

Abstract
Introduction
Mutations in gap junction protein beta 1 (GJB1) on the X chromosome represent one of the most common causes of hereditary neuropathy. We assessed manifestations associated with a rare 3′ untranslated region mutation (UTR) of GJB1 in a large family with X-linked Charcot-Marie-Tooth (CMTX).
Methods
Clinical, electrophysiological, and molecular genetic analyses of an eight-generation family with CMTX.
Results
There we 22 affected males and 19 symptomatic females, including an 83-year old woman followed for 40 years. Electrophysiologic studies showed a primarily axonal neuropathy. The c.*15C>T mutation in the GJB1 3′UTR was identified in four branches of the family with a log of odds (LOD) of 4.91. This created a BstE-II enzyme recognition site that enabled detection by restriction digestion.
Discussion
The c.*15C>T mutation in the GJB1 3′UTR segregates with CMTX1 in eight generations. Penetrance in males and females is essentially complete. A straightforward genetic method to detect this mutation is described.
Introduction
Charcot-Marie-Tooth hereditary motor and sensory neuropathy (CMT) is primarily an autosomal dominant disease most often caused by a duplication in the peripheral myelin protein-22 gene.1 Shortly after the first descriptions of CMT, Harringham in England reported an affected family having X-linked inheritance2. This X-linked pattern, the second most common type of CMT, is usually caused by mutations in the GJB1 gene encoding a gap junction protein (connexin-32)3. More than 400 mutations in GJB1 have been reported, the overwhelming majority of which are missense mutations.4–6 Recently, Tomaselli et al reported a variant (c.*15C>T) in the 3´UTR of GJB1 in two small nuclear families.7 We evaluated a large family with this 3´UTR mutation in which we confirmed its pathogenicity and for which we provide details of clinical manifestations.
Methods
The eight-generation family (Figure 1) has been followed for more than 40 years by one author (TDB). With informed consent, under protocols approved by the University of Washington (UW) institutional review board, medical records were reviewed and blood or saliva samples collected from 5 and 11 family members, respectively.
Figure 1.
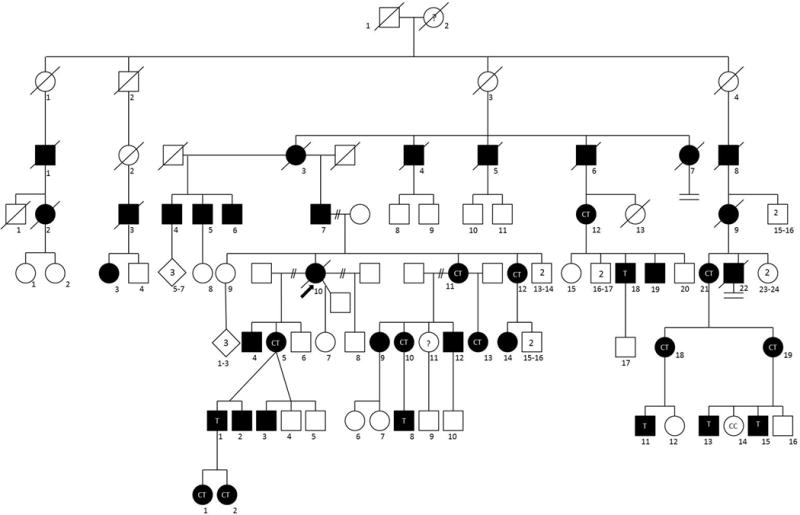
Eight generation pedigree of family with X-linked inheritance of CMT (CMTX). Squares are males. Circles are females. Black indicates affected persons with symptoms by exam, medical record and/or family history. Diagonal line through symbol indicates death. Arrow indicates the index family member. Genotypes within symbols indicate C for wild type and T for mutation as described in Figure 2.
Motor nerve conduction studies (MNCS) were performed on the upper limbs with surface electrodes, recording median and ulnar compound muscle action potentials (CMAPs) from the abductor pollicis brevis and abductor digiti minimi muscles, respectively. Some patients also underwent MNCS of the lower limbs, where fibular and tibial CMAPs were recorded from the extensor digitorum brevis and abductor hallucis muscles, respectively. We did not have consistent records for sensory nerve conduction or needle electromyography data.
