

Abstract
Background
Next-generation sequencing (NGS) studies of matched pairs of primary and metastatic tumors in renal cell carcinoma (RCC) have been limited to small cohorts.
Objective
To evaluate the discordance in somatic mutations between matched primary and metastatic RCC tumors.
Design, setting, and participants
Primary tumor (P), metastasis (M), and germline DNA from 60 patients with RCC was subjected to NGS with a targeted exon capture–based assay of 341 cancer-associated genes. Somatic mutations were called using a validated pipeline.
Outcome measurements and statistical analysis
Mutations were classified as shared (S) or private (Pr) in relation to each other within individual P-M pairs. The concordance score was calculated as (S − Pr)/(S + Pr). To calculate enrichment of Pr/S mutations for a particular gene, we calculated a two-sided p value from a binomial model for each gene with at least ten somatic mutation events, and implemented a separate permutation test procedure. We adjusted p values for multiple hypothesis testing using the Benjamini-Hochberg procedure. The mutation discordance was calculated using Mann-Whitney U tests according to gene mutations or metastatic sites.
Results and limitations
Twenty-one pairs (35%) showed Pr mutations in both P and M samples. Of the remaining 39 pairs (65%), 14 (23%) had Pr mutations specific to P samples, 12 (20%) had Pr mutations to M samples, and 13 (22%) had identical somatic mutations. No individual gene mutation was preferentially enriched in either P or M samples. P-M pairs with SETD2 mutations demonstrated higher discordance than pairs with wild-type SETD2. We observed that patients who received therapy before sampling of the P or M tissue had higher concordance of mutations for P-M pairs than patients who did not (Mann-Whitney p = 0.088).
Conclusions
Our data show mutation discordance within matched P-M RCC tumor pairs. As most contemporary precision medicine trials do not differentiate mutations detected in P and M tumors, the prognostic and predictive value of mutations in P versus M tumors warrants further investigation.
Patient summary
In this study we evaluated the concordance of mutations between matched primary and metastatic tumors for 60 kidney cancer patients using a panel of 341 cancer genes. Forty-seven patients carried nonidentical cancer gene mutations within their matched primary-metastatic pair. The mutation profile of the primary tumor alone could compromise precision in selecting effective targeted therapies and result in suboptimal clinical outcomes.
Keywords: Renal cell carcinoma, Metastasis, Primary-metastasis tumor pairs, Genomics, Next-generation sequencing, Discordance, Convergent evolution, Spatiotemporal divergence
1. Introduction
The prognosis for renal cell carcinoma (RCC) is directly related to histology, and survival is dictated by clinical stage. Metastatic disease is present in 25–30% of patients at the time of diagnosis, and 20–40% of patients who initially present with localized disease develop local or distant recurrence following resection of the primary tumor [1]. Advances in surgical and medical management over the past decade have improved the 5-yr survival for patients with metastatic RCC (mRCC). However, most mRCC patients eventually succumb to their disease [2].
The main cause of cancer-related morbidity and mortality in RCC is distant metastasis, but most of the molecular profiling undertaken to characterize genetic events driving RCC has been performed on primary tumor tissues. Landmark papers by The Cancer Genome Atlas consortium, which exclusively utilizes primary tumors, have detailed the molecular landscape, including somatic mutations, copy number alterations, mRNA expression, miRNA expression, and methylation changes for the top three prevalent RCC subtypes, clear cell RCC (ccRCC), papillary RCC (pRCC), and chromophobe RCC (chRCC) [3–5]. ccRCC is characterized by nearly ubiquitous biallelic loss of the tumor suppressor gene VHL, frequent concomitant loss or mutation of several other tumor suppressors on chromosome 3p, including PBRM1, BAP1, and SETD2 [6], and clustered mutations of the oncogene MTOR [7]. Similarly, distinct critical genetic driver events have been identified for pRCC, chRCC, and unclassified RCCs, which are often grouped clinically as non–clear cell RCC (nccRCC) [1,8–11].
Owing to the paucity of direct genomic analysis of RCC metastasis, somatic alterations driving RCC metastasis and therapeutic responses to targeted therapies are largely inferred from knowledge obtained from studying primary tumors [12]. Thus far, evaluation of genetic divergence among primary and metastatic tumors in RCC remains limited to small or unmatched cohorts [13–15]. In spite of limitations in sample size, Gerlinger and colleagues [13] provided a seminal description of parallel, convergent evolution involving loss-of-function mutations to SETD2, KDM5C, and PTEN within the same tumor. In a distinct but equally important study, Gerlinger and colleagues[14] described the stark prevalence of subclonal driver mutations in RCC tumors, emphasizing the importance of comprehensive multiregion sequencing of these cancers. Such intratumoral heterogeneity is particularly important for metastatic studies, as a rare but particularly aggressive subclone in a primary tumor may be the most likely to seed distant metastases. Hence, understanding mutation differences between paired primary and metastatic RCC tumors may provide further molecular and therapeutic insights.
