

Abstract
Background
Bioactive peptides, including biological sources-derived peptides with different biological activities, are protein fragments that influence the functions or conditions of organisms, in particular humans and animals. Conventional methods of identifying bioactive peptides are time-consuming and costly. To quicken the processes, several bioinformatics tools are recently used to facilitate screening of the potential peptides prior their activity assessment in vitro and/or in vivo. In this study, we developed an efficient computational method, SpirPep, which offers many advantages over the currently available tools.
Results
The SpirPep web application tool is a one-stop analysis and visualization facility to assist bioactive peptide discovery. The tool is equipped with 15 customized enzymes and 1–3 miscleavage options, which allows in silico digestion of protein sequences encoded by protein-coding genes from single, multiple, or genome-wide scaling, and then directly classifies the peptides by bioactivity using an in-house database that contains bioactive peptides collected from 13 public databases. With this tool, the resulting peptides are categorized by each selected enzyme, and shown in a tabular format where the peptide sequences can be tracked back to their original proteins. The developed tool and webpages are coded in PHP and HTML with CSS/JavaScript. Moreover, the tool allows protein-peptide alignment visualization by Generic Genome Browser (GBrowse) to display the region and details of the proteins and peptides within each parameter, while considering digestion design for the desirable bioactivity. SpirPep is efficient; it takes less than 20 min to digest 3000 proteins (751,860 amino acids) with 15 enzymes and three miscleavages for each enzyme, and only a few seconds for single enzyme digestion. Obviously, the tool identified more bioactive peptides than that of the benchmarked tool; an example of validated pentapeptide (FLPIL) from LC-MS/MS was demonstrated. The web and database server are available at http://spirpepapp.sbi.kmutt.ac.th.
Conclusion
SpirPep, a web-based bioactive peptide discovery application, is an in silico-based tool with an overview of the results. The platform is a one-stop analysis and visualization facility; and offers advantages over the currently available tools. This tool may be useful for further bioactivity analysis and the quantitative discovery of desirable peptides.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2143-0) contains supplementary material, which is available to authorized users.
Keywords: SpirPep, Genome, Bioactive peptides, In silico, Bioactive peptide discovery, GBrowse
Background
Bioactive peptides (BP) are protein fragments or peptides that play a significant role in human and animal health [1] and can be classified as endogenous; natural synthesis in the organisms or exogenous; by food-derived digestive processes using gastrointestinal (GI) enzymes in the GI-tract [2, 3]. These peptides can be found in many sources, such as dairy products [4], land animals [5], marine animals [6, 7], plants [8] and cyanobacteria [9]. The bioactivities of these peptides are classified according to their primary structure and ability to form products. The majority of peptides derived from food proteins are generated by enzymatic hydrolysis. Examples include “IQP”, an antihypertensive peptide from Spirulina platensis after digestion with alcalase from Bacillus licheniformis [10], and “YAEERYPIL”, an angiotensin I-converting enzyme (ACE) inhibitor and antioxidant peptide from the hydrolysate of ovalbumin with pepsin [11]. Although, enzymatic hydrolysis is a preferred method, several peptide-containing food products available in markets are generated from microbial fermentation. For example, sour milk and fermented milk which contain lactotripeptides such as isoleucine-proline-proline (IPP) and valine-proline-proline (VPP) [12, 13], were reported by European Food Safety Authority (EFSA) to have no significant effect in maintaining normal blood pressure [14]. However, other reports based on meta-analysis reported that IPP and VPP lactotripeptides could significantly reduce systolic blood pressure in Japanese subjects [13, 15].
Conventionally, the discovery of bioactive peptides is a time-consuming and costly process. The classical approach consists of enzyme selection, peptide production from protein hydrolysis, peptide-purification, peptide-identification, and in vivo or in vitro assays. Recently, the use of computational tools offers relief as they shorten time for the screening of peptide candidate prior to purification and the biochemical validation process in the laboratory. Currently available online tools can be classified into three groups: (i) in silico peptide digestion tools, e.g., PeptideCutter, a tool with a single protein sequence input, various enzymes and chemical options, can generate peptide sequences, allowing users to manually search for bioactivities against other databases [16], (ii) bioactive peptide prediction tools, e.g., PeptideLocator that directly predicts possible bioactive peptide sequence locations in protein sequences from the input of UniProt IDs [17] and PeptideRanker, which computes the bioactive peptide probability from input peptide sequence [18], and (iii) tools that combine in silico digestion and bioactive peptide prediction, e.g., mMass and BIOPEP tools. The mMass tool allows only one protein sequence and one enzyme as inputs, with an option of miscleavage [19], while BIOPEP tool allows one protein sequence and up to three enzymes as inputs, with an option to search against one bioactive peptide database [20]. Practically, a protein is hydrolyzed with a protease at the cleavage sites accord with cleavage rules, mentioned as ‘no miscleavage’. However, there is often found ‘miscleavage’, a lack of cleavage by the designated protease at one or both ends of the peptide, but cleavage at the other, resulting from an incomplete protein digestion (see Additional file 1: Figure S1) based on factors including protein structure, digestion technique, source of enzyme and enzyme kinetics [21–24]. The cleavable site(s) can be skipped due to configuration and sequence of amino acid residues leading to sterically inaccessible for the enzyme and/or slow kinetics [22], as example of Keil rule for miacleavage by trypsin [25].
Due to the advantages of bioinformatics tools, the in silico methodology is used for in silico peptide digestion and predicting bioactive peptides; for example, dipeptidyl peptidase-IV and angiotensin I-converting enzyme inhibitory peptides were identified by combining online in silico tools (BIOPEP, PeptideCutter tool and PeptideRanker) as an in-house tool [26]. However, these tools are incapable of the input of multiple protein sequences and have no miscleavage option.