DNA was extracted from buffy coat fractionated from blood. DNA from saliva samples collected via the Orange Discover ORG-500 kit (DNAGenotek, Ottawa, Canada) was extracted using prep IT L2P reagent (DNAGenotek). Exome sequencing was obtained from the UW Center for Precision Diagnostics on one subject. Molecular and parametric linkage analyses methods are provided in the Supplement.
Results
Family and Clinical Findings
Descendants of the original couple in this family emigrated from Scotland to the United States in the 19th century. By pedigree analysis individual I-2 was a likely mutation carrier. There are 22 affected males, at least 20 obligate carrier females and no male-to-male transmission of the disorder, highlighted by individuals III-4, 5, and 8. The subtle onset, slow progression and disease manifestations are typical of CMT. The most common findings are pes cavus, bilateral mild to moderate foot drop, hypoactive or absent tendon reflexes, and decreased vibratory sensation in the feet. Weakness and atrophy of hand muscles often develops. Several persons require ankle-foot orthoses (AFOs) but no use of a wheelchair. Two individuals have had surgical correction of pes cavus.
Manifestations in carrier females varied greatly. The index subject (V-10) was one of the more severely affected persons. At age 20 she noted “dragging” of her feet. By 28 she had bilateral foot drop, bilateral hand atrophy, mild pes cavus and marked loss of vibatory sensation in the feet and hands. She required AFOs by age 30. At age 39 electrodiagnostic (EDX) studies showed a demyelinating neuropathy (Table). The youngest examined female carrier (VIII-2) was 10 years old and asymptomatic, but showed mild distal muscle weakness of hands and feet and slight difficulty with heel walking. The oldest examined female carrier (IV-12) was 83 years old. At age 8 she had high arches, difficulty running and frequent ankle sprains. At age 43 she showed depressed tendon reflexes, pes cavus, mild atrophy of hand muscles and decreased vibratory sensation in her feet. EDX studies showed an axonal neuropathy. At age 83 she remained ambulatory with a walker, and had mild bilateral footdrop. Hand atrophy was more pronounced and vibration sensation was absent to the knees. Five women (IV-2, V-11, V-12 V-21, VI-5) in their 40s and 50s have had mildly unsteady gait with weakness of ankle dorsiflexion, high arched feet, and atrophy of hand muscles. Two women (V-10, VI-5) have used AFOs. One has had carpal tunnel surgery. Two women (V-12, VI-10) have had symptoms of neuropathic pain in the hands and/or feet. Several females have been asymptomatic, but on examination have had depressed tendon reflexes and mild loss of vibratory sensation. If mild findings on examination are included, the disease is fully penetrant in males and females.
Table 1.
Electrophysiologic Studies on an Extended Family with CMTX1
| Pedigree # | Sex | Age | Median | Ulnar | Peroneal | Tibial | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| DML (<4.4) | Amp (>5) | CV (>50) | DML (<3.7) | Amp (>8) | CV (>50) | DML (<6.5) | Amp (>2.5) | CV (>40) | DML (<6.1) | Amp (>4) | CV (>40) | |||