In this study we used targeted, deep sequencing of 341 cancer genes to profile matched primary and metastatic kidney tumors from 60 mRCC patients. We sought to evaluate mutational differences and infer clonal relationships between paired primary and metastatic RCC tumors. To the best of our knowledge, this study represents the largest primary tumor-metastatic tumor (P-M) RCC pair cohort reported to date.
2. Patients and methods
2.1. Patients and DNA samples
This study was conducted following approval by the institutional review board of the Memorial Sloan Kettering Cancer Center (MSKCC). All specimens were obtained from patients evaluated at MSKCC. Patients with mRCC were eligible if they provided informed consent for tumor molecular characterization and had histologically confirmed RCC. Patients who had both their primary and metastatic tumors sequenced at our institution were included in the study. Demographic and clinical characteristics were reviewed for all patients. Metastases were classified as synchronous when they were detected via preoperative radiograph screening or within 3 mo of the initial RCC diagnosis; otherwise, tumors were judged as metachronous. The size of the metastatic tumors was obtained by reviewing pathology reports; for cases for which the size was not available, radiologic studies were reviewed. All tumor samples were reviewed and classified by an MSKCC genitourinary pathologist upon DNA extraction for sequencing. Out of the 60 primary tumors, 58 sample (96.7%) were obtained from surgically resected specimens; the reminding samples were obtained from core needle biopsies. For metastases, 47 samples (78.3%) were obtained from surgically resected specimens and the rest were core needle biopsies. Core needle biopsies of primary tumors were performed only in patients unfit for surgery.
2.2. Next-generation sequencing (NGS)
Genomic profiling of DNA from tumors and matched normal tissue was carried out via analysis on NGS platforms using our custom Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay, a targeted-exon capture assay with ultra-deep sequencing coverage (median, 570) on an Illumina HiSeq 2000 system. Target-specific probes were used for hybrid selection to capture all protein-coding exons and selected introns for 341 oncogenes (Supplementary Table 1). Details of our genomic pipeline have been published elsewhere [16].
2.3. Statistical analysis
A measure of concordance/discordance was determined for each tumor pair by first counting the number of shared (S) and private (Pr) mutations, and the concordance score was calculated as (S − Pr)/(S + Pr). When primary and metastatic tumors have an identical mutation profile, the score is 1. When primary and metastatic tumors have no shared mutations, the score is −1.
Enrichment of Pr/S mutations was calculated separately for ccRCC and nccRCC samples, as well as for the whole cohort. To calculate enrichment of Pr/S mutations for a particular gene, we first calculated the proportion of all mutations in our data set that were private (PPr = 0.41). For a null model, we parametrized a binomial model with the probability of success equal to PPr, the number of trials equal to the total number of mutations in a gene (counting each shared mutation once), and the number of successes equal to the number of Pr mutations. We calculated a two-sided p value from this binomial model for each gene with at least ten mutations. We adjusted p values for multiple hypothesis testing using the Benjamini-Hochberg procedure and reported as q values.
We also developed a separate permutation test procedure to determine enrichment of Pr/S mutations for a particular gene. For each patient, we identified a list of mutation events and labeled them as either Pr or S, so that each S mutation was counted once (because it occurs in the ancestral tumor cell) and each Pr mutation was also counted once. To generate a reference/null distribution, we randomly shuffled the Pr and S labels for mutation events within each patient. After shuffling the labels for all patients, we counted the total number of Pr mutation events for the gene of interest. This procedure was repeated for n = 2000 distinct permutations. For a gene for which we observed P Pr mutations, left-sided p values were calculated by counting the number of permutations with less than or equal to the true number P of Pr mutations observed. Similarly, for right-sided p values, we counted the number of permutations with greater than or equal to P Pr mutations. These one-sided p values were converted to two-sided p values by taking their minimum and then multiplying by two. We then adjusted p values for multiple hypothesis testing using the Benjamini-Hochberg procedure. Association between mutation discordance and a specific gene or metastatic site was determined using a Mann-Whitney U test.
3. Results
3.1. Patient demographics and mutation landscapes of the paired P-M cohort
Targeted NGS sequencing (MSK-IMPACT) was performed on 60 pairs of P-M RCC tumor samples (a total of 120 tumor samples) along with their matched normal samples (blood; Supplementary Table 2). Patient demographics and tumor characteristics are listed in Table 1. Forty-nine pairs (82%) were ccRCC and 11 (18%) were nccRCC, including four chRCC, four pRCC, and three epithelioid angiomyolipomas. Metastatic tissues for profiling were obtained from various sites: retroperitoneal lymph nodes (n = 14), lung (n = 13), bone (n = 12), adrenal gland (n = 7), liver (n = 3), distant lymph nodes (n = 3), brain (n = 2), muscle (n = 2), pleura (n = 2), stomach (n = 1), and thyroid (n = 1). A total of 558 nonsynonymous somatic mutations were identified with a mean (SD, standard deviation) of 4.6 (2.5) and 4.7 (2.8) mutations per primary and metastatic tumor, respectively. Gene mutation frequencies are shown in Figure 1. Of the 558 total mutations, 206 (37%) were shared mutations present in both the primary and the metastatic sample, and 352 (63%) were private mutations (Fig. 2). In ccRCC samples only, 132/312 (42%) mutations were Pr, whereas in nccRCC samples, 14/40 (35%) mutations were Pr.
