

Abstract
Classical homocystinuria (HCU) is the most common inherited disorder of sulfur amino acid metabolism caused by deficiency in cystathionine beta-synthase (CBS) activity and characterized by severe elevation of homocysteine in blood and tissues. Treatment with dietary methionine restriction is not optimal, and poor compliance leads to serious complications. We developed an enzyme replacement therapy (ERT) and studied its efficacy in a severe form of HCU in mouse (the I278T model). Treatment was initiated before or after the onset of clinical symptoms in an effort to prevent or reverse the phenotype. ERT substantially reduced and sustained plasma homocysteine concentration at around 100 μM and normalized plasma cysteine for up to 9 months of treatment. Biochemical balance was also restored in the liver, kidney, and brain. Furthermore, ERT corrected liver glucose and lipid metabolism. The treatment prevented or reversed facial alopecia, fragile and lean phenotype, and low bone mass. In addition, structurally defective ciliary zonules in the eyes of I278T mice contained low density and/or broken fibers, while administration of ERT from birth partially rescued the ocular phenotype. In conclusion, ERT maintained an improved metabolic pattern and ameliorated many of the clinical complications in the I278T mouse model of HCU.
Keywords: cystathionine beta-synthase, homocysteine, preclinical studies, metabolomics, PEGylation, inborn error of metabolism, eye defect, alopecia, bone density, enzyme replacement
Graphical Abstract
Majtan et al. studied efficacy of a novel enzyme replacement therapy for homocystinuria using transgenic mouse model with a severe form of the disease. The treatment maintained and improved metabolic balance in the face of an unrestricted diet and ameliorated multitude of clinical symptoms, including the disease-characteristic ocular phenotype.
Introduction
Cystathionine beta-synthase (CBS; EC# 4.2.1.22) deficiency, also known as classical homocystinuria (HCU; OMIM# 236200), is a recessive inborn error of metabolism caused by mutation in the CBS gene encoding the enzyme, which catalyzes the condensation of homocysteine with serine to cystathionine.1 Homocysteine (Hcy) is a non-proteinogenic intermediary amino acid derived from the metabolism of the essential amino acid methionine (Met) in the methionine cycle2 with two metabolic fates. To conserve sulfur in the methionine cycle, Hcy can be converted back to Met via action of two independent enzymes: either universally distributed methionine synthase or liver- and kidney-localized betaine-homocysteine methyltransferase. Alternatively, Hcy enters the transsulfuration pathway, where it is irreversibly converted by CBS to cystathionine (Cth) followed by its hydrolysis into cysteine (Cys) by the action of cystathionine gamma-lyase (CGL). Consequently, the lack of CBS results in highly elevated Hcy and Met and low concentrations of Cth and Cys in plasma of HCU patients. This chemical imbalance leads to numerous clinical complications, including dislocation of the ocular lenses (ectopia lentis), connective tissue defects, thromboembolism, stroke, osteoporosis, and mental retardation.3 Limited treatment options are implemented based on a clinical assessment of disease severity. HCU exhibits a marked genotype/phenotype relationship. The most common panethnic I278T mutations, which accounts for almost 25% of all reported alleles, is responsible for a mild phenotype, which can be managed by up to a few hundreds of milligrams of vitamin B6 (pyridoxine) per day in humans, although not in mice.4, 5 Pyridoxine is a precursor of the catalytic cofactor of CBS, pyridoxal-5′-phosphate, and thus, such treatment results in stimulation of residual activity of some mutants. On the other hand, pyridoxine non-responsive mutations, such as the Spanish T191M or Irish G307S, usually confer a severe phenotype, which is managed by a combination of low-methionine/low-protein diet, methionine-free formula, and cystine and/or betaine supplementation.6, 7 The therapeutic goal is to normalize Hcy plasma concentration or, failing that, to reduce it to below 100 μM. It is also important that plasma Hcy concentration is kept stable, as concentrations above 100 μM threshold are often associated with severe clinical complications.8 These therapeutic approaches showed efficacy in small cohorts of HCU patients.9, 10 However, dietary compliance is particularly challenging in late-diagnosed patients or once patients reach adolescence, typically leading to impaired biochemical control and development of some clinical complications.8, 11
To address the core enzyme deficiency in HCU and alleviate the burdensome dietary requirements, we developed an enzyme replacement therapy (ERT) based on human CBS lacking its C-terminal regulatory domain and carrying the C15S mutation (htCBS C15S).12 When injected into a mouse model of HCU, htCBS was cleared rapidly, and there was little effect on plasma Hcy. However, modification of the enzyme with polyethylene glycol (PEG) led to a substantial increase in its half-life and corrected the underlying biochemical imbalance in the HCU mouse.12, 13 Additional in vivo evaluation and characterization of various PEG htCBS C15S conjugates yielded the most promising candidate with superior biological efficacy and suitable for commercial production.13 The lead candidate is an htCBS C15S conjugate modified with on average of five 20 kDa linear N-hydroxysuccinamide PEG moieties (henceforth referred to as PEG-CBS). When administered to severely affected CBS KO mice,14 PEG-CBS rescued neonatal mortality and prevented liver disease and osteoporosis.15
Here, we studied how the long-term administration of PEG-CBS affects biochemical biomarkers and clinical symptoms of HCU in the I278T mouse model.16 In contrast to the CBS KO mouse,14 I278T mice do not suffer from neonatal lethality or severe liver disease (despite exhibiting a similar biochemical profile to the KO mice). Instead, the disease typically manifests as facial alopecia, osteoporosis, and low fat content.16, 17, 18, 19 We began the treatment of the I278T mice either prior to the development of clinical symptoms or after they had become apparent, allowing us to assess the efficacy of PEG-CBS in preventing or reversing the phenotype. We correlated the clinical findings with multiple biomarkers in plasma and tissues, such as amino acid and aminothiol profiles, thioethers as markers of hydrogen sulfide biogenesis, liver enzymes, lipids, and various cytokines. We also report the evidence of ocular manifestations in I278T mice, which share some of the features with the ectopia lentis phenotype commonly observed in human HCU patients.
