

Abstract
Background
Neuromuscular junction (NMJ) development is a multistep process mediated by coordinated interactions between the nerve terminal, target muscle, and perisynaptic Schwann cell that require constant back-and-forth communication. Retrograde and anterograde growth and differentiation factors have been postulated to participate in this communication. While neuregulin1 (NRG1) has been shown to be potent anterograde signal that activates acetylcholine receptor (AChR) transcription and clustering in vitro, its roles in NMJ development in vivo remain elusive.
Results
Using the model of chicken embryo, we measured the effects of NRG1 signaling during NMJ development in ovo using quantitative, sequential measures of AChR cluster size and density, pre- and post-synaptic apposition, and the alignment of perisynaptic Schwann cells. Using in ovo electroporation at early stages and a targeted soluble neuregulin antagonist through all developmental stages, we found soluble NRG1 regulates AChR cluster density and size at the earliest stage prior to nerve-AChR cluster contact. Once the nerve contacts with muscle AChRs, NRG1 has pronounced effects on presynaptic specialization and on the alignment of perisynaptic Schwann cells at endplates.
Conclusion
These findings suggest that, while NRG1 may not be critical for overall development, it appears to be important in fine-tuning pre-, post-, and peri-synaptic development of the NMJ.
Keywords: Neuregulin1, neuromuscular junction, acetylcholine receptor
Introduction
To build a functional neuromuscular junction, a precisely organized structure is required between motor neuron axons, muscle fibers, and perisynaptic Schwann cells in a highly dynamic and activity-dependent process (Sanes and Lichtman, 1999). This requires close communication between these different cell types using soluble and matrix-bound growth and differentiation factors that can signal bi-directionally (Loeb, 2003). One of these is neuregulin1 (NRG1). The NRG1 gene produces a large repertoire of growth and differentiation factors involved in both development and disease (Falls, 2003, Mei and Xiong, 2008). Most NRG1 isoforms are synthesized as transmembrane precursors to produce either soluble or membrane-bound forms through proteolytic shedding. A common feature shared by all isoforms is an EGF-like domain that is necessary to activate the EGF-family tyrosine kinase receptors ErbB2, ErbB3 and ErbB4. Soluble NRG1 forms have a unique, N-terminal heparin-binding domain (Loeb and Fischbach, 1995) that targets them to cell surfaces rich in heparan-sulfate proteoglycans (HSPGs) (Loeb et al., 1999, Loeb and Fischbach, 1995, Pankonin et al., 2005). NRG1-HSPG interactions have been shown to be important to produce sustained receptor signaling (Li and Loeb, 2001) and selective targeting of NRG1 to various cell types (Li et al., 2004, Ma et al., 2011).
In vitro, NRG1 has long been known to be a highly potent factor that induces transcription and membrane insertion of acetylcholine receptors (AChRs) in muscle (Falls, 2003). NRG1 is highly expressed in spinal motor neurons and sensory ganglia neurons shortly after their birth and is efficiently transported along the axon to reach endplates where it becomes highly concentrated in the synaptic basal lamina (Loeb et al., 1999). ErbB receptors are highly expressed in muscle and Schwann cells (Moscoso et al., 1995, Zhu et al., 1995, Trinidad et al., 2000, Adler et al., 2012). NRG1 has also been shown to work synergistically with agrin, another synaptic organizing proteoglycan, to increase AChR cluster size through direct interactions between NRG1’s heparin-binding domain and agrin (Li et al., 2004, Ngo et al., 2012).
The role of NRG1 in NMJ formation in vivo has been more elusive (Sanes and Lichtman, 2001). While motor neuron-derived NRG1 has been postulated to increase the expression of AChR subunit genes from subsynaptic muscle nuclei by activating ErbB receptors (Schaeffer et al., 2001), mice with disrupted NRG1-ErbB signaling by selectively knocking down ErbB2 and ErbB4 in muscle display roughly normal expression of AChRs and normal appearing NMJs (Escher et al., 2005). More recently, however, NRG1 has been shown to improve stability of postsynaptic AChRs at mature NMJs (Schmidt et al., 2011) and increase the size of AChR clusters after direct injection of NRG1 in muscle (Ngo et al., 2012).
More definitive roles for NRG1 in development have been shown for Schwann cell survival and differentiation (Ma et al., 2011). NRG1 has been shown to affect the highly specialized perisynaptic Schwann cell that has important roles in regeneration and participates in modulating synaptic activity at the NMJ (Trachtenberg and Thompson, 1997). Application of type II NRG1 results in perisynaptic Schwann cell process extension and migration away from synapses, producing subsequent nerve terminal retraction and regrowth. At late stages of development type III NRG1 is important for peripheral nerve myelination (Stassart et al., 2013, Nave and Salzer, 2006). We have recently shown distinct differences in the transcriptional regulation of type I versus type III neuregulin isoforms by neurotrophins and axon-target interaction that adds further diversity to this complex intercellular, regulatory machinery (Wang et al., 2015).
Here, we have developed quantitative measures of NMJ development to show multiple effects of soluble NRG1 at multiple stages of NMJ development in the chicken embryo by either increasing or reducing NRG1 function. Prior to nerve-muscle contact, NRG1 promotes AChR cluster density; and at later stages NRG1 promotes the apposition of pre- and post-synaptic nerve and muscle together with the ingrowth of the perisynaptic Schwann cell.
Results
Quantitative measures reveal sequential steps in neuromuscular junction development
Although there have been a number of studies on NMJ development in the chicken embryo focusing on the density and size of synaptic versus perisynaptic AChR clusters, few have detailed quantitative measures capable of assessing the effects of NRG1 (Burden, 1977, Dahm and Landmesser, 1988). Here, we developed three quantitative parameters to investigate NMJ development. The first measure includes both AChR cluster size and density using fluorescently labeled α-bungarotoxin (BTX). The other two measures quantify the degree of apposition of motor nerve endings and perisynaptic Schwann cells with AChR clusters. Fig. 1 shows the developmental time course for each of these 3 measures from embryonic day 7 (E7) through E18 of the hindlimb muscle posterior iliotibialis (PITIB), a focally innervated, homogeneous, fast-twitch muscle that receives, like most skeletal muscles in higher vertebrates, a focal innervation with an “en plaque” endplate (Dahm and Landmesser, 1988). We found that, at E7, the majority of AChR clusters are small (median size <2 μm2) and are not yet associated with nerve terminals and that AChR cluster size increases at the greatest rate between E7 and E10 with a slower rate of increase to around 15 μm2 by E18 (Figs. 1A, 1B, and 1D).
Figure 1. Quantitative analysis of NMJ development.