Table 1.
Baseline patient and tumor characteristics in clear cell renal cell carcinoma (ccRCC) and non–clear cell renal cell carcinoma (nccRCC)
| ccRCC (n = 49) |
nccRCC (n = 11) a |
Total (n = 60) |
|
|---|---|---|---|
| Median a ge, yr (range) | 55 (38 – 71) | 21 (22 – 66) | 55 (22 – 71) |
| Sex, n (%) | |||
| Male | 34 (69) | 7 (64) | 41 (68) |
| Female | 15 (31) | 4 (36) | 19 (32) |
| Race, n (%) | |||
| White | 41 (84) | 7 64) | 48 (80) |
| African American | 8 (16) | 4 (36) | 12 (20) |
| Histologic subtype, n (%) | |||
| Clear cell | 49 (100) | – | 49 (81) |
| Chromophobe | – | 4 (36) | 4 (7) |
| Papillary | – | 4 (36) | 4 (7) |
| Epithelioid angiomyolipoma | – | 3 (28) | 3 (5) |
| American Joint Committee on Cancer stage, n (%) | |||
| Stage I | 8 (16) | – | 8 (13) |
| Stage II | 13 (27) | – | 13 (22) |
| Stage III | 6 (12) | 3 (27) | 9 (15) |
| Stage IV | 22 (45) | 8 (73) | 30 (50) |
| T stage, n (%) | |||
| T1 | 13 (27) | 3 (27) | 16 (27) |
| T2 | 14 (29) | 1 (9) | 15 (25) |
| T3 | 15 (31) | 3 (27) | 18 (30) |
| Not known | 7 (14) | 4 (36) | 11 (18) |
| Metastasis, n (%) | |||
| Synchronous | 24 (49) | 4 (36) | 28 (470 |
| Metachronous | 25 (51) | 7 (64) | 32 (53) |
| Metastatic tissue, n (%) | |||
| Adrenal | 7 (14) | – | 7 (12) |
| Bone | 12 (24) | – | 12 (20) |
| Brain | 2 (4) | – | 2 (3) |
| Liver | 1 (2) | 2(18) | 3 (5) |
| Retroperitoneal lymph node | 8 (16) | 6 (55) | 14 (23) |
| Lymph node | 1 (2) | 2 (18) | 3 (5) |
| Lungs | 12 (24) | 1 (9) | 13 (22) |
| Muscle | 2 (4) | – | 2 (3) |
| Pleura | 2 (4) | – | 2 (3) |
| Stomach | 1 (2) | – | 1 (2) |
| Thyroid | 1 (2) | – | 1 (2) |
| Treatment before primary tumor resection, na (%) | |||
| Yes | 2 (4) | 1 (9) | 3 (5) |
| No | 46 (93) | 7 (64) | 53 (88) |
| Not known | 1 (2) | 3 (27) | 4 (7) |
| Treatment Before metastatic tumor resection, n (%)b | |||
| Yes | 12 (24) | 2 (18) | 14 (23) |
| No | 36 (74) | 6 (55) | 42 (70) |
| Not known | 1 (2) | 3 (27) | 4 (7) |
Systemic therapy before resection/biopsy of the primary tumor from which the sample for sequencing was obtained.
Systemic therapy before resection/biopsy of the metastatic tumor from which the sample for sequencing was obtained.
Fig. 1.
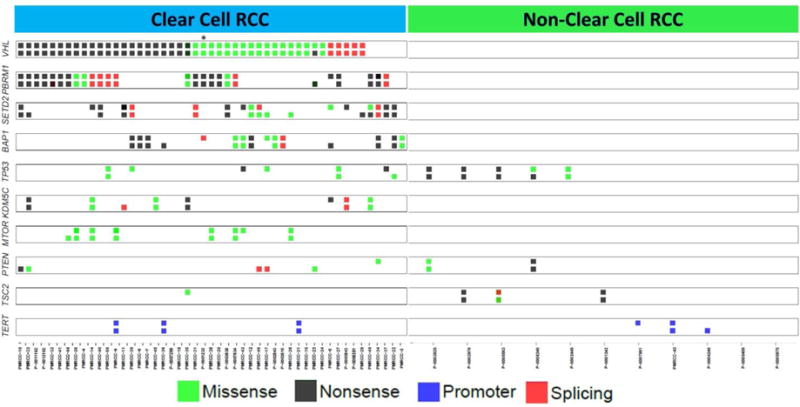
Distribution of mutations across the cohort for known cancer genes with prevalent mutations in renal cell carcinoma (RCC). For each gene, the top row represents a mutation in the primary tumor and the bottom row represents a mutation in the metastatic lesion.
