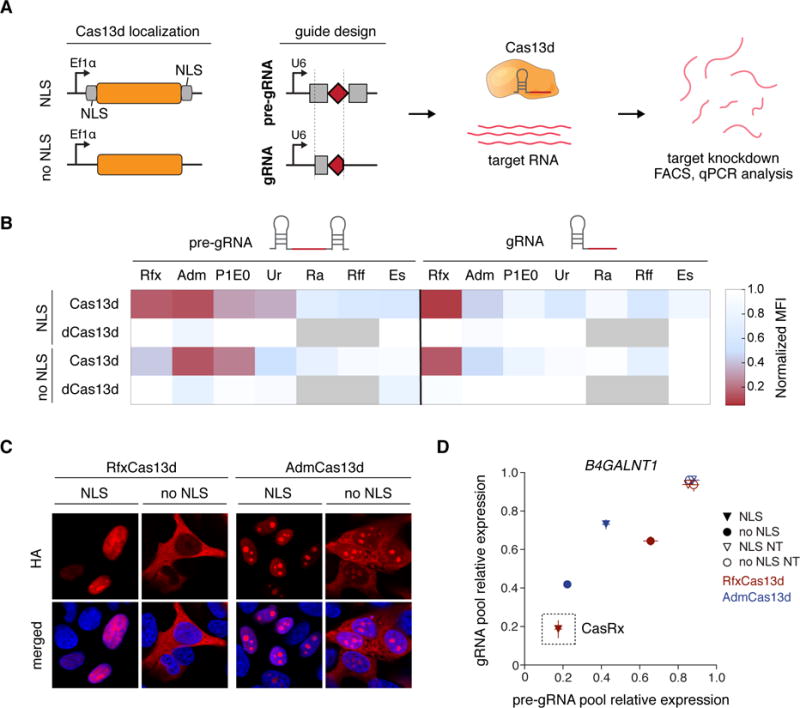
(A) Schematic for mammalian expression constructs encoding for engineered Cas13d effectors and guides. NLS, nuclear localization signal. pre-gRNA, artificial unprocessed guide RNA containing a single 30 nt spacer sequence flanked by 2 full length 36 nt DRs. gRNA, predicted mature guide RNA with a single 30 nt processed DR and 22 nt spacer sequence (B) Heatmap of mCherry protein knockdown in a Cas13d ortholog activity screen in HEK 293FT cells using pools of 4 pre-gRNAs or gRNAs. Normalized MFI, median fluorescent intensity relative to non-targeting condition. Positions in gray were not tested, with n = 3. (C) Immunocytochemistry of Cas13d showing localization and expression of engineered constructs. Scale bar, 10 μm. Blue pseudocolor, DAPI staining of nuclei. (D) Comparison of Adm and Rfx Cas13d ortholog constructs for knockdown of endogenous B4GALNT1 mRNA reveals RfxCas13d-NLS (CasRx) to be most effective for both guide RNA architectures. Pools of 4 guides were used for targeting. NT, non-targeting. Values are mean ± SEM with n = 3.
48 hours post-transfection, flow cytometry indicated that RfxCas13d and AdmCas13d efficiently knocked down mCherry protein levels by up to 92% and 87% (P<0.0003), respectively, relative to a non-targeting control guide (Figure 4B). In contrast, EsCas13d along with RaCas13d and RffCas13d exhibited limited activity in human cells. Furthermore, none of the HEPN-inactive Rfx-dCas13d constructs significantly affected mCherry fluorescence, suggesting HEPN-dependent knockdown (P>0.43 for all cases). Robust nuclear translocation of the Rfx and AdmCas13d NLS fusion constructs was observed via immunocytochemistry, while the wild-type effectors remain primarily extra-nuclear (Figure 4C).
Proceeding with RfxCas13d and AdmCas13d as lead candidates, we next compared their ability to knockdown endogenous transcripts. To determine the optimal ortholog and guide architecture, we systematically assayed the capability of Rfx and AdmCas13d construct variants to target β-1,4-N-acetyl-galactosaminyl transferase 1 (B4GALNT1) transcripts. In each condition, we again pooled four guides containing distinct spacer sequences tiling the B4GALNT1 transcript. We found that the RfxCas13d-NLS fusion targeted B4GALNT1 more efficiently than wild-type RfxCas13d and both variants of AdmCas13d, with both the gRNA and pre-gRNA mediating potent knockdown (~82%, P<0.0001) (Figure 4D). We therefore chose Cas13d-NLS from Ruminococcus flavefaciens strain XPD3002 for the remaining experiments (CasRx).
Programmable RNA knockdown in human cells with CasRx
Because Cas13d is capable of processing its own CRISPR array, we next leveraged this property for the simultaneous delivery of multiple targeting guides in a simple single-vector system (Figure 5A). Arrays encoding four spacers that each tile the transcripts of mRNAs (B4GALNT1 and ANXA4) or nuclear localized lncRNAs (HOTTIP and MALAT1) consistently facilitated robust (>90%) RNA knockdown by CasRx (P<0.0001) (Figure 5B).
Figure 5. CasRx mediates efficient and specific knockdown of diverse human coding and noncoding transcripts.
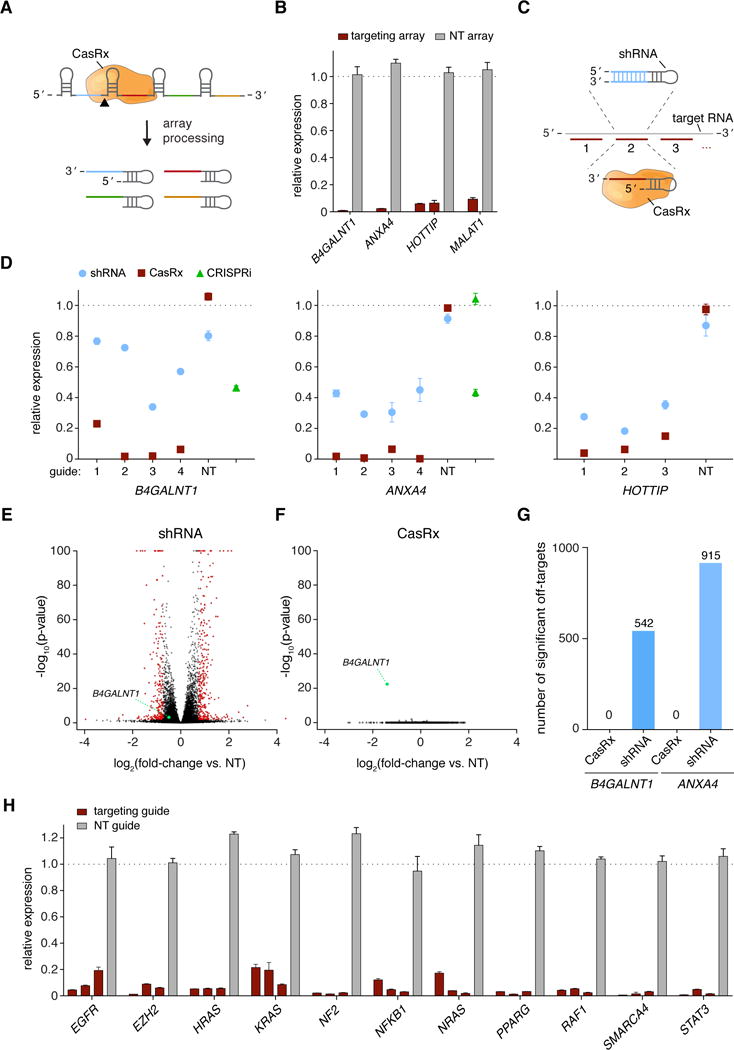
(A) Multiple guide RNAs tiling a target transcript can be expressed as a single array and processed by RfxCas13d-NLS (CasRx) into individual gRNAs within the same cell. (B) Arrays of 4 guides each mediate target knockdown by CasRx in 293FT cells via transient transfection. Knockdown relative to GFP vehicle control was determined by qPCR, with n = 3. (C) Schematic of CasRx target sequences and spacer position-matched shRNAs. (D) Relative target RNA knockdown by individual position-matched shRNAs and CasRx gRNAs. NT, non-targeting. CRISPRi, dCas9-mediated transcriptional repression (n = 3) (E) Volcano plot of differential transcript levels between B4GALNT1 targeting and non-targeting (NT) shRNAs as determined by RNA sequencing (n = 3). 542 non-specific transcript changes were identified. (F) Volcano plot of differential transcript levels between B4GALNT1 -targeting CasRx and non-targeting (NT) guide. Targeting guide position is matched to the shRNA shown in (E). B4GALNT1 was the only transcript exhibiting a significant change, with n = 3. (G) Summary of significant off-target transcript perturbations by matched shRNAs and CasRx guides. (H) CasRx targeting of 11 endogenous transcripts, each with 3 guides and a non-targeting (NT) guide in 293FT cells. Transcript levels are relative to GFP vehicle control, mean ± SEM with n = 3.