In this work, we, therefore, develop a web-based application tool, SpirPep, which is a one-stop analysis and visualization pipeline for bioactive peptide discovery. Our pipeline is able to rapidly predict putative peptides by the in silico digestion of protein(s) up to a genome-wide level with the choice of 15 restriction enzymes and the number of miscleavages as input parameters. The resulting peptides are then searched against our in-house database collected from 13 public bioactive peptide databases for bioactive peptide identification. All peptides are kept temporarily in the database and are used to visualize protein-peptide alignment and display the region as well as details of the proteins and peptides listed separately for each input parameter by the Generic Genome Browser (GBrowse) [27]. In addition, we developed the GBrowse-based visualizer to generate a location overview of the bioactive peptides on their original protein, thus, it could provide potential decision-making information to assist users in re-designing the digestive systems, of which enzymes, miscleavages and proteins are considered to achieve the desirable bioactive peptide. The key performance comparison of the bioactive peptide identification tools is shown in detail in Table 1. Therefore, our SpirPep provides a shortcut for efficient screening and identifying target bioactive peptides and is applicable for other organisms of interest.
Table 1.
Key performance comparison of ‘SpirPep’ to other bioactive peptide identification tools
| Category | Bioactive peptide identification tool | Input | In silico peptide digestion | Bioactive peptide prediction | Result of in silico peptide digestion/bioactive peptide prediction tool | Accessibility | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bioactivity | No. of known bioactive peptide sequence | ||||||||||
| No. of protein per analysis | No. of Enzyme selection | No. of Miscleavage | Yes/No | Identification | Original protein track back | Resulting peptide categorized by enzyme | Protein-peptide alignment visualization by GBrowse | ||||
| In silico peptide digestion tool | PeptideCutter tool | 1 protein | 1–29 | N/A | No | N/A | N/A | N/A | √ | N/A | http://web.expasy.org/peptide_cutter/ |
| Bioactive peptide prediction tool | PeptideLocator | 1 Uniprot ID (≤10,000 aa) | N/A | N/A | Yes | N/A | Unknown | N/A | N/A | N/A | http://bioware.ucd.ie/~compass/biowareweb/Server_pages/biopred.php |
| PeptideRanker | Up to 150 peptides | N/A | N/A | Yes | N/A | Unknown | N/A | N/A | N/A | http://bioware.ucd.ie/~compass/biowareweb/Server_pages/peptideranker.php | |
| In silico peptide digestion and bioactive peptide prediction tool | mMass | 1 protein | 1 | As user input | Yes | √ | 612 sequences | N/A | N/A | N/A | Standalone version (http://www.mmass.org/download/) |
| BIOPEP | 1 protein | 1–3 | N/A | Yes | √ | 3587 sequences | N/A | N/A | N/A | http://www.uwm.edu.pl/biochemia/index.php/en/biopep | |
| SpirPep (In this study) | Thousands proteins or the whole genome | 1–15 | 0–3 | Yes | √ | 28,892 sequences | √ | √ | √ | http://spirpepapp.sbi.kmutt.ac.th/SpirPep/SpirPepTool | |
√ = Available; N/A = Not available
Construction and content
SpirPep was constructed as a web-based application tool for bioactive peptide discovery by the in silico peptide digestion of protein sequences derived from protein-coding genes such as single sequences, multiple sequences, and genome-wide scales. This tool consists of a database server and a web server for bioactive peptide discovery.
Databases
Four databases were constructed for storing data and putative peptides from each process as well as to integrate the in silico peptide results with bioactive peptide sequences retrieved from 13 public bioactive peptide databases, which contained both naturally synthesized and hydrolysed proteins (Table 2 and Fig. 1). The peptides were screened for bioactivity redundancy, thereby resulting in bioactive peptides with single or multi-functional bioactivity whose information source could be tracked. The first database, FrontendDB, stores queries from user input, which consists of user information, job title, enzyme and miscleavage of choice, and statistical analysis time (Fig. 1a). The second, CoreDB, contains the restriction site rules of the enzymes (by SpirPep default, 15 enzymes from the PeptideCutter tool [16]) and details the non-redundant bioactive peptide sequences from the retrieved bioactive peptide databases (Fig. 1b). The third, SpirPepApps, stores the input protein sequences and the putative peptide sequences from in silico peptide digestion, non-redundant peptide sequences, and the exact matching results from the comparison with retrieved bioactive peptide sequences (Fig. 1c). The fourth, GBrowseDB, stores the data generated in the GFF file format containing the matched results for protein-peptide alignment visualization (Fig. 1d).
Table 2.
Names and descriptions of online bioactive peptide databases (Accessed 11 January 2018)
| No. | Database name | Biological Function | Database Description | Last Update | Reference |
|---|---|---|---|---|---|
| 1. | APD | Antimicrobial | Antimicrobial and anticancer peptides | Jan 2018 | [31] |
| 2. | BACTIBASE | Antibacterial | Antibacterial peptides (Bacteriocins) | May 2017 | [32] |
| 3. | BAGEL3 | Antibacterial | Antibacterial peptides (Bacteriocins) | a/2018 | [33] |
| 4. | CAMP | Antimicrobial | Antimicrobial peptides and proteins | Apr 2010b | [34] |
| 5. | Defensins knowledgebase | Defensin, Antimicrobial | Antimicrobial peptides from the defensin family | Aug 2010b | [35] |
| 6. | EROP-Moscow | - | Endogenous Regulatory oligopeptides | Nov 2016 | [36] |
| 7. | Hmrbase | Hormone | Hormones & receptors | May 2009 | [37] |
| 8. | PenBase | Antimicrobial | Database of antimicrobial peptides from penaeid shrimps | Now not available (Jul 2008) | [38] |
| 9. | PeptideDB | Various bioactivities | Biologically active peptides, peptide precursors and motifs in Metazoa. | Apr 2008 | [39] |
| 10. | PhytAMP | Antimicrobial | Antimicrobial peptides and proteins of plant origin | Jan 2012 | [40] |
| 11. | RAPD | Antimicrobial | Recombinant antimicrobial peptides | Now not available (Mar 2010) | [41] |
| 12. | ACEpepDB | Antihypertensive | Food derived antihypertensive peptides that are available in the literature | Now not available (a/2011) | [42] |
| 13. | BIOPEP | Various bioactivities | Food proteins | a/2008b | [20] |
arepresents an unavailable month
bThe database is updated but not shown the date on the website
Fig. 1.