Results
Long-Term Treatment with PEG-CBS Results in Sustained Improvements in Plasma Sulfur Amino Acids
Clinical symptoms of the I278T mice, such as facial alopecia or osteoporosis, develop only gradually and, having developed, respond slowly to treatment. Consequently, potential treatment regimens must be capable of improving or normalizing the underlying metabolic imbalance over prolonged periods. Accordingly, we first tested if treatment with PEG-CBS was able to substantially correct the biochemical imbalance in I278T mice and whether any such improvement was sustained (Figure 1). In asymptomatic mice, treatment had begun at 3 weeks of age and continued for a further 9 months. This regimen resulted in a significant improvement in plasma Hcy, Cth, and Cys concentrations (p < 0.001 for all three metabolites) 24 hr after the first dose, which was maintained throughout the study (Figure 1A). Similarly, commencing the treatment in 6-month-old animals (which, by that point, already exhibited multiple disease symptoms) and continuing treatment for a further 6 months resulted in a correction of plasma metabolites that was sustained throughout the remainder of the study (Figure 1B). Taken together, regular subcutaneous (s.c.) injections of PEG-CBS yielded a sustained correction of plasma metabolite profile in the I278T mouse, permitting efficacy studies requiring long-term administration.
Figure 1.

Long-Term Treatment of the I278T Mice with PEG-CBS Results in a Sustained Improvement in Plasma Sulfur Amino Acid Profiles
Plasma concentrations of Hcy (solid line, squares), Cth (dashed line, circles), and Cys (dotted line, diamonds) are corrected in asymptomatic 21-day-old mice by three weekly s.c. injections of 7.5 mg/kg PEG-CBS (n = 3; A). The improvement is sustained over the subsequent ∼9-month treatment period. A similar, sustained improvement is observed in symptomatic 26-week-old mice (n = 10; B) treated with PEG-CBS for ∼6 months. For each metabolite, the initial data point represents the concentration prior to treatment, while subsequent data points represent concentrations of plasma metabolites 24 hr post-injection.
PEG-CBS Treatment Corrects Tissue Metabolites
In addition to improvements in plasma metabolites, administration of PEG-CBS resulted in correction of tissue metabolites in adult I278T mice. Figure 2 shows concentrations of sulfur-containing metabolites in liver, kidney, and brain homogenates from untreated and treated mice compared to those in negative controls. Non-protein-bound Hcy (Figure 2A) was markedly elevated in all the tissues of the untreated mice compared to both the negative controls and, more importantly, the treated mice. Hcy in tissues of the treated I278T mice was normalized to the concentration observed in negative controls. Interestingly, non-protein-bound Cys (Figure 2B) in tissue homogenates of the untreated mice was similar compared to both the treated mice and negative controls. As anticipated, Cth in tissues of untreated mice was diminished (Figure 2C). Treatment with PEG-CBS resulted in marked elevation of Cth in kidney homogenates, likely due to PEG-CBS activity and accumulation of Cth in plasma and subsequent clearance of Cth via glomerular filtration. Substantially lower elevation of Cth in liver and brain of treated mice is most likely caused by limited cellular uptake of Cth20 from blood compared to its massive elimination via kidney. Interestingly, tissue concentrations of Met and reduced glutathione (GSH) were found to be similar in all groups (Figures 2D and 2E). However, the S-adenosylmethionine/S-adenosylhomocysteine (SAM/SAH) ratio (Figure 2F) of the untreated mice was substantially reduced in tissues and, except liver, significantly ameliorated with the treatment close to the concentrations observed in negative controls.
Figure 2.

Administration of PEG-CBS to I278T Mice Normalizes or Improves Sulfur Metabolites in Tissues
Individual panels show tissue concentrations of non-protein-bound total homocysteine (Hcy; A), non-protein-bound total cysteine (Cys; B), cystathionine (Cth; C), methionine (Met; D), non-protein-bound total glutathione (GSH; E), SAM/SAH ratio (F), homolanthionine (Hlth; G), and lanthionine (Lth; H). Newborn I278T mice (n = 3, hatched bars) were treated from day 2 for up to ∼4 months of age with PEG-CBS (three times a week, s.c., 7.5 mg/kg) and compared to age-matched untreated positive (n = 3, white bars) and negative controls (n = 3, black bars). Columns that are significantly different from each other (p < 0.05) are indicated by having a different letter above the column, with no letter indicating non-significance.
We also determined thioethers homolanthionine (Hlth) and lanthionine (Lth) as emerging surrogate markers of H2S biogenesis.21, 22 The concentration of Hlth (Figure 2G) was markedly elevated in tissues of untreated mice, and the PEG-CBS administration resulted in significant correction in treated mice. Although improved, Htlh tissue concentrations remained significantly different from those in healthy controls. Interestingly, tissue concentration of Lth (Figure 2H) remained decreased compared to healthy controls in both untreated and treated I278T mice. Taken together, administration of PEG-CBS improved or restored metabolic balance in tissues of the treated I278T mice.