See also Figure S5 and S6.
We next sought to benchmark CasRx against more established technologies for transcript knockdown or repression, comparing CasRx-mediated RNA interference to dCas9-mediated CRISPR interference (Gilbert et al., 2014; Gilbert et al., 2013) and spacer sequence-matched shRNAs via transient transfection (Figure 5C). For CRISPRi-based repression, we included the most potent dCas9 guide for B4GALNT1 from previous reports (Gilbert et al., 2014; Zalatan et al., 2015). Across 3 endogenous transcripts, CasRx outperformed shRNAs (11/11) and CRISPRi (4/4) in each case (Figure 5D), exhibiting a median knockdown of 96% compared to 65% for shRNA and 53% for CRISPRi after 48 hours. In addition, we compared knockdown by CasRx to two recently described Cas13a and Cas13b effectors (Abudayyeh et al., 2017; Cox et al., 2017) (Figure S5A). Across three genes and eight guide RNAs, CasRx mediated significantly greater transcript knockdown than both LwaCas13a-msfGFP-NLS and PspCas13b-NES (median: 97% compared to 80% and 66% respectively, P< 0.0001) (Figure S5B).
RNAi has been widely used to disrupt any gene of interest due to a combination of simple retargeting principles, scalable synthesis, knockdown potency, and ease of reagent delivery. However, widespread off-target transcript silencing has been a consistent concern (Jackson et al., 2003; Sigoillot et al., 2012), possibly due to the entry of RNAi reagents into the endogenous miRNA pathway (Doench et al., 2003; Smith et al., 2017). Consistent with these reports, upon RNA sequencing of human cells transfected with a B4GALNT1-targeting shRNA, we observed widespread off-target transcriptional changes relative to a non-targeting shRNA (>500 significant off-target changes, P<0.01, Figure 5E, 5G). In contrast, transcriptome profiling of spacer-matched CasRx guide RNAs revealed no significant off-target changes other than the targeted transcript (Figure 5F). This suggests that the moderate bystander cleavage observed in vitro (Figure 2C) may not result in observable off-target transcriptome perturbation in mammalian cells. We observed a similar pattern when targeting ANXA4 (Figure S6), with over 900 significant off-target changes resulting from shRNA targeting compared to zero with CasRx (Figure 5G).
To confirm that CasRx interference is broadly applicable, we selected a panel of 11 additional genes with diverse roles in cancer, cell signaling, and epigenetic regulation and screened 3 guides per gene. CasRx consistently mediated high levels of transcript knockdown across genes with a median reduction of 96% (Figure 5H). Each tested guide mediated at least 80% knockdown, underscoring the consistency of the CasRx system for RNA interference.
Splice isoform engineering with dCasRx
Our experiments on RNA targeting with CasRx revealed that target RNA and protein knockdown is dependent on the catalytic activity of the HEPN domains (Figure 4B, 2B). The same guide sequences mediating efficient knockdown with CasRx failed to significantly reduce mCherry levels when paired with catalytically inactive dCasRx (Figure 4B), indicating that targeting of dCasRx to the coding portion of mRNA does not necessarily perturb protein translation. This observation suggested the possibility of utilizing dCasRx for targeting of specific coding and non-coding elements within a transcript to study and manipulate RNA. To validate this concept, we sought to expand the utility of the dCasRx system by creating a splice effector.
Alternative splicing is generally regulated by the interaction of cis-acting elements in the pre-mRNA with positive or negative trans-acting splicing factors, which can mediate exon inclusion or exclusion (Matera and Wang, 2014; Wang et al., 2015). We reasoned that dCasRx binding to such motifs may be sufficient for targeted isoform perturbation. For proof-of-concept, we identified distinct splice elements in a bichromatic splicing reporter containing DsRed upstream of mTagBFP2 in two different reading frames following an alternatively spliced exon (Orengo et al., 2006) (Figure 6A). Inclusion or exclusion of this second exon toggles the reading frame and resulting fluorescence, facilitating quantitative readout of splicing patterns by flow cytometry. To mediate exon skipping, four guide RNAs were designed to target the intronic branchpoint nucleotide, splice acceptor site, putative exonic splice enhancer, and splice donor of exon 2.
Figure 6. AAV delivery of catalytically inactive dCasRx splice effectors to manipulate alternative splicing.
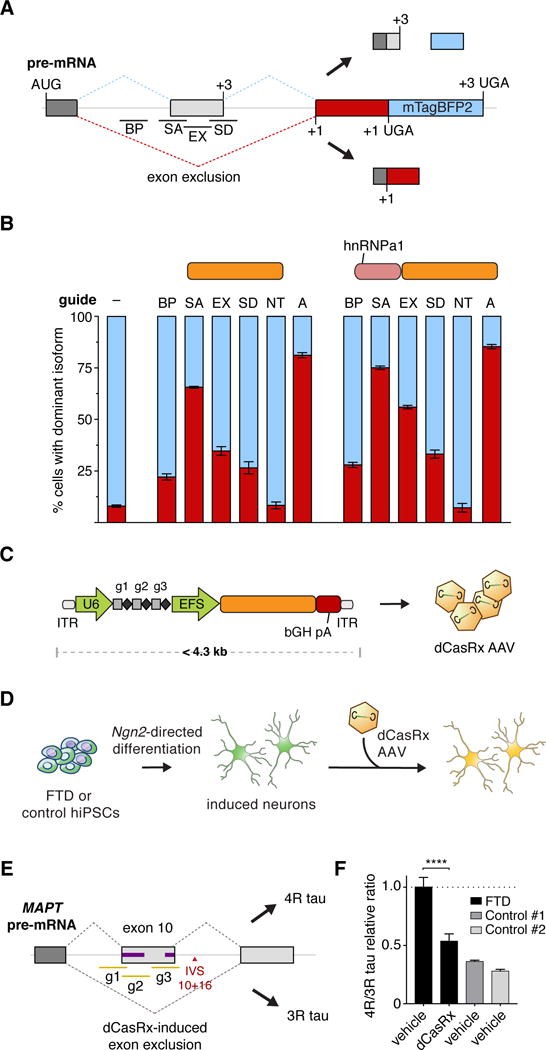
(A) Schematic of bichromatic exon skipping reporter. +1 and +3, reading frame. BP, intronic branch point-targeting guide. SA, splice acceptor site-overlapping guide. EX, exonic guide. SD, splice donor site-overlapping guide. AUG, start codon. UGA, stop codon. Inclusion of the second exon leads to an out-of-frame (+3), non-fluorescent translation of dsRed followed by in-frame mTagBFP2. Exclusion of the targeted exon leads to an in frame translation of dsRed (+1) followed by a stop codon. (B) Induced exon exclusion by dCasRx and an N-terminal hnRNPa1-dCasRx fusion protein targeted to pre-mRNA. The Gly-rich C-terminal domain of hnRNPa1 is used as the effector domain. Exon skipping efficiency is depicted as a relative percentage of cells carrying primarily the dsRed or BFP isoform, determined through flow cytometry. ‘A’, CRISPR array carrying all 4 guides. Values are mean ± SEM with n = 3. (C) AAV design carrying dCasRx and a three-guide array with total transgene size < 4.3 kb, including AAV inverted terminal repeats (ITRs). (D) Schematic of frontotemporal dementia (FTD) disease modeling. Neurons are generated via Neurogenin-2 (Ngn2) directed differentiation of patient-derived and control iPSCs followed by transduction with dCasRx or vehicle control AAV (EFS-mTagBFP2). (E) FTD is associated with SNPs in a putative intronic splice enhancer following exon 10 of the MAPT transcript encoding for tau. Alternative splicing of MAPT exon 10 results in 4R tau (by inclusion) and 3R tau (by exclusion). SNPs in the intronic splice enhancer including the indicated IVS 10 + 16 mutation result in increased exon inclusion and higher levels of 4R tau. gRNAs contained in a dCasRx array were targeted to the exon 10 splice acceptor (g1) as well as two putative exonic splice enhancers indicated in purple (g2, g3). (F) Relative 4R/3R tau transcript ratios in differentiated neurons were assayed via qPCR at 14 days following transduction with AAV. FTD, frontotemporal dementia cells carrying IVS 10+16. Values are mean ± S.D. with n = 3. ****indicates P < 0.0001.