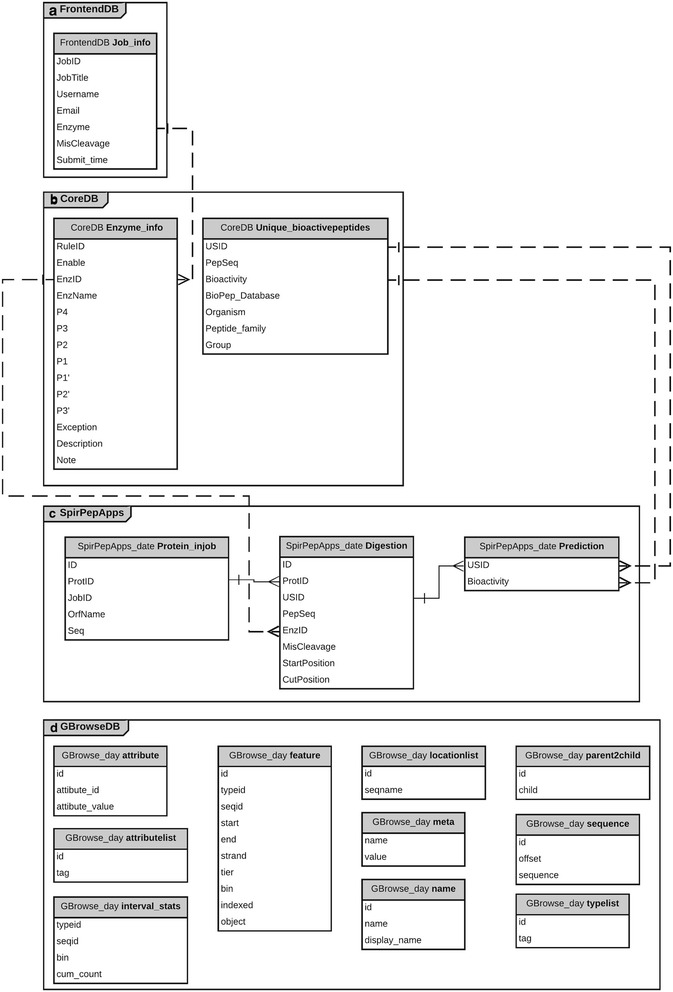
Database schematic diagram for all databases in the SpirPep web application: a FrontendDB, b CoreDB, c SpirPepApps and d GBrowseDB
To manage input and output data, we created both SpirPepApps and GBrowseDB by date and deleted the entire database information within 3 days due to storage space limitation.
SpirPep web application
The SpirPep web-based application tool has been developed to facilitate the use of SpirPep workflow (Fig. 2). It is useful for discovering bioactive peptides from protein-coding genes in genomes by in silico peptide digestion. It is a three-tier system containing front-end (left), queuing system (middle), and back-end (right) sites, as shown in the sequence diagram (see Additional file 2: Figure S2). The front-end site, which is written in PHP, accepts input queries from users, then encapsulates the user’s queries into jobs, and sends them through the back-end site. Using the PHP-Resque queuing framework on the Redis server [28], each Resque worker on the back-end server takes the job from the queue and proceeds into the SpirPep workflow.
Fig. 2.