Fig. 2.
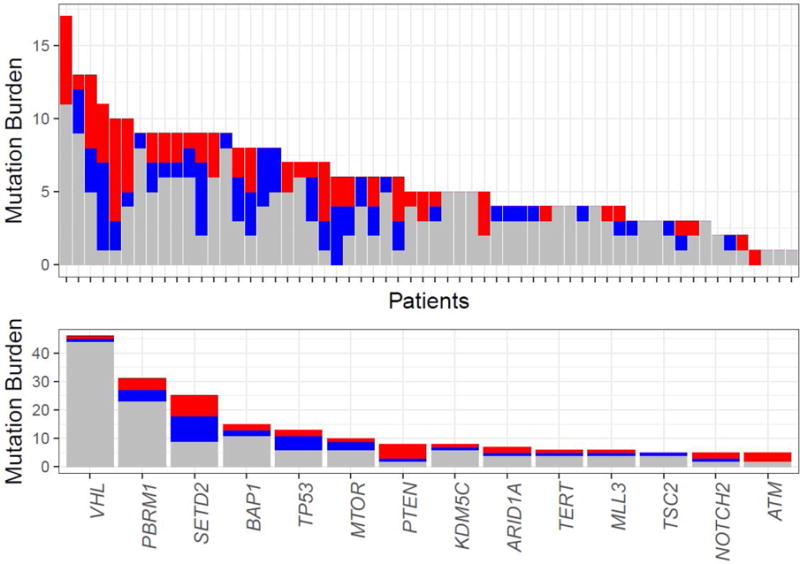
Discordance of mutation patterns between primary and metastatic tumors. Shared mutations are in gray and private mutations in the primary tumor and metastasis in red and blue, respectively. (A) Shared and private mutations among the paired primary and metastatic samples. Every column represents a patient and the overall height of the column depicts the number of mutations in that pair. (B) Most commonly mutated genes across our cohort and events as shared or private alterations.
3.2. Mutations enriched in metastatic samples
In other disease settings it has been found that specific genes are mutated at higher rates in metastatic compared to primary samples (eg, androgen receptor mutations in metastatic prostate cancers) [16]. Using our matched-pair data set, we evaluated whether such enrichment was evident in RCC. No single gene mutation exhibited significant enrichment (q value <0.05) in either primary or metastatic samples, which could be because of the known low mutation burden of RCC, the cancer gene panel used, and the size of our cohort. Nevertheless, ATM (2 mutations in primary samples, 5 in metastatic samples) and PTEN (3 mutations in primary samples, 7 mutations in metastatic samples) mutations were enriched more than twofold in metastatic samples. Furthermore, no significant enrichment/depletion of mutations in metastases was identified when restricting the analysis to ccRCC or nccRCC samples.
3.2. Enrichment of Pr or S mutations in P-M pairs
We next evaluated whether specific gene mutations were enriched for Pr or S mutations in our cohort using two complementary models, a binomial model and a permutation model. In brief, for the binomial model we empirically determined the probability of a Pr mutation in our data set (PPr = 0.41), and used this to parameterize a null model. For the permutation model, we shuffled the labels for Pr and S mutation events within each patient, and used the frequency of private mutations within these permuted samples to generate a null reference distribution. We then investigated whether genes with at least ten mutations exhibited more Pr or S mutations than expected by chance. Consistent with its role as a truncal, founding mutation in ccRCC, we found that nearly all VHL mutations were shared between primary and metastatic samples (44/46 mutations shared, binomial q < 10−8, permutation q < 10−3).
By contrast, SETD2 mutations were enriched for Pr mutations under the binomial model, but not the permutation model (16/25 mutations private, binomial q = 0.077, permutation p = 0.25; Fig. 3), which requires a larger sample size and higher statistical power to more accurately determine statistical significance. Notably, when repeating the analysis separately for ccRCC and nccRCC samples, PTEN was found to be enriched in Pr (binomial q = 0.02, permutation p = 0.047). In particular, all six PTEN mutations in ccRCC samples were Pr (1 in a primary tumor and 5 in metastatic tumors). By contrast, PTEN mutations in nccRCC samples were S mutations.
Fig. 3.