See also Figure S5.
One widespread family of negative splice factors are the highly conserved heterogeneous nuclear ribonucleoproteins (hnRNPs), which typically inhibit exon inclusion via a C-terminal, glycine-rich domain (Wang et al., 2015). We targeted the splicing reporter with dCasRx and engineered fusions to the Gly-rich C-terminal domain of hnRNPa1, one of the most abundant hnRNP family members (Figure 6B).
Guide position appears to be a major determinant of the efficiency of engineered exon skipping. While each guide position mediated a significant increase in exon exclusion (P<0.0001 in all cases) relative to the non-targeting guide, targeting the splice acceptor resulted in the most potent exon exclusion (increase from 8% basal skipping to 65% for dCasRx alone and 75% with hnRNPa1 fusion). By comparison, dLwaCas13a-msfGFP-NLS mediated significantly lower levels of exon skipping across all four positions (19% skipping for splice acceptor guide) (Figure S5C and D, P<0.0001).
Targeting all 4 positions simultaneously with a CRISPR array achieved higher levels of exon skipping than individual guides alone (81% for dCasRx and 85% for hnRNPa1 fusion, P<0.006 compared to SA guide) (Figure 6B). These results indicate that dCasRx allows for tuning of isoform ratios through varying guide placement and suggest that it can be leveraged as an efficient RNA binding module in human cells for targeting and manipulation of specific RNA elements.
Viral delivery of dCasRx to a neuronal model of frontotemporal dementia
The Cas13d family averages 930 amino acids in length, in contrast to Cas9 (~1100 aa to ~1400 aa depending on subtype, with compact outliers such as CjCas9 or SaCas9), Cas13a (1250 aa), Cas13b (1150 aa), and Cas13c (1120 aa) (Figure S2B) (Chylinski et al., 2013; Cox et al., 2017; Hsu et al., 2014; Kim et al., 2017; Shmakov et al., 2015; Smargon et al., 2017). Although adeno-associated virus (AAV) is a versatile vehicle for transgene delivery and gene therapy due to its broad range of capsid serotypes, low levels of insertional mutagenesis, and lack of apparent pathogenicity, its limited packaging capacity (~4.7kb) makes it challenging to effectively deliver many single effector CRISPR enzymes (Abudayyeh et al., 2017; Ran et al., 2015; Swiech et al., 2015). The remarkably small size of Cas13d effectors render them uniquely suited for all-in-one AAV delivery with a CRISPR array, an optional effector domain, and requisite expression or regulatory elements (Figure 6C).
Frontotemporal Dementia with Parkinsonism linked to Chromosome 17 (FTDP-17) is an autosomal dominant major neurodegenerative disease caused by diverse point mutations in MAPT, the gene encoding for tau. Tau exists as two major isoforms in human neurons, 4R and 3R, which are distinguished by the presence or absence of tau exon 10 and thus contain 4 or 3 microtubule binding domains. The balance of these two isoforms is generally perturbed in FTDP-17 as well as other tauopathies, driving the progression of neurodegeneration (Boeve and Hutton, 2008). Some forms of FTD are caused by mutations in the intron following MAPT exon 10 which disrupt an intronic splice silencer and elevate the expression of 4R tau (Kar et al., 2005), thereby inducing pathological changes (Schoch et al., 2016).
We reasoned that dCasRx targeted to MAPT exon 10 could induce exon exclusion to alleviate dysregulated 4R/3R tau ratios. Patient-derived human induced pluripotent stem cells (hiPSCs) were differentiated into cortical neurons via Neurogenin-2 directed differentiation for 2 weeks (Zhang et al., 2013). Postmitotic neurons were then transduced with AAV1 carrying dCasRx (Figure 6D) paired with a repeat array containing 3 spacers that target the exon 10 splice acceptor and two putative exonic splice enhancers (Figure 6E). dCasRx-mediated exon exclusion was able to reduce the relative 4R/3R tau ratio by nearly 50% relative to a BFP vehicle control (Figure 6F) and to a level similar to unaffected control neurons, suggesting that CasRx can be exploited for transcriptional modulation in primary cell types via AAV delivery.
Discussion
Class 2 CRISPR systems are found throughout diverse bacterial and archaeal life. Using a minimal definition of the CRISPR locus for bioinformatic mining of prokaryotic genome and metagenome sequences, which requires only a CRISPR repeat array and a nearby protein, we report the identification of an uncharacterized, remarkably compact family of RNA-targeting class 2 CRISPR systems that we designate Type VI CRISPR-Cas13d.
Because CRISPR systems generally exist as a functional operon within 20 kilobases of genome sequence, even fragmented metagenome reads may be sufficient to recover useful Cas enzymes for bioengineering purposes. CRISPR genome mining strategies described here and by others (Shmakov et al., 2015), combined with ongoing efforts to profile microbial populations via next-generation sequencing, should be anticipated to contribute mechanistically diverse additions to the genome engineering toolbox.
We biochemically characterized two distinct ribonuclease properties of the Cas13d effector, which processes a CRISPR repeat array into mature guides via a HEPN domain-independent mechanism followed by guide sequence-dependent recognition of a complementary activator RNA. This triggers HEPN-mediated RNase activity, enabling Cas13d to cleave both activator and bystander RNAs, a property shared by other RNA-targeting CRISPR systems. Cas13d additionally exhibits no apparent flanking sequence requirements and was found to be active across crRNAs tiling a target RNA, suggesting the ability to target arbitrary single-stranded RNA sequences.
A comprehensive activity reporter screen in human cells of Cas13d orthologs sampled from distinct branches of the Cas13d family revealed that NLS fusions to Cas13d from Ruminococcus flavefaciens strain XPD3002 (CasRx) can be engineered for programmable RNA targeting in a eukaryotic context (Figure 4D). CasRx fusions knocked down a diverse set of 14 endogenous mRNAs and lncRNAs, consistently achieving >90% knockdown with favorable efficiency relative to RNA interference, dCas9-mediated CRISPR interference, and other members of the Cas13 superfamily (Figure S5). Additionally, CasRx interference is markedly more specific than spacer-matching shRNAs, with no detectable off-target changes compared with hundreds for RNA interference.
CasRx is a minimal two-component platform, consisting of an engineered CRISPR-Cas13d effector and an associated guide RNA, and can be fully genetically encoded. Because CasRx is an orthogonally delivered protein, HEPN-inactive dCasRx can be engineered as a flexible RNA-binding module to target specific RNA elements. Importantly, because CasRx uses a distinct ribonuclease activity to process guide RNAs, dCasRx can still be paired with a repeat array for multiplexing applications. We demonstrated the utility of this concept by creating a dCasRx splice effector fusion for tuning alternative splicing and resulting protein isoform ratios, applying it in a neuronal model of frontotemporal dementia.