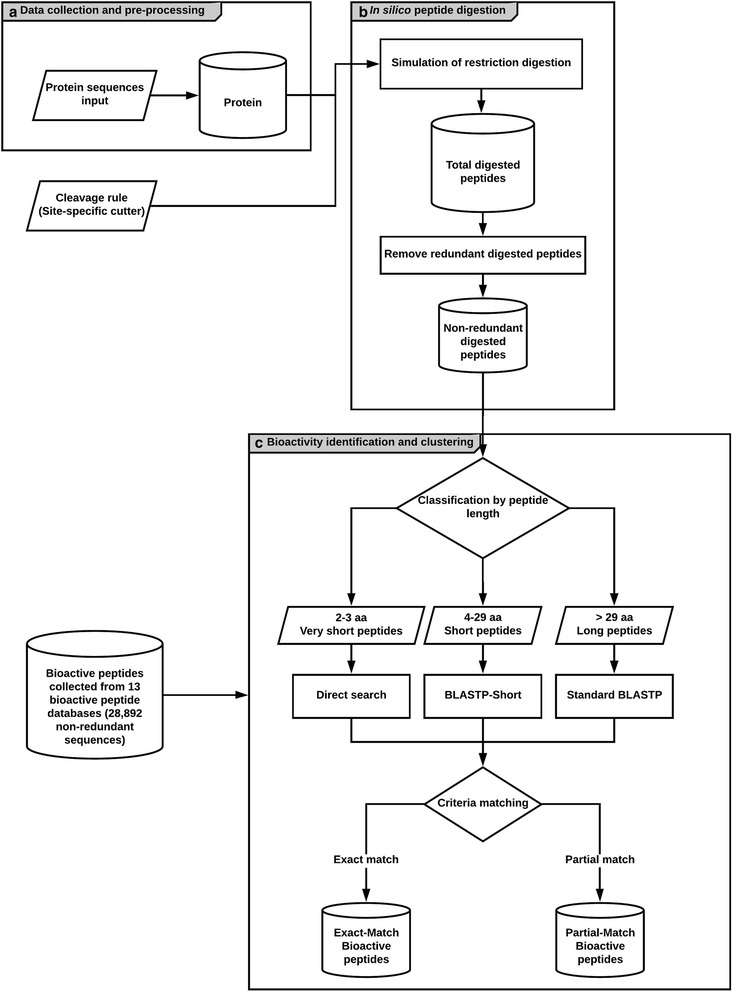
SpirPep workflow: This workflow is based on the in silico peptide digestion for bioactive peptide discovery. There were three modules: a data collection and pre-processing: the protein sequences were sent to the Protein database; b in silico peptide digestion: protein sequences were digested with the selected enzymes and the miscleavage number and non-redundant digested peptides were removed to classify them into three groups by peptide length (very short, short, and long peptides); and c bioactivity identification and clustering where these peptide groups were compared against the bioactive peptide sequences with the different methods with 100% in both identity and query or subject coverage
First, it invokes the in silico peptide digestion module with the given proteins and parameters (by default, using the enzyme trypsin with no miscleavage allowed). Then, all non-redundant digested peptides are compared against the in-house bioactive peptide database, which is gathered from both naturally synthesized and hydrolysed proteins (Table 2). The matched results are then generated in the GFF file format for protein-peptide alignment visualization. The completed results are next sent back to the front end in order to send a notification email to users with link to the results page. In the results page, the exact matched data, bioactive peptide sequences, and protein-peptide alignment visualization from SpirPepApps, CoreDB, and GBrowseDB databases, respectively, are retrieved (Additional file 2: Figure S2).
Utility
The SpirPep web application contains five parts: Home, SpirPep Tool, Bioactive Peptide Database, User Guide, and Contact Us.
Home; presents an introduction to bioactive peptide discovery, motivation of the computational method for bioactive peptide prediction and advantages of the SpirPep web application.
SpirPep Tool; here the user can input multiple protein sequences of interest, whole organism genome, or upload files in FASTA format with the required parameters. User replies are sent via email and displayed in the response page. Afterwards, SpirPep Tool predicts the candidate bioactive peptide sequences and then sends the output to the user by email.
Bioactive Peptide Database; stores the number and biological function of retrieved bioactive peptides from 13 online bioactive peptide databases and their respective tree maps, grouped by size (very short peptides with di- and tri-peptides, short peptides with 4–29 amino acids, and long peptides with more than 29 amino acids).
User Guide; assists user’s to familiarize themselves with the contents and functionalities embedded in the SpirPep Tool. It demonstrates the processes involved from inputting queries into SpirPep Tool and accessing the results from the email notification. Additional file 3: Figure S3 shows an example of all of the steps, which can be divided into three major parts: (i) “Query input” for inputting three protein sequences with the trypsin enzyme and the miscleavage limit set to one miscleavage (Additional file 3: Figure S3a); (ii) “Notification” after the submission as a response page and email with the submission information and email notification when the results are completed with the link to the two results pages (Additional file 3: Figure S3b); and (iii) “Result pages” for showing the screening results of candidate bioactive peptides from proteins and selected parameters (Additional file 3: Figure S3c). On the results page, the “List all” provides details of the bioactive peptides from all of the digested proteins. The example shows three di-bioactive peptide sequences (FK, QK and KK) from protein SPLC1_S010010 digested with the trypsin enzyme with zero and one miscleavage. These peptide sequences can be tracked back to their original proteins separated by a comma (,). The “Summary” presents the list and number of predicted bioactive peptides within the proteins sequences. The number can be linked to the bioactive peptide sequence information, as derived from bioactive peptide databases and organisms. The visualization page in GBrowse tool shows the position of the predicted bioactive peptides on an individual protein. The user-interface feature displays three graphical panels namely “Overview”, “Region,” and “Detail” providing access to the regions and detailed overview of the protein and peptide alignment. In the “Detail” view, the protein and derived peptide sequences from individual enzymes and miscleavages are organized into the “Protein” and “Enzyme name” tracks, respectively. The “Enzyme name” tracks are organized with different colours for convenient viewing, while an individual is divided into the miscleavage number as subtracks. The features are organized as glyphs with tracks and subtracks that display all of the peptide sequences with bioactivity as popup balloon tooltips. For this example, three derived peptide sequences (FK and QK from zero miscleavage and KK from one miscleavage) of SPLC1_S010010 protein are aligned on the “trypsin enzyme track” with the deep sky-blue colour and defined with the miscleavage number subtracks (Additional file 3: Figure S3c). In the case where more than one enzyme with the same protein sequence is produced, users can overview all enzyme tracks by selecting the enzyme(s) on “Select Tracks” to show the optional track in the “Browser”. The region can be scrolled and zoomed with the buttons in the header for overviewing the available peptide sequences along the proteins. For viewing all enzymes, users can consider re-designing the single digestion to double digestion to retrieve more desirable bioactive peptide sequences. The results are presented in tabular format, which users can copy, export as Excel, CSV, or PDF files, or even print by clicking the buttons in the table header and then search for the desired data and filter the results. Our bioinformatics tool allows users to quickly screen candidate bioactive peptides from a set of proteins before validation in laboratory.
Contact Us; provides the contact information of the SpirPep Team from KMUTT for consulting, suggestions, or usage problems.