| IV-2 | F | 56 | 4.8 | 2 | 50 | 3.9 | 8 | 58 | 4.6 | 0.7 | 40 | |||
| IV-12 | F | 43 | 4.4 | 8 | 48 | 3.9 | 8 | 47 | Absent | 4 | 34 | |||
| IV-14 | F | 52 | 43 | 49 | 34 | 31 | ||||||||
| V-10 | F | 39 | 5.3 | 0.8 | 28 | 4 | 0.7 | 29 | 8.5 | 0.4 | 26 | |||
| V-11 | F | 39 | 3.9 | 8 | 44 | 2.6 | 15 | 55 | 6.4 | 6 | 45 | 6.3 | 9 | 32 |
| V-12 | F | 33 | 4.2 | 10 | 48 | 3.2 | 12 | 49 | 6 | 5 | 35 | 6.1 | 12 | 32 |
| VI-5 | F | 56 | 7.1 | 3 | 28 | 8.6 | 2.6 | |||||||
| 56 | 4.3 | 7.4 | 40 | 3.6 | 5.3 | 42 | ||||||||
| 60 | 4.3 | 3.1 | 36 | 3.2 | 5.1 | 47 | ||||||||
| VI-9 | F | 18 | 47 | 42 | 45 | |||||||||
| VI-10 | F | 17 | 3.9 | 13 | 51 | 2.8 | 12 | 64 | 3.3 | 4 | 45 | |||
| 46 | 4.8 | 11 | 50 | 3.6 | 8 | 47 | 5.8 | 5 | 42 | 6.4 | 8 | 40 | ||
| VI-13 | F | 7 | 2.7 | 8 | 50 | 2 | 8 | 45 | 3.3 | 8 | 47 | |||
| 12 | 3.4 | 8 | 47 | 5.1 | 3.5 | 34 | ||||||||
| VI-14 | F | 13 | 3.4 | 10 | 44 | 3 | 10 | 42 | 5.6 | 10 | 46 | |||
|
| ||||||||||||||
| V-19 | M | 15 | 5.9 | 1.2 | 39 | Absent | 6 | 0.25 | 24 | |||||
| VI-4 | M | 23 | 3.4 | 9 | 57 | 3.1 | 10 | 55 | 5.2 | 8 | 44 | |||
| VI-12 | M | 13 | 4.5 | 2.5 | 39 | 3.9 | 6 | 45 | 5.9 | 1.2 | 44 | |||
| VII-8 | M | 16 | 3.2 | 64 | 4.9 | 2 | 61 | |||||||
Normal values listed in parenthesis. Abnormal values in italics. M=male; F=female; DML=distal motor latency; AMP= amplitude; CV=conduction velocity
EDX studies were available for 15 persons and showed no consistent difference between males and females (Table). MNCS varied from normal, to mildly slow, to occasionally within the “demyelinating” range. CMAPs were often normal but sometimes reduced. Three females had repeat studies over 4–24 years with relatively little change.
Genetic Analysis
Given the X-linked inheritance pattern in the pedigree, GJB1 was analyzed by commercial laboratories on two separate occasions but no mutation was identified. With the report of GJB1 *15C>T, we reevaluated the available exome sequence of affected subject VII-8 and found this variant.7
Sanger sequencing confirmed the variant and identified it in four other affected relatives. The mutation creates a BstE-II restriction enzyme recognition site making genetic screening for this mutation relatively simple (Figure 2). Using genotypes from eighteen family members (Figure 1), two-point parametric linkage analysis under an X-linked dominant model provided strong evidence of linkage between the GJB1 c.*15C>T allele and CMTX, with a LOD = 4.91.
Figure 2.
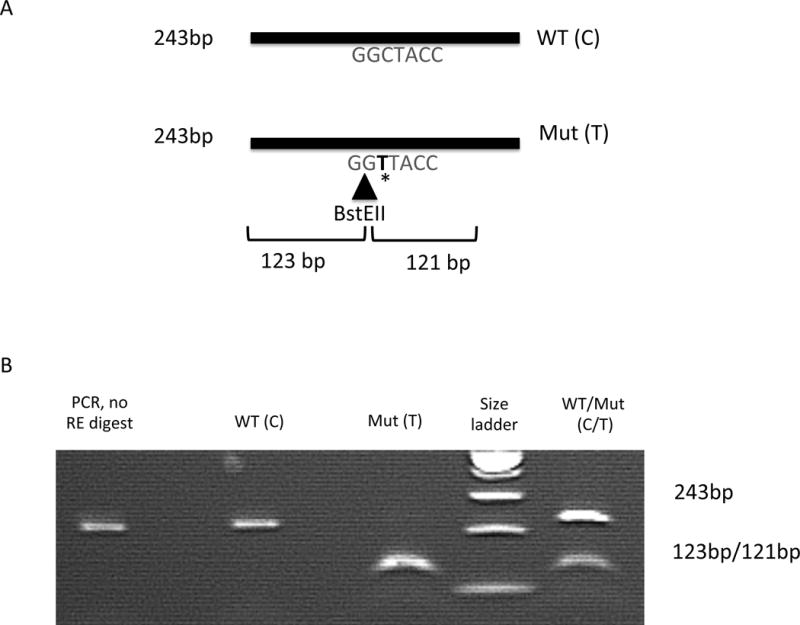
RFLP analysis to screen for the 3′ UTR non-coding region mutation in GJB1. (A) The mutation creates a restriction enzyme digestion site by BstEII that cuts the 243bp PCR amplification products into two Fragments of 123bp and 121bp. (B) shows the digestion results in patients with wild-type (WT, C allele), hemizygous mutation (Mut, T allele) in an affected male or heterozygous mutation (C and T alleles) in a carrier female. The two digested fragments of 123bp and 121bp are unable to be distinguished on the 2% agrose gel and appears as a single line.
