

Abstract
Lentiviral vectors (LVs) have been successfully used in clinical trials showing long term therapeutic benefits. Studying the role of cellular proteins in lentivirus HIV-1 life cycle can help understand virus assembly and budding, leading to improvement of LV production for gene therapy. Lentiviral vectors were purified using size exclusion chromatography (SEC). The cellular protein composition of LVs produced by two different methods was compared: the transient transfection system pseudotyped with the VSV-G envelope, currently used in clinical trials, and a stable producer cell system using a non-toxic envelope derived from cat endogenous retrovirus RD114, RDpro. Proteins of LVs purified by size exclusion chromatography were identified by tandem mass spectrometry (MS/MS). A smaller number of cellular protein species were detected in stably produced vectors compared to transiently produced vector samples. This may be due to the presence of co-purified VSV-G vesicles in transiently produced vectors. AHNAK (Desmoyokin) was unique to RDpro-Env vectors. The potential role in LV particle production of selected proteins identified by MS analysis including AHNAK was assessed using shRNA gene knockdown technique. Down-regulation of the selected host proteins AHNAK, ALIX, and TSG101 in vector producer cells did not result in a significant difference in vector production.
Keywords: Lentiviral vectors, Host proteins, Mass spectrometry, ALIX, AHNAK
1. Introduction
Lentiviral vectors (LVs) have been applied in several clinical trials for the treatment of monogenic disorders such as beta-thalassaemia [1], Wiscott-Aldrich syndrome [2] and Metachromatic Leukodystrophy (MLD) [3]. Besides ex vivo therapy of hematopoietic stem cells (HSC), LVs can efficiently transduce dendritic cells [[4], [5], [6]] given them the potential to be used as vaccines in larger groups of patients. LVs are also widely used as experimental tools for stable genetic modifications of cells in research laboratories, e.g. shRNA libraries [7] and gene editing [8].
Most commonly lentiviral vectors are produced by transient transfection. This method has several disadvantages as vectors can only be produced over a short time and production is labour-intensive as well as difficult to scale up. Furthermore the presence of DNA plasmids in produced vectors requires their purification, complicating downstream-processing. With the increasing need for large amounts of LVs a packaging cell line stably expressing LVs is desirable. Packaging cell lines such as tetracycline-dependent systems [[9], [10], [11]] have been developed, with variable vector production abilities. The producer cell line STAR stably expresses high levels of a codon-optimised HIV-1 gag-pol introduced by transduction with murine leukaemia virus (MLV) derived vectors as well as rev and tat [12]. STAR cell produced vectors are pseudotyped with a gammaretroviral envelope RDpro as it can efficiently transduce HSC as shown for vectors produced by STAR-RDpro cells [13] and shown for the next generation of stable RDpro-pseudotyped producer cells (WinPac) in comparison to transiently produced VSV-G pseudotyped vectors [14] and is not cytotoxic [15], unlike VSV-G which has been used in transiently produced LVs in current clinical trials.
Studies of wild-type HIV-1 virus showed that proteins such as the transcription factor 1-alpha (EEF1A1), programmed cell death 6-interacting protein (ALIX or AIP1), annexin A2 or 5 and alpha-enolase [[16], [17], [18]] were associated with virus particles. These proteins were also identified in studies on crude or purified lentiviral vectors [19,20]. Several groups [[19], [20], [21]] have used mass spectrometry (MS) to identify vector associated host cell proteins. Further analysis of vector-associated host proteins can point the way towards fundamental virus-host interaction in viral vector assembly, in particular, the protein-protein and protein-RNA interactions during viral particle assembly and formation. Ultimately this knowledge can be applied to improve vector production.
In this study we purified lentiviral vectors using size exclusion chromatography (SEC) a purification method that was used during processing of a HIV-1 based VSV-G pseudotyped lentiviral vector in a phase I clinical trial [22] and then used LC-MS/MS to analyse the protein composition in the purified vectors generated by transient production pseudotyped with VSV-G and by stable producer cell line, STAR, pseudotyped with the RDpro envelope protein.
We further analysed the effect of these proteins on vector production using small hairpin RNA (shRNAs) -mediated gene regulation. Our results showed insignificant effects of knock-down expression on vector production levels.
2. Materials and methods
2.1. Lentiviral vectors
Lentiviral vectors were either produced by transient transfection of HEK 293T cells [23] or by a stable producer cell line [12]. Six different vector samples were prepared. Transiently produced vectors were 1) VSV-G-GFP: transfection of DNA plasmids coding for VSV-G Env, structural protein HIV-1 Gag/Pol, Rev and a SIN-vector genome with GFP marker gene driven by a human phosphoglycerate kinase 1 (PGK) promoter; 2) VSV-G-Empty: transfection of DNA plasmids coding for VSV-G Env, HIV-1 Gag-Pol and Rev, 3) Gag/Pol-GFP: transfection of DNA plasmids coding for Gag/Pol, a vector genome and Rev. 4) VSV-G only: a sample produced by transfection of only the VSV-G envelope and the SIN-LV genome plasmid. Stably produced vectors were packaged by STAR-RDpro-HV or STAR-RDpro cells [12]. STAR-RDpro-HV, derived from HEK 293T cells, are continuously expressing RDpro envelope protein, Gag/Pol, a non-SIN vector genome with marker protein GFP, driven by a spleen focus-forming virus (SFFV) promoter and Rev, producing vector sample ‘RDpro-GFP’ (sample 5). STAR-RDpro express RDpro, Gag/Pol and Rev but no vector genome and produced vector sample ‘RDpro-Empty’ (sample 6). For vector sample preparation, cells were seeded on 15 cm dishes. For vector samples 1) to 4) cells were transiently transfected 24 h later. Following a medium change 24 h later, vector containing supernatant was collected 24 h and 48 h post transfection. Supernatants of each plate were concentrated by ultracentrifugation to a final volume of 900 μl. Transiently produced vector samples were concentrated 40 fold and stably produced vectors 240 fold.
2.2. Lentiviral vector purification
For vector purification, a Gilson liquid chromatography system was used, consisting of a pump (model 306), an autosampler (model 231XL) as well as a fraction collector (model FC203B) connected to a temperature controlled rack. Sample injection was controlled by the software Trilution LC 2.1 and sample elution and detection were controlled by Unipoint 5.11 (all parts and software by Gilson, Middleton, WI, USA). Samples were separated by a cooling jacked XK16/70 column packed with Sephacryl-500–HR medium (both by GE Healthcare, Little Chalfont, UK). Sephacryl-500–HR medium consists of cross-linked copolymer beads of allyl dextran and N,N′-methylene bisacrylamide with an average particle size of 47 μm, allowing the separation of macromolecules in the range of 4 × 104 to 2 × 107 relative molecular mass (Mr). TEN buffer containing 150 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl pH 7.4 was used as sample running buffer (Sigma-Aldrich, UK). For size exclusion chromatography 900 μl of crude vector sample were purified per column at a flow rate of 0.8 ml/min. Void peak fractions from a total of 8 runs of 900 μl crude vector samples were pooled and dialysed using slide-A-Lyzer dialysis cassettes (3–12 ml capacity, molecular cut-off 10 000 Da, Thermo Fisher Scientific, UK) overnight in 3 L of 10 mM ammonium bicarbonate (ABC), pH 8.0 (Sigma-Aldrich). Dialysed samples were freeze-dried using an Edwards E2M2 High Vacuum Pump and resuspended in distilled water. Total protein amounts were quantified by Bradford Protein Assay (BioRad, Hercules, CA, USA) using BSA protein standard (Thermo Fisher Scientific).
2.3. Purified lentiviral vector characterisation using LC-MS/MS
Purified and concentrated lentiviral vector samples were digested with trypsin in the presence of 0.05% Rapigest (an enzyme-compatible detergent to ensure protein denaturing; Waters, Milford, MA, USA) and 50 mM ammonium bicarbonate, pH 8.5 for 3 h at 37 °C. HCl was added to terminate digestion and ensure breakdown of Rapigest.