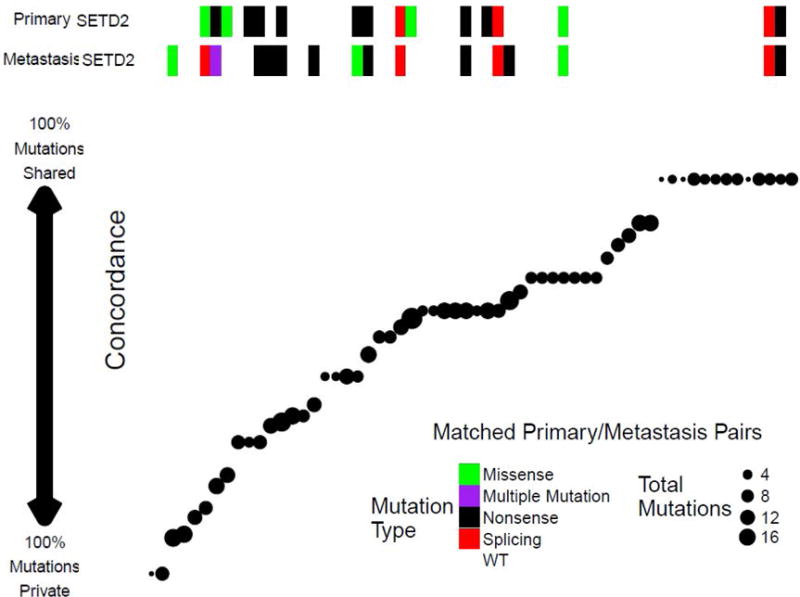
Cases with SETD2 mutations in primary and/or metastatic lesions had lower concordance than cases with a wild-type SETD2 pair. The concordance score for each pair, ranging from −1 (all mutations were private) to +1 (all mutations were shared for both samples), is shown. Every point represents a pair sorted by concordance. To the left of the figure, pairs have predominantly private mutations, while predominantly shared mutations are shown to the right.
While mutations in a gene may be preferentially Pr or S, it is also possible that certain gene mutations might result in a propensity for developing additional mutations, thereby manifesting with higher P-M discordance. Accordingly, we tested whether samples with mutations of an individual gene were more discordant than samples with wild-type alleles. We found that tumors with either SETD2 or KDM5C mutations demonstrated higher discordance between primary and metastatic tumors than tumors with wild-type SETD2 (q = 0.068) or KDM5C (q = 0.078). When restricting the analysis to either ccRCC or nccRCC histology, the findings became marginally stronger for enrichment of SETD2 (q = 0.048) and KDM5C (q = 0.062) mutations in discordant samples. These findings for SETD2 mutations are consistent with prior studies demonstrating that spatially separated regions of RCC tumors tend to harbor distinct SETD2 mutations [14] and promote ccRCC progression [17].
3.3. Discordance of mutations between P-M RCC tumor pairs
When evaluating the mutation concordance of 60 matched P-M pairs, we noted that 13 pairs (22%) exhibited identical mutation profiles. By contrast, 21 pairs (35%) showed Pr mutations in both the primary and metastatic samples, suggesting that both primary and metastatic clones further evolved, with addition of new mutations after the initial spread to the metastatic site tested. Two of these samples (P-0007981 and P-0005409) showed no common mutations between the primary and metastasis samples, suggesting that metastasis arose from a primary tumor clone that was not captured by our sequencing. Both primary and metastatic tissues for sample P-0007981 were obtained via core needle biopsy, suggesting that the comparatively small volume of tissue obtained in this way may have led to the discrepancy in mutations between the primary and metastatic samples. To evaluate whether the method of tissue acquisition (surgical excision vs core needle biopsy) affected P-M mutational differences, we tested whether levels of discordance varied between patients who did and did not have tissue acquired via core needle biopsy. Notably, we observed no significant difference in discordance between the two groups (Mann-Whitney p = 0.77, Supplementary Fig. 2).
Of the remaining samples, we found 14 pairs (24%) with Pr mutations restricted to the primary tumor, and 12 (20%) with Pr mutations restricted to the metastatic site. Patient tumors with a lower mutation burden exhibited significantly higher mutation concordance (Spearman correlation −0.41, p = 0.001). There was no significant difference in mutation concordance when comparing synchronous and metachronous metastases (Supplementary Fig. 1). Interestingly, tumor size was positively correlated with concordance (Spearman correlation 0.31, p = 0.02), indicating that larger metastases were likely to have a larger proportion of shared mutations in comparison to smaller metastases.
3.4. Mutation discordance between regional and distant lymph node metastasis
Metastatic spread of RCC can occur at regional and distant lymph nodes and has prognostic significance [1]. We evaluated whether gene mutations detected in regional (retroperitoneal) and distant lymph nodes exhibited different degrees of concordance when compared to the corresponding primary tumors. We observed a trend towards higher concordance in samples obtained from local lymph node metastases (p = 0.052), which probably corresponds to the chronological sequence of lymphatic spread and favors the regional to distant lymphatic dissemination of RCC.