(A) Representative images of chicken E7 dorsal muscle mass of the hindlimbs and the PITIBs from E10 to E18 at a two-day interval show the growth in size of AChR clusters, and the apposition of AChR clusters (BTX, green) with nerve terminals (SV2, red). Scale bar =25μm. (B) The normalized distribution (percentage of the number of observations per bin of the total number within the group) of AChR cluster size and (C) the percentage of pre- and post-synaptic overlap are shown as a function of development from at least 200 NMJs. Arrows indicate the median occupancy. (D, E) The medians of the two measures (n=6, 6, 6, 5, 4, 4 for each stage, respectively) are plotted against developmental stages. (F) Representative images of the PITIBs from E12 to E18 at a two-day intervals show the ingrowth of Schwann cells (1E8, red) at the neuromuscular junctional area (BTX, green). (G) The median values of perisynaptic Schwann cell and postsynaptic AChR cluster appositions (n=4 for each stage, respectively) are plotted against developmental stages.
Pre- and post-synaptic apposition occurs at later stages, becoming significant between E10 and E12. By E18 most AChR clusters are fully associated with axonal terminals (Figs. 1A, 1C, and 1E). In the final stages of NMJ development, perisynaptic Schwann cells migrate into the NMJ starting at E14 and progressively engulf maturing NMJs through E18 (Figs. 1F, 1G). These successive stages of chicken NMJ embryonic development provide a series of quantitative measures to ask when and how NRG1 signaling might modulate the NMJ throughout development.
NRG1 in motor neurons regulates AChR cluster density at early stages
In the chicken hindlimb, the first appearance of AChR clusters is coincident with the ingrowth of nerve trunks at E4. By E7, numerous AChR clusters can be observed in a broad region on developing muscle fibers that are still not yet in direct contact with nerve endings (Fig. 1A). Previous studies have shown that those AChR clusters contacted by nerve terminals enlarge, while those that do not are eventually eliminated (Dahm and Landmesser, 1988). We used in ovo electroporation to over-express soluble NRG1 isoforms in lumbar motor neurons at E2.5 with a full-length type I proNRG1β1a cDNA with a c-Myc tag at the C terminus co-transfected with GFP, to track its distribution. Over-expression of NRG1 was confirmed by in situ hybridization and at the protein level using anti-c-Myc antibodies (Fig. 2A). Compared to controls electroporated with empty vector, over-expression of NRG1 in motor neurons resulted in a significant increase in AChR cluster density at E7. Morphometric analysis, however, did not show a significant change in AChR cluster size (Figs. 2C, 2D). Consistently, reducing NRG1 signaling using the plasmid expressing the NRG antagonist HBD-S-H4 in motor neurons led to a decrease of AChR cluster density, but not size. This antagonist is a fusion protein that delivers a dummy receptor decoy to the same heparan sulfate-rich surfaces which endogenous NRG1 binds since it is fused to NRG1’s own heparin-binding domain and has previously been used in ovo to suppress NRG1 meditated Schwann cell precursor survival and differentiation (Ma et al., 2011, Ma et al., 2009). Over-expression of NRG antagonist HBD-S-H4 was confirmed using antibodies against the fusion protein (Fig. 2B). This suggests that, prior to nerve-AChR cluster contact, for this muscle type, diffusible forms of NRG1 promote the total number of AChR clusters without regulating their size. Together, these results suggest that while NRG1 is not required for initial cluster formation or size, the amount of NRG1 released from nerve endings promotes the density of AChR clusters in the vicinity of the invading nerve.
Figure 2. Soluble NRG1 increases the density of AChR clusters.

(A) In situ hybridization shows increased levels of NRG1 mRNA in the ventral spinal cord (circled area) on the electroporated side but not the contralateral side. Scale bar =50μm. Over-expression of c-Myc tagged proNRG1 protein was demonstrated by c-Myc expression (red) in GFP (green)-positive cells. Scale bar =25μm. (B) Expression of HBD-S-H4 (antibody targeted to HBD, anti-HBD, red; antibody targeted to human type I NRG1, ADO3, blue) in GFP (green)-positive cells on the electroporated side only. Scale bar =25μm. (C) Increased AChR cluster (BTX, red) density was observed at E7 after electroporation with proNRG1 (GFP, green; 3A10, blue) compared with vector alone whereas overexpression of the NRG antagonist HBD-S-H4 led to decreased AChR cluster density. (D) Data are reported as the mean ± SEM of n = 8, 7, 6 for each condition, respectively; * p<0.05, **p<0.01, one-way ANOVA followed by Dunnett’s post hoc test by comparison to control. No detectable change of AChR cluster size was seen in the animals with either proNRG1 over-expression or HBD-S-H4 expression.
NRG1 signaling has minimal effects on AChR cluster size in later developmental stages
NRG1 expression initiates soon after motor neuron birth and increases throughout development in both chickens and mice (Loeb et al., 1999, Meyer et al., 1997). To determine the function of NRG1 at NMJ at later stages, we applied the NRG antagonist HBD-S-H4 for 2-day periods throughout development. To ask whether NRG1 plays roles in postsynaptic AChR clusters once nerve contact has been made, we examined the AChR cluster size in both the PITIB and anterior latissimus dorsi (ALD), a multi-innervated, slow-tonic muscle that possesses multiple ‘en grappe’ endplates along the muscle fibers (Hess, 1967, Kwong and Gauthier, 1987). The two muscle types provide unique opportunity to study how neuregulin affects NMJ development in chicken embryos. NRG antagonist HBD-S-H4 addition between E8-E10 led to a significant reduction of AChR cluster size on E10 ALD muscles. Compared with the control group with the mean value of 13.30 μm2, the AChR cluster size in NRG antagonist treated animals decreased to 11.85 μm2 (201 clusters from 4 animals (n=4) with saline control and 177 clusters from 4 animals (n=4) with NRG antagonist; p<0.05, Mann-Whitney U-test) (Figs. 3A, 3B). However, this effect was not observed in later stages of development in ALD muscles (E12-E18) or in PITIB muscles (E10-E18) (Figs. 3A, 3B and data not shown). Hence, NRG1 appears to contribute to AChR cluster size for the multi-innervated ALD muscle only during early development.
Figure 3. Reducing NRG1 decreases AChR cluster size early in development.

(A) Representative stacked confocal images show that treatment with the NRG antagonist HBD-S-H4 on E8 and E9 resulted in the reduction of AChR cluster size (BTX, green) in E10 ALD muscles. The same treatment at E10 and E11 did not affect the AChR cluster size on E12. Scale bar =25μm. (B) This histogram summarizes the normalized distribution (percentage of the number of observations per binned AChR cluster size of total number within the group) between Saline (control) and NRG antagonist treated embryos and shows a significant decrease in AChR cluster size on E10 (p<0.05, control n=201, NRG antagonist n=177, Mann-Whitney U-test), but not at E12 (p>0.05, control n=321, NRG antagonist n=336, Mann-Whitney U-test).