LC-MS/MS was carried out using a MS system (Thermo Fisher Scientific, Hemel Hampstead, Herts, UK) equipped with a nano-electrospray ion source and two mass detectors i.e. linear trap (LTQ) and orbitrap, coupled with an Ultimate 3000 nano-LC system, comprising a solvent degasser, a loading pump, a nano-pump, and a thermostated autosampler. After an automated injection, the extracted peptides from each digestion were desalted in a trapping cartridge (PepMap reversed phase C18, 5 μm 100 Å, 300μ id x 5 mm length; Thermo Fisher Scientific), eluted on to a C18 reversed phase nano-column (3 μm, 100 Å, 5 cm length; Thermo Fisher Scientific) followed by a 60 min separation under a column flow rate of 0.3 μl/min using linear gradient of 5–70% acetonitrile and 0.1% formic acid (both by Sigma-Aldrich). Separated and eluted peptides were ionised by electrospray ionisation followed by a MS survey scan (mass-to charge-ratio, m/z 400–2000) in the LTQ, sequentially selecting the five most abundant ions of peptides eluting from the LC at that time, before being passed on to the Orbi-trap. The total cycle time for each MS/MS was approximately 30 milliseconds. The Orbi-trap took accurate mass measurement with the resolution of 30 000 parts per million (ppm) and ions were then fragmented in the linear ion trap by collision induced dissociation at collision induced energy of 35%. Subsequently, fractionated ions were separated according to their m/z ratio. Data was collected in data dependent MS/MS mode with dynamic exclusion set to 2 counts. Data analysis including mass spectra processing and database searching was carried out using Thermo Proteome Discoverer 1.2. with built-in Sequest (Thermo Fisher Scientific). Initial mass tolerances for protein identification by MS were set to 10 ppm. Up to two missed tryptic cleavages were considered and methionine oxidation was set as dynamic modification. Peptide sequences by MS/MS were only included when Xcorrelation scores were greater than 1.5, 2 or 2.2 for charge states 1, 2 and 3, respectively. An unambiguous identification was considered when at least two peptides matched to the protein. The protein FASTA databases were downloaded from www.uniprot.org, release 2012-03 including the complete entries from homo sapiens (taxon identifier 9606), bos taurus (9913), human immunodeficiency virus type 1 group M subtype B (isolate HXB2) (HIV-1) (11706), vesicular stomatitis indiana virus (strain San Juan) (VSIV) (11285), RD114 virus (11834), RDpro Env (Ikeda et al., 2003) AA sequence: (Bell et al., 2010, Ikeda et al., 2003), and GFP (P42212).
2.4. Knock-down of protein expression in vector producer cells using shRNA
Selected host proteins were knocked-down using lentiviral vector particles carrying the pGIPZ vector genome (LVs-GIPZ). PGIPZ target sequences were: TSG101- TGCAATAACTTATTCTGGG, ALIX- TAATCTGCAGCCTGATTAG, AHNAK- TAGATCAGGAGCTCCTACG) (Thermo Fisher Scientific). Transduced 293T cells were selected in DMEM with puromycin (10 μg/ml) twenty-four hours after transduction. After a minimum of 10 days in selection medium vector samples and producer cells were analysed for host protein knock-down levels and vector levels by western blotting.
2.5. Vector titration
For titration of infectious vector titers by FACS 293T cells were transduced with serial dilutions of vector then cultured for 48 h. Cells were harvested, washed, and analysed by FACS (BD FACSCanto II™) to determine the percentage of GFP-expressing cells and calculate the transduction titre. For titration of physical titre, the p24 ELISA kit ‘Lenti-X p24 Rapid Titer Kit’ (Clontech, Mountain View, CA, USA) was used to measure p24 protein levels in vector harvests from STAR, transiently transfected 293T cells and STAR-GIPZ and 293T-GIPZ LV producer cells. The manufacturers' protocol was followed. Standard curve dilutions of the kit supplied p24 control were prepared in complete DMEM [Dulbecco's modified Eagle medium (DMEM) (Sigma-Aldrich, Gillingham, UK) completed by the addition of 10% fetal calf serum (Appleton Woods, Birmingham, UK), 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (all by Sigma-Aldrich)] and samples measured in one or two dilutions (in complete DMEM) depending on levels of infectious titer (1:4 up to 1:5000).
Q-RT-PCR was used to quantify copies of the vector genome RNA in LV knock-down producer cells and LV harvests. Q-PCR was performed using QuantiTect® SYBR® Green PCR kit (Qiagen, Limburg, Netherlands) targeting the SFFV promoter (forward primer 5′ CGATAAGCTTGATATCGAATTCCT3′, reverse primer TGCGGTGACCATCTGTTC. The reference gene was human beta-actin.
2.6. Silver staining and western blotting
Proteins were separated on sodiumdodecyl sulfate (SDS)-polyacrylamide gels (10% acrylamide) and either visualised by silver staining using the Silver Stain PlusOne kit (GE Healthcare, Little Chalfont, UK) or SilverXpress kit (Invitrogen) or transferred to nitrocellulose membranes (Hybond enhanced chemiluminescence, ECL, Amersham). Antibodies used were: anti-p24 (1:100, mouse monoclonal, ARP 365, CFAR, NIBSC, UK; binds sequence NPPIPVGEIY in p24 of HIV-1 Gag), anti-VSV-G (1:2000, mouse monoclonal, Sigma-Aldrich), anti-RDpro pg70 (1:2000, goat polyclonal, Quality Biotech Inc., Camden, NJ, USA), anti-AHANAK (1:500, mouse-monoclonal, Abcam, Cambridge, UK), anti-TSG101 (1:500, rabbit-polyclonal, Sigma-Aldrich), anti-ALIX (1:1000, rabbit polyclonal), anti-EEF1A (1:500, mouse-monoclonal), anti-ENO1 (1:500, mouse-monoclonal), anti-MARCKSL1 (1:1000, rabbit-polyclonal) and anti-GAPDH (1:5000, mouse-monoclonal, all by Millipore, Billerica, MA, USA).
2.7. Functional category and canonical pathway analysis of LC-MS/MS data
IPA (‘Interactive Pathway analysis’ of complex ‘omics data by Ingenuity® Systems; [www.ingenuity.com]; Mountain View, CA, USA) was used to determine the most significant molecular and cellular functions and cellular pathways in the LC-MS/MS datasets. Datasets of UniProt identifiers were uploaded into the application. For core analysis of a dataset default settings were applied and only direct relationships between proteins that have been experimentally confirmed in research publications were considered.
2.8. Statistical analysis
Statistical analysis was carried out using GraphPad Prism5 (GraphPad Software Inc., La Jolla, CA). Data were analysed using a two-way analysis of variance. Differences were accepted to be statistically significant at p < 0.05.
3. Results
3.1. Vector production and purification by size exclusion chromatography
LV samples were produced either by transient transfection or by STAR-derived LV packaging cells. In addition to full vectors, samples that lack 1 or 2 viral components, e.g. lacking envelope and/or transgene, were also produced. Average transduction titer obtained for full GFP vectors produced transiently and stably were 34.3 ± 8.97 × 106 TU/ml and 3.4 ± 2.9 × 106 TU/ml, respectively; indicating 10 fold higher transduction titers in transiently produced vectors than in stably produced vectors. The level of HIV p24, a surrogate measurement of vector particle concentration, was 6 fold higher in transiently produced vectors than stably produced vectors. As a result, some stably produced LV were concentrated 240 fold by ultracentrifugation; whilst the majority of vectors were concentrated 40 fold. The concentrated samples were then subjected to size exclusion chromatography and the void peaks were collected for MS/MS analysis.