At an average size of 930 aa, Cas13d is to our knowledge the smallest class 2 CRISPR effector characterized in mammalian cells. This allows CasRx effector domain fusions to be paired with a CRISPR array encoding multiple guide RNAs while remaining under the packaging size limit of the versatile adeno-associated virus (AAV) delivery vehicle (Naldini, 2015) for primary cell and in vivo delivery. Further, targeted AAV delivery of CasRx to specific postmitotic cell types such as neurons has the potential to mediate long-term expression of a corrective payload that avoids permanent genetic modifications or frequent re-administration (Chiriboga et al., 2016), complementing other nucleic acid targeting technologies such as DNA nuclease editing or antisense oligonucleotides. RNA mis-splicing diseases have been estimated to account for up to 15% of genetic diseases (Hammond and Wood, 2011), highlighting the potential for engineered splice effectors capable of multiplexed targeting. We envision diverse applications to complement RNA targeting for knockdown and splicing, such as live cell labeling and genetic screens to transcript imaging, trafficking, or regulation. CRISPR-Cas13d and engineered variants such as CasRx collectively enable flexible nucleic acid engineering, transcriptome-related study, and future therapeutic development, expanding the genome editing toolbox beyond DNA to RNA.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Patrick D. Hsu (patrick@salk.edu). Key plasmids described in this study will be distributed to the research community via the Addgene plasmid repository under a standard MTA.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture of Human Embryonic Kidney (HEK) cell line 293FT
Human embryonic kidney (HEK) cell line 293FT (female, Thermo Fisher) was maintained in DMEM (4.5 g/L glucose), supplemented with 10% FBS (GE Life Sciences) and 10 mM HEPES at 37°C with 5% CO2. Upon reaching 80–90% confluency, cells were dissociated using TrypLE Express (Life Technologies) and passaged at a ratio of 1:2. This cell line was purchased directly from the manufacturer and was not otherwise authenticated.
Cell culture of human bone osteosarcoma epithelial cell line U2OS
Human bone osteosarcoma epithelial U2OS (female) were maintained in DMEM (4.5 g/L glucose) supplemented with 10% FBS and 10 mM HEPES at 37°C with 5% CO2. Cells were passaged at a 1:3 ratio upon reaching 70% confluence. This cell line was not authenticated.
Maintenance of induced pluripotent stem cells and neuronal differentiation
Stable human iPSC lines containing the FTDP-17 IVS10+16 mutation or age- and sex-matched control lines were obtained from the laboratory of Fen-Biao Gao (Biswas et al., 2016). Briefly, cells obtained from one male patient with the MAPT IVS10 + 16 mutation and two separate lines from one male control patient were reprogrammed into hiPSCs (Almeida et al., 2012). iPSCs were transduced with lentivirus containing a doxycycline-inducible Ngn2 cassette. Lentiviral plasmids were a gift from S. Schafer and F. Gage. iPSCs were then passaged with Accutase and plated into a Matrigel-coated 6-well plate with mTESR media containing ROCK inhibitor Y-27632 (10 μM, Cayman) at 500,000 cells per well. On day 1, media was changed with mTESR. On day 2, media was changed to mTESR containing doxycycline (2 μg/ml, Sigma) to induce Ngn2 expression. On day 3, culture media was replaced with Neural Induction media (NIM, DMEM/F12 (Life Technologies) containing BSA (0.1 mg/ml, Sigma), apo-transferrin (0.1 mg/ml, Sigma), putrescine (16 μg/ml, Sigma), progesterone (0.0625 μg/ml, Sigma), sodium selenite (0.0104 μg/ml, Sigma), insulin (5 μg/ml, Roche), BDNF (10 ng/ml, Peprotech), SB431542 (10 μM, Cayman), LDN-193189 (0.1 μM, Sigma), laminin (2 μg/ml, Life Technologies), doxycycline (2 μg/ml, Sigma) and puromycin (Life Technologies)). NIM media was changed daily. Following 3 days of puromycin selection, immature neuronal cells were passaged with Accumax (Innovative Cell Technologies) and plated onto 96-well plates coated with poly-D-lysine and Matrigel in Neural Maturation media (NMM; 1:1 Neurobasal/DMEM (Life Technologies) containing B27 (Life Technologies), BDNF (10 ng/ml, Peprotech), N-Acetylcysteine (Sigma), laminin (2 μg/ml, Life Technologies), dbcAMP (49 μg/ml, Sigma) and doxycycline (2 μg/ml, Sigma). Media was replaced the next day (day 7) with NMM containing AraC (2 μg/ml, Sigma) to eliminate any remaining non-differentiated cells. On day 8, AraC was removed and astrocytes were plated on top of neurons to support neuron cultures in NMM containing hbEGF (5 ng/ml, Peprotech). Cells were transduced with AAV on day 10 and assayed on day 24.
METHOD DETAILS
Computational pipeline for Cas13d identification
We obtained whole genome, chromosome, and scaffold-level prokaryotic genome assemblies from NCBI Genome in June 2016 and compared CRISPRfinder, PILER-CR, and CRT for identifying CRISPR repeats. The 20 kilobase flanking regions around each putative CRISPR repeat was extracted to identify nearby proteins and predicted proteins using Python. Candidate Cas proteins were required to be >750 aa in length and within 5 proteins of the repeat array, and extracted CRISPR loci were filtered out if they contained Cas genes associated with known CRISPR systems such as types I-III CRISPR. Putative effectors were clustered into families via all-by-all BLASTp analysis followed by single-linkage hierarchical clustering where a bit score of at least 60 was required for cluster assignment. Each cluster of at least 2 proteins was subjected to BLAST search against the NCBI non-redundant (nr) protein database, requiring a bit score >200 to assign similarity. The co-occurrence of homologous proteins in each expanded cluster to a CRISPR array was analyzed and required to be >70%. Protein families were sorted by average amino acid length and multiple sequence alignment for each cluster was performed using Clustal Omega and the Geneious aligner with a Blosum62 cost matrix. The RxxxxH HEPN motif was identified in the Cas13d family on the basis of this alignment. TBLASTN was performed on all predicted Cas13d effectors against public metagenome whole genome shotgun sequences without predicted open reading frames (ORFs). The Cas13d family was regularly updated via monthly BLAST search on genome and metagenome databases to identify any newly deposited sequences. New full-length homologs and homologous fragments were aligned using Clustal Omega and clustered using PhyML 3.2. CRISPRDetect was used to predict the direction of direct repeats in the Cas13d array and DR fold predictions were performed using the Andronescu 2007 RNA energy model at 37°C (Andronescu et al., 2007). Sequence logos for Cas13d direct repeats were generated using Geneious 10.
Protein expression and purification
Recombinant Cas13d proteins were PCR amplified from genomic DNA extractions of cultured isolates or metagenomic samples and cloned into a pET-based vector with an N-terminal His-MBP fusion and TEV protease cleavage site. The resulting plasmids were transformed into Rosetta2(DE3) cells (Novagen), induced with 200 μM IPTG at OD600 0.5, and grown for 20 hours at 18°C. Cells were then pelleted, freeze-thawed, and resuspended in Lysis Buffer (50 mM HEPES, 500 mM NaCl, 2 mM MgCl2, 20 mM Imidazole, 1% v/v Triton X-100, 1 mM DTT) supplemented with 1X protease inhibitor tablets, 1 mg/mL lysozyme, 2.5U/mL Turbo DNase (Life Technologies), and 2.5U/mL salt active nuclease (Sigma Aldrich). Lysed samples were then sonicated and clarified via centrifugation (18,000 × g for 1 hour at 4°C), filtered with 0.45 μM PVDF filter and incubated with 50 mL of Ni-NTA Superflow resin (Qiagen) per 10 L of original bacterial culture for 1 hour. The bead-lysate mixture was applied to a chromatography column, washed with 5 column volumes of Lysis Buffer, and 3 column volumes of Elution Buffer (50 mM HEPES, 500 mM NaCl, 300 mM Imidazole, 0.01% v/v Triton X-100, 10% glycerol, 1 mM DTT). The samples were then dialyzed overnight into TEV Cleavage Buffer (50mM Tris-HCl, 250 mM KCl, 7.5% v/v glycerol, 0.2 mM TCEP, 0.8 mM DTT, TEV protease) before cation exchange (HiTrap SP, GE Life Sciences) and gel filtration (Superdex 200 16/600, GE Life Sciences). Purified, eluted protein fractions were pooled and frozen at 4 mg/mL in Protein Storage Buffer (50 mM Tris-HCl, 1M NaCl, 10% glycerol, 2 mM DTT).