NRG1 signaling is required in presynaptic differentiation
In contrast to the subtle postsynaptic findings above, reducing NRG1 signaling at later stages had a greater effect on presynaptic NMJ development. Using the synaptic vesicle marker synaptic vesicle protein 2 (SV2), synaptic vesicles can be shown to be initially distributed throughout the axon, but become concentrated at synaptic sites when pre- and post-synaptic apposition consolidates (Fig. 1A). Suppressing NRG1 signaling between E12–14 decreased the normal concentration of SV2 at synaptic sites in ALD muscles compared to controls. The average percentage of extra-synaptic to total synaptic vesicle was 46.24 ± 5.14 (n=4) for control and 69.69 ± 2.87 (n=4) for the NRG antagonist treatment. A similar effect was observed between E14–16, but was less striking with an average extra-synaptic vesicle concentration of 43.85 ± 1.68 (n=4) for controls compared to 54.75 ± 4.22 (n=4) for NRG antagonist treatment. No effects of the antagonist were see between E16–18 with mean values of 36.73 ± 3.40 (n=4) and 35.89 ± 5.29 (n=4) for controls versus antagonist treatment, respectively (Figs. 4A, 4B). These results demonstrate a clear effect of NRG1 signaling on presynaptic maturation of NMJs. Given that ErbB receptors are concentrated postsynaptically, the observed effects presynaptically suggests an indirect action of NRG1 signaling mediated through postsynaptic-induced effects. Interestingly, unlike agrin knockout animals that showed marked presynaptic effects on nerve-fiber branching (Burgess et al., 1999, Gautam et al., 1996), the NRG antagonist treated animals did not show any significant changes in nerve branching pattern (data not shown).
Figure 4. Reducing NRG1 delays synaptic vesicle concentration at NMJs.

(A) Representative stacked confocal images show that treatment with NRG antagonist at E12 and E13, or E14 and E15 synaptic vesicles aggregation (SV2, red) in the axonal area distal to junction sites (BTX, green) in ALDs at E14 or E16, respectively. The synaptic vesicle distribution was not affected at E18 with the antagonist treatment from E16 to E17. Scale bar =25μm. (B) Quantification of the percentage of synaptic vesicles failing to concentrate in nerve endings over total synaptic vesicle staining shows significant increase of synaptic vesicle residing in the axonal area. Data are reported as the mean ± SEM of n=4 for both control and NRG antagonist group,* p<0.05, ** p<0.01, unpaired two-tailed t-test.
NRG1 signaling promotes pre- and post-synaptic apposition
In addition to the presynaptic maturation of terminal axons, the precise apposition of the presynaptic nerve terminals with postsynaptic AChR clusters is required to generate robust neurotransmission. This apposition increases steadily from E10 until most AChR clusters are fully associated with axonal terminals by E18 (Figs. 1A, 1B, and 1D). To ask whether NRG1 signaling effects pre- and post-synaptic apposition, we measured the overlap ratio of presynaptic SV2 and postsynaptic AChR clusters in PITIBs treated with and without NRG antagonist. While there was little effect at earlier stages, the antagonist significantly reduced the degree of pre- and post-synaptic apposition at E18 (361 clusters from 4 animals (n=4) with saline control and 334 clusters from 4 animals (n=4) with antagonist; p<0.05, Mann-Whitney U-test). The decrease of pre- and post-synaptic apposition was not observed in any other stages (E10-E16), as shown in Fig. 5, suggesting that NRG1 signaling is involved in fine-tuning the final stages of pre- and post-synaptic apposition when the NMJ matures.
Figure 5. Reducing NRG1 function decreases pre- and post-synaptic apposition late in development.

(A) Representative stacked confocal images of E18 PITIBs with or without NRG antagonist treatment on E16 and E17 show decreased degree of postsynaptic AChR clusters (BTX, green) territory association with nerve terminals (SV2, red). Scale bar =25μm. (B) The histogram summarizes the normalized distribution (percentage of the number of observations per binned AChR cluster size of total number within the group) between Saline (control) and NRG antagonist treated embryos and shows significant decrease of the association between AChR clusters territory and axonal terminals (p< 0.05, control n=361, NRG antagonist n=334, Mann-Whitney U-test). (C) Bottom panel shows zoomed-in images of the arrowed NMJ.
NRG1 signaling promotes perisynaptic Schwann cell engulfment
Once a stable, fully-functioning NMJ is formed, figure 1 shows that the last stage of development involves the encapsulation of the synapse with perisynaptic Schwann cells. These highly specialized cells are thought to be important for NMJ formation, maintenance of structural integrity, and signal transduction (Auld and Robitaille, 2003). Treatment with the antagonist significantly decreased the degree of Schwann cell alignment with AChR clusters at E14, E16, and E18 (Fig. 6). The mean value of percentage of AChR cluster area associated with Schwann cells decreased from 14.96 with Saline to 10.91 with NRG antagonist at E14 (p<0.0001, 584 AChR clusters across three animals (n=3), and 680 AChR clusters (n=3), Mann-Whitney U test); 27.66 versus 19.18 at E16 (p<0.0001, 489 AChR clusters (n=4), and 463 AChR clusters (n=4), respectively, Mann-Whitney U test); and 38.87 versus 29.96 at E18 (p<0.0001, 627 AChR clusters (n=4), and 571 AChR clusters (n=4), respectively, Mann-Whitney U test). Given the important role of NRG1 in Schwann cell development and myelination (Ma et al., 2011), it is not surprising that it also is important for perisynaptic Schwann cell development.
Figure 6. Reducing NRG1 affects perisynaptic Schwann cell alignment at NMJs.

(A) Representative epifluorescent images show that treatment with the NRG antagonist resulted in a reduced overlap between postsynaptic AChR clusters (BTX, green) and Schwann cells (P0, red) in PITIB muscles at E14, E16, and E18. Arrows denote representative individual NMJs at different stages. Scale bar =25μm. (B) The histogram summarizes the normalized distribution of the percent overlap of Schwann cell immunoreactivity for each AChR cluster (percentage of the number of observations per bin of total number within the group) between Saline (control) and NRG antagonist treated embryos and shows a significant decrease of the overlap of Schwann cell immunoreactivity overlying AChR clusters seen both as a reduction in AChR clusters with 100% overlap and an increase in clusters with no overlap (E14, E16, E18, p<0.0001, Mann-Whitney U-test).
Discussion
Effects of NRG1 on postsynaptic NMJ development
NMJ development is a highly regulated process that requires pre- and post-synaptic communication from the earliest stages, when the motor axon initially extends towards muscle targets, to later stages, when pre-, post-, and peri-synpatic components coalesce to form a precise synaptic structure leading to focal concentration of presynaptic vesicles with postsynaptic AChRs that are engulfed by a perisynaptic Schwann cell (Sanes and Lichtman, 1999). Here, we used a series of quantitative measures that implicate NRG1 as an important mediator that helps to bring these precise structures together at the appropriate stages of NMJ development (See summary of findings in Fig. 7).