Silver staining of purified vectors collected from SEC void peaks showed dominant bands at the positions correlating with viral proteins pr55Gag (55 kDa) and capsid p24 (24 kDa), (Fig. 1). Increasing concentration factor of stably produced samples to 240 fold resulted in an increased level of p24 proteins in purified samples, generating a comparable numbers of vector particles to that of transiently produced vector samples. As shown in Fig. 1 the intensity of capsid protein p24 is comparable between 240 fold concentrated RDpro-pseudotypes and 40 fold VSV-G-pseudotypes. The 57 kDa proteins, correlating to the size of VSV-G Env, gave a strong signal in transiently produced samples, whereas RDpro Env (76 kDa) is below detection limit. A significant number of protein bands shown in crude samples are not detectable in the samples collected from SEC void peaks (Fig. 1), demonstrating the fidelity of the adopted purification method.
Fig. 1.

Comparable amounts of p24 in vector samples for LC-MS/MS. Silver staining of SDS-Page of vector samples after SEC purification and preparation for LC-MS/MS analysis. Transiently and stably produced samples show dominant bands at the position of pr55Gag (55 kDa) and capsid p24 (24 kDa), the latter being of comparable signal strength. Signals at position equivalent of 57 kDa is stronger in transiently produced samples, correlating to the size of VSV-G Env, whereas RDpro Env (76 kDa) is below detection limit. (V+ = VSV-G-GFP, V- = VSV-G-Empty, RD+ = RDpro-GFP, RD- = RDpro-Empty); 100 or 50 ng total protein/lane loaded as indicated.
3.2. Identification of viral proteins in purified LVs by LC-MS/MS
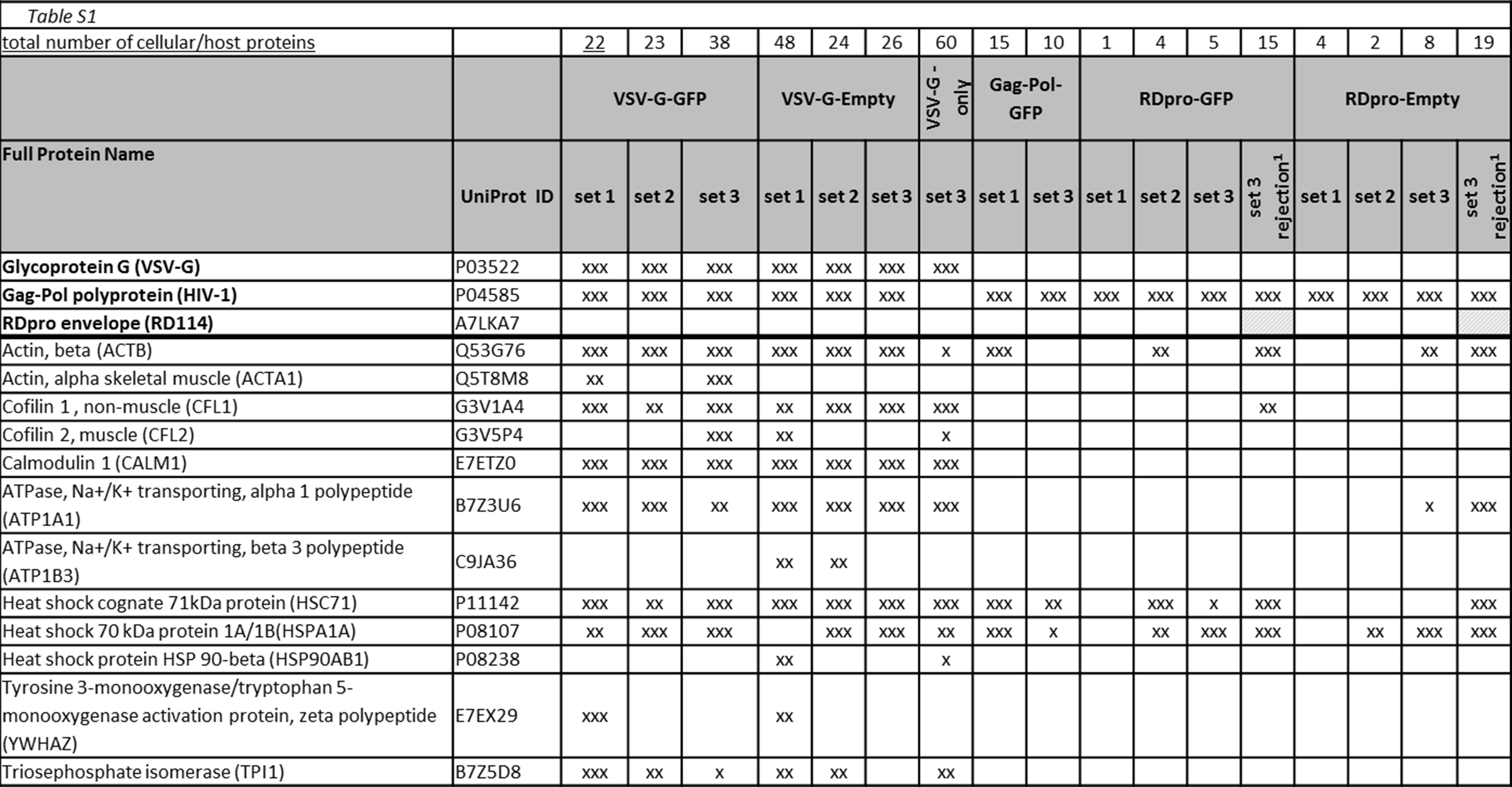
Transiently and stably produced vector samples for set 1 and 2 MS analysis were prepared from a comparable number of producer cells and concentrated 40 fold by ultracentrifugation and purification before MS analysis. Samples for set 3 MS analysis contain a comparable level of p24, thus comparable physical titres of vectors produced stably and transiently. Peptides of viral proteins, including HIV Gag protein domains (matrix (MA), capsid (CA) and p2), protease (PR), reverse transcriptase (RT), integrase (IN) and VSV-G Env were detected in all 3 sets of purified vectors by LC-MS/MS. The percentage of coverage in protein sequence is shown in Table 1 for MS set 3 samples, produced transiently (40 fold concentrated) and stably (240 fold concentrated). Surprisingly, the RDpro Env was not detectable in RD pseudotyped vectors in the initial study. This may be due to the fact that during a default MS survey scan, five most abundant ions are dynamically chosen for peptide fragmentation to generate a tandem MS spectrum of fragmented ions for subsequent identification of their corresponding peptide sequences; therefore, the ions and their corresponding peptides at low abundance, e.g. RDpro Env, would not be identified. To increase the detection of less abundant proteins in a sample, MS data acquisition was modified, in which, ions that had been detected and selected in the first MS survey scan would be rejected from selection in the next MS survey scan in favour of selecting ions of other peptides. Using this method (from now on referred to as ‘rejection method’) to analyse RDpro pseudotyped vectors resulted in the identification of RDpro Env protein sequence and significantly more peptide sequences in the MS set 3 samples (Table 1, column ‘MS set 3 rejection’). The percentage of protein sequences identified using the rejection method in stably produced samples is also displayed in Table 1. RDpro Env sequences identified using the rejection method cover 4%–7% of the RDpro amino acid (AA) sequence in RDpro-GFP and RDpro-Empty vector samples, respectively. Gag-p2 and PR were also only detected using the rejection method in stably produced samples. Coverage of CA protein sequence was the highest ranging between 57% and 79%, equivalent to 122 to 169 AA out of capsid's 215 AA. Peptides of Gag-p6 were never detected in stably produced samples but in transiently produced samples VSV-G-GFP and VSV-G-Empty. As expected no peptides of VSV-G Env were detected in Gag/Pol-GFP samples but in all other transiently produced samples prepared by co-transfection including VSV-G Env. No peptides originating from Gag or Gag-Pol protein were detected in the VSV-G-only sample.
Table 1.