We hypothesized the mutation would lead to RNA instability or affect transcript decay rate. Other than fresh blood, no tissues were available from patients. The expression databases lack consensus regarding GJB1 expression in blood; some show very low expression by RNAseq or RNA microarray (BioGPS, GTEx). Given the high sensitivity of PCR to detect RNAs present in very low copy number, we tried to amplify the GJB1 cDNA fragment from isolated blood RNA from a patient and normal controls. Despite using multiple primer sets in different regions of GJB1, we were unable to obtain an RT-PCR product.
Discussion
The majority of mutations in GJB1 are missense but rare ones have been described in the 5′ UTR.8 Recently, a GJB1 mutation (c.*15C>T) in the 3´UTR was reported in two nuclear families each with only a single affected male, precluding proof of cosegregation with disease.7 The present study documents cosegregation of the c.*15C>T GJB1 mutation in four branches of a large family, with confirmation by linkage analysis. Affected males have a typical CMT syndrome and female carriers show mild manifestations of the disease with great variability.
Results of EDX studies are also variable typically showing normal or mildly slow motor NCV and sometimes low CMAP. These characteristics are similar to the CMT syndrome associated with other mutations in GJB1.4–6 Variability of manifestations in carrier females is presumably the result of random X inactivation although Siskind et al were not able to document this phenomenon.9
Prior EDX studies in CMTX have demonstrated motor conduction slowing in the intermediate range (30-40 m/s in the upper limb), which we observed in 2 males and 1 female.10 Pathological studies have shown both demyelination and axon loss in CMTX. In the present family, we observed a correlation between conduction slowing and CMAP amplitude reduction and in subsequent studies we observed axon loss over time.
Without special attention to the 3´UTR of GJB1 this mutation would be missed by many commercial genetic tests because it lies outside the normal coding region of the gene. Thus, the frequency of this particular mutation remains unknown. We have shown that the mutation creates a new DNA restriction digest site providing a simple way to screen for the mutation.
GJB1 codes for the gap junction protein connexin-32 (Cx32). How mutations in GJB1 produce peripheral neuropathy in CMTX1 is not entirely clear.11–13 Transgenic mouse models indicate that altered synthesis or trafficking and loss of gap junction function is likely most important. Also impaired cytoskeletal organization and axonal transport defects appear to precede demyelination, suggesting the early occurrence of an axonopathy.14
The biological consequences of this UTR mutation remain unknown. The 3´UTR of other genes plays an important role in gene expression by effects on polyadenylation, translation efficiency, localization, transport and stability of mRNA.15 Tomeselli et al speculated that this variant may create a 5´ donor splice site leading to aberrant splicing within the 3´UTR.7 This could affect mRNA stability leading to decreased GJB1 protein Cx32. Patients with UTR mutations (including reported mutations in the 5´UTR) present typical phenotypes of CMTX1 as seen in cases with missense mutations in GJB1. In vitro and in vivo models of the disease indicate that most GJB1 missense mutations likely cause loss of function of Cx32.14, 16 If the effect of the UTR mutations is mediated through RNA instability, this would be the equivalent of a loss of function. Because we were unable to identify GJB1 RNA in peripheral white blood cells in either normal individuals or mutation carriers, further investigation of this question will require additional techniques and other tissues.
Supplementary Material
Acknowledgments
Veterans Affairs research funds and NIH (R01 NS069719). We thank the members of this family who have provided decades of valuable participation.
Abbreviations
- GJB1
gap junction protein beta 1
- CMTX
X-linked Charcot-Marie-Tooth
- LOD
Log of odds
- CMT
Charcot-Marie-Tooth hereditary motor and sensory neuropathy
- MNCS
Motor nerve conduction studies
- CMAP
Compound muscle action potentials
- AFO
Ankle-foot orthoses
- UTR
untranslated region mutation
Footnotes
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Drs. Chen, Raskind and Bird receive licensing fees from Athena Diagnostics, Inc. The remaining authors have no conflicts of interest to disclose.