3.5. Association between mutation discordance and therapy
It is known that the mutational landscape of a tumor changes in response to therapy. Given the limited treatment information available for patients, we evaluated whether administration of systemic therapy before sampling of either primary or metastatic tissue affected mutational concordance. We observed that patients who received therapy before sampling of the primary or metastatic tissue (n = 15) had higher P-M mutation concordance (Mann-Whitney p = 0.088) when compared to patients who did not (n = 41). Four patients were excluded from the analysis because of incomplete treatment information. All available information on treatment is provided in the Supplementary material. Because of the relatively small sample size, the heterogeneity in treatment received, and the intratumoral heterogeneity of RCC in general, we would caution that this finding remains preliminary and should be evaluated in an independent cohort. Nevertheless, the observation that of higher mutation concordance for specimens from patients who received treatment before sequencing suggests that systemic therapy may induce a contraction of the mutational landscape in a patient.
3.6. Convergent evolution of mutations in a spatiotemporally divergent manner in RCC
The convergent evolution of gene mutations and pathway activation reported for ccRCC was interrogated using our P-M cohort. We examined this phenomenon and identified convergent evolution of the same individual genes between primary and metastatic tumors in 15 P-M pairs (Fig. 4). Of these, eight were ccRCC and one was chRCC. Furthermore, three different spatiotemporally divergent patterns of convergent evolution were observed. Six pairs showed Pr mutations on the same gene in both the primary and metastatic samples, suggesting parallel convergent evolution. Nine pairs carried the same mutation of an individual gene, yet six had additional Pr mutations of the same gene in the primary tumor (typical cases of convergent evolution on key tumor genes seen on multiregion sequencing) and three had additional Pr mutations of the same gene in the metastatic tumor, implying subclone reseeding. Of note, these cases were highly enriched for mutations in SETD2 (n = 4) and PBRM1 (n = 4), all in ccRCC.
Fig. 4.
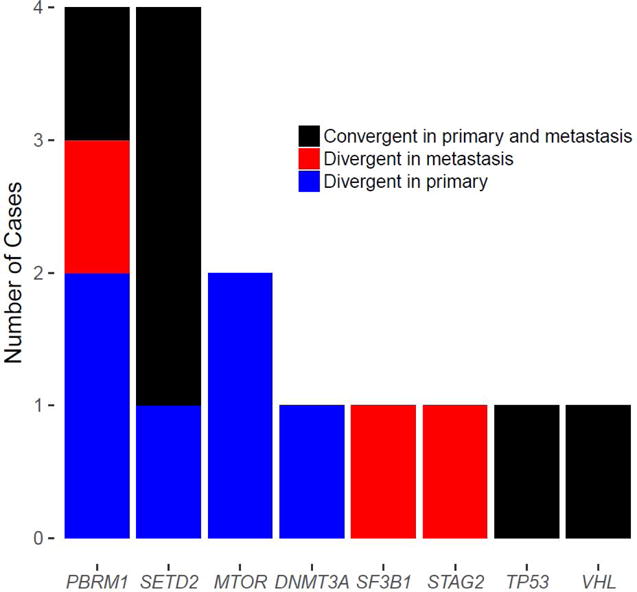
Three spatiotemporally distinct patterns of convergent evolution through loss of function on the same individual genes. Six primary tumor-metastasis pairs carry private mutations on the same individual gene in both the primary and metastatic samples (black). Nine pairs not only have shared mutations on the same individual genes but also carry private mutations in either the primary tumor (n = 6, blue) or the metastatic tumor (n = 3, red).
4. Discussion
Molecular profiling of primary tumors has dramatically increased our understanding of the biological mechanisms driving cancer, and has become a bona fide diagnostic and prognostic tool, has influenced the therapeutic course in several cancer types, and has facilitated precision medicine [18]. In the RCC setting, studies of somatic alterations in hundreds of primary RCC tumors by TCGA and others [19–21] have transformed our understanding of the molecular landscape of RCC. Furthermore, tumor genomic profiling of exceptional responders to and randomized clinical trial patients on targeted therapies have shed light on the how we might use genomic mutations as prognostic or predictive biomarkers [22–25]. However, much improvement is needed to fulfill the promise of precision RCC cancer treatment. The known high tumor heterogeneity in RCC represents a hurdle [14,26], especially in the area of patient-matched primary-metastatic tumor pairs [12].