Coverage of viral protein sequences by LC-MS/MS in purified vectors.
| Viral protein | Protein length (amino acids) | MS set 3 |
MS set 3 rejection |
MS set 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RDpro-GFP (240x) | RDpro-Empty (240x) | RDpro-GFP (240x) | RDpro-Empty (240x) | VSV-G GFP (40x) | VSV-G Empty (40x) | Gag/Pol-GFP (40x) | VSV-G only (240x) | |||
| Gag | MA | 130 AA | 45% | 46% | 63% | 66% | 73% | 96% | 67% | ND |
| CA | 215 AA | 57% | 57% | 79% | 61% | 49% | 50% | 52% | ND | |
| p2 | 13 AA | ND | ND | 100% | 100% | ND | ND | ND | ND | |
| p6 | 56 AA | ND | ND | ND | ND | 27% | 18% | ND | ND | |
| Pol | PR | 98 AA | ND | ND | 21% | ND | 17% | ND | 18% | ND |
| RT | 559 AA | 3% | 5% | 33% | 21% | 17% | 17% | 17% | ND | |
| IN | 287 AA | ND | ND | 15% | 9% | 15% | 9% | 1% | ND | |
| VSV-G Env | 411 AA | ND | ND | ND | ND | 20% | 22% | ND | 41% | |
| RDpro Env | 565 AA | ND | ND | 4% | 7% | ND | ND | ND | ND | |
3.3. Identified host cell proteins in purified LVs by LC-MS/MS
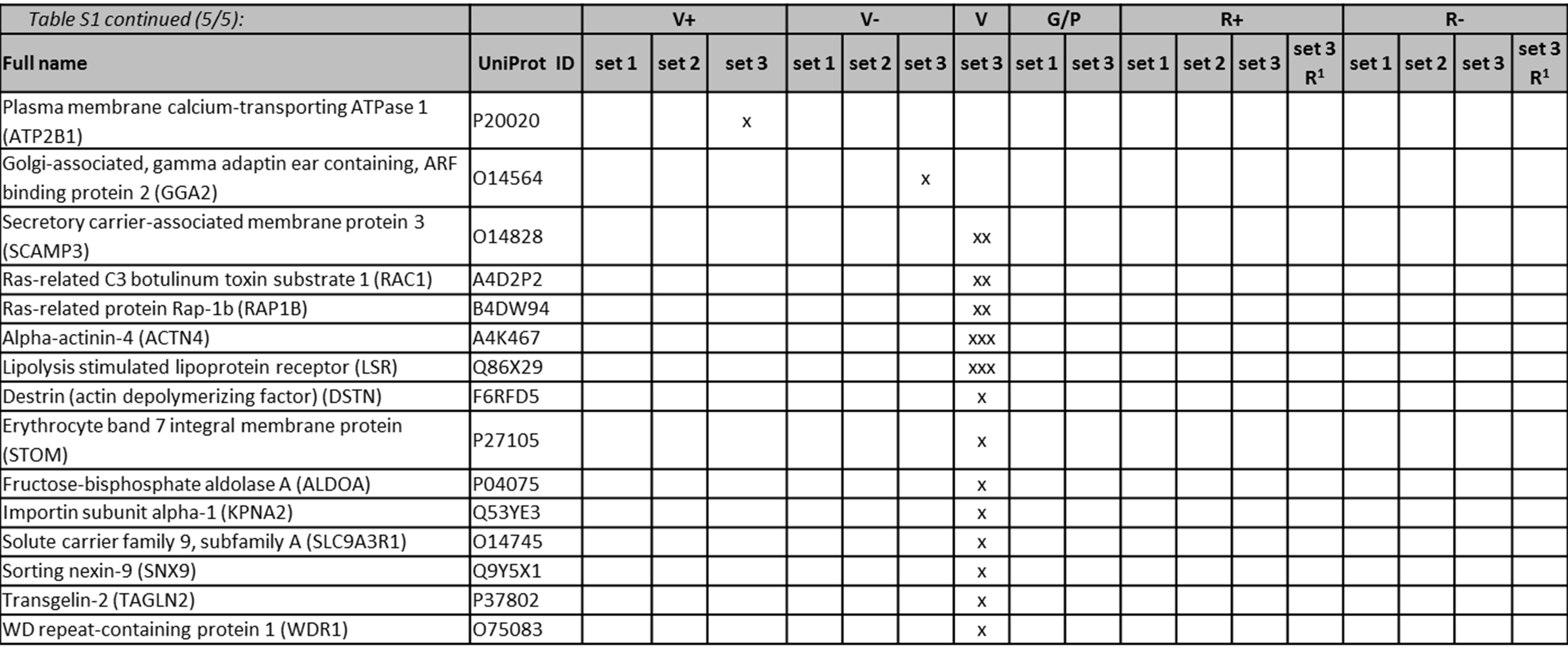
The list of MS detected cellular proteins is presented in supplementary Table 1. Of each type of transiently and stably produced samples three batches were made and analysed by MS (MS set 1 to 3), with the exception of the VSV-G-only sample, which was produced and analysed once. A total of 93 different cellular proteins were identified from all samples analysed. In Gag/Pol-GFP samples that did not carry any envelope proteins, 10 and 15 cellular proteins were detected in MS set 1 and 3, respectively. Five cellular proteins were shared between these two sets. The cellular proteins that were common to all five stably and transiently produced vector samples include Beta-Actin, Heat shock cognate 71, Heat shock protein 70, Histone cluster 1- H2ah, Clathrin heavy chain 1, Alpha-enolase (ENO1), ALIX, Elongation factor 1-alpha (EEF1A) and Cyclophilin A (CypA). VSV-G-GFP and VSV-G-Empty shared 39 cellular proteins. MARCKSL1 was common to VSV-G-GFP, VSV-G-Empty as well as RDpro-GFP and RDpro-Empty LV samples. A summary of the proteins detected in transiently and stably produced samples is shown in Fig. 2A, showing that transiently produced VSV-G-pseudotyped vectors contain considerably more host protein species compared to stably produced RDpro-pseudotyped vectors. A number of bovine proteins were detected in this study, including bovine serum albumin, hemoglobin foetal subunit alpha and beta. These may have potentially been carried over from foetal calf serum used in cell culture and vector production and are excluded from Fig. 2 and Supplementary Table 1. No cellular proteins were identified that are unique to vector samples carrying a vector genome (GFP transfer vector), suggesting that none of the detected cellular proteins is solely associated with viral RNA. All cellular proteins that were identified in stably produced RD-pseudotyped vectors were also identified in transiently produced VSV-G pseudotyped vectors with the exception of AHNAK (or Desmoyokin; Neuroblast differentiation-associated protein), that is unique to RDpro-GFP and RDpro-Empty samples (Fig. 2).
Fig. 2.

A). Venn diagram illustrating the number of protein species identified by LC-MS/MS in VSV-G pseudotyped transiently produced vectors or RDpro-pseudotyped stably produced vectors, as well as commonly found in both vector preparations. B). Venn diagram of top molecular and cellular functions of LC-MS/MS identified proteins classified by functional annotation of the Ingenuity® Knowledge Database of IPA by Ingenuity® Systems. Functional overlap of proteins is shown. The IPA software maps the proteins in the dataset to the information in the Ingenuity® Knowledge Base and then places the identified cellular proteins into well-established signalling or metabolic pathways, termed “canonical pathways”. Canonical pathways have been defined as “idealised or generalised pathways that represent common properties of a particular signalling module or pathway” (Science magazine (http://stke.sciencemag.org/about/help/cm.dtl) as opposed to specific pathways in which components are known to act together in a particular organism, tissue or cell type.
3.4. Functions of identified host proteins in cellular assembly and organisation
Functional characterisation and analysis of LC-MS/MS identified proteins were carried out using the data analysis software IPA (Ingenuity® Systems Mountain View, CA, USA) based on functional annotations in the web-based Ingenuity® Knowledge Database. The total of 93 identified cellular proteins were mapped by IPA, of which 52 are located in the cytoplasm, 15 in the nucleus, 24 at the plasma membrane and two proteins were classified as being located in the extracellular space (Lactadherin and WD repeat-containing protein 1). Identified proteins were further categorised into functional families. Out of the 93 proteins identified, 23 were classified as enzymes, 12 as transporters, 5 as transcription regulators and the remaining 53 proteins were not classified. About 51% of all identified proteins fall in at least one of the functional categories, e.g. cellular assembly and organisation, cell function and maintenance and cell death (supplementary Table 2). Some of the proteins have more than one specific function as shown in Fig. 2B, being annotated to function in processes such as the organisation of the cytoplasm and cytoskeleton, in microtubule dynamics, formation of cellular protrusions and in necrosis and apoptosis.