Administration of PEG-CBS Improves Liver Metabolism of Glucose and Lipids
In addition to sulfur-containing metabolites in liver, kidney, and brain, we complemented these results with liver nuclear magnetic resonance (NMR) metabolomics. Figures 3A and 3B shows selected liver metabolites of hydrophilic and lipophilic fractions, respectively. While liver concentrations of reduced GSH were found similar in all three groups (and thus correlating with values obtained using a different technique shown in Figure 2E), the significantly decreased total glutathione pool in untreated mice compared to controls was normalized by the treatment. Interestingly, glucose was found elevated in untreated mice and was normalized to negative control values by the treatment. On the other hand, the glycogen concentration moved in the opposite direction to that of glucose and thus was significantly reduced, while administration of ERT to I278T mice normalized glycogen entirely. As Figure 3B illustrates, the liver lipid metabolism of untreated mice was characterized by decreased concentration of multiple lipophilic compounds, such as triacylglycerides (TAG), polyunsaturated fatty acids (PUFA), total unsaturated fatty acids (TUFA), and total fatty acids (TFA). Treatment with PEG-CBS resulted in its normalization. Interestingly, liver cholesterol and phospholipids, such as phosphatidylcholine or phosphatidylinositol, were significantly different among groups (data not shown). Therefore, NMR metabolomics on livers showed that metabolism of glucose and lipids was substantially affected in I278T mice and that ERT normalized it.
Figure 3.

Administration of PEG-CBS Improves or Normalizes Disturbed Glucose and Lipid Liver Metabolism in I278T Mice
The hydrophilic (A) and lipophilic (B) metabolites were determined by NMR metabolomics in livers from mice treated from day 2 for up to ∼4 months of age with PEG-CBS (three times a week, s.c., 7.5 mg/kg; n = 3, hatched bars) and compared to age-matched untreated positive (n = 3, white bars) and negative controls (n = 3, black bars). (A) Reduced (GSH) and total glutathione, glucose, and glycogen, while (B) compares triacylglycerides (TAG), polyunsaturated fatty acids (PUFA), total unsaturated fatty acids (TUFA), and total fatty acids (TFA). Columns that are significantly different from each other (p < 0.05) are indicated by having a different letter above the column, with no letter indicating non-significance.
The I278T Mice Suffer from Disturbed Lipid Metabolism, Inflammation, and Oxidative Stress, but No Liver Damage
Similar disturbances in lipid metabolism as seen from the NMR metabolomics were confirmed in untreated mice by analysis of plasma lipids (Figure 4A). Total/high-density lipoprotein (HDL)-bound/low-density lipoprotein (LDL)-bound cholesterols were elevated, while triglycerides were markedly decreased in untreated mice. Long-term treatment resulted in a significant correction of plasma cholesterol fractions as well as triglycerides, which were essentially normalized to their concentrations detected in negative controls.
Figure 4.

Administration of PEG-CBS Improves or Normalizes Multiple Plasma Biomarkers of Lipid Metabolism, Liver Function, Oxidative Stress, and Inflammation in I278T Mice
Plasma lipids (A), biomarkers of liver function and oxidative stress (B), and various cytokines, chemokines, and growth factors (C) were determined in samples from ∼10–13-month-old I278T mice injected with PEG-CBS for a period of ∼6–9 months prior to the study end (three times a week, s.c., 7.5 mg/kg; n = 13, hatched bars) and compared to age-matched untreated positive (n = 13, white bars) and negative controls (n = 13, black bars). (A) Plasma concentrations of total, HDL-bound, and LDL-bound cholesterols and triglycerides. (B) Comparison of plasma activities of alanine and aspartate aminotransferases (ALT, AST), alkaline phosphatase (ALP), superoxide dismutase (SOD), and catalase (CAT). (C) Six out of 23 determined cytokines, chemokines, and growth factors: IL-12 (p40), IL-12 (p70), IL-13, G-CSF, MCP-1, and TNF-α. Columns that are significantly different from each other (p < 0.05) are indicated by having a different letter above the column, with no letter indicating non-significance. Two letters indicate that the column is not significantly different from the single-letter-designated columns.
Despite the clearly disturbed glucose and lipid liver metabolism, the plasma markers of liver function, such as alanine and aspartate aminotransferases (ALT, AST) and alkaline phosphatase (ALP), were similar to those in negative controls (Figure 4B). Treatment with PEG-CBS resulted in a significant decrease compared to the concentrations in healthy controls; however, the ALT, AST, and ALP plasma activities in all the analyzed samples were within the reference ranges for mouse. On the other hand, activities of liver antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT) in plasma of untreated mice were slightly elevated compared to negative controls, while PEG-CBS treatment resulted in their significant reduction. Increased plasma activities of SOD and CAT suggest the presence of oxidative stress.
We also determined the concentrations of a panel of 23 cytokines, chemokines, and growth factors. The subset showing variable expression between mouse groups is shown in Figure 4C. The most notable were marked elevation of IL-12 (p40), IL-13, and TNF-α in untreated mice, all of which were normalized with the treatment. These results suggest that the I278T mice suffer from an increased systemic inflammation.