The design of our study has several limitations. The cohort was relatively small in size, and metastatic specimens were collected at various time points throughout the disease course. The samples were collected as standard practice and therefore the patients were undergoing different courses of therapy. Our sequencing technology was targeted at identification of somatic alterations in a panel of known cancer-associated genes. Furthermore, difficulties with sequencing low-purity metastatic samples prevented us from evaluating differences in copy-number alterations between primary and metastatic samples. Perhaps most importantly, only a single region of each tumor was genetically profiled, which could significantly confound our estimates of concordance between primary and metastatic tumors. With limited resources, we elected to boost statistical power (and trade off some confidence in discordant mutation calls) by sequencing samples from more patients at a single site per tumor sample rather than completing multiregional sequencing for a smaller cohort of patients. As demonstrated by Gerlinger et al [14] and others, RCC tumors exhibit a high degree of intratumoral heterogeneity, and each subclone within a tumor has the potential to seed a distant metastasis. By evaluating concordance between a metastasis and only a single site in a tumor, we are ignoring the possibility that the metastasis was seeded from a distinct (but related) subclone of the primary tumor. Therefore, further efforts, perhaps using multiregional profiling (which could be carried out in a cost-effective way by multiplexing samples), exome sequencing, and larger sample sizes, are needed to identify genomic differences between primary and metastatic tumors.
Using high-resolution NGS for a small cohort (n = 4), Gerlinger et al [13] established that intratumoral heterogeneity drives substantial mutational differences between primary and metastatic RCC tissues. Thus far, evaluation of genetic divergence between primary and metastatic tumors in RCC using NGS platforms has remained limited to small or unmatched cohorts. These studies demonstrated that clonal evolution is prevalent in RCC, supporting a branched rather than linear model of tumor evolution [27]. However, when protein expression of PBRM1 and BAP1 was examined using immunohistochemistry for 97 matched P-M ccRCC pairs, high concordance for loss of PBRM1 (90%) and BAP1 (98%) was observed [28].
Our study confirms that at coarse resolution, RCC metastases are genomically quite distinct from matched primary tumors. Overall, 41% of the mutations we identified in our cohort were Pr mutations in the primary or metastatic tumor. As shown in prostate cancer, potential mechanisms include (1) parallel genetic evolution of primary and metastatic tumors and (2) seeding of metastases by additional subclones from the primary tumor or other metastases [29]. In some cases, we were able to identify evolutionary convergence in the form of parallel acquisition of distinct, independent mutations to genes in the primary and metastatic samples (eg, PBRM1 and SETD2) in various spatiotemporally divergent patterns. Furthermore, we found evidence of an association between SETD2 mutations and higher P-M mutation discordance.
Our results suggest that it could be of clinical value to sequence both primary tumor and metastatic tissues for a more comprehensive tally of the genetic alterations in individual patients. Such sequencing could reveal the presence of targetable mutations that would influence therapeutic decisions for a subset of patients. In fact, two patients in our cohort harbored mutations in TSC1 or TSC2 that were evident in the primary but not the matched metastatic sample, suggesting that treatment with everolimus might not have been effective in the metastatic setting. Furthermore, the primary tumor from patient P-0001232 (labeled with asterisk in Fig. 1) harbored mutations in both PBRM1 and BAP1, which predict poor clinical outcome; nevertheless, this patient had a relatively indolent clinical course and responded to targeted therapies. Sequencing of the metastatic tumor from patient P-0001232 revealed that it carried only the PBRM1 mutation [12]. Observations such as these emphasize the value of additional information gained from comprehensive knowledge of the genomic landscape of a patient’s cancer.
Finally, we also observed that treatment with systemic therapy was associated with a contraction of the mutational landscape. However, care should be taken in interpreting these results. Because our study was based on a relatively limited set of cancer genes (341 in total) and a relatively small sample of patients (n = 56 with treatment information), further profiling should be completed on an independent, larger cohort of patients at the exome scale, and potentially with multiregional profiling to account for intratumoral heterogeneity. Such an effort would not only increase the statistical power for confirming/disaffirming our observation but would also add increased resolution for estimating mutational discordance and identifying metastasis-specific mutations.
5. Conclusions
Our data suggest low concordance in the mutational profiles of matched primary and metastatic RCC samples. Although it is not clear whether these differences correspond to intratumoral heterogeneity [13,26] or evolution of the tumors [27,30], discordance is highly relevant for personalized medicine approaches, which often rely on molecular profiling of archived primary tissue. In summary, study of primary RCC tumors alone is probably insufficient for therapeutic decision-making in the treatment of metastatic kidney cancer.
Supplementary Material
Take Home Message.
Using next-generation sequencing, we determined mutation discordance for cancer genes between matched primary and metastatic tumor samples from patients with renal cell carcinoma. As most precision cancer medicine administers targeted agents on the basis of mutations detected in primary tumors, further investigation into the genomic discordance between primary and metastatic tumor pairs is warranted.
Acknowledgments
Funding/Support and role of the sponsor: This work and James J. Hsieh were supported by the J. Randall and Kathleen L. MacDonald Family Research Fund and the Jill and Jeffrey Weiss Family Fund for the cure of metastatic kidney cancer. This work was supported in part by the Sidney Kimmel Center for Prostate and Urologic Cancers and NIH/NCI Cancer Center support grant P30 CA008748. Robert J. Motzer and Mazyar Ghanaat were supported by Ruth L. Kirschstein National Research Service award T3to2CA082088. Jozefina Casuscelli was supported by German Research Foundation grant CA1403/1-1. Maria I. Carlo was supported by a Kidney Cancer Association Young Investigator Award. The sponsors played no direct role in the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosures: James J. Hsieh certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
Author contributions: James J. Hsieh had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Hsieh.