Preparation of guide and target RNAs
Oligonucleotides carrying the T7 promoter and appropriate downstream sequence were synthesized (IDT) and annealed with an antisense T7 oligo for crRNAs and PCR-amplified for target and array templates. Homopolymer target RNAs were synthesized by Synthego. The oligo anneal and PCR templates were in vitro transcribed with the Hiscribe T7 High Yield RNA Synthesis kit (New England Biolabs) at 31°C for 12 hours. For labeled targets, fluorescently labelled aminoallyl-UTP atto 680 (Jena Biosciences) was additionally added at 2 mM. Guide RNAs were purified with RNA-grade Agencourt AMPure XP beads (Beckman Coulter) and arrays and targets were purified with MEGAclear Transcription Clean-Up Kit (Thermo Fisher) and frozen at −80°C. For ssDNA and dsDNA targets, corresponding oligonucleotide sequences were synthesized (IDT) and either gel purified, or PCR amplified and then subsequently gel purified respectively.
Biochemical cleavage reactions
Purified EsCas13d protein and guide RNA were mixed (unless otherwise indicated) at 2:1 molar ratio in RNA Cleavage Buffer (25mM Tris pH 7.5, 15mM Tris pH 7.0, 1mM DTT, 6mM MgCl2). The reaction was prepared on ice and incubated at 37°C for 15 minutes prior to the addition of target at 1:2 molar ratio relative to EsCas13d. The reaction was subsequently incubated at 37°C for 45 minutes and quenched with 1 μL of enzyme stop solution (10 mg/mL Proteinase K, 4M Urea, 80mM EDTA, 20mM Tris pH 8.0) at 37°C for 15 minutes. The reaction was then denatured with 2X RNA loading buffer (2X: 13mM Ficoll, 8M Urea, 25 mM EDTA), at 85°C for 10 minutes, and separated on a 10% TBE-Urea gel (Life Technologies). Gels containing labeled targets were visualized on the Odyssey Clx Imaging System (Li-Cor); unlabeled array or target cleavage gels were stained with SYBR Gold prior to imaging via Gel Doc EZ system (Bio-Rad).
Transient transfection of human cell lines
Engineered Cas13 coding sequences were cloned into a standardized plasmid expression backbone containing an EF1a promoter and prepared using the Nucleobond Xtra Midi EF Kit (Machery Nagel) according to the manufacturer’s protocol. NLS-LwaCas13a-msfGFP and PspCas13b-NES-HIV were PCR amplified from Addgene #103854, and #103862, respectively, a gift from Feng Zhang. Cas13d pre-gRNAs and gRNAs were cloned into a minimal backbone containing a U6 promoter. shRNAs and guides for LwaCas13a were cloned into the same backbone and position matched to their corresponding guide RNA at the 3′ of the target sequence. Matched gRNAs for PspCas13b were moved to the closest 5′-G nucleotide.
For transient transfection, HEK 293FT cells were plated at a density of 20,000 cells per well in a 96-well plate and transfected at >90% confluence with 200 ng of Cas13 expression plasmid and 200 ng of gRNA expression plasmid using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Transfected cells were harvested 48–72 hours post-transfection for flow cytometry, gene expression analysis, or other downstream processing.
For reporter assays, HEK 293FT cells were transfected in 96-well format with 192ng of Cas13d expression plasmid, 192ng of guide expression plasmid, and 12ng of mCherry expression plasmid with Lipofectamine 2000 (Life Technologies). Cells were harvested after 48 hours and analyzed by flow cytometry.
U2OS cells were plated at a density of 20,000 cells per well in a 96-well plate and transfected at >90% confluence with 100 ng of Cas13d expression plasmid using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s protocol and processed for immunocytochemistry after 48h.
Flow cytometry
Cells were dissociated 48 hours post-transfection with TrypLE Express and resuspended in FACS Buffer (1X DPBS−/−, 0.2% BSA, 2 mM EDTA). Flow cytometry was performed in 96-well plate format using a MACSQuant VYB (Miltenyi Biotec) and analyzed using FlowJo 10. RG6 was a gift from Thomas Cooper (Addgene plasmid # 80167) and modified to replace EGFP with mTagBFP2. All represented samples were assayed with three biological replicates. In the mCherry reporter assay, data is representative of at least 20,000 gated events per condition. In the splicing reporter assay, data is representative of at least 2,500 gated events per condition.
Gene expression analysis
Cells were lysed 48 hours post-transfection with DTT-supplemented RLT buffer and total RNA was extracted using RNeasy Mini Plus columns (Qiagen). 200 ng of total RNA was then reverse transcribed using random hexamer primers and Revertaid Reverse Transcriptase (Thermo Fisher) at 25°C for 10 min, 37°C for 60 min, and 95°C for 5 min followed by qPCR using 2X Taqman Fast Advanced Master Mix (Life Technologies) and Taqman probes for GAPDH and the target gene as appropriate (Life Technologies and IDT). Taqman probe and primer sets were generally selected to amplify cDNA across the Cas13 or shRNA target site position to prevent detection of cleaved transcript fragments (Table S4). qPCR was carried out in 5 μL multiplexed reactions and 384-well format using the LightCycler 480 Instrument II (Roche). Fold-change was calculated relative to GFP-transfected vehicle controls using the ddCt method. One-way or two-way ANOVA with multiple comparison correction was used to assess statistical significance of transcript changes using Prism 7.
Immunohistochemistry
For immunohistochemical analysis, U2OS cells were cultured on 96-well optically clear plates (Greiner Bio-One), transfected as previously described, then fixed in 4% PFA (Electron Microscopy Sciences) diluted in PBS (Gibco) and washed with 0.3M glycine (Sigma) in PBS to quench PFA. Samples were blocked and permeabilized in a PBS solution containing 8% donkey serum (Jackson ImmunoResearch), 8% goat serum (Cell Signaling Technologies), and 0.3% Triton-X 100 (Sigma) for one hour, followed by primary antibody incubation in 1% BSA (Fisher Bioreagents), 1% goat serum, and 0.25% Triton-X overnight at 4°C. Samples were washed 3 times with PBS containing 0.1% BSA and 0.1% Triton-X 100 before incubating with fluorophore-conjugated secondary antibodies in PBS with 0.05% Triton-X 100 and 1% BSA at room temperature for one hour. Cells were washed with PBS with 0.1% Triton-X, stained with DAPI, and then covered with Mounting Media (Ibidi) before imaging. Primary antibody, HA-Tag 6E2 (Cell Signaling, 2367), was used at a 1:100 dilution as per manufacturer’s instructions. Secondary antibodies used were goat anti-mouse IgG1-Alexa-Fluor 647 (Thermo Fisher, A21240) and Anti-Mouse IgG1 CF 633 (Sigma, SAB4600335). Confocal images were taken using a Zeiss Airyscan LSM 880 followed by image processing in Zen 2.3 (Zeiss).
Bacterial small RNA sequencing and analysis
E. coli DH5a cells were transformed with pACYC184 carrying the CRISPR-Cas13d locus derived from an uncultured Ruminococcus sp. strain. Cells were harvested in stationary phase, rinsed in PBS, resuspended in TRIzol (Life Technologies), transferred to Lysing Matrix B tubes containing 0.1 mm silica beads (MP Biomedicals), and homogenized on a Bead Mill 24 (Fisher Scientific) for three 30-second cycles. Total RNA was isolated by phenol-chloroform extraction, then purified using the DirectZol Miniprep Kit (Zymo Research). RNA quality was assessed on an Agilent 2200 Tapestation followed by Turbo DNase treatment (Ambion). Total RNA was treated with T4 Polynucleotide Kinase (NEB) and rRNA-depleted using the Ribo-Zero rRNA Removal Kit for bacteria (Illumina). RNA was treated with RNA 5′ polyphosphatase, poly(A)-tailed with E. coli poly(A) polymerase, and ligated with 5′ RNA sequencing adapters using T4 RNA ligase 1 (NEB). cDNA was generated via reverse transcription using an oligo-dT primer and M-MLV RT/RNase Block (AffinityScript, Agilent) followed by PCR amplification and barcoding. Resulting libraries were sequenced on Illumina MiSeq, demultiplexed using custom Python scripts, and aligned to the Cas13d CRISPR locus using Bowtie 2. Alignments were visualized with Geneious.