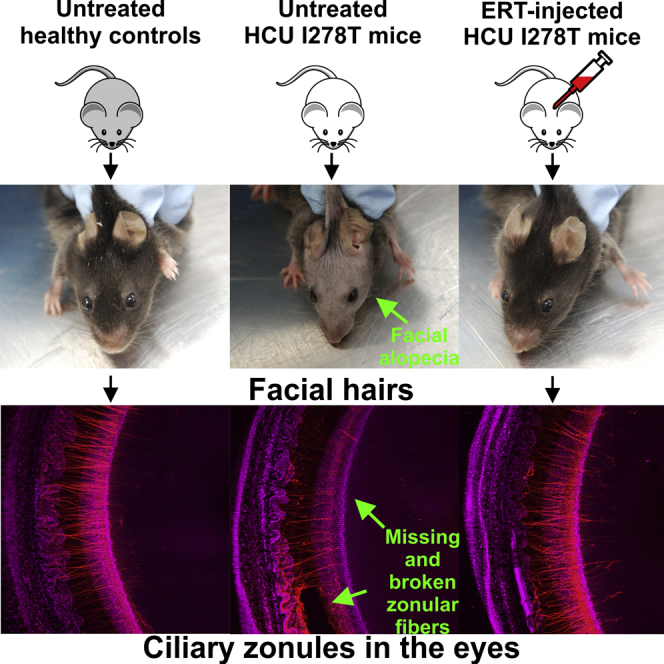
Administration of PEG-CBS Rescues Facial Alopecia
Facial alopecia, decreased glossiness, and patchy coat are characteristics of I278T mice,16 somewhat resembling the appearance of human HCU patients who often have fair, thin, and fragile hair. With successful correction of various biochemical markers with PEG-CBS treatment, we were interested to determine whether facial alopecia (which usually has an onset of 105–120 days in I278T mice17) might be prevented or reversed. Figure 5A shows normal facial hair in a negative control, while Figure 5B illustrates facial alopecia typical for an adult I278T mouse. Continuous treatment of I278T mice from weaning prevented alopecia entirely, resulting in facial hair indistinguishable from that of a negative control (compare mouse headshot at W35 in Figure 5C with that of the healthy control at W34 in Figure 5A). We were also interested whether the alopecia phenotype can be reversed once it has fully developed. The treatment of a mouse with fully developed facial alopecia resulted in regrowth of facial hair (Figure 5D), although the glossiness and thickness of the coat was less than seen in negative controls (Figure 5A). Taken together, treatment of I278T mouse with PEG-CBS prevents or reverses facial alopecia.
Figure 5.

Administration of PEG-CBS Rescues Facial Alopecia in I278T Mice
Headshots of representative mice from each study group are shown. (A) Untreated negative controls at 19 and 34 weeks of age (W19, W34). (B) Untreated positive controls with fully developed facial alopecia at age of 24 and 38 weeks. (C) Alopecia is prevented by continuous treatment of I278T mice from weaning age onward. (D) Fully developed facial alopecia is reversed by instigating continuous PEG-CBS treatment at 27 weeks.
The PEG-CBS Treatment Prevents Disruption of Zonular Fibers in the Eyes of I278T Mice
Myopia and ectopia lentis are characteristic clinical symptoms of human HCU.3 However, the occurrence of ocular phenotype has not been noted previously in mouse model of the disease. To determine if the structure of the ciliary zonule was compromised in I278T mice and, if so, whether treatment with PEG-CBS could prevent damage to the zonule, mice were treated from birth with PEG-CBS. We first confirmed that, as expected, the I278T mice suffered from sulfur metabolite imbalance compared to negative controls and that this was substantially corrected by PEG-CBS treatment (Figure 6A). Administration of PEG-CBS resulted in a marked decrease of plasma Hcy and normalization of plasma Cys and SAM/SAH ratio in treated versus untreated I278T mice.
Figure 6.

Administration of PEG-CBS Rescues the Structure of the Ciliary Zonule in the Eyes of I278T Mice
(A) Plasma concentrations of total homocysteine (Hcy), cystathionine (Cth), total cysteine (Cys), methionine (Met), and SAM/SAH ratio of the PEG-CBS-treated mice from 2 days of age up to ∼4 months of age (three times a week, s.c., 7.5 mg/kg; n = 3, hatched bars) compared to age-matched untreated mice (n = 3, white bars) and negative controls (n = 3, black bars). Columns that are significantly different from each other (p < 0.05) are indicated by having a different letter above the column, with no letter indicating non-significance. (B) A portion of the lens and adjacent eye wall imaged from the posterior aspect by confocal microscopy. In negative controls (left), zonular fibers (red) extend from the wall of the eye (white arrow) to attachment points near the lens equator (yellow arrow). Fibers then run for a few hundred micrometers across the lens surface toward the posterior pole. Untreated I278T mice (center) serve as positive controls. In these animals, zonular immunofluorescence is generally reduced, fiber density decreased, and fibers are absent from the posterior lens surface (orange arrow). In some regions, groups of fibers have broken (red arrow). In treated mice (right), staining intensity is largely restored, fiber integrity is preserved, and fibers are present on the posterior lens surface.