Discussion
In comparison to other available bioactive peptide prediction tools (Table 1), SpirPep contains several unique features that differ from the online tools mentioned above. First, it has the capability for completing the entire process of in silico digestion and bioactive peptide prediction from multiple protein sequences or whole genome input, desirable enzyme selection with the miscleavage option and searching against in-house bioactive peptide databases (28,892 sequences) for bioactive peptide identification. Second, SpirPep output allows back-tracking of the resulting peptides to their original proteins, categorizes them by enzyme and miscleavage parameters, and performs protein-peptide alignment visualization by GBrowse, which presents alignment overview of derived peptide sequences on their original protein separately, based on the selected parameters with the bioactivity popup balloon tooltips and also helps in re-designing digestion for double digestion to meet user’s research needs. Moreover, the entire process of SpirPep takes less than 20 min for the digestion of 3000 proteins (751,860 amino acids) with 15 enzymes and three miscleavages for each enzyme or only a few seconds for single enzyme digestion. However, in silico peptide digestion tools are protein digestion simulations with selected parameters that may be different from in vitro digestion. From the structure of the Enzyme_info table of the CoreDB database (Fig. 1), we can easily modify the restriction site rules of enzymes that are commercially available and also add more rules for other enzymes. Our ongoing study focuses on the discovery of novel bioactive peptides by in vitro digestion using the enzyme obtained from SpirPep prediction which yields the peptides of interest.
For benchmarking, we compared SpirPep to another tool classified in the same category, BIOPEP (Table 1), using the same protein dataset, and selecting trypsin enzyme with no miscleavage allowed. Results showed the number of bioactive peptides identified by SpirPep were more than that of BIOPEP, although the generated peptides are less (Table 3 and Additional file 4: Table S1). The difference in the number of generated peptide sequences of each tool is in accord with the different cleavage rules. Cleavage sites of trypsin in ‘BIOPEP’ tool are at the C-terminal of lysine or arginine residues, whereas the cleavage rules in ‘SpirPep’ are referred from the PeptideCutter tool. In our tool, cleavage sites of trypsin are at the C-terminal of lysine or arginine residues with no proline at the C-terminal of lysine or arginine. However, this blocking of cleavage exerted by proline is negligible when methionine is at the N-terminal of arginine or tryptophan at the N-terminal of lysine with some exceptions. SpirPep identified more bioactive peptides on account of the ‘miscleavage’ option and the capacity of our in-house bioactive peptide database as shown in Tables 3 and 4. The generated peptides from BIOPEP were annotated with our in-house bioactive peptide database resulting in a higher number of identified bioactive peptides compared to that of BIOPEP, which demonstrates the capacity of our in-house bioactive peptide database. However, we found that prediction success was also dependent on the selected enzyme, for example, no bioactive peptide was found by trypsin; whereas for thermolysin digestion, bioactive peptides were found (Table 4).
Table 3.
Output comparison between SpirPep and BIOPEP, the identified bioactive peptides obtained from temperature stress – expressed protein [30] digested with trypsin and no miscleavage
| In silico peptide digestion tool | No. of peptide | No. of bioactive peptide (Percentage of No. of peptides) | |
|---|---|---|---|
| SpirPep | BIOPEP | ||
| SpirPep | 4333 | 418 (9.65) | 371 (8.56) |
| BIOPEP | 4556 | 450 (9.88) | 399 (8.76) |
Table 4.
Output comparison of identified bioactive peptides of genomic proteins of Arthrospira platensis strain C1 from SpirPep by selecting trypsin and thermolysin enzyme with miscleavage allowed up to three miscleavages against SpirPep in-house bioactive peptide and BIOPEP
| Selected enzymes | Miscleavage option | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |||||||||
| No. of peptide | No. of bioactive peptidea | No. of peptide | No. of bioactive peptidea | No. of peptide | No. of bioactive peptidea | No. of peptide | No. of bioactive peptidea | |||||
| SpirPep | BIOPEP | SpirPep | BIOPEP | SpirPep | BIOPEP | SpirPep | BIOPEP | |||||
| Trypsin | 99,905 | 298 (0.3%) | 247 (0.25%) | 135,899 | 48 (0.04%) | 39 (0.03%) | 137,309 | 1 (0%) | 1 (0%) | 133,145 | 0 (0%) | 0 (0%) |
| Thermolysin | 115,846 | 302 (0.26%) | 249 (0.21%) | 285,983 | 182 (0.06%) | 140 (0.05%) | 403,052 | 56 (0.01%) | 32 (0.01%) | 449,652 | 15 (0%) | 10 (0%) |
a The number in bracket is the percentage of bioactive peptide
Additionally, in our testing (unpublished data), 6108 Spirulina proteins were input into ‘SpirPep’, the enzyme thermolysin was selected, and up to three miscleavage was allowed (see example of our obtained result in Additional file 1: Figure S1b). The output showed that 1,379,992 peptides were obtained from three groups of peptides (very short, short and long peptides) with 572 bioactive peptides (0.04% of the total peptides), of 2–7 amino acids peptide length. For validation, the protein extracted from Spirulina cells grown at optimal condition was in vitro digested with thermolysin enzyme and sequentially isolated and analyzed by Liquid chromatography-tandem mass spectrometry (LC-MS/MS, Dionex UltiMate™ 3000 RSLCnano System (Thermo Fisher Scientific, Waltham, MA, USA) and MaXis II Mass Spectrometer Detector (Bruker, Germany)). The identified results from LC-MS/MS contained 3372 proteins (55.21% of the total genomic proteins) according to protein expression and 7366 peptides (0.53% of in silico peptides). The sample digested by thermolysin prior to LC-MS/MS were 6233 in common with the peptides found in the in silico ‘SpirPep’. Due to the limitation of LC-MS/MS detection, there are the peptides predicted by SpirPep but not present in the results from LC-MS/MS, which consist of only 5–24 amino acids peptide length. Hence, we could find only 76 (5–7 amino acids peptide length), but lost 496 (including di-, tri- and tetrapeptides) bioactive peptides. Fortunately, a pentapeptide (FLPIL, with a neuropeptide property) from LC-MS/MS (0.17% of the total in silico bioactive peptides, 572) was also detected in ‘SpirPep’ (1.32% of the in silico found, 76), but not in ‘BIOPEP’ (see additional related information (Table 3) for the output comparison between ‘SpirPep’ and ‘BIOPEP’).