References
- 1.Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013 Oct;9(10):562–71. doi: 10.1038/nrneurol.2013.179. [DOI] [PubMed] [Google Scholar]
- 2.Harringham WP. Muscular atrophy of the peroneal type affecting many members of a family. Brain. 1888;11(2):230–236. [Google Scholar]
- 3.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MV, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie_tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Yin F. A review of X-linked Charcot-Marie-Tooth Disease. J Child Neurol. 2016;31(6):761–72. doi: 10.1177/0883073815604227. [DOI] [PubMed] [Google Scholar]
- 5.Lu YY, Lyu H, Jin SQ, Zuo YH, Lui J, Wang ZX, Zhang W, Yuan Y. Clinical and Genetic Features of Chinese X-linked Charot-Marie-Tooth Type 1 Disease. Chinese Med J. 2017;130:1049–1054. doi: 10.4103/0366-6999.204925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong YB, Park JM, Yu JS, Yoo DH, Nam DE, Park HJ, Lee JS, Hwang SH, Chung KW, Choi BO. Clinical characterization and genetic analysis of Korean patients with X-linked Charcot-Marie-Tooth disease type 1. J Peripher Nerv Syst. 2017 Apr 27; doi: 10.1111/jns.12217. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Tomaselli PJ, Rossor AM, Horga A, Jaunmuktane Z, Carr A, Saveri P, Piscosquito G, Pareyson D, Laura M, Blake JC, Poh R, Polke J, Houlden H, Reilly MM. Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology. 2017 Apr 11;88(15):1445–1453. doi: 10.1212/WNL.0000000000003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulshrestha R, Burton-Jones S, Antoniadi T, Rogers M, Jaunmuktane Z, Brandner S, Kiely N, Manuel R, Willis T. Deletion of P2 promoter of GJB1 gene a cause of Charcot-Marie-Tooth disease. Neuromuscul Disord. 2017 May 4; doi: 10.1016/j.nmd.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Siskind CE, Murphy SM, Ovens R, Polke J, Reilly MM, Shy ME. Phenotype expression in women with CMTX1. J Peripher Nerv Syst. 2011;16(2):102–7. doi: 10.1111/j.1529-8027.2011.00332.x. [DOI] [PubMed] [Google Scholar]
- 10.Berciano J, Garcia A, Gallardo E, Peeters K, Pelayo-Negro AL, Alvarez-Paradelo S, Gazulla J, Martinez-Tames M, Infante J, Jordanova A. Intermediate Charcot-Marie-Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol. 2017 Mar 31; doi: 10.1007/s00415-017-8474-3. [DOI] [PubMed] [Google Scholar]
- 11.Shy ME, Siskind C, Swan ER, Krajewski KM, Doherty T, Fuerst DR, Ainsworth PJ, Lewis RA, Scherer SS, Hahn AF. CMTX1 phenotypes represent loss of GJB1 gene function. Neurology. 2007;68:849–855. doi: 10.1212/01.wnl.0000256709.08271.4d. [DOI] [PubMed] [Google Scholar]
- 12.Kleopa KA, Abrams CK, Scherer SS. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 2012;1487:198–205. doi: 10.1016/j.brainres.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrams CK, Freidin M. GJB1-associated X-linked Charcot-Marie-Tooth disease, a disorder affecting the central and peripheral nervous systems. Cell Tissue Res. 360(3):659–73. doi: 10.1007/s00441-014-2014-6. [DOI] [PubMed] [Google Scholar]
- 14.Vavlitou N, Sargiannidou I, Markoullis K, Kyriacou K, Scherer SS, Kleopa KA. Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy. J Neuropathol Exp Neurol. 2010 Sep;69(9):945–58. doi: 10.1097/NEN.0b013e3181efa658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012 May;9(5):563–76. doi: 10.4161/rna.20231. [DOI] [PubMed] [Google Scholar]
- 16.Kleopa KA, Sargiannidou I. Connexins, gap junctions and peripheral neuropathy. Neurosci Lett. 2015 Jun 2;596:27–32. doi: 10.1016/j.neulet.2014.10.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.