In 4-month-old negative control (+/−) animals, the zonule was visible as a meshwork of thin fibers that extended from the inner wall of the eye and attached to the lens near its equator (Figure 6B). From their initial attachment on the lens equatorial surface, zonular fibers ran for a few hundred micrometers along the posterior surface of the lens in the direction of the posterior pole. Thus, the arrangement of fibers in control animals was indistinguishable from that described previously for wild-type (WT; C57BL6) mice.23 The organization of the ciliary zonule was markedly disrupted in the eyes of untreated I278T mice. Fiber density was reduced, and the staining intensity of individual fibers was lessened considerably. In some areas, the fibers had broken completely. The projections of the fibers along the posterior lens surface were absent. In PEG-CBS-treated animals, fiber density and staining intensity were increased, although in only a few cases they were restored to the extent observed in the negative controls. The number and extent of posterior projections of the zonular fibers on the lens surface were also substantially increased over untreated positive controls. These results show that ERT substantially rescued the zonular structure of the I278T mice eyes.
Discussion
Our study was designed to determine whether enzyme replacement with PEG-CBS could reverse or prevent biochemical and clinical symptoms of CBS deficiency. The I278T mouse is a suitable model to address these issues, because it exhibits multiple symptoms including disturbed plasma concentrations of sulfur amino acids, low weight, low fat content, facial alopecia and thin fur, osteoporosis,16, 17, 18 and disruption of zonular fibers as shown in this study. As shown here, administration of PEG-CBS was broadly efficacious in preventing or reversing many of these phenotypes in I278T mice, in spite of the relatively low number of test subjects (n = 3) in some studies.
To alter the clinical course of a disease, potential therapies must be able to effectively correct the underlying biochemical imbalances over a sustained period. Among other metabolites, the lack of CBS activity in HCU results in markedly elevated Hcy and decreased Cys in plasma. Regular and long-term administration of PEG-CBS to I278T mice via s.c. injection resulted in substantially decreased Hcy (from over 300 μM to ∼100 μM) and normalized Cys plasma concentrations (from ∼120 μM to ∼250 μM), which were maintained for up to 9 months (i.e., during the entire course of the longest study presented here; Figure 1). Notably, despite the significant correction of all the assayed clinical symptoms, the treated mice still had elevated plasma Hcy compared to heterozygous controls (50–100 μM versus 12 μM). This finding is consistent with previous reports showing that the clinical symptoms of murine HCU are either less severe with lower plasma Hcy (∼180 μM) in CBS knockout (KO) mice expressing the human CBS WT transgene24, 25 or entirely prevented in I278T mice on low-methionine diet while still yielding elevated plasma Hcy (∼80 μM).18 Interestingly, while plasma Cys was decreased compared to controls in the former case (200 μM versus 290 μM), serum Cys was normalized in the latter (∼260 μM). Taken together, the data suggest that the severity of phenotype is proportional to plasma Hcy concentration, which, importantly, does not need to be normalized to achieve a significant positive impact on the phenotype. The plasma Hcy concentrations achieved by the treatment correspond with a limit of 100 μM recently recommended in guidelines for HCU management as the threshold level at which patients are unlikely to develop serious complications.8 More importantly, the improved plasma metabolic profile led to a correction of the metabolic balance in tissues (Figure 2). This correction was likely made possible by creating a concentration gradient, where the enzyme in bloodstream served as a metabolic sink. Not all the studied metabolites were affected by the disease and impacted by ERT to the same extent. As an example, Hcy in tissues was elevated in untreated mice and entirely normalized with the treatment. Met and Cys or GSH as important metabolites up- and downstream of the enzymatic block, respectively, were not significantly different in tissues of both the untreated and treated I278T mice compared to controls. Production of thioether Hlth and thus H2S21, 22 was substantially decreased in the treated mice, particularly in the liver compared to kidney and brain. The differential correction of Hlth could be explained by the amount of CGL activity in the studied tissues. CGL is a sole source of Hlth by condensing two molecules of Hcy.26 CGL is six times more abundant in liver than in kidney,27 while its activity in brain is over 100-fold lower than in liver.2 We showed previously that CBS does not form Hlth.22 Thus, the decreased availability of Hcy in PEG-CBS-treated mice might have a much bigger impact on CGL-catalyzed Hlth and H2S production in liver than in kidney and brain.
The reduction of an excessive H2S biogenesis from Hcy by CGL in treated mice could explain normalization of the glucose and glycogen metabolism in the liver (Figure 3A). It was shown that NaHS (an H2S donor) reduced glucose consumption and glycogen concentration in hepatocyte cells via decreased glucokinase activity.28 Insulin resistance induced by elevated Hcy may also contribute to the increase in glucose and decrease of glycogen in livers of the I278T mice.29 However, oral glucose tolerance test performed in HCU patients did not show signs of impaired endocrine pancreatic function.30 On the other hand, an association among CBS deficiency, Cys, and lipid metabolism is better established.31, 32 The CBS KO mice suffer from liver fibrosis and steatosis14 with severely elevated triglycerides in the liver.33 However, the livers of the I278T mice showed only slight to moderate focal steatosis.16 Our results are in contrast to those reports showing markedly decreased triglycerides and fatty acids in the livers of untreated mice (Figure 3B), further supported by an overall decreased fat content in these mice34. Decreased fat content in I278T mice was observed earlier17 and has been associated with a reduced expression of stearoyl-CoA desaturase-1 (SCD-1), a key enzyme involved in lipogenesis.35 Interestingly, plasma triglycerides, but not cholesterol correlated with findings in the livers (Figure 4B). Treatment of I278T mice with PEG-CBS completely normalized their plasma Cys, liver glucose, and glycogen, as well as triglycerides and fatty acids and plasma lipids. Similar effect on fat content was observed in I278T mice fed with methionine-restricted diet,18 but not with similar mice, whose standard diet was supplemented with N-acetylcysteine.17 Taken together, these results suggest that the normalization of Cys in I278T mice leads to correction of lipid metabolism only when accompanied with reduction in Hcy by either methionine-restricted diet or the ERT.