Conclusions
SpirPep is a one-stop web application that offers rapid identification and efficient analysis of bioactive peptides with six key features: (i) genome-wide scale inputs, (ii) a miscleavage option, (iii) the output of a number of known bioactive peptides for bioactivity identification, (iv) the resulting peptide categorized by enzyme, (v) the original protein tracked, and (vi) a GBrowse-based visualizer. Hence, SpirPep is a promising alternative pipeline for efficient screening and identification of bioactive peptides and their proteins of origin.
Availability and requirements
Project name: SpirPep: A web-based database for bioactive peptide discovery. Project home page: http://spirpepapp.sbi.kmutt.ac.th. Operation system(s): Web based, Platform independent. Programming language: HTML, CSS, JavaScript, MySQL, PHP.
Additional files
Figure S1. Schematic diagram of miscleavage. a) Example of miscleavage occurrence, b) Example of peptides resulting of thermolysate of molecular chaperone DnaK (SPLC1_S010870) from LC-MS/MS at amino acid 1–26 and 123–134 (Our unpublished data). (TIFF 8492 kb)
{kind=link}
Figure S2. SpirPep sequence diagram: SpirPep was designed into a three-tier system (front-end, queuing, and back-end). The front-end sends the queries to SpirPepDB (FrontendDB and SpirPepApps) and responds to the users. The queries are queued by the Redis server and sent to Resque worker(s) in the back-end. When the analysis is complete, the system will send an email notification with the link to the results page to users. The results will be stored in the database temporary table and exported to the GFF file format, which can be internally used by the SpirPep visualizer. (JPEG 466 kb)
{kind=link}
Figure S3. Example of webpage snapshots from the SpirPep tool: a) query input from user submission (protein sequences, user’s name and email, job title, desired enzyme(s) and allowed miscleavage number), b) notification of received query from users as the response page and submission information email when the analysis complete, and c) results pages that contain the list all and summary pages, which show the predicted bioactive peptide sequences derived from input proteins and parameters. This provides a valuable decision to users for their re-designing the digestion system to obtain desirable bioactive peptides and enzymes before validation in a laboratory. (JPEG 2333 kb)
Table S1. Dataset and output comparison between SpirPep and BIOPEP, the identified bioactive peptides obtained from temperature stress – expressed protein [30] digested with trypsin and no miscleavage. (XLSX 18 kb)
Acknowledgements
We acknowledge Dr. Teeraphan Laomettachit and Dr. Weerayuth Kittichotirat for their suggestions and discussions along with Mr. Craig Robert Butler, Dr. Tenzan Eaghll and Mr. Oscar Nnaemeka from the School of Bioresources and Technology, KMUTT, for their editing and proofreading of the manuscript.
Funding
We would like to express our gratitude to Bioinformatics and Systems Biology HRD project (project No. P-11-01089), the National Center for Genetic Engineering and Biotechnology (BIOTEC), Thailand, and the National Research University Project, KMUTT (project No. 58000492) for the financial support and provision computational facilities for this study.
Availability of data and materials
The four databases, FrontendDB, CoreDB, SpirPepApps and GBrowseDB, were implemented on a MySQL database management system (MySQL Database Version 5.6.27-0ubuntu0.14.04.1 – (Ubuntu)). phpMyAdmin was used to manage the database via a web interface, and the webpages were based on the Apache system and PHP programming language [29]. The GBrowse tool was preferred to visualize the protein and peptide alignments from the GBrowseDB database. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CSS
Cascading style sheet
- CSV
Comma separated values
- GBrowse
Generic genome browser
- HTML
Hypertext markup language
- MySQL
My structured query language
Portable document format
- PHP
Hypertext preprocessor
- URL
Uniform resource identifier
Authors’ contributions
KA performed the bioactive peptide sequence data analysis. KA and JS designed the database scheme and web interface and implemented the web tools. AH, and MR were involved in designing the web interface. KA, MR, JS, and AH all contributed to writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2143-0) contains supplementary material, which is available to authorized users.
Contributor Information
Krittima Anekthanakul, Email: krittima.anek@mail.kmutt.ac.th.
Apiradee Hongsthong, Email: apiradee@biotec.or.th.
Jittisak Senachak, Email: jittisak.sen@biotec.or.th.
Marasri Ruengjitchatchawalya, Email: marasri.rue@kmutt.ac.th.