Figure 7. A stage-dependent model of NRG1 signaling functions in NMJ development.
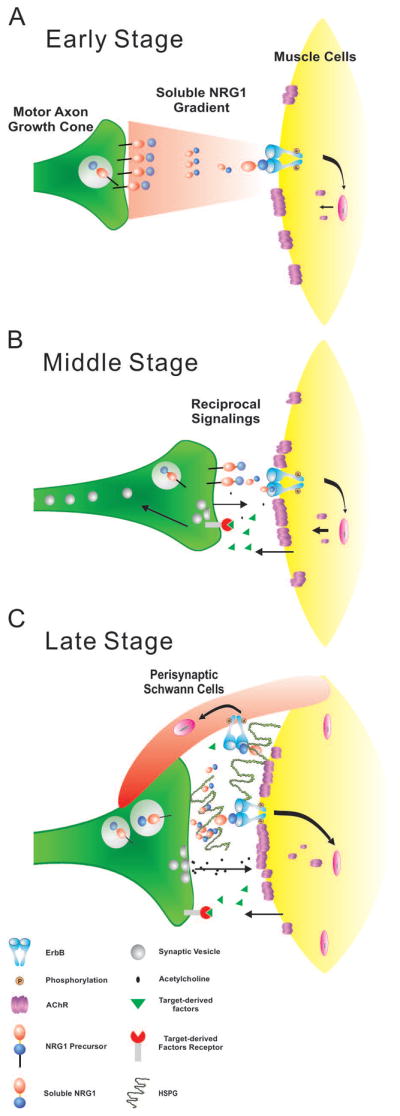
During early stages when motor axons first extend toward muscle targets soluble NRG1 induce AChR cluster density and in the ALD muscle also increase AChR cluster size. Once nerve muscle contact is made (middle stage), NRG1 supports the differentiation of the pre- and post-synaptic components of the NMJ leading to the proper alignment. In late stage NMJ development, NRG1 promotes the engulfment of the NMJ by perisynaptic Schwann cells. Previous studies suggest that this may be coincident with the deposition of heparan-sulfate proteoglycans (HSPGs) in the basal lamina providing a sustained source of NRG1 to the synapse (Loeb et al., 1999).
NRG1 is expressed in motor neurons and transported down motor axons shortly after their birth in the chicken at E4, however, immunoreactivity at the NMJ is only first detected in the synaptic basal lamina after E16, concurrent with the concentration of heparan-sulfate proteoglycans in the basal lamina that interact with soluble forms of NRG1 through its heparin-binding domain (Loeb et al., 1999, Loeb and Fischbach, 1995, Pankonin et al., 2005). At the earliest developmental time points, prior to synapse formation, NRG1 promotes both AChR cluster density and AChR cluster size, which varies by muscle type. Subsequently, when pre- and post-synaptic apposition occurs (after E10), NRG1 expression promotes the apposition of pre- and post-synaptic structures as well as the alignment of the perisynaptic Schwann cell at the NMJ once the synapse is nearly fully mature (by E18).
We found that over-expression of NRG1 in the hind limb muscle mass resulted in an increase of AChR cluster density, without regulating AChR cluster size. However, in the multi-innervated ALD muscle at later stages NRG1 had a modest effect on cluster size. Consistently, Ngo et al. reported that exogenous NRG1 increased the size of developing AChR clusters when injected into muscles of embryonic mice (Ngo et al., 2012). Schmidt et al. also reported that loss of neuromuscular NRG1-ErbB signaling destabilized anchoring of AChRs in the postsynaptic muscle membrane in adult animals (Schmidt et al., 2011). Despite the difference in model organisms, methods of drug administration, duration of drug action, and usage of agonist versus antagonist, all of these studies show that during early development and in maturity, NRG1 plays continuous roles in regulating AChR cluster size and density in vivo. This is not surprising, given the profound effects of NRG1 shown in vitro (Fischbach and Rosen, 1997). Previous studies showed different roles of NRG1-ErbB signaling in cultured myotubes as it can either potentiate AChR clustering (Ngo et al., 2004) or inhibit AChR aggregation (Trinidad and Cohen, 2004). In genetically modified mammalian models, it can either stabilize AChRs anchoring (Schmidt et al., 2011) or disrupt NMJ formation (Ponomareva et al., 2006). Here, we add to this literature showing that NRG1 also promotes AChR cluster size and density in vivo in different muscle types in the developing chickenin a way that would be difficult to achieve in rodent models. Exactly why there is this dissociation between NRG1’s effect on density, but not size at early developmental stages is not clear. One possibility is that prior to significant pre- and post-synaptic alignment NRG1 is free to diffuse and leads to new AChR cluster formation, independent of nerve contact. At later stages, the presence of nerve terminals with agrin brings together a dense heparan-sulfate matrix that keeps NRG1 concentrated at sites of nerve contact and could thus facilitate the formation of larger AChR clusters.
While the detailed downstream mechanisms remain to be determined, the changes observed appear directly linked to the level of neuronal NRG1 expression, Consistently, activity blockade during late embryonic development with curare leads to a lack of NRG1 protein expression at the NMJ, massive sprouting of new axons that reversed once activity is restored, and a significant reduction in AChR cluster size (Loeb et al., 2002). Interestingly, both NRG1 expression and pre- and post-synaptic structures could be maintained in the absence of activity by providing exogenous BDNF, suggesting a feedback loop between presynaptic NRG1 and postsynaptic (or perisynaptic) BDNF.
Another important neural-derived factor that promotes AChR clustering and NMJ development is agrin (Sanes and Lichtman, 2001, Glass et al., 1996). In fact, the absence of agrin, or either component of its receptor MuSK-Lrp4 complex has dramatic phenotypic changes leading to a failure of NMJ development (Gautam et al., 1996, Lin et al., 2001, Weatherbee et al., 2006, DeChiara et al., 1996). Agrin is a large proteoglycan that has both heparin and laminin-binding sites as well as alternatively spliced small domains that are critical for AChR clustering activity (Ruegg and Bixby, 1998). While the effects of NRG1 in vivo are more subtle than those of agrin, in vitro studies have demonstrated direct interactions between agrin and NRG1 that produce synergistic effects on AChR clustering (Li et al., 2004, Ngo et al., 2012). Thus, in ‘fine-tuning’ NMJ development, NRG1 works in concert with other factors to achieve this complex, yet important structure.