The enzyme replacement reversed or entirely prevented facial alopecia characteristic of the I278T mice.16 The mechanism responsible for hair loss in these mice is not understood and may relate to impaired keratin formation; however, this phenotype was not observed in other mouse models of HCU.14, 25
Connective tissue impairment is a classical complication of human CBS deficiency and includes osteoporosis, myopia, ectopia lentis, and marfanoid features in some patients. Decreased bone mass is a shared phenotypic trait of CBS KO mice,15 the I278T mice,24 and human patients.36 The PEG-CBS treatment completely reversed low bone mass in adult I278T mice characterized by reduced bone mineral density (BMD) and bone mineral content (BMC) compared to controls.34 Similar impact was observed after the I278T mice were fed with a methionine-restricted diet.18 On the contrary, betaine supplementation did not rescue bone mineralization of these mice37 suggesting that betaine as an Hcy-lowering agent often used by HCU patients may not be as effective as ERT or methionine restriction for rescuing osteoporosis.
Ophthalmic findings, particularly ectopia lentis and/or severe myopia, are among the most prevalent and characteristic clinical symptoms of HCU.38 The lens is suspended in the eye by fibers of the ciliary zonule. In species that accommodate, such as humans, the zonule transmits the forces that flatten the lens allowing the eye to focus on distant objects. Cys-rich fibrillin-1 accounts for approximately 70% of the zonule by mass, although accessory proteins such as microfibril associated protein-1 (MAGP1) and Ltbp2 are also relatively abundant.39 The disintegration of zonular fibers is the underlying cause of lens dislocation,40 but the precise pathogenic mechanism remains unclear. Here, we showed that zonular fibers are less numerous in I278T mice and prone to breakage. Treatment of I278T mice from birth with PEG-CBS largely ameliorated this phenotype (Figure 6B). Normalized Cys metabolism in treated animals may serve to stabilize the conformation of the bundles of fibrillin-rich microfibrils that constitute the zonular fibers. This is an encouraging result, as similar outcomes might be expected in HCU patients. However, studies on animals suggest that zonule synthesis is probably completed during early childhood. If this is also true in humans, then it is unlikely that ERT would restore sight in individuals who have already developed ocular symptoms and might argue that treatment should be initiated as early as possible, before the onset of ocular symptoms.
In conclusion, enzyme replacement with PEG-CBS successfully corrected a range of symptoms manifesting in the I278T mouse model, despite imperfect metabolic balance. Some symptoms, such as low bone mass and damage to the ciliary zonule, are shared with HCU patients, and we hope that once PEG-CBS reaches the clinic, it may have a similarly beneficial effect on those aspects of the human disease. At present, it is unclear how the ERT will be immunologically tolerated by patients, what will be the efficacious dose and route of administration in humans, whether there will be any safety or toxicity concerns particularly due to high PEG content and grossly elevated Cth plasma concentrations, although inherited cystathioninuria is considered a benign condition,41 or what will be the cost and accessibility of ERT for HCU patients. It is noteworthy that a broad correction of the I278T phenotype was achieved solely by ERT in the absence of any dietary restrictions or Cys and/or betaine supplementation. If this carries over to human patients, the PEG-CBS has the potential to supplant current therapies and significantly improve the quality of life of HCU patients.
Materials and Methods
Chemicals
Unless stated otherwise, materials were purchased from Sigma or Fisher Scientific. L-[U-14C]-serine was obtained from Perkin Elmer Life Sciences.
Protein Purification and PEGylation
Purification and PEGylation of human truncated CBS carrying the C15S mutation (htCBS C15S) were carried out as described.12, 13, 15
Animals
Animal procedures were approved under animal protocol# B-49414(03)1E by the University of Colorado Denver (UCD) IACUC. UCD is an AAALAC-accredited (#00235), Public Health Service-assured (#A 3269-01), and USDA-licensed (#84-R-0059) institution. A breeding pair of heterozygous transgenic I278T mice on the C57BL6 background was provided by Dr. Warren Kruger (Fox Chase Cancer Center, Philadelphia, PA, USA). Mice were propagated and genotyped at our facility, as described.16 Breeding pairs were maintained on water containing 25 mM ZnCl2 to induce transgene expression and thus rescue the homozygous I278T pups from neonatal death. After weaning at 21 days of age, mice switched to a regular water supply and were maintained on extruded standard diet 2920X (Envigo, CA, USA). A single-use lancet for submandibular bleeding was used for blood collection into Capiject T-MLHG lithium heparin (12.5 IU) tubes with gel (Terumo Medical, NJ, USA). Tubes were then centrifuged at 1,200 × g for 10 min, followed by transfer of plasma to 1.5 mL tubes and storage at −80°C.