Ngn2 lentivirus preparation
Low passage HEK 293FT cells were transfected with Polyethylenimine Max (PEI, Polysciences) and Ngn2 target plasmid plus pMDG.2 and psPAX2 packaging plasmids (a gift from Didier Trono, Addgene #12259 and #12260) in DMEM + 10% FBS media during plating. The following day, media was changed to serum-free chemically defined minimal medium (Ultraculture supplemented with Glutamax, Lonza). Viral supernatant was harvested 48h later, clarified through a 0.45 micron PVDF filter (Millipore) and concentrated using ultracentrifugation.
AAV preparation
Low passage HEK 293FT cells were transfected with Polyethylenimine Max (PEI, Polysciences) and AAV target plasmid plus AAV1 serotype and pAdDeltaF6 helper packaging plasmids (UPenn Vector Core) in DMEM + 10% FBS media during plating. The following day, 60% of the media was changed to chemically defined minimal medium (Ultraculture supplemented with Glutamax, Lonza). 48h later, AAV-containing supernatant was harvested and clarified through a 0.45μm PVDF filter (Millipore) and concentrated using precipitation by polyethylene glycol (PEG virus precipitation kit #K904, Biovision) following the manufacturer’s protocol.
RNA-seq library preparation and sequencing
48h after transfection, total RNA was extracted from 293FT cells using the RNeasy Plus Mini kit from Qiagen. Stranded mRNA libraries were prepared using the NEBNext II Ultra Directional RNA Library Prep Kit from New England Biolabs (Cat# E7760S) and sequenced on an Illumina NextSeq500 with 42 nt paired end reads. ~15M total reads were demultiplexed per condition.
RNA-seq analysis
Sequenced reads were quality-tested using FASTQC and aligned to the hg19 human genome using the 2.5.1b STAR aligner (Dobin et al., 2013). Mapping was carried out using default parameters (up to 10 mismatches per read, and up to 9 multi-mapping locations per read). The genome index was constructed using the gene annotation supplied with the hg19 Illumina iGenomes collection (Illumina) and sjdbOverhang value of 100. Uniquely mapped reads were quantified across all gene exons using the top-expressed isoform as proxy for gene expression with the HOMER analysis suite (Heinz et al., 2010), and differential gene expression was carried out with DESeq2 v 1.14.1 (Love et al., 2014) using triplicates to compute within-group dispersion and contrasts to compare between targeting and non-targeting conditions. Significant differentially expressed genes were defined as having a false discovery rate (FDR) <0.01 and a log2 fold change >0.75. Volcano plots were generated in R 3.3.2 using included plotting libraries and the alpha() color function from the scales 0.5.0 package.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
All values are reported as mean ± SD or mean ± SEM as indicated in the appropriate figure legends. For comparing two groups, a one-tailed student’s t-test was used and statistical significance was determined using the Holm-Sidak method with alpha = 0.05. A one-way ANOVA with Tukey multiple hypothesis correction was used to assess significance between more than two groups. Two-way ANOVA was used when comparing across two factors (i.e. RNA targeting modality and guide position) and adjusted for multiple hypothesis correction by Sidak’s multiple comparisons test. For comparing groups that were found to not meet the assumption of a normal distribution by a D’Agostino and Pearson normality test, the non-parametric Friedman test with Dunn’s multiple comparison adjustment was performed. PRISM 7.0 was used for all statistical analysis. Sample sizes were not determined a priori. At least three biological replicates were used for each experiment, as indicated specifically in each figure.
DATA AND SOFTWARE AVAILABILITY
Sequencing data reported in this paper can be found in the NCBI Gene Expression Omnibus under GEO Series accession number GSE108519.
Supplementary Material
Figure S1. Bioinformatic pipeline for the identification of the RNA-targeting class 2 CRISPR system Cas13d. Related to Figure 1. (A) Schematic describing a computational pipeline for CRISPR system identification. A minimal definition for a putative class 2 CRISPR locus was used, requiring only a CRISPR repeat array and a nearby protein >750 aa in length. As per Methods, initial search was performed on prokaryotic genome assemblies derived from NCBI Genome, and later expanded via TBLASTN of predicted Cas13d proteins against public metagenome sequences without predicted open reading frames. DR, direct repeat. (B) Phylogenetic classification and alignment of full-length Cas13d effectors and metagenomic fragments. Cas13d effectors and metagenomic Cas13d protein fragments cluster into several distinct branches, which are colored for ease of interpretation. Shading indicates residue conservation using the Blosum62 matrix. Full-length Cas13d effectors used in this study were sampled from distinct branches of the Cas13d family. Alignment of Cas13d proteins and protein fragments was performed using ClustalOmega 1.2.4 and maximum-likelihood tree building was performed with PhyML 3.2.
Figure S2. Phylogenetic classification of RNA-targeting class 2 CRISPR effectors and sequence conservation within the Cas13d family. Related to Figure 1. (A) HEPN motif conservation in Cas13d effectors used in this study with conserved residues shaded according to Blosum62. The RxxxxH HEPN motif is highlighted. (B) Maximum-likelihood tree of type VI CRISPR-Cas families. Average amino acid lengths of Type VI Cas13 superfamily effectors are indicated in red. Alignment of previously described class 2 CRISPR RNA-targeting proteins (Abudayyeh et al., 2017; Cox et al., 2017; East-Seletsky et al., 2017; East-Seletsky et al., 2016; Smargon et al., 2017) and Cas13d effectors was performed using MAFFT 7.38 and maximum-likelihood tree building was performed with PhyML 3.2. Branch labels and scale bar indicate substitutions per site. (C) Predicted Cas13d direct repeat RNA secondary structure. (D) Sequence logo of full length 36 nt Cas13d direct repeats.
Figure S3. Purification of recombinant Cas13d protein. Related to Figure 1. EsCas13d was expressed as N-terminal His-MBP fusion and purified by successive affinity, cation exchange, and size exclusion chromatography. The His-tag was removed by TEV protease cleavage. (A) Chromatogram from Superdex 200 column for EsCas13d. (B) SDS-PAGE gel of size exclusion chromatography fractions for E. siraeum Cas13d. (C) SDS-PAGE gel of purified E. siraeum Cas13d and dCas13d (R295A, H300A, R849A, H854A mutations of predicted catalytic residues in both HEPN motifs).
Figure S4. In vitro characterization of Cas13d properties. Related to Figures 2 and 3. (A) Schematic showing the length and sequence of gRNA spacer truncations and spacer position relative to the complementary ssRNA target. (B) Denaturing gel depicting EsCas13d cleavage activity of target RNA with different spacer lengths. (C) Denaturing gel depicting EsCas13d cleavage reactions paired with 12 guides from Figure 3A tiling a complementary ssDNA version of the ssRNA target. (D) Denaturing gel depicting cleavage reactions using EsCas13d paired with the same 12 guides tiling a dsDNA version of the complementary target. (E) Quantification of cleavage efficiency from Figure 3A. Each PFS base is the average of 3 different spacer sequences tiling a complementary target RNA. Cleavage percentage is determined by the ratio of cleaved band intensity divided by total lane intensity. Mean is depicted ± SD with each data point representing an independent replicate. (F) Cas13d-mediated cleavage of target RNA carrying different PFS bases given an invariant spacer sequence. Quantification of Cas13d cleavage efficiency and a representative denaturing gel depicting EsCas13d cleavage activity are shown. Differences are not significant (one-way ANOVA, P=0.768). Cleavage percentage is determined as above, and mean is depicted ± SD with n = 3. (G) and (H) Optimal temperature range for Cas13d activity. Denaturing gels depicting EsCas13d cleavage activity at temperatures ranging from 16–62°C for two different target RNAs.