The top five canonical pathways each with at least 10 associated proteins of the dataset are displayed in supplementary Figure 1. These pathways include actin cytoskeleton signalling, epithelial adherence junction signalling and remodelling (supplementary Figure 1B), integrin signalling and clathrin mediated endocytosis (supplementary Figure 1C). Proteins associated with mechanisms of viral host cell exit and their association with the top five pathways is illustrated (supplementary Figure 1B and C). Several proteins act in more than one pathway as depicted in the Venn diagrams.
3.5. Functions of proteins exclusive to samples containing VSV-G
The functional categories and pathways of 19 proteins that were exclusive to the three VSV-G samples, i.e. VSV-G-GFP, VSV-G-Empty and VSV-G-only, were further analysed using the Ingenuity® Pathway Knowledge Base. Ten out of the 19 proteins are located at the plasma membrane, eight in the cytoplasm and one in the nucleus. As shown in supplementary Table 2, these 19 proteins are involved in cellular assembly and organisation, such as organisation of the cytoskeleton, microtubules dynamics and formation of cellular protrusions but also molecular transport. In particular, Actin-alpha, Cofilin 2, Radixin, Ezrin, Tubulin-beta class I, Protein kinase C and casein kinase substrate and Talin 1 are part of the cytoskeleton or function in its organisation. Four of the proteins found in VSV-G Enveloped vectors serve as transporters, including transmembrane transporters (SLC1A5, SLC3A2 and Basigin) and proteins helping vesicular transport, such as Vamp3.
3.6. Functions of host proteins common to transiently and stably produced LV samples
Eight cellular proteins common to all produced vector samples include Beta actin, Heat shock cognate 71kDa protein (HSC71), heat shock protein 1 alpha (HSPA1A), HIST1H2AH, Clathrin heavy chain 1, Alpha-enolase, ALIX and Cyclophilin A. Categorising these proteins using IPA software shows that six of them are functioning in cellular proliferation. HSPA1A and Histone H2ah (HIST2AHA) are not categorised by IPA but with chaperone activity and DNA structure organisational functions, respectively, they are also essential for cell maintenance. Three chaperones proteins HSC71, HSPA1A and CypA are common to all LC-MS/MS analysed LV samples. Beta actin and ALIX are involved in viral exit from cells. ALIX is a factor of the Endosomal Sorting Complexes Required for Transport (ESCRT) and functions in multivesicular body (MVB) biogenesis. It has to be noted all of the above proteins, apart from Clathrin heavy chain 1, were also detected in the 240 fold VSV-G-only sample. Elongation factor 1 alpha and MARCKSL1 were common to VSV-G-GFP, VSV-G-Empty as well as RDpro-GFP and RDpro-Empty LV samples and their individual functions are shown in supplementary Table 3.
3.6.1. AHNAK - cellular protein unique to RDpro-VSV-G and RDpro-empty
AHNAK or Neuroblast differentiation-associated protein has been identified only in stably produced LV samples pseudotypes with RDpro Env. Analysis of the IPA database shows that it has been found in several subcellular locations such as the nucleus, cytoplasmic vesicles and intercellular junctions.
3.6.2. Effect of AHANK, ALIX or TSG101 knock-down on vector production
To identify proteins that might be involved in vector assembly in 293T or 293T-derived STAR-RDpro cells, 3 proteins, i.e. AHANK, ALIX or TSG101, were selected for gene knocked down in producer cells using shRNAs. AHNAK was the only host protein identified in RDpro-GFP and RDpro-Empty samples and thus interesting for knock-down analysis. ALIX (ALG-2-interacting protein X or programmed cell death 6-interacting protein PDCDI6P) is one of the proteins common to all vector samples tested. TSG101 was not identified in our MS analysis, but has shown to be important in HIV-1 budding; thus was included in the gene knock down study, as detailed below.
LVs-GIPZ particles to deliver ALIX-, AHNAK- or TSG101-specific shRNA were prepared as detailed in the Methods and Materials section and were used to transduce 293T and STAR-GFP producer cells. TSG101, ALIX and AHNAK protein expression were compared before and after shRNA gene knockdown in both 293T and STAR cells (Fig. 3A and B). Based on western blotting analysis, a complete knockdown was achieved for AHNAK, ALIX and nearly complete knock down achieved for TSG101. Lentiviral vectors were further produced from the gene knock-down cells to assess the effects of gene regulation on vector production. Fig. 3C showed that the relative transduction titer of STAR-GIPZ-AHNAK and STAR-GIPZ-TSG101 produced vectors to the control ‘scramble‘ was between 0.7-2.1 fold and 0.6–1.0 fold, respectively. STAR-GIPZ-ALIX produced vectors had an infectious titer of 1.1–2.4 fold compared to the scramble control (not shown). Overall no significant difference in infectious titers compared to the scramble control was observed in STAR-GIPZ-ALIX produced vectors (Fig. 3C). Vector titers of 293T-GIPZ-AHNAK and 293T-GIPZ-TSG101 cells (Fig. 3D) were 1.2 fold and between 0.9 and 2.1 fold different, respectively, from the titer of vectors produced by 293T-GIPZ-scramble. 293T-GIPZ-ALIX produced particles had an infectious titer of 0.6–0.8 fold relative to the control scramble. Overall no significant difference in infectious titers compared to the scramble control was observed (Fig. 3D).
Fig. 3.

A). Knock-down efficiency of AHNAK and TSG101 by shRNA in 293T cells by western blotting. B). Knock-down efficiency of ALIX and AHNAK by shRNA is similar in STAR and 293T cells and baseline expression levels in both cell lines are comparable; western blotting allows direct comparison of protein knock-down efficiency in STAR-GIPZ and 293T-GIPZ 7 days post-GIPZ transduction. Equivalent loading of total protein was verified by western blotting of GAPDH (10 μg of total protein of cell lysates per lane). Negative control was 293T cells without transduced GIPZ-LV (‘no shRNA’). Protein molecular seizes are indicated. C). No significant effect of shRNA mediated knock-down of AHNAK and TSG101 on infectious particle production from STAR-GIPZ cells was seen. Infectious particle production from STAR-GIPZ cells from four sets of samples (transducing units/ml vector harvest) are shown and are the average of 3 wells/sample per set. Bars on columns indicate SD. D). No significant effect of shRNA mediated knock-down of AHNAK, ALIX and TSG101 on infectious particle production in 293T-GIPZ cells (p > 0.05). Infectious particle numbers in 293T-GIPZ vectors of two sets of samples (transducing units/ml vector harvest), shown is the average of 3 wells/sample per set.
3.6.3. Effect of AHANK, ALIX or TSG101 knock-down on the expression of vector components
Effects of selected gene knock-down on vector production, in particular the expression of vector genome RNA and capsid protein p24 were also analysed. No significant difference compared to the control scramble was observed in viral genome RNA levels (supplementary Figure 2 and 4) and in the expression of viral protein p24 (supplementary Figure 3) of vectors produced from any of the three gene knockdown STAR-GIPZ and 293T-GIPZ producer cells. Furthermore, the incorporation of viral genome RNA (supplementary Figure 5) or p24 (supplementary Figure 6) into vector particles, produced by STAR-GIPZ after knock-down of AHNAK and TSG101, was measured and showed no significant changes compared to the controls. Interestingly, western blotting of the structural proteins showed, that p55 Gag protein in vectors of STAR-GIPZ-TSG101 cells are 15 to 31 fold higher in three out of four experiments compared to scramble control and STAR-GIPZ-AHNAK Gag precursor levels (supplementary Figure 7). These data suggest that Gag precursor processing to p24 is impaired when TSG101 protein expression is reduced.