Design of Animal Studies
Three cohorts of I278T mice were treated thrice a week with s.c. injections of PEG-CBS (7.5 mg/kg). Treated animals were compared with age-matched untreated I278T mice and healthy heterozygotes, which served as positive and negative controls, respectively. In one study, just-weaned mice (n = 3) were treated for a period of ∼9 months and compared to equal numbers of positive and negative controls. Similarly, another cohort of I278T mice (n = 10), with fully developed phenotypes at the age of ∼6 months, was treated for a period of 6 months and compared to equal numbers of positive and negative controls. Plasma samples from the treated mice were collected every 2 weeks 24 hr post-injection for the time course of sulfur metabolites, while terminal bleedings from all study mice were used for determination of plasma lipids, liver function panel, antioxidant enzymes, and cytokines. Facial alopecia, bone mineralization, and body composition were assessed prior to termination. In another study, newborn mice (n = 3) were treated from day 2 up to ∼4 months in an attempt to prevent irreversible changes due to the disease and compared to equal numbers of positive and negative controls. At the end of this study, all the mice were exsanguinated, euthanized using CO2 followed by a cervical dislocation, and perfused via intracardiac catheter with PBS. Livers, kidneys, and brains were harvested, frozen immediately in liquid nitrogen, and later processed for determination of various metabolites using liquid chromatography/tandem mass spectrometry (LC-MS/MS) and NMR metabolomics. The eyes were extracted and processed for analysis of the ciliary zonule.
Plasma Biomarkers and Metabolomics
Plasma sulfur amino acid metabolites were determined by stable-isotope-dilution gas chromatography mass spectrometry, as described.42 Sulfur metabolites in tissues and the matching plasma values were determined using high-performance liquid chromatography (HPLC) and LC-MS/MS, as described elsewhere.21, 43, 44 Plasma lipids were determined using Beckman AU480 chemistry analyzer. Plasma activities of ALT, AST, and ALP were measured on the Roche COBAS Mira Plus automated chemistry analyzer, while activities of SOD and CAT were determined using ELISA assay kits (Cayman Chemical, Ann Arbor, MI, USA). Concentrations of cytokines, chemokines, and growth factors in plasma samples were determined using Bio-Plex Pro mouse cytokine 23-plex assay (Bio-Rad, Hercules, CA, USA). The extraction of hydro- and lipophilic fractions from the liver extracts and subsequent NMR metabolomics was performed as described elsewhere.45 All analyses were performed in a blinded fashion, without knowledge of the animal genotype or treatment.
Ocular Analyses
The ocular lens is centered and suspended in the eye by a series of extracellular fibers, known collectively as the ciliary zonule. In human HCU patients, weakening and breakage of zonular fibers commonly leads to lens dislocation (ectopia lentis). To visualize the organization and integrity of zonular fibers in treated and control I278T mice, we used an antibody raised against MAGP1, an abundant zonule protein.39 In brief, eyes were removed and placed in 4% paraformaldehyde (PFA) in PBS (pH 7.4) (4% PFA). A small hole was made in the posterior globe, immediately below the optic nerve, to facilitate penetration of the fixative. After overnight fixation at 4°C, samples were washed three times with PBS, and the posterior portion of the eye globe was removed, exposing the ocular lens and the ciliary zonule. Non-specific antibody binding was minimized by blocking with 8% BSA solution for 2 hr. Samples were incubated overnight at 4°C with MAGP1 antibody (Sigma, diluted 1:50 in 4% BSA). Samples were then washed six times with PBS and incubated with Alexa 488-conjugated secondary antibody (Invitrogen, dilution 1:200) for 2 hr at room temperature. Methylgreen was used as a nuclear counterstain. Samples were imaged in three dimensions using a Zeiss LSM510 microscope, as described.23 Final images were presented as maximum intensity projections of the collapsed image stack.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical comparisons of two groups were conducted using an unpaired, two-tailed Students t test. Statistical analysis of three or more groups or factors was conducted by ANOVA, followed by Tukey’s multiple comparison test to determine significance. For all the tests, a value of p < 0.05 was considered significant. Significance in figures is designated using letters at the top of the error bars, with no letter indicating non-significance. Columns that are significantly different from each other (p < 0.05) are indicated by having a different letter. Two letters indicate that the column is not significantly different from the single-letter-designated columns.
Author Contributions
T.M. designed and performed animal studies, prepared and analyzed PEG-CBS conjugates, analyzed data, prepared figures, wrote the initial draft, and coordinated the collaborations. W.J. and S.B. performed analyses of the eyes and interpreted the results. J.K. and V.K. determined sulfur amino acid metabolites in tissues and matching plasmas. I.P. took care of animal colony and executed animal studies. W.D.K. generously provided the I278T mice. E.M.B. co-designed the studies and reviewed the data. J.P.K. conceived the idea, reviewed the data, and coordinated the project. All authors reviewed the draft, contributed to its revisions, and approved the final form of the manuscript.
Conflicts of Interest
The research was funded by Orphan Technologies, Ltd., a private pharmaceutical company developing an enzyme replacement therapy for CBS-deficient homocystinuria. T.M., E.M.B., and J.P.K. are inventors on patents related to the processes and products referred here (US patents 9,034,318 and 9,243,239).
Acknowledgments
The authors would like to acknowledge Richard Carrillo for htCBS C15S purifications and assistance in preparation of various PEG-CBS conjugates and Carla Ray, Linda Farb, Sally Stabler, and Robert Allen for determination of plasma sulfur amino acid metabolites. In addition, we would like to acknowledge Natalie Serkova and Denise Davis from UC Denver Animal Imaging Shared Resources core for performing NMR metabolomics. The core receives support from the NCI through a Cancer Center Support Grant (P30CA046934) and the NIH through a CCTSI Grant (UL1TR001082). We also acknowledge the National Mouse Metabolic Phenotyping Centers at UC Davis and Yale University (supported by NIDDK grants U24DK092993 and U24DK059635, respectively) for analysis of plasma and tissue samples for selected biomarkers. S.B. and W.J. are supported by NIH grants EY024607, P30 EY02687, and T32-EY013360 and the National Marfan Foundation. Institutional support to J.K. and V.K. was provided by Charles University project PROGRES Q26 and the Ministry of Health of the Czech Republic (program RVO-VFN 64165/2012). T.M. is a recipient of the American Heart Association Scientist Development Grant (16SDG30040000). This work was supported by a research grant from Orphan Technologies, Ltd. (to J.P.K.).