Indirect effects of NRG1 on presynaptic NMJ development
The NRG1 gene is highly complex with multiple alternatively spliced forms enabling it to have many functions both inside and outside the nervous system (Falls, 2003, Esper et al., 2006). Functionally, these forms can be subdivided into two groups: those that are secreted (types I/II) and those that remain tethered to the membrane (type III). Since NRG1 is primarily expressed in motor neurons and their axons (Loeb et al., 1999), the mechanisms by which NRG1 affects muscle and nearby Schwann cells is likely through these soluble forms that do not require direct cell-cell contact and can form gradients that surround developing motor axons (Ma et al., 2011). We have previously shown that the transcription (Wang et al., 2015) and release (Esper and Loeb, 2004, Ma et al., 2011) of soluble NRG1 forms are tightly regulated by postsynaptic- and Schwann cell-produced neurotrophic factors, including BDNF and GDNF, that produce a regulatory loop between the motor axons and these interacting cell types. At the NMJ, BDNF-TrkB signaling has been studied with well-characterized effects on neuromuscular synaptic structure and function (Pitts et al., 2006). We have also shown that this feedback loop is highly dependent on the degree of synaptic activity (Loeb et al., 2002). These findings provide a number of possible mechanisms to help explain why changes in NRG1 expression and activity has many diverse effects on both postsynaptic (muscle), perisynaptic (Schwann cell), and presynaptic (nerve terminal) components of the NMJ.
Specifically we found that down-regulating NRG1 signaling with a soluble, heparin-binding NRG antagonist led to significant presynaptic defects in vesicle accumulation and alignment with postsynaptic AChRs at NMJs. This could well be an indirect effect due to this feedback loop between neuron-derived NRG1 and target/perisynaptic Schwann cell-derived growth factors as described above. In addition to BDNF and GDNF, a similar role for presynaptic organization has been ascribed fibroblast growth factors (FGFs) (Fox et al., 2007), that could also be part of this indirect signaling mechanism.
Effects of NRG1 on the perisynaptic Schwann cell
NRG1 signaling plays important roles in Schwann cell migration, maturation, and myelination (Birchmeier and Nave, 2008, Ma et al., 2011). Part of these roles takes advantage of NRG1’s highly specific interactions with heparan-sulfate proteoglycans. A recent study showed that membrane-bound type III NRG1 in the mouse modulates synapse elimination and NMJ remodeling in the adult through its effects on perisynpatic Schwann cells (Lee et al., 2016). While prior to NMJ formation, the NRG antagonist is most likely suppressing soluble forms of NRG1, at later stages it may also suppress membrane-bound forms expressed on motor axons. Consistently, loss of NRG1 or ErbB receptors in Schwann cells results in Schwann cell death and loss of muscle innervation (Jaworski and Burden, 2006, Lin et al., 2000); and, following peripheral nerve injury, perisynaptic Schwann cells have NRG1-dependent functions that mediate regeneration of nerves and NMJ synapses (Fricker et al., 2011, Trachtenberg and Thompson, 1997).
A potential role of NRG1 in neuromuscular disorders
Understanding the mechanisms of how NRG1 regulates NMJ development could have potential therapeutic ramifications to recover from nerve injuries and neuropathies. In fact a recent study showed that in an animal model of Charcot-Marie-Tooth disease 1A (CMT1A), treatment using soluble NRG1 during early postnatal development effectively overcomes impaired peripheral nerve development and restores axon survival into adulthood (Fledrich et al., 2014). Emerging evidence also supports a potential role for NRG1 signaling is in the pathogenesis and progression of both chronic pain after nerve injury (Calvo et al., 2010) and amyotrophic lateral sclerosis (ALS) by activating microgila in the CNS (Song et al., 2012, Song et al., 2014). In the case of nerve injury, reducing NRG1 signaling after nerve injury using the same NRG antagonist used here (HBD-S-H4) significantly blocked both microglial activation and manifestation of chronic pain that followed.
Experimental Procedures
Chicken eggs and in ovo treatment
Fertilized chicken eggs were obtained from Michigan State University Poultry Farms and incubated in a Kuhl rocking incubator at 50% humidity. Daily treatments of the NRG antagonist (HBD-S-H4) on chicken embryos were performed as described previously (Loeb et al., 2002, Winseck et al., 2002, Ma et al., 2009, Ma et al., 2011). In brief, 20 μg of NRG antagonist HBD-S-H4 were each prepared in saline containing 0.2% BSA and added onto the chorioallantoic membrane through a small hole in the air sac without damaging underlying blood vessels for 2 consecutive days. HBD-S-H4 was prepared, purified, and tested for its ability to block NRG1 activity as described (Ma et al., 2011). Staging of chicken embryos was determined according to Hamburger-Hamilton (HH) stage series (Hamburger and Hamilton, 1951); E2.5 (Stage 15–16); E7 (stage 30–31); E8 (stage 34); E10 (stage 36); E12 (stage 38); E14 (stage 40); E16 (stage 42); E18 (stage 44).
In ovo electroporation
Chicken type I proNRG1β1a cDNA with a c-Myc tag at the C terminus was subcloned into the pMES vector downstream from the chicken β-actin promoter with IRES–EGFP (Krull, 2004). This promoter has been widely used for ventral horn transfection. While the promoter is not specific, by aiming the electroporation ventrally at E2.5 mostly motor neurons are transfected. Similarly, a c-Myc tag was added at the C terminus of the coding sequence of HBD-S-H4, which was subsequently subcloned into the pMES vector. The empty vector of pMES was used as control. The final concentration of each injection was 3 μg/μl. The plasmid DNAs were electroporated unilaterally into the ventral part of the neural tube at the lumbar level at E2.5 as described previously (Ma et al., 2009). Electrodes were placed ventrodorsal across the neural tube and pulsed for five times at 35 V for 50 ms with a square-wave pulse generator (Intracept TSS10; Intracel). Embryos were collected at indicated times and only those with strong GFP expression were processed for additional analysis.
In situ hybridization
Chicken embryos were fixed in 4% paraformaldehyde in PBS at 4°C overnight, washed in PBS, equilibrated in 30% sucrose, and mounted in OCT (Tissue-Tek). Frozen sections were cut transversely at 14 μm and placed on Superfrost plus slides (VWR). Non-radioactive in situ hybridization was performed using Digoxigenin (Dig)-labeled RNA probes. In brief, sense and antisense Dig-labeled RNA probes were generated from linearized plasmids, containing the chicken NRG1 heparin binding domain, by in vitro transcription (Roche). Probes were hydrolyzed to optimum length of ~250 bp, and then purified on Sephadex G-50 quick spin columns for RNA purification (Roche). Sections were treated for 10 min at room temperature with 1 μg/ml proteinase K (Sigma) and acetylated with 0.25% acetic anhydride in 0.1 M triethanolamine, pH 8.0 for 15 min. Afterward, the sections were incubated with 2 μg/ml probe in hybridization buffer at 60°C overnight. Hybridization buffer contained 50% formamide, 0.3 M NaCl, 0.5 mg/ml yeast tRNA, 1x Denhardt’s solution (Sigma), 20 mM Tris, pH 7.4, 10% Dextran Sulphate, 10 mM NaPO4 and 5 mM EDTA. After hybridization, the sections were sequentially washed in 5x SSC, 50% formamide in 1x SSC, 2x SSC, and 0.2x SSC for 10, 30, 20, and 20 min at 65°C sequentially. To remove excess cRNA, the sections were incubated with 20 μg/ml RNase A (Sigma) in TNE buffer (10 mM Tris pH 7.5, 0.5 M NaCl, 1 mM EDTA), at 37°C for 30 min before they were washed by 2x SSC. The sections then were blocked with Blocking solution (Roche) for 1 h at room temperature and incubated with sheep anti-DIG conjugated with alkaline phosphatase (AP) (1:1000) (Roche) in blocking solution at 4°C overnight. Finally, sections were washed in washing buffer (Roche), incubated in detection buffer (Roche), and then visualized by reacting with BM purple (nitroblue-tetrazolium-chloride (NBT)/5-bromo-4-chlor-indolyl-phosphate (BCIP) ready-to-use solution) (Roche) in the dark for 2 h.