A general assessment of vector production levels in STAR-GIPZ and 293T-GIPZ based on various characteristics of harvested particles was undertaken in order to elucidate any potential differences between both producer cells. The ratio of infectious particles over physical particles based on vector genome RNA copy numbers and p24 levels (TU:RNA:p24) was calculated. Physical particle numbers are based on the assumption that one particle contains two copies of viral RNA genomes and 1 ng p24 equals 3.33 × 107 vector particles. The ratios of TU:RNA:p24 in STAR-GIPZ and 293T-GIPZ produced vectors show an interesting difference. RNA copies of STAR-GIPZ produced particles are on average 43 fold higher and corresponding physical particles based on p24 levels are on average 1000 fold higher compared to infectious particles. In contrast, vectors from 293T-GIPZ cells contain 3 fold higher physical particles based on RNA copies and 100 fold higher physical particles based on p24 levels compared to infectious particles meaning they are 10 times more infectious compared to STAR produced vectors (supplementary Figure 8 and 9). Overall none of the titration assays showed that protein knock-down of AHNAK, TSG101 or ALIX had a significant, consistent impact in replicate sets of samples on vector particle production regarding infectious or physical particle numbers. In all assays the titer of the control ‘no shRNA’ varied between 0.5 and 2.0 fold of the titer of the scramble control with no significant difference in any of the experiments.
4. Discussion
The outcome of recent clinical trials shows that the demand for a large scale of LV production, e.g. in the form of a stable producer cell line, is increasing [2,3,25]. A better understanding of vector particle formation during a production cycle, specifically the interaction between host and viral proteins for particle assembly and budding would no doubt be beneficial for an improvement in vector production. In this study, using mass spectrometry (MS) we identified and compared host cell proteins associated with viral components of the transiently produced lentiviral vectors, pseudotyped with VSV-G envelope currently used in clinical trials to stably produced lentiviral vectors, pseudotyped with a non-toxic alternative envelope to VSV-G.
The most significant difference observed was that RDpro pseudotyped vectors contained substantially fewer protein species than VSV-G pseudotyped vectors, indicating that stable STAR-derived producer cells may provide a better production system than the conventional transient production system using VSV-G as a vector envelope, in terms of obtaining a higher purity and quality. A similar number of cellular proteins were detected in envelope free Gag/Pol samples to stably produced RDpro-pseudotyped vectors, indicating that VSV-G envelope proteins have a far higher potential than RD envelope proteins to interact or aggregate with host cell proteins. The larger number of identified cellular proteins is exclusive to SEC-purified samples that contain VSV-G envelope proteins and independent of LV production method, indicating that VSV-G proteins may form VSV-G vesicles of similar size to vector particles and thus could be eluted in SEC void peaks as full LV particles. VSV-G vesicles have been previously reported to be present in VSV-G pseudoyped lentiviral vectors [26]. The significantly smaller number of cellular proteins detected in RDpro pseudotyped vectors may suggest that in contrast to VSV-G proteins, RDpro envelope may form RDpro vesicles at a much lower levels or not at all, which may provide an explanation to the high purity of RDpro pseudotyped vectors observed in this study. It is however possible that differences in cell characteristics between 293T cells and STAR cells (clonal cells derived from 293T cells) is responsible for the difference in cellular protein co-purification. In order to confirm that envelope characteristics are dictating this difference, RDpro pseudotypes transiently produced from 293T cells may be analysed alongside. A method to eliminate VSV-G vesicles could be protease digest of vectors, e.g. using subtilisin, which has been used for protein digestion on the surface but not inside the HIV-1 virion [27] followed by sucrose-gradient centrifugation to separate microvesicles from vector particle samples [28].
To distinguish proteins associated with the viral envelope or potentially present microvesicles from proteins that are associated with the viral core, a transiently produced RDpro-pseudotyped vector sample should be analysed.
RDpro-pseudotyped vectors showed a 10 fold lower transduction efficiency. This has been shown in other experiments with both stable and transient LV productions using RD114 derived envelopes in this project as well as by others [15,24]. This could be due to less abundant RDpro envelope proteins on the vector surface or fewer envelope receptors for RDpro on host cells compared to VSV-G envelope proteins.
The predominant functions of MS-identified cellular proteins are cytoskeleton organisation, vesicular transport and cell death. Some of the identified proteins are known to assist in trafficking of viral proteins to assembly site e.g. actin, tubulin as well as budding from cell e.g. ALIX and Clathrin. Other detected proteins, such as MARCKSL1 and ENO, that have not been shown to function in HIV-1 assembly, could be associated with purified vectors due to a role in vector assembly and budding. The presence of proteins functioning in necrosis and apoptosis could be due to induced stress of vector production in the cells. Many of the proteins, such as proteins of the cytoskeleton or cytoskeleton regulators as well as proteins associated with vesicular trafficking and transmembrane proteins, that were identified in our study have been found in mass spectrometry analysis of various sucrose gradient purified enveloped viruses in other publications [16,[29], [30], [31], [32], [33]] suggesting similarities in assembly and budding processes of different enveloped viruses resulting in the association of similar cellular proteins, e.g. secretory pathway trafficking of envelope proteins. Taking into account the MS-results from this study using 293T cells and those from a a study on HIV-1 virions with a different cell type, MDMs, it suggests that at least a subset of host cell proteins is not randomly incorporated [16].
Knock down of gene expression of AHNAK, ALIX and TSG101 in producer cells in STAR-RDpro as well as 293T cells showed that infectious titers as well as physical titers of vectors were comparable to cells in which gene expression was not knocked-down; hence reduced protein levels did not have a measurable effect on LV production. One explanation could be that these cellular proteins do not influence LV assembly or budding in producer cells. Depletion of ALIX in HIV-1 infected cells by siRNA resulted only in a infectivity reduction of approximately 2 fold compared to about 17 fold reduction in infectivity after knock-down of ALIX in EIAV infected cells [34]. ALIX may only play a minor role in HIV-1 budding and may be more important in EIAV budding. Increasing the efficiency of TSG101 knock-down in 293T or STAR-RDpro cells used in our study could potentially result in a better knock-down reducing residual TSG101 mRNA levels. Alternatively, only a simultaneous knock-down of TSG101 and ALIX protein expression might result in reduced vector release and, as each protein binds to a different L-domain during viral assembly, PTAP [35] and YPXL [36].
For complete elimination of a specific protein, gene expression can be knocked out by using sequence specific nucleases such as transcription activator-like effector nucleases (TALENs) [37], clustered regularly interspaced short palindromic repeats (CRISRP) [38] or zinc finger nucleases (ZFNs) which can be delivered by viral vectors or directly by transfection [39], disrupting gene expression or resulting in dysfunctional protein expression. Knock-down of ALIX and TSG101 could potentially affect assembly and budding of VSV-G and RDpro-pseudotypes differently compared to wild type HIV-1 virions with the gp120 envelope protein [34]. Both, ALIX and TSG101 bind HIV-1 Gag L-domains suggesting that they recruit the viral protein to the assembly side, suggesting that binding of ALIX to p6-Gag compensates for any functional loss regarding virus release when TSG101 expression levels are reduced. This could also be the case vice versa, i.e. TSG101 could compensate in ALIX knock-down cell in vector particle budding.
Since there are a significant numbers of cellular proteins identified in this study, it is considered that the majority of these proteins may still be product of associated impurities rather than functional components. Therefore, the presence of these cellular proteins may compromise product safety and efficacy, may interfere in dose determination and potential related toxicity and unwanted immunogenicity to patients. Current protocols used for vector production and purification in clinical trials include steps that eliminate contaminants including DNA derived from transfected DNA plasmids or genomic DNA from lysed producer cells, using benzonase [40] as well as purification steps such as anion-exchange chromatography for the removal of serum proteins derived from cell culture medium or proteins derived from producer cells [41]. However these methods are not able to remove potentially present impurities that have similar physical properties to those of vector particles. Anion-exchange chromatography is used to separate vector particles from impurities based on the negative charge of vectors binding to the positively charged chromatographic supports such as diethylaminoethanol (DEAE) anion-exchangers [2,3,42], suggesting that protein complexes of similar charge could co-purify with vectors during AExc. Electron microscopy can be performed [26,43] to visualise the presence of potentisal microvesicles in purified vectors. To analyse if specific proteins are located on the surface or inside purified vector particles, immunogold staining of vectors can be used [21].