Figure S5. Comparison of engineered Cas13 superfamily effectors for targeted knockdown and splicing. Related to Figures 5 and 6. (A) Relative target RNA knockdown by individual position-matched gRNAs for CasRx, NLS-LwaCas13a-msfGFP (Abudayyeh et al., 2017) and PspCas13b-NES (Cox et al., 2017) in HEK 293FT cells. NT, non-targeting. Values are mean ± SEM with n = 3. (B) Comparison of Cas13 median knockdown efficiencies. n = 3 per guide RNA. **** indicates P<0.0001 according to Friedman’s test. (C) Exon exclusion by catalytically inactive NLS-dCas13a-msfGFP on the bichromatic splicing reporter. Guides are position-matched to those reported in Figure 6B for CasRx. Values are mean ± SEM with n = 3. (D) Comparison of splicing modulation by NLS-dCas13a-msfGFP and CasRx. Fold change in targeted exon exclusion relative to non-targeting guide is shown. **** indicates P<0.0001 according to two-way ANOVA.
Figure S6. RNA sequencing from CasRx and shRNA targeting of ANXA4 in human cells. Related to Figure 5. (A) Volcano plots of differential transcript levels between ANXA4 targeting and non-targeting (NT) shRNAs as determined by RNA sequencing (n = 3). 915 non-specific transcript changes were identified. (B) Volcano plot of differential transcript levels for an ANXA4 targeting CasRx array used in Figure 5B containing a guide position matched to the shRNA shown in (A) and a non-targeting (NT) array. ANXA4 was the only transcript exhibiting significant downregulation with n = 3. HIST2HBE was the only transcript identified to exhibit significant upregulation. H2B is a dimer partner of H2AX (Du et al., 2006) which has been shown to interact with ANXA4 (Yang et al., 2010).
Table S1. Sequences used in this study for biochemical characterization. Related to Figures 1–3 and S4.
Table S2. Sequences used in this study for mammalian RNA targeting. Related to Figures 4–6 and S5–6.
Table S3. shRNAs used in this study. Related to Figure 5 and S6.
Table S4. qPCR probes for gene expression analysis used in this study. Related to Figures 4–6 and S5.
Table S5. CRISPR effectors used in this study for mammalian RNA targeting. Related to Figures 4–6.
Computational pipeline identifies the RNA-targeting Type VI-D CRISPR-Cas family
Ortholog screen and protein engineering yields the programmable ribonuclease CasRx
CasRx RNA knockdown exhibits favorable efficiency and specificity relative to RNAi
Neuronal AAV delivery of dCasRx splice effectors alleviates tau mis-splicing
Acknowledgments
We thank the entire Hsu laboratory for support and advice; T. Hunter and W. Eckhart for helpful comments; F. Gao for the gift of FTDP-17 patient-derived and control iPSC lines; A. Hsu for assistance with data analysis; S. Tyagi for guidance with protein purification; S. Schafer for the gift of Neurogenin-2 plasmid, F. Zhang for the gift of His-MBP-TEV plasmid; J. Karlseder, M. Montminy, J. Ayres, and D. Lyumkis for instrument access; C. Pourcel for CRISPRFinder source code; and K. Reilly, S. Leahy, P. Kougias, P. Weimer, F. Farquharson, J. Dore, and F. Levenez for gifts of genomic DNA samples from Ruminococcus flavefaciens XPD3002, Rum. albus, Rum. bicirculans, Rum. sp. CAG:57, Rum. flavefaciens FD1, and Anaerobic digester metagenome. This work was additionally supported by the Waitt Advanced Biophotonics Core Facility of the Salk Institute with funding from NIH-NCI CCSG P30 014195, NINDS Neuroscience Core Grant NS072031 and the Waitt Foundation; the Stem Cell, NGS, Razavi Newman Integrative Genomics and Bioinformatics, and the Flow Cytometry Core Facilities of the Salk Institute with funding from the Helmsley Trust, NIH-NCI CCSG: P30 014195, and the Chapman Foundation. S.K. is supported by a Catharina Foundation Fellowship, the Howard Hughes Medical Institute Hannah Gray Fellowship, and the Salk Women & Science Special Award. P.D.H. is supported by the NIH through the Office of the Director (5 DP5 OD021369-02) and the National Institutes on Aging (5 R21 AG056811-02), and the Helmsley Charitable Trust. Plasmids described in this study will be conveniently distributed to the academic community through the nonprofit repository Addgene.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.K. and P.D.H. conceived this study, developed the CRISPR identification pipeline, and participated in the design of all experiments. N.J.B., S.K., and P.D.H. led the biochemical characterization of Cas13d enzymes. S.K., P.L., and P.D.H. performed cell-based activity screens and knockdown experiments. J.O. performed microscopy experiments and P.L. cloned most constructs. S.K. and P.D.H. performed RNA sequencing experiments. M.N.S. and S.K. analyzed the sequencing data. S.K. J.O., P.L., and P.D.H. led the splicing experiments. P.D.H. wrote the manuscript with input from S.K. and P.L. and help from all authors.
Declaration of Interests
P.D.H. is a founder and scientific advisor for Spotlight Therapeutics. S.K. and P.D.H. are coinventors on U.S. provisional patent application no. 62/572,963 relating to CRISPR-Cas13 and CasRx, as well as other patents on CRISPR technology.
References
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. RNA targeting with CRISPR-Cas13. Nature. 2017 doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, Geschwind MD, Tartaglia MC, Gao F, Gianni D, et al. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012;2:789–798. doi: 10.1016/j.celrep.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct. 2013;8:15. doi: 10.1186/1745-6150-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronescu M, Condon A, Hoos HH, Mathews DH, Murphy KP. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23:i19–28. doi: 10.1093/bioinformatics/btm223. [DOI] [PubMed] [Google Scholar]
- Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell. 2017;170:899–912.e810. doi: 10.1016/j.cell.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- Biswas MHU, Almeida S, Lopez-Gonzalez R, Mao W, Zhang Z, Karydas A, Geschwind MD, Biernat J, Mandelkow EM, Futai K, et al. MMP-9 and MMP-2 Contribute to Neuronal Cell Death in iPSC Models of Frontotemporal Dementia with MAPT Mutations. Stem Cell Reports. 2016;7:316–324. doi: 10.1016/j.stemcr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN) Arch Neurol. 2008;65:460–464. doi: 10.1001/archneur.65.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong CG, Hall TM. Engineering RNA sequence specificity of Pumilio repeats. Proc Natl Acad Sci U S A. 2006;103:13635–13639. doi: 10.1073/pnas.0606294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–737. doi: 10.4161/rna.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Du YC, Gu S, Zhou J, Wang T, Cai H, Macinnes MA, Bradbury EM, Chen X. The dynamic alterations of H2AX complex during DNA repair detected by a proteomic approach reveal the critical roles of Ca(2+)/calmodulin in the ionizing radiation-induced cell cycle arrest. Mol Cell Proteomics. 2006;5:1033–1044. doi: 10.1074/mcp.M500327-MCP200. [DOI] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Burstein D, Knott GJ, Doudna JA. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol Cell. 2017;66:373–383.e373. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, Doudna JA. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–521. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Wood MJ. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011;27:196–205. doi: 10.1016/j.tig.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Heidrich N, Dugar G, Vogel J, Sharma CM. Investigating CRISPR RNA Biogenesis and Function Using RNA-seq. Methods Mol Biol. 2015;1311:1–21. doi: 10.1007/978-1-4939-2687-9_1. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A, Kuo D, He R, Zhou J, Wu JY. Tau alternative splicing and frontotemporal dementia. 2005;19(Alzheimer Dis Assoc Disord)(Suppl 1):S29–36. doi: 10.1097/01.wad.0000183082.76820.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500. doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li X, Wang J, Wang M, Chen P, Yin M, Li J, Sheng G, Wang Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell. 2017;168:121–134.e112. doi: 10.1016/j.cell.2016.12.031. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516:263–266. doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo JP, Bundman D, Cooper TA. A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res. 2006;34:e148. doi: 10.1093/nar/gkl967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peabody DS. The RNA binding site of bacteriophage MS2 coat protein. EMBO J. 1993;12:595–600. doi: 10.1002/j.1460-2075.1993.tb05691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, Dawson HN, Bennett CF, Rigo F, Miller TM. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron. 2016;90:941–947. doi: 10.1016/j.neuron.2016.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, Waterston RH. DNA sequencing at 40: past, present and future. Nature. 2017 doi: 10.1038/nature24286. advance online publication. [DOI] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon AA, Cox DB, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol Cell. 2017;65:618–630.e617. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, Tirosh I, Narayan R, Root DE, Golub TR, Subramanian A, et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017;15:e2003213. doi: 10.1371/journal.pbio.2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treu L, Kougias PG, Campanaro S, Bassani I, Angelidaki I. Deeper insight into the structure of the anaerobic digestion microbial community; the biogas microbiome database is expanded with 157 new genomes. Bioresour Technol. 2016;216:260–266. doi: 10.1016/j.biortech.2016.05.081. [DOI] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu J, Huang BO, Xu YM, Li J, Huang LF, Lin J, Zhang J, Min QH, Yang WM, et al. Mechanism of alternative splicing and its regulation. Biomed Rep. 2015;3:152–158. doi: 10.3892/br.2014.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AV, Doudna JA. Protecting genome integrity during CRISPR immune adaptation. Nat Struct Mol Biol. 2016;23:876–883. doi: 10.1038/nsmb.3289. [DOI] [PubMed] [Google Scholar]
- Yang X, Zou P, Yao J, Yun D, Bao H, Du R, Long J, Chen X. Proteomic dissection of cell type-specific H2AX-interacting protein complex associated with hepatocellular carcinoma. J Proteome Res. 2010;9:1402–1415. doi: 10.1021/pr900932y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Strecker J, Abudayyeh OO, Gootenberg JS, Scott DA, Zhang F. A Survey of Genome Editing Activity for 16 Cpf1 orthologs. bioRxiv. 2017 doi: 10.2302/kjm.2019-0009-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Graham S, Tello A, Liu H, White MF. Multiple nucleic acid cleavage modes in divergent type III CRISPR systems. Nucleic Acids Res. 2016;44:1789–1799. doi: 10.1093/nar/gkw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Bioinformatic pipeline for the identification of the RNA-targeting class 2 CRISPR system Cas13d. Related to Figure 1. (A) Schematic describing a computational pipeline for CRISPR system identification. A minimal definition for a putative class 2 CRISPR locus was used, requiring only a CRISPR repeat array and a nearby protein >750 aa in length. As per Methods, initial search was performed on prokaryotic genome assemblies derived from NCBI Genome, and later expanded via TBLASTN of predicted Cas13d proteins against public metagenome sequences without predicted open reading frames. DR, direct repeat. (B) Phylogenetic classification and alignment of full-length Cas13d effectors and metagenomic fragments. Cas13d effectors and metagenomic Cas13d protein fragments cluster into several distinct branches, which are colored for ease of interpretation. Shading indicates residue conservation using the Blosum62 matrix. Full-length Cas13d effectors used in this study were sampled from distinct branches of the Cas13d family. Alignment of Cas13d proteins and protein fragments was performed using ClustalOmega 1.2.4 and maximum-likelihood tree building was performed with PhyML 3.2.
Figure S2. Phylogenetic classification of RNA-targeting class 2 CRISPR effectors and sequence conservation within the Cas13d family. Related to Figure 1. (A) HEPN motif conservation in Cas13d effectors used in this study with conserved residues shaded according to Blosum62. The RxxxxH HEPN motif is highlighted. (B) Maximum-likelihood tree of type VI CRISPR-Cas families. Average amino acid lengths of Type VI Cas13 superfamily effectors are indicated in red. Alignment of previously described class 2 CRISPR RNA-targeting proteins (Abudayyeh et al., 2017; Cox et al., 2017; East-Seletsky et al., 2017; East-Seletsky et al., 2016; Smargon et al., 2017) and Cas13d effectors was performed using MAFFT 7.38 and maximum-likelihood tree building was performed with PhyML 3.2. Branch labels and scale bar indicate substitutions per site. (C) Predicted Cas13d direct repeat RNA secondary structure. (D) Sequence logo of full length 36 nt Cas13d direct repeats.
Figure S3. Purification of recombinant Cas13d protein. Related to Figure 1. EsCas13d was expressed as N-terminal His-MBP fusion and purified by successive affinity, cation exchange, and size exclusion chromatography. The His-tag was removed by TEV protease cleavage. (A) Chromatogram from Superdex 200 column for EsCas13d. (B) SDS-PAGE gel of size exclusion chromatography fractions for E. siraeum Cas13d. (C) SDS-PAGE gel of purified E. siraeum Cas13d and dCas13d (R295A, H300A, R849A, H854A mutations of predicted catalytic residues in both HEPN motifs).
Figure S4. In vitro characterization of Cas13d properties. Related to Figures 2 and 3. (A) Schematic showing the length and sequence of gRNA spacer truncations and spacer position relative to the complementary ssRNA target. (B) Denaturing gel depicting EsCas13d cleavage activity of target RNA with different spacer lengths. (C) Denaturing gel depicting EsCas13d cleavage reactions paired with 12 guides from Figure 3A tiling a complementary ssDNA version of the ssRNA target. (D) Denaturing gel depicting cleavage reactions using EsCas13d paired with the same 12 guides tiling a dsDNA version of the complementary target. (E) Quantification of cleavage efficiency from Figure 3A. Each PFS base is the average of 3 different spacer sequences tiling a complementary target RNA. Cleavage percentage is determined by the ratio of cleaved band intensity divided by total lane intensity. Mean is depicted ± SD with each data point representing an independent replicate. (F) Cas13d-mediated cleavage of target RNA carrying different PFS bases given an invariant spacer sequence. Quantification of Cas13d cleavage efficiency and a representative denaturing gel depicting EsCas13d cleavage activity are shown. Differences are not significant (one-way ANOVA, P=0.768). Cleavage percentage is determined as above, and mean is depicted ± SD with n = 3. (G) and (H) Optimal temperature range for Cas13d activity. Denaturing gels depicting EsCas13d cleavage activity at temperatures ranging from 16–62°C for two different target RNAs.
Figure S5. Comparison of engineered Cas13 superfamily effectors for targeted knockdown and splicing. Related to Figures 5 and 6. (A) Relative target RNA knockdown by individual position-matched gRNAs for CasRx, NLS-LwaCas13a-msfGFP (Abudayyeh et al., 2017) and PspCas13b-NES (Cox et al., 2017) in HEK 293FT cells. NT, non-targeting. Values are mean ± SEM with n = 3. (B) Comparison of Cas13 median knockdown efficiencies. n = 3 per guide RNA. **** indicates P<0.0001 according to Friedman’s test. (C) Exon exclusion by catalytically inactive NLS-dCas13a-msfGFP on the bichromatic splicing reporter. Guides are position-matched to those reported in Figure 6B for CasRx. Values are mean ± SEM with n = 3. (D) Comparison of splicing modulation by NLS-dCas13a-msfGFP and CasRx. Fold change in targeted exon exclusion relative to non-targeting guide is shown. **** indicates P<0.0001 according to two-way ANOVA.
Figure S6. RNA sequencing from CasRx and shRNA targeting of ANXA4 in human cells. Related to Figure 5. (A) Volcano plots of differential transcript levels between ANXA4 targeting and non-targeting (NT) shRNAs as determined by RNA sequencing (n = 3). 915 non-specific transcript changes were identified. (B) Volcano plot of differential transcript levels for an ANXA4 targeting CasRx array used in Figure 5B containing a guide position matched to the shRNA shown in (A) and a non-targeting (NT) array. ANXA4 was the only transcript exhibiting significant downregulation with n = 3. HIST2HBE was the only transcript identified to exhibit significant upregulation. H2B is a dimer partner of H2AX (Du et al., 2006) which has been shown to interact with ANXA4 (Yang et al., 2010).
Table S1. Sequences used in this study for biochemical characterization. Related to Figures 1–3 and S4.
Table S2. Sequences used in this study for mammalian RNA targeting. Related to Figures 4–6 and S5–6.
Table S3. shRNAs used in this study. Related to Figure 5 and S6.
Table S4. qPCR probes for gene expression analysis used in this study. Related to Figures 4–6 and S5.
Table S5. CRISPR effectors used in this study for mammalian RNA targeting. Related to Figures 4–6.