Immunofluorescence staining
Muscle whole-mounts were prepared as described previously (Dahm and Landmesser, 1988, Loeb et al., 2002). Briefly, embryos were decapitated and eviscerated. Skin and connective tissue were removed to expose the underlying muscles. Anterior latisimusdorsi (ALD) muscles were exposed and fixed in situ by fresh 4% paraformaldehyde in PBS for 40 min to maintain normal muscle shape. Whole-mount staining was adapted from previously described (Dong et al., 2006). Briefly, muscles were incubated in 0.1 M glycine in PBS for 1 h, and rinsed with 0.5% Triton X-100 in PBS. After incubation in the blocking buffer (3% BSA, 5% goat serum, and 0.5% Triton X-100 in PBS) for 2 h at room temperature, muscles were then incubated with primary antibodies (SV2, 1:100; 3A10, 1:100; Developmental Studies Hybridoma Bank) and α-bungarotoxin conjugated with Alexa Fluro 488 (1:500, Life Technologies) in the blocking buffer overnight at 4°C. After washing three times for 1 h each in 0.5% Triton X-100 in PBS, the muscles were incubated with goat-anti-mouse IgG conjugated with Alexa Fluro 546 (1:500, Life Technologies) for 2 h at room temperature. After three times washing for 1 h each in 0.5% Triton X-100 in PBS, muscles were rinsed with PBS, and flat-mounted in mounting medium.
To obtain posterior iliotibialis (PITIB), thighs were fixed in 4% paraformaldehyde in PBS overnight and rinsed with PBS at room temperature, PITIB were then dissected off all connective tissue and the sciatic trunk. For PITIB and ALD after E14, muscles were mounted in OCT and sectioned into 20 μm sections. Frozen embryo sections were the same as used for in situ hybridization. After incubation with blocking solution (10% goat serum, 0.1% Triton X-100 in PBS) at room temperature for 1 h, sections were incubated with α-bungarotoxin conjugated with Alexa Flour 488 (1:500) and primary antibodies in blocking solution overnight at 4°C, followed by incubation with the corresponding goat-anti-mouse or goat-anti-rabbit IgG conjugated with Alexa Fluor antibodies for visualization. Primary antibodies were used at the following dilutions: P0 (1:5; 1E8), SV2 (1:100, SV2), c-Myc (1:100, 9E10) and neurofilament (1:10; 3A10) (Developmental Studies Hybridoma Bank); GFP (1:100; ab6662, Abcam).
Imaging and quantitative analysis
Confocal stacks of images of whole mount muscle staining were acquired with a z-step of 1.05 μm using a Nikon Eclipse microscope with a D-Eclipse C1 confocal system. 20 consecutive images were combined into a single image processed by MetaMorph image analysis software (Molecular Devices) using Maximum-Stack Arithmetic. Epifluorescent images were obtained with a Nikon Eclipse 600 epifluorescence microscope with a Princeton Instruments Micromax 5 MHz cooled CCD camera, and analyzed using MetaMorph. Quantitative, and when possible, blinded methods were developed so as not to introduce any bias. Uniformity across different sections and animals was maintained by manually adjusting the threshold for each nonsaturated image for the following types of measurements:
AChR Clusters. Regions of interest (ROIs) were selected as objects with a nearest-neighbor filter and a size filter for α-bungarotoxin signal above the threshold. The size and total grey value of AChR clusters were determined based on ROI measurements. AChR cluster size was converted from pixels into micrometers squared. To quantify the density of AChR cluster at early stages, the dorsal muscle mass regions with adjacent GFP+ motor axons were defined as the ROI. The number of AChR clusters and total grey value of each cluster were measured within each ROI. The density was calculated after pixel-to-area conversion.
Pre-, post-, and peri-synaptic Schwann cell apposition. The ratio of pre- and post-synaptic apposition was determined as the percentage of area positive for nerve terminal (antibody SV2) within each ROI. The ratio of perisynaptic Schwann cell apposition was determined as the percentage of area positive for Schwann cell P0 (antibody 1E8). Extra-synaptic synaptic vesicle signal was determined by subtraction of terminal SV2 positive areas that were overlapped by α-bungarotoxin signal, from the total signal for SV2. The percentage of extra-synaptic synaptic vesicle signal was calculated.
Statistical Analysis
Frequency histograms were produced in Prism 5.0 (GraphPad Software Inc.). Numerical datasets presented in histograms were tested for statistical significance by means of the nonparametric, two-sided Mann-Whitney U-test. For comparison between groups for in ovo electroporation, statistical differences were tested using one-way ANOVA followed by Dunnett’s post hoc test. Unpaired two-tailed t-tests were used for analyses between animals with and without antagonist treatment. The median was used to represent the measurement of each image. The mean of the medians of 3–5 images was used to represent each animal and subsequent data were presented as the mean ± SEM with 3–5 animals for each group. Statistical significance was presented as * p<0.05, ** p<0.01.
Key Findings.
We developed quantitative measures of postsynaptic, presynaptic, and perisynaptic (Schwann cell) development.
We used a novel neuregulin antagonist to probe the effects of neuregulin1 during multiple embryonic stages of chicken synaptic development.
Rather than an all-or-none effect, neureguiln1 appears to play multiple direct effects that ‘fine-tune’ muscle AChR clusters and Schwann cell ingrowth as well as indirect effects on apposition of presynaptic nerve terminals.
Acknowledgments
This work was supported by NIH Grant RO1 NS059947. The authors declare no competing financial interests.
Footnotes
Author Contributions:
JW designed research, performed research, analyzed data, and wrote the paper; FS and JL designed research, analyzed data, and wrote the paper.
Conflict of Interest:
The authors declare no competing financial interests.