In conclusion, this study shows that 1) the cellular composition in Lentiviral products can vary significantly due to the pseudotyped envelopes, e.g. VSV-G vs RD114 pseudo-typed LV and the production system used, e.g. transient transfection vs stable production and 2) mass spectrometry analysis may be explored as one of the future characterisation methods for quality control of gene therapy products.
Funding
This work was funded by the UK Department of Health.
Author disclosure statement
MC and YT are inventors of the STAR packaging system. No competing financial interests exist for all other authors.
Acknowledgments
The Authors would like to thank Natalie Werling and Carl Dolman for their technical support.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.biologicals.2017.12.005.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
LC-MS/MS identified proteins listed in the order of their detection in MS set 1 of vector samples, then grouped into for example heat shock proteins, ribosomal proteins. UniProt accession number for each protein is given (http://www.uniprot.org/). Between 1 and 3 preparations per vector sample were analysed (MS sets). Identified proteins in RDpro-GFP and RDpro-Empty samples MS set 3 using the rejection method are indicated (‘set 3R’). Colour coding: a protein was detected in one (x), two (xx) or three runs (xxx) out of a total three runs of one set. Striped shading of RDpro Env in MS set 3R indicates only one peptide of this protein was detected at high confidence or 2 or more at low confidence.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proteins identified in all three samples containing VSV-G with UniProt identifier and cellular location are shown; proteins were detected in at least one MS sample, functional annotation taken from NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/gene/) or other sources as indicated.
{kind=link}
{kind=link}
Proteins identified in all five VSVG-G as well as RDpro-pseudotyped vector samples with UniProt identifier and cellular location; proteins were detected in at least one replicate of each vector sample, functional annotation taken from NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/gene/).
{kind=link}
Fig. S1.

LC-MS/MS Identified Proteins of the Top Five Canonical Pathways. A) Top five canonical pathways, at least 10 of the identified host proteins are associated to the listed pathways. Furthermore four proteins were identified out of 45 proteins that are associated with mechanisms of viral exit from host cells stored in the Ingenuity® Pathways Knowledge Base. B & C) Venn diagrams of proteins that are associated with at least one of the top five canonical pathways.
Fig. S2.

No effect on vector genome copy numbers in STAR-GIPZ vectors (reverse transcribed pHV RNA copies/ml vector harvest); four sets of samples were analysed. Quantification of reverse transcribed HV RNA copies in vector harvests and HV DNA plasmid standard using Q-RT-PCR is shown. Duplicate wells A and B for each sample; vector genome copies in vectors produced by STAR-GIPZ-AHNAK in well A of set I day 16 are 0.5 log higher compared to other samples, however this result did not repeat and was considered to be a result of variable.
Fig. S3.

No considerable effect of shRNA mediated knock-down of AHNAK and TSG101 on Gag precursor and p24 levels in STAR-GIPZ cells; western blot of STAR-GIPZ-cell lysates in four sets of samples showed comparable levels of p24 in three out of four set; equivalent loading of total protein was verified by western blotting of GAPDH, 10 μg of total protein of cell lysates per lane.
Fig. S4.

No consistent effect of shRNA mediated knock-down of AHNAK, ALIX and TSG101 on the number of vector genome RNA copies (HV) in 293T-GIPZ cells compared to 293T-GIPZ-scramble. Comparative quantification of HV RNA fold difference in cell lysates using Q-RT-PCR is shown. Results of vector genome RNA copy levels in two sets of samples; normalised to β-actin copies per cell; negative control 293T-GIPZ-scramble and ‘no shRNA’ are comparable.
Fig. S5.

No effect on vector genome copy numbers in STAR-GIPZ vectors (reverse transcribed pHV RNA copies/ml vector harvest); four sets of samples were analysed. Quantification of reverse transcribed HV RNA copies in vector harvests and HV DNA plasmid standard using Q-RT-PCR is shown. Duplicate wells A and B for each sample; vector genome copies in vectors produced by STAR-GIPZ-AHNAK in well A of set I day 16 are 0.5 log higher compared to other samples, however this result did not repeat and was considered to be a result of variable.
Fig. S6.

No consistent effect on viral protein p24 levels in STAR-GIPZ vectors; p24 in STAR-GIPZ vectors are shown grouped by STAR-GIPZ producer cells, p24 ng/ml of one well of vector harvests for each set was measured; in three out of four sets STAR-GIPZ-AHNAK vectors contained significantly higher numbers of vector particles compared to STAR-GIPZ-TSG101 and scramble produced vectors (p-value 0.0151 and 0.0035, respectively), this did not repeat in the fourth set of samples; no significant difference between STAR-GIPZ-AHNAK and ‘no shRNA’ control was measured.
Fig. S7.

No effect of shRNA mediated knock-down of host protein AHNAK and TSG101 on p24 levels in STAR-GIPZ vectors; western blot using anti-p24 monoclonal antibody, proteins p24 and pre-cursor were detected; Gag precursor levels in STAR-GIPZ-TSG101 cells are significantly higher in three out of four sets of samples compared to STAR-GIPZ-AHNAK and scramble control (15–31 fold compared to scramble) however comparable in the fourth set; 45 μl of vector harvest per lane.
Fig. S8.

STAR-GIPZ - ratio of RNA copies and p24 in vectors relative to infectious particles per ml of vector harvest.
Fig. S9.

293T-GIPZ - ratio of RNA copies and p24 in vectors relative to infectious particles per ml of vector harvests, samples, ND: not detected.