Acquisition of data: Becerra, Redzematovic, Tennenbaum, Kashan, Ghanaat, Casuscelli, Manley, Jonsson, DiNatale, Blum, Durack, Solomon, Arcila, Bourque, Socci, Carlo, Lee, Voss, Feldman, Motzer, Coleman, Russo, Hakimi.
Analysis and interpretation of data: Becerra, Reznik, Redzematovic, Tennenbaum, Cheng, Hakimi, Hsieh.
Drafting of the manuscript: Becerra, Reznik, Hsieh.
Critical revision of the manuscript for important intellectual content: Becerra, Reznik, Hsieh.
Statistical analysis: Reznik.
Obtaining funding: Hsieh.
Administrative, technical, or material support: Redzematovic.
Supervision: Hsieh.
Other: None.
References
- 1.Hsieh JJ, Purdue MP, Signoretti S, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choueiri TK, Motzer RJ. Systemic therapy for metastatic renal-cell carcinoma. N Engl J Med. 2017;376:354–66. doi: 10.1056/NEJMra1601333. [DOI] [PubMed] [Google Scholar]
- 3.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–30. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakimi AA, Pham CG, Hsieh JJ. A clear picture of renal cell carcinoma. Nat Genet. 2013;45:849–50. doi: 10.1038/ng.2708. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Pham CG, Albanese SK, et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest. 2016;126:3526–40. doi: 10.1172/JCI86120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shuch B, Amin A, Armstrong AJ, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67:85–97. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 9.Sankin A, Hakimi AA, Hsieh JJ, Molina AM. Metastatic non-clear cell renal cell carcinoma: an evidence based review of current treatment strategies. Front Oncol. 2015;5:67. doi: 10.3389/fonc.2015.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez-Pello S, Hofmann F, Tahbaz R, et al. A systematic review and meta-analysis comparing the effectiveness and adverse effects of different systemic treatments for non-clear cell renal cell carcinoma. Eur Urol. 2017;71:426–36. doi: 10.1016/j.eururo.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 11.Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131. doi: 10.1038/ncomms13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh JJ, Manley BJ, Khan N, Gao J, Carlo MI, Cheng EH. Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: a braided cancer river model of kidney cancer. Semin Cell Dev Biol. 2017;64:98–106. doi: 10.1016/j.semcdb.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voss MH, Hakimi AA, Pham CG, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20:1955–64. doi: 10.1158/1078-0432.CCR-13-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hakimi AA, Ostrovnaya I, Reva B, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. 2013;19:3259–67. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen FJ, Zhang YQ, Senbabaoglu Y, et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016;14:2476–89. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–7. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 21.Durinck S, Stawiski EW, Pavia-Jimenez A, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21. doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voss MH, Hsieh JJ. Therapeutic guide for mTOuRing through the braided kidney cancer genomic river. Clin Cancer Res. 2016;22:2320–2. doi: 10.1158/1078-0432.CCR-16-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwiatkowski DJ, Choueiri TK, Fay AP, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22:2445–52. doi: 10.1158/1078-0432.CCR-15-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh JJ, Chen D, Wang PI, et al. Genomic biomarkers of a randomized trial comparing first-line everolimus and sunitinib in patients with metastatic renal cell carcinoma. Eur Urol. 2017;71:405–14. doi: 10.1016/j.eururo.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim SM, Park HS, Kim S, et al. Next-generation sequencing reveals somatic mutations that confer exceptional response to everolimus. Oncotarget. 2016;7:10547–56. doi: 10.18632/oncotarget.7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sankin A, Hakimi AA, Mikkilineni N, et al. The impact of genetic heterogeneity on biomarker development in kidney cancer assessed by multiregional sampling. Cancer Med-Us. 2014;3:1485–92. doi: 10.1002/cam4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatesan S, Swanton C. Tumor evolutionary principles: how intratumor heterogeneity influences cancer treatment and outcome. Am Soc Clin Oncol Educ Book. 2016;35:e141–9. doi: 10.1200/EDBK_158930. [DOI] [PubMed] [Google Scholar]
- 28.Eckel-Passow JE, Serie DJ, Cheville JC, et al. BAP1 and PBRM1 in metastatic clear cell renal cell carcinoma: tumor heterogeneity and concordance with paired primary tumor. BMC Urol. 2017;17:19. doi: 10.1186/s12894-017-0209-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei EY, Hsieh JJ. A river model to map convergent cancer evolution and guide therapy in RCC. Nat Rev Urol. 2015;12:706–12. doi: 10.1038/nrurol.2015.260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.