References
- ADLER T, EISENBARTH I, HIRSCHMANN MT, MULLER-GERBL M, FRICKER R. Can clinical examination cause a Stener lesion in patients with skier’s thumb?: a cadaveric study. Clin Anat. 2012;25:762–6. doi: 10.1002/ca.22005. [DOI] [PubMed] [Google Scholar]
- AULD DS, ROBITAILLE R. Perisynaptic Schwann cells at the neuromuscular junction: nerve- and activity-dependent contributions to synaptic efficacy, plasticity, and reinnervation. Neuroscientist. 2003;9:144–57. doi: 10.1177/1073858403252229. [DOI] [PubMed] [Google Scholar]
- BIRCHMEIER C, NAVE KA. Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia. 2008;56:1491–7. doi: 10.1002/glia.20753. [DOI] [PubMed] [Google Scholar]
- BURDEN S. Development of the neuromuscular junction in the chick embryo: the number, distribution, and stability of acetylcholine receptors. Dev Biol. 1977;57:317–29. doi: 10.1016/0012-1606(77)90218-4. [DOI] [PubMed] [Google Scholar]
- BURGESS RW, NGUYEN QT, SON YJ, LICHTMAN JW, SANES JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- CALVO M, ZHU N, TSANTOULAS C, MA Z, GRIST J, LOEB JA, BENNETT DL. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J Neurosci. 2010;30:5437–50. doi: 10.1523/JNEUROSCI.5169-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAHM LM, LANDMESSER LT. The regulation of intramuscular nerve branching during normal development and following activity blockade. Dev Biol. 1988;130:621–44. doi: 10.1016/0012-1606(88)90357-0. [DOI] [PubMed] [Google Scholar]
- DECHIARA TM, BOWEN DC, VALENZUELA DM, SIMMONS MV, POUEYMIROU WT, THOMAS S, KINETZ E, COMPTON DL, ROJAS E, PARK JS, SMITH C, DISTEFANO PS, GLASS DJ, BURDEN SJ, YANCOPOULOS GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–12. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- DONG XP, LI XM, GAO TM, ZHANG EE, FENG GS, XIONG WC, MEI L. Shp2 is dispensable in the formation and maintenance of the neuromuscular junction. Neurosignals. 2006;15:53–63. doi: 10.1159/000094484. [DOI] [PubMed] [Google Scholar]
- ESCHER P, LACAZETTE E, COURTET M, BLINDENBACHER A, LANDMANN L, BEZAKOVA G, LLOYD KC, MUELLER U, BRENNER HR. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 2005;308:1920–3. doi: 10.1126/science.1108258. [DOI] [PubMed] [Google Scholar]
- ESPER RM, LOEB JA. Rapid axoglial signaling mediated by neuregulin and neurotrophic factors. J Neurosci. 2004;24:6218–27. doi: 10.1523/JNEUROSCI.1692-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESPER RM, PANKONIN MS, LOEB JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev. 2006;51:161–75. doi: 10.1016/j.brainresrev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- FALLS DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- FISCHBACH GD, ROSEN KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–58. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- FLEDRICH R, STASSART RM, KLINK A, RASCH LM, PRUKOP T, HAAG L, CZESNIK D, KUNGL T, ABDELAAL TA, KERIC N, STADELMANN C, BRUCK W, NAVE KA, SEREDA MW. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med. 2014;20:1055–61. doi: 10.1038/nm.3664. [DOI] [PubMed] [Google Scholar]
- FOX MA, SANES JR, BORZA DB, ESWARAKUMAR VP, FASSLER R, HUDSON BG, JOHN SW, NINOMIYA Y, PEDCHENKO V, PFAFF SL, RHEAULT MN, SADO Y, SEGAL Y, WERLE MJ, UMEMORI H. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–93. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- FRICKER FR, LAGO N, BALARAJAH S, TSANTOULAS C, TANNA S, ZHU N, FAGEIRY SK, JENKINS M, GARRATT AN, BIRCHMEIER C, BENNETT DL. Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci. 2011;31:3225–33. doi: 10.1523/JNEUROSCI.2568-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUTAM M, NOAKES PG, MOSCOSO L, RUPP F, SCHELLER RH, MERLIE JP, SANES JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–35. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- GLASS DJ, BOWEN DC, STITT TN, RADZIEJEWSKI C, BRUNO J, RYAN TE, GIES DR, SHAH S, MATTSSON K, BURDEN SJ, DISTEFANO PS, VALENZUELA DM, DECHIARA TM, YANCOPOULOS GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–23. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- HAMBURGER V, HAMILTON HL. A series of normal stages in the development of the chick embryo. J Morph. 1951;88:49–92. [PubMed] [Google Scholar]
- HESS A. The structure of vertebrate slow and twitch muscle fibers. Invest Ophthalmol. 1967;6:217–28. [PubMed] [Google Scholar]
- JAWORSKI A, BURDEN SJ. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1. J Neurosci. 2006;26:655–61. doi: 10.1523/JNEUROSCI.4506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRULL CE. A primer on using in ovo electroporation to analyze gene function. Dev Dyn. 2004;229:433–9. doi: 10.1002/dvdy.10473. [DOI] [PubMed] [Google Scholar]
- KWONG WH, GAUTHIER GF. Neuromuscular junctions in adult and developing fast and slow muscles. Anat Rec. 1987;219:409–19. doi: 10.1002/ar.1092190412. [DOI] [PubMed] [Google Scholar]
- LEE YI, LI Y, MIKESH M, SMITH I, NAVE KA, SCHWAB MH, THOMPSON WJ. Neuregulin1 displayed on motor axons regulates terminal Schwann cell-mediated synapse elimination at developing neuromuscular junctions. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1519156113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI Q, ESPER RM, LOEB JA. Synergistic effects of neuregulin and agrin on muscle acetylcholine receptor expression. Mol Cell Neurosci. 2004;26:558–69. doi: 10.1016/j.mcn.2004.04.009. [DOI] [PubMed] [Google Scholar]
- LI Q, LOEB JA. Neuregulin-heparan-sulfate proteoglycan interactions produce sustained erbB receptor activation required for the induction of acetylcholine receptors in muscle. J Biol Chem. 2001;276:38068–75. doi: 10.1074/jbc.M104485200. [DOI] [PubMed] [Google Scholar]
- LIN W, BURGESS RW, DOMINGUEZ B, PFAFF SL, SANES JR, LEE KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–64. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- LIN W, SANCHEZ HB, DEERINCK T, MORRIS JK, ELLISMAN M, LEE KF. Aberrant development of motor axons and neuromuscular synapses in erbB2- deficient mice. Proc Natl Acad Sci U S A. 2000;97:1299–304. doi: 10.1073/pnas.97.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOEB JA. Neuregulin: an activity-dependent synaptic modulator at the neuromuscular junction. J Neurocytol. 2003;32:649–64. doi: 10.1023/B:NEUR.0000020640.84708.35. [DOI] [PubMed] [Google Scholar]