References
- 1.Sánchez A, Vazquez A. Bioactive peptides: a review. Food Qual Saf. 2017;1(1):29–46. doi: 10.1093/fqsafe/fyx006. [DOI] [Google Scholar]
- 2.Karra E, Chandarana K, Batterham RL. The role of peptide YY in appetite regulation and obesity. J Physiol. 2009;587(1):19–25. doi: 10.1113/jphysiol.2008.164269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonow RO. New insights into the cardiac natriuretic peptides. Circulation. 1996;93:1946–1950. doi: 10.1161/01.CIR.93.11.1946. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto N. Antihypertensive peptides derived from food proteins. Biopolymers. 1997;43(2):129–134. doi: 10.1002/(SICI)1097-0282(1997)43:2<129::AID-BIP5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 5.Meisel H, Walsh DJ, Murray BA, FitzGerald RJ. ACE inhibitory peptides. In: Mine Y, Shahidi F, editors. Nutraceutical proteins and peptides in health and disease. New York: CRC Press, Taylor and Francis Group; 2006. pp. 269–315. [Google Scholar]
- 6.Wang YK, He HL, Wang GF, Wu H, Zhou BC, Chen XL, Zhang YZ. Oyster (Crassostrea gigas) hydrolysates produced on a plant scale have antitumor activity and immunostimulating effects in BALB/c mice. Mar Drugs. 2010;8(2):255–268. doi: 10.3390/md8020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suarez-Jimenez GM, Burgos-Hernandez A, Ezquerra-Brauer JM. Bioactive peptides and depsipeptides with anticancer potential: sources from marine animals. Mar Drugs. 2012;10(5):963–986. doi: 10.3390/md10050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guang C, Phillips RD. Plant food-derived angiotensin I converting enzyme inhibitory peptides. J Agric Food Chem. 2009;57(12):5113–5120. doi: 10.1021/jf900494d. [DOI] [PubMed] [Google Scholar]
- 9.Welker M, von Döhren H. Cyanobacterial peptides – nature’s own combinatorial biosynthesis. FEMS Microbiol Rev. 2006;30(4):530–563. doi: 10.1111/j.1574-6976.2006.00022.x. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, Ren DF, Xue YL, Sawano Y, Miyakawa T, Tanokura M. Isolation of an antihypertensive peptide from alcalase digest of Spirulina platensis. J Agric Food Chem. 2010;58(12):7166–7171. doi: 10.1021/jf100193f. [DOI] [PubMed] [Google Scholar]
- 11.Dávalos A, Miguel M, Bartolomé B, López-Fandiño R. Antioxidant activity of peptides derived from egg white proteins by enzymatic hydrolysis. J Food Prot. 2004;67(9):1939–1944. doi: 10.4315/0362-028X-67.9.1939. [DOI] [PubMed] [Google Scholar]
- 12.Beltrán-Barrientos LM, Hernández-Mendoza A, Torres-Llanez MJ, González-Córdova AF, Vallejo-Córdoba B. Invited review: fermented milk as antihypertensive functional food. J Dairy Sci. 2016;99(6):4099–4110. doi: 10.3168/jds.2015-10054. [DOI] [PubMed] [Google Scholar]
- 13.Chanson-Rolle A, Aubin F, Braesco V, Hamasaki T, Kitakaze M. Influence of the lactotripeptides isoleucine–proline–proline and valine–proline–proline on systolic blood pressure in Japanese subjects: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2015;10(11):0142235. doi: 10.1371/journal.pone.0142235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA) Scientific opinion on the substantiation of a health claim related to isoleucine-proline-proline (IPP) and valine-proline-proline (VPP) and maintenance of normal blood pressure (ID 661, 1831, 1832, 2891, further assessment) pursuant to article 13(1) of regulation (EC) No 1924/2006. EFSA J. 2012;10(6):2715. [Google Scholar]
- 15.Chanson-Rolle A, Braesco V, Aubin F, Hamasaki T, Kitakaze M. Influence of the lactotripeptides isoleucine-proline-proline and valine-proline-proline on blood pressure in Asian subjects: a systematic review and meta-analysis of randomized controlled trials. PROSPERO 2014 CRD42014014322. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. New York: Humana Press; 2005. pp. 571–607. [Google Scholar]
- 17.Mooney C, Haslam NJ, Holton TA, Pollastri G, Shields DC. PeptideLocator: prediction of bioactive peptides in protein sequences. Bioinformatics. 2013;29(9):1120–1126. doi: 10.1093/bioinformatics/btt103. [DOI] [PubMed] [Google Scholar]
- 18.Mooney C, Haslam NJ, Pollastri G, Shields DC. Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS One. 2012;7(10):0045012. doi: 10.1371/journal.pone.0045012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niedermeyer THJ, Strohalm M. mMass as a software tool for the annotation of cyclic peptide tandem mass spectra. PLoS One. 2012;7(9):0044913. doi: 10.1371/journal.pone.0044913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minkiewicz P, Dziuba J, Iwaniak A, Dziuba M, Darewicz M. BIOPEP database and other programs for processing bioactive peptide sequences. J AOAC Int. 2008;91(4):965–980. [PubMed] [Google Scholar]
- 21.Chiva C, Ortega M, Sabidó E. Influence of the digestion technique, protease, and missed cleavage peptides in protein quantitation. J Proteome Res. 2014;13(9):3979–3986. doi: 10.1021/pr500294d. [DOI] [PubMed] [Google Scholar]
- 22.Šlechtová T, Gilar M, Kalíková K, Tesarová E. Insight into trypsin miscleavage: comparison of kinetic constants of problematic peptide sequences. Anal Chem. 2015;87(45):7636–7643. doi: 10.1021/acs.analchem.5b00866. [DOI] [PubMed] [Google Scholar]
- 23.Walmsley SJ, Rudnick PA, Liang Y, Dong Q, Stein SE, Nesvizhskii AIJ. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. J Proteome Res. 2013;12(12):5666–5680. doi: 10.1021/pr400611h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen CY, Russell S, Mendoza AM, Meyer-Arendt K, Sun S, Cios KJ, Ahn NG, Resing KA. Improving sensitivity in shotgun proteomics using a peptide-centric database with reduced complexity: protease cleavage and SCX elution rules from data mining of MS/MS spectra. Anal Chem. 2006;78(4):1071–1084. doi: 10.1021/ac051127f. [DOI] [PubMed] [Google Scholar]
- 25.Vandermarliere E, Mueller M, Martens L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom Rev. 2013;32(6):453–465. doi: 10.1002/mas.21376. [DOI] [PubMed] [Google Scholar]
- 26.Lafarga T, O’Connor P, Hayes M. Identification of novel dipeptidyl peptidase-IV and angiotensin-I-converting enzyme inhibitory peptides from meat proteins using in silico analysis. Peptides. 2014;59:53–62. [DOI] [PubMed]
- 27.Stein LD. Using GBrowse 2.0 to visualize and share next-generation sequence data. Brief Bioinform. 2013;14(2):162–171. doi: 10.1093/bib/bbt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haynes M. php-resque. 2013. [Google Scholar]
- 29.PHP Document Group. PHP Manual. In: Cowburn P, editor. 2018. http://php.net/manual/en/index.php. Accessed 27 Mar 2018.