References
- 1.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science (New York, NY) 2013;341(6148):1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science (New York, NY) 2013;341(6148):1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 4.Lopes L., Dewannieux M., Takeuchi Y., Collins M.K. A lentiviral vector pseudotype suitable for vaccine development. J Gene Med. 2011;13(3):181–187. doi: 10.1002/jgm.1553. [DOI] [PubMed] [Google Scholar]
- 5.Yang H.G., Hu B.L., Xiao L., Wang P. Dendritic cell-directed lentivector vaccine induces antigen-specific immune responses against murine melanoma. Canc Gene Ther. 2011;18:370–380. doi: 10.1038/cgt.2011.13. England: 2011 Nature America, Inc. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez M., Sarry E., Bejanariu A., Casaban L., Abdalla S., Sabbah-Petrover E. Design and development of a new lentiviral based anti-HIV therapeutic vaccine. Retrovirology. 2012;9(Suppl 2) [Google Scholar]
- 7.Pang S., Pokomo L., Chen K., Kamata M., Mao S.H., Zhang H. High-throughput screening of effective siRNAs using luciferase-linked chimeric mRNA. PLoS One. 2014;9(5) doi: 10.1371/journal.pone.0096445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi J.G., Dang Y., Abraham S., Ma H., Zhang J., Guo H. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016;23(7):627–633. doi: 10.1038/gt.2016.27. [DOI] [PubMed] [Google Scholar]
- 9.Broussau S., Jabbour N., Lachapelle G., Durocher Y., Tom R., Transfiguracion J. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol Ther: J Am Soc Gene Ther. 2008;16(3):500–507. doi: 10.1038/sj.mt.6300383. [DOI] [PubMed] [Google Scholar]
- 10.Stewart H.J., Leroux-Carlucci M.A., Sion C.J.M., Mitrophanous K.A., Radcliffe P.A. Development of inducible EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther. 2009;16(6):805–814. doi: 10.1038/gt.2009.20. [DOI] [PubMed] [Google Scholar]
- 11.Stewart H.J., Fong-Wong L., Strickland I., Chipchase D., Kelleher M., Stevenson L. A stable producer cell line for the manufacture of a lentiviral vector for gene therapy of Parkinson's disease. Hum Gene Ther. 2011;22(3):357–369. doi: 10.1089/hum.2010.142. [DOI] [PubMed] [Google Scholar]
- 12.Ikeda Y., Takeuchi Y., Martin F., Cosset F.L., Mitrophanous K., Collins M. Continuous high-titer HIV-1 vector production. Nat Biotechnol. 2003;21(5):569–572. doi: 10.1038/nbt815. [DOI] [PubMed] [Google Scholar]
- 13.Relander T., Johansson M., Olsson K., Ikeda Y., Takeuchi Y., Collins M. Gene transfer to repopulating human CD34+ cells using amphotropic-, GALV-, or RD114-pseudotyped HIV-1-based vectors from stable producer cells. Mol Ther: J Am Soc Gene Ther. 2005;11(3):452–459. doi: 10.1016/j.ymthe.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Sanber K.S., Knight S.B., Stephen S.L., Bailey R., Escors D., Minshull J. Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep. 2015;5:9021. doi: 10.1038/srep09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell A.J., Fegen D., Ward M., Bank A. RD114 envelope proteins provide an effective and versatile approach to pseudotype lentiviral vectors. Exp Biol Med. 2010;235(10):1269–1276. doi: 10.1258/ebm.2010.010053. [DOI] [PubMed] [Google Scholar]
- 16.Chertova E., Chertov O., Coren L.V., Roser J.D., Trubey C.M., Bess J.W., Jr. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol. 2006;80(18):9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saphire A.C., Gallay P.A., Bark S.J. Proteomic analysis of human immunodeficiency virus using liquid chromatography/tandem mass spectrometry effectively distinguishes specific incorporated host proteins. J Proteome Res. 2006;5(3):530–538. doi: 10.1021/pr050276b. [DOI] [PubMed] [Google Scholar]
- 18.Ott D.E. Cellular proteins detected in HIV-1. Rev Med Virol. 2008;18(3):159–175. doi: 10.1002/rmv.570. [DOI] [PubMed] [Google Scholar]
- 19.Wheeler J.X., Jones C., Thorpe R., Zhao Y. Proteomics analysis of cellular components in lentiviral vector production using Gel-LC-MS/MS. Proteonomics Clin Appl. 2007;1(2):224–230. doi: 10.1002/prca.200600522. [DOI] [PubMed] [Google Scholar]
- 20.Denard J., Rundwasser S., Laroudie N., Gonnet F., Naldini L., Radrizzani M. Quantitative proteomic analysis of lentiviral vectors using 2-DE. Proteomics. 2009;9(14):3666–3676. doi: 10.1002/pmic.200800747. [DOI] [PubMed] [Google Scholar]
- 21.Segura M.M., Garnier A., Di Falco M.R., Whissell G., Meneses-Acosta A., Arcand N. Identification of host proteins associated with retroviral vector particles by proteomic analysis of highly purified vector preparations. J Virol. 2008;82(3):1107–1117. doi: 10.1128/JVI.01909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine B.L., Humeau L.M., Boyer J., MacGregor R.R., Rebello T., Lu X. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103(46):17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutner R.H., Zhang X.-Y., Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc. 2009;4(4):495–505. doi: 10.1038/nprot.2009.22. [DOI] [PubMed] [Google Scholar]
- 24.Strang B.L., Ikeda Y., Cosset F.L., Collins M.K.L., Takeuchi Y. Characterization of HIV-1 vectors with gammaretrovirus envelope glycoproteins produced from stable packaging cells. Gene Ther. 2004;11(7):591–598. doi: 10.1038/sj.gt.3302189. [DOI] [PubMed] [Google Scholar]
- 25.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pichlmair A., Diebold S.S., Gschmeissner S., Takeuchi Y., Ikeda Y., Collins M.K. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via toll-like receptor 9. J Virol. 2007;81(2):539–547. doi: 10.1128/JVI.01818-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ott D.E., Coren L.V., Johnson D.G., Sowder R.C., 2nd, Arthur L.O., Henderson L.E. Analysis and localization of cyclophilin A found in the virions of human immunodeficiency virus type 1 MN strain. AIDS Res Hum Retrovir. 1995;11(9):1003–1006. doi: 10.1089/aid.1995.11.1003. [DOI] [PubMed] [Google Scholar]
- 28.Ott D.E., Coren L.V., Kane B.P., Busch L.K., Johnson D.G., Sowder R.C., 2nd Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J Virol. 1996;70(11):7734–7743. doi: 10.1128/jvi.70.11.7734-7743.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu F.X., Chong J.M., Wu L., Yuan Y. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol. 2005;79(2):800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varnum S.M., Streblow D.N., Monroe M.E., Smith P., Auberry K.J., Pasa-Tolic L. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol. 2004;78(20):10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johannsen E., Luftig M., Chase M.R., Weicksel S., Cahir-McFarland E., Illanes D. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A. 2004;101(46):16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. Cellular proteins in influenza virus particles. PLoS Pathog. 2008;4(6) doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moerdyk-Schauwecker M., Hwang S.-I., Grdzelishvili V.Z. Analysis of virion associated host proteins in vesicular stomatitis virus using a proteomics approach. Virol J. 2009;6:166. doi: 10.1186/1743-422X-6-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin-Serrano J., Yarovoy A., Perez-Caballero D., Bieniasz P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci Unit States Am. 2003;100(21):12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrus J.E., von Schwedler U.K., Pornillos O.W., Morham S.G., Zavitz K.H., Wang H.E. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 36.Strack B., Calistri A., Craig S., Popova E., Göttlinger H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 37.Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S. Breaking the code of DNA binding specificity of TAL-type III effectors. Science (New York, NY) 2009;326(5959):1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 38.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaj T., Guo J., Kato Y., Sirk S.J., Barbas C.F., 3rd Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Br J Pharmacol. 2012;9(8):805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sastry L., Xu Y., Cooper R., Pollok K., Cornetta K. Evaluation of plasmid DNA removal from lentiviral vectors by benzonase treatment. Hum Gene Ther. 2004;15(2):221–226. doi: 10.1089/104303404772680029. [DOI] [PubMed] [Google Scholar]
- 41.Yamada K., McCarty D.M., Madden V.J., Walsh C.E. Lentivirus vector purification using anion exchange HPLC leads to improved gene transfer. Biotechniques. 2003;34(5):1074–1078. doi: 10.2144/03345dd04. 80–8, 80. [DOI] [PubMed] [Google Scholar]
- 42.Merten O.-W., Charrier S., Laroudie N., Fauchille S., Dugué C., Jenny C. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011;22(3):343–356. doi: 10.1089/hum.2010.060. [DOI] [PubMed] [Google Scholar]
- 43.Transfiguracion J., Jaalouk D.E., Ghani K., Galipeau J., Kamen A. Size-exclusion chromatography purification of high-titer vesicular stomatitis virus G glycoprotein-pseudotyped retrovectors for cell and gene therapy applications. Hum Gene Ther. 2003;14(12):1139–1153. doi: 10.1089/104303403322167984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LC-MS/MS identified proteins listed in the order of their detection in MS set 1 of vector samples, then grouped into for example heat shock proteins, ribosomal proteins. UniProt accession number for each protein is given (http://www.uniprot.org/). Between 1 and 3 preparations per vector sample were analysed (MS sets). Identified proteins in RDpro-GFP and RDpro-Empty samples MS set 3 using the rejection method are indicated (‘set 3R’). Colour coding: a protein was detected in one (x), two (xx) or three runs (xxx) out of a total three runs of one set. Striped shading of RDpro Env in MS set 3R indicates only one peptide of this protein was detected at high confidence or 2 or more at low confidence.
Proteins identified in all three samples containing VSV-G with UniProt identifier and cellular location are shown; proteins were detected in at least one MS sample, functional annotation taken from NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/gene/) or other sources as indicated.
Proteins identified in all five VSVG-G as well as RDpro-pseudotyped vector samples with UniProt identifier and cellular location; proteins were detected in at least one replicate of each vector sample, functional annotation taken from NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/gene/).