- LOEB JA, FISCHBACH GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. Journal of Cell Biology. 1995;130:127–35. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOEB JA, HMADCHA A, FISCHBACH GD, LAND SJ, ZAKARIAN VL. Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci. 2002;22:2206–14. doi: 10.1523/JNEUROSCI.22-06-02206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOEB JA, KHURANA TS, ROBBINS JT, YEE AG, FISCHBACH GD. Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development. 1999;126:781–91. doi: 10.1242/dev.126.4.781. [DOI] [PubMed] [Google Scholar]
- MA Z, LI Q, AN H, PANKONIN MS, WANG J, LOEB JA. Targeting HER singaling with neuregulin’s heparin-binding domain. J Biol Chem. 2009 doi: 10.1074/jbc.M109.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA Z, WANG J, SONG F, LOEB JA. Critical period of axoglial signaling between neuregulin-1 and brain-derived neurotrophic factor required for early Schwann cell survival and differentiation. J Neurosci. 2011;31:9630–40. doi: 10.1523/JNEUROSCI.1659-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEI L, XIONG WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–52. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEYER D, YAMAAI T, GARRATT A, RIETHMACHER-SONNENBERG E, KANE D, THEILL LE, BIRCHMEIER C. Isoform-specific expression and function of neuregulin. Development. 1997;124:3575–86. doi: 10.1242/dev.124.18.3575. [DOI] [PubMed] [Google Scholar]
- MOSCOSO LM, CHU GC, GAUTAM M, NOAKES PG, MERLIE JP, SANES JR. Synapse-associated expression of an acetylcholine receptor-inducing protein, ARIA/heregulin, and its putative receptors, ErbB2 and ErbB3, in developing mammalian muscle. Developmental Biology. 1995;172:158–69. doi: 10.1006/dbio.1995.0012. [DOI] [PubMed] [Google Scholar]
- NAVE KA, SALZER JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- NGO ST, BALKE C, PHILLIPS WD, NOAKES PG. Neuregulin potentiates agrin-induced acetylcholine receptor clustering in myotubes. Neuroreport. 2004;15:2501–5. doi: 10.1097/00001756-200411150-00014. [DOI] [PubMed] [Google Scholar]
- NGO ST, COLE RN, SUNN N, PHILLIPS WD, NOAKES PG. Neuregulin-1 potentiates agrin-induced acetylcholine receptor clustering through muscle-specific kinase phosphorylation. J Cell Sci. 2012;125:1531–43. doi: 10.1242/jcs.095109. [DOI] [PubMed] [Google Scholar]
- PANKONIN MS, GALLAGHER JT, LOEB JA. Specific structural features of heparan sulfate proteoglycans potentiate neuregulin-1 signaling. J Biol Chem. 2005;280:383–8. doi: 10.1074/jbc.M402645200. [DOI] [PubMed] [Google Scholar]
- PEARSON RJ, JR, CARROLL SL. ErbB transmembrane tyrosine kinase receptors are expressed by sensory and motor neurons projecting into sciatic nerve. J Histochem Cytochem. 2004;52:1299–311. doi: 10.1177/002215540405201006. [DOI] [PubMed] [Google Scholar]
- PITTS EV, POTLURI S, HESS DM, BALICE-GORDON RJ. Neurotrophin and Trk-mediated signaling in the neuromuscular system. Int Anesthesiol Clin. 2006;44:21–76. doi: 10.1097/00004311-200604420-00004. [DOI] [PubMed] [Google Scholar]
- PONOMAREVA ON, MA H, VOCK VM, ELLERTON EL, MOODY SE, DAKOUR R, CHODOSH LA, RIMER M. Defective neuromuscular synaptogenesis in mice expressing constitutively active ErbB2 in skeletal muscle fibers. Mol Cell Neurosci. 2006;31:334–45. doi: 10.1016/j.mcn.2005.10.004. [DOI] [PubMed] [Google Scholar]
- RUEGG MA, BIXBY JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–7. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- SANES JR, LICHTMAN JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–42. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- SANES JR, LICHTMAN JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- SCHAEFFER L, DE KERCHOVE D’EXAERDE A, CHANGEUX JP. Targeting transcription to the neuromuscular synapse. Neuron. 2001;31:15–22. doi: 10.1016/s0896-6273(01)00353-1. [DOI] [PubMed] [Google Scholar]
- SCHMIDT N, AKAABOUNE M, GAJENDRAN N, MARTINEZ-PENA Y VALENZUELA I, WAKEFIELD S, THURNHEER R, BRENNER HR. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J Cell Biol. 2011;195:1171–84. doi: 10.1083/jcb.201107083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG F, CHIANG P, RAVITS J, LOEB JA. Activation of microglial neuregulin1 signaling in the corticospinal tracts of ALS patients with upper motor neuron signs. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:77–83. doi: 10.3109/21678421.2013.853802. [DOI] [PubMed] [Google Scholar]
- SONG F, CHIANG P, WANG J, RAVITS J, LOEB JA. Aberrant neuregulin 1 signaling in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2012;71:104–15. doi: 10.1097/NEN.0b013e3182423c43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STASSART RM, FLEDRICH R, VELANAC V, BRINKMANN BG, SCHWAB MH, MEIJER D, SEREDA MW, NAVE KA. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat Neurosci. 2013;16:48–54. doi: 10.1038/nn.3281. [DOI] [PubMed] [Google Scholar]
- TRACHTENBERG JT, THOMPSON WJ. Nerve terminal withdrawal from rat neuromuscular junctions induced by neuregulin and Schwann cells. J Neurosci. 1997;17:6243–55. doi: 10.1523/JNEUROSCI.17-16-06243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRINIDAD JC, COHEN JB. Neuregulin inhibits acetylcholine receptor aggregation in myotubes. J Biol Chem. 2004;279:31622–8. doi: 10.1074/jbc.M400044200. [DOI] [PubMed] [Google Scholar]
- TRINIDAD JC, FISCHBACH GD, COHEN JB. The Agrin/MuSK signaling pathway is spatially segregated from the neuregulin/ErbB receptor signaling pathway at the neuromuscular junction. J Neurosci. 2000;20:8762–70. doi: 10.1523/JNEUROSCI.20-23-08762.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J, HMADCHA A, ZAKARIAN V, SONG F, LOEB JA. Rapid transient isoform-specific neuregulin1 transcription in motor neurons is regulated by neurotrophic factors and axon-target interactions. Mol Cell Neurosci. 2015;68:73–81. doi: 10.1016/j.mcn.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEATHERBEE SD, ANDERSON KV, NISWANDER LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- WINSECK AW, CALDERO J, CIUTAT D, PREVETTE D, SCOTT SA, WANG G, ESQUERDA JE, OPPENHEIM RW. In vivo analysis of Schwann cell programmed cell death in the embryonic chick: Regulation by axons and glial growth factor. Journal of Neuroscience. 2002;22:4509–4521. doi: 10.1523/JNEUROSCI.22-11-04509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU X, LAI C, THOMAS S, BURDEN SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO Journal. 1995;14:5842–8. doi: 10.1002/j.1460-2075.1995.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]