- 30.Senachak J, Cheevadhanarak S, Hongsthong A. SpirPro: a Spirulina proteome database and web-based tools for the analysis of protein-protein interactions at the metabolic level in Spirulina (Arthrospira) platensis C1. BMC Bioinformatics. 2015;16:233. doi: 10.1186/s12859-015-0676-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44(D1):1087–1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammami R, Zouhir A, Le Lay C, Ben Hamida J, Fliss I. BACTIBASE second release: a database and tool platform for bacteriocin characterization. BMC Microbiol. 2010;10:22. doi: 10.1186/1471-2180-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Heel AJ, de Jong A, Montalbán-López M, Kok J, Kuipers OP. BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41(W1):448–453. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waghu FH, Barai RS, Gurung P, Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016;44(D1):1094–1097. doi: 10.1093/nar/gkv1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seebah S, Suresh A, Zhuo S, Choong YH, Chua H, Chuon D, Beuerman R, Verma C. Defensins knowledgebase: a manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007;35(Suppl 1):265–268. doi: 10.1093/nar/gkl866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamyatnin AA, Borchikov AS, Vladimirov MG, Voronina OL. The EROP-Moscow oligopeptide database. Nucleic Acids Res. 2006;34(Suppl 1):261–266. doi: 10.1093/nar/gkj008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rashid M, Singla D, Sharma A, Kumar M, Raghava GP. Hmrbase: a database of hormones and their receptors. BMC Genomics. 2009;10:307. doi: 10.1186/1471-2164-10-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gueguen Y, Garnier J, Robert L, Lefranc MP, Mougenot I, de Lorgeril J, et al. PenBase, the shrimp antimicrobial peptide penaeidin database: sequence-based classification and recommended nomenclature. Dev Comp Immunol. 2006;30(3):283–288. doi: 10.1016/j.dci.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 39.Liu F, Baggerman G, Schoofs L, Wets G. The construction of a bioactive peptide database in Metazoa. J Proteome Res. 2008;7(9):4119–4131. doi: 10.1021/pr800037n. [DOI] [PubMed] [Google Scholar]
- 40.Hammami R, Ben Hamida J, Vergoten G, Fliss I. PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009;37(Suppl 1):963–968. doi: 10.1093/nar/gkn655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Chen Z. RAPD: a database of recombinantly-produced antimicrobial peptides. FEMS Microbiol Lett. 2008;289(2):126–129. doi: 10.1111/j.1574-6968.2008.01357.x. [DOI] [PubMed] [Google Scholar]
- 42.Jimsheena VK, Gowda LR. Arachin derived peptides as selective angiotensin I-converting enzyme (ACE) inhibitors: structure-activity relationship. Peptides. 2010;31(6):1165–1176. doi: 10.1016/j.peptides.2010.02.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic diagram of miscleavage. a) Example of miscleavage occurrence, b) Example of peptides resulting of thermolysate of molecular chaperone DnaK (SPLC1_S010870) from LC-MS/MS at amino acid 1–26 and 123–134 (Our unpublished data). (TIFF 8492 kb)
Figure S2. SpirPep sequence diagram: SpirPep was designed into a three-tier system (front-end, queuing, and back-end). The front-end sends the queries to SpirPepDB (FrontendDB and SpirPepApps) and responds to the users. The queries are queued by the Redis server and sent to Resque worker(s) in the back-end. When the analysis is complete, the system will send an email notification with the link to the results page to users. The results will be stored in the database temporary table and exported to the GFF file format, which can be internally used by the SpirPep visualizer. (JPEG 466 kb)
Figure S3. Example of webpage snapshots from the SpirPep tool: a) query input from user submission (protein sequences, user’s name and email, job title, desired enzyme(s) and allowed miscleavage number), b) notification of received query from users as the response page and submission information email when the analysis complete, and c) results pages that contain the list all and summary pages, which show the predicted bioactive peptide sequences derived from input proteins and parameters. This provides a valuable decision to users for their re-designing the digestion system to obtain desirable bioactive peptides and enzymes before validation in a laboratory. (JPEG 2333 kb)
Table S1. Dataset and output comparison between SpirPep and BIOPEP, the identified bioactive peptides obtained from temperature stress – expressed protein [30] digested with trypsin and no miscleavage. (XLSX 18 kb)
Data Availability Statement
The four databases, FrontendDB, CoreDB, SpirPepApps and GBrowseDB, were implemented on a MySQL database management system (MySQL Database Version 5.6.27-0ubuntu0.14.04.1 – (Ubuntu)). phpMyAdmin was used to manage the database via a web interface, and the webpages were based on the Apache system and PHP programming language [29]. The GBrowse tool was preferred to visualize the protein and peptide alignments from the GBrowseDB database. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.