Contributor Information
Tomas Majtan, Email: tomas.majtan@ucdenver.edu.
Jan P. Kraus, Email: jan.kraus@ucdenver.edu.
References
- 1.Kraus J.P., Janosík M., Kozich V., Mandell R., Shih V., Sperandeo M.P., Sebastio G., de Franchis R., Andria G., Kluijtmans L.A. Cystathionine β-synthase mutations in homocystinuria. Hum. Mutat. 1999;13:362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 2.Finkelstein J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 3.Mudd S.H., Levy H.L., Kraus J.P. Disorders of transsulfuration. In: Scriver C.R., Sly W.S., Childs B., Beaudet A.L., Valle D., Kinzler K.W., Vogelstein B., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; 2001. pp. 2007–2056. [Google Scholar]
- 4.Barber G.W., Spaeth G.L. Pyridoxine therapy in homocystinuria. Lancet. 1967;289:337. [Google Scholar]
- 5.Chen X., Wang L., Fazlieva R., Kruger W.D. Contrasting behaviors of mutant cystathionine beta-synthase enzymes associated with pyridoxine response. Hum. Mutat. 2006;27:474–482. doi: 10.1002/humu.20320. [DOI] [PubMed] [Google Scholar]
- 6.Komrower G.M., Lambert A.M., Cusworth D.C., Westall R.G. Dietary treatment of homocystinuria. Arch. Dis. Child. 1966;41:666–671. doi: 10.1136/adc.41.220.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smolin L.A., Benevenga N.J., Berlow S. The use of betaine for the treatment of homocystinuria. J. Pediatr. 1981;99:467–472. doi: 10.1016/s0022-3476(81)80352-6. [DOI] [PubMed] [Google Scholar]
- 8.Morris A.A., Kožich V., Santra S., Andria G., Ben-Omran T.I., Chakrapani A.B., Crushell E., Henderson M.J., Hochuli M., Huemer M. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017;40:49–74. doi: 10.1007/s10545-016-9979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap S., Naughten E. Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J. Inherit. Metab. Dis. 1998;21:738–747. doi: 10.1023/a:1005445132327. [DOI] [PubMed] [Google Scholar]
- 10.Yap S., Boers G.H., Wilcken B., Wilcken D.E., Brenton D.P., Lee P.J., Walter J.H., Howard P.M., Naughten E.R. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler. Thromb. Vasc. Biol. 2001;21:2080–2085. doi: 10.1161/hq1201.100225. [DOI] [PubMed] [Google Scholar]
- 11.Walter J.H., Wraith J.E., White F.J., Bridge C., Till J. Strategies for the treatment of cystathionine β-synthase deficiency: the experience of the Willink Biochemical Genetics Unit over the past 30 years. Eur. J. Pediatr. 1998;157(Suppl 2):S71–S76. doi: 10.1007/pl00014308. [DOI] [PubMed] [Google Scholar]
- 12.Bublil E.M., Majtan T., Park I., Carrillo R.S., Hůlková H., Krijt J., Kožich V., Kraus J.P. Enzyme replacement with PEGylated cystathionine β-synthase ameliorates homocystinuria in murine model. J. Clin. Invest. 2016;126:2372–2384. doi: 10.1172/JCI85396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majtan T., Park I., Carrillo R.S., Bublil E.M., Kraus J.P. Engineering and characterization of an enzyme replacement therapy for classical homocystinuria. Biomacromolecules. 2017;18:1747–1761. doi: 10.1021/acs.biomac.7b00154. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M., Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M.R., Maeda N. Mice deficient in cystathionine β-synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majtan T., Hůlková H., Park I., Krijt J., Kožich V., Bublil E.M., Kraus J.P. Enzyme replacement prevents neonatal death, liver damage, and osteoporosis in murine homocystinuria. FASEB J. 2017;31:5495–5506. doi: 10.1096/fj.201700565R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L., Chen X., Tang B., Hua X., Klein-Szanto A., Kruger W.D. Expression of mutant human cystathionine beta-synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum. Mol. Genet. 2005;14:2201–2208. doi: 10.1093/hmg/ddi224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta S., Kruger W.D. Cystathionine beta-synthase deficiency causes fat loss in mice. PLoS ONE. 2011;6:e27598. doi: 10.1371/journal.pone.0027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta S., Melnyk S.B., Kruger W.D. Cystathionine β-synthase-deficient mice thrive on a low-methionine diet. FASEB J. 2014;28:781–790. doi: 10.1096/fj.13-240770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perła-Kajan J., Utyro O., Rusek M., Malinowska A., Sitkiewicz E., Jakubowski H. N-Homocysteinylation impairs collagen cross-linking in cystathionine β-synthase-deficient mice: a novel mechanism of connective tissue abnormalities. FASEB J. 2016;30:3810–3821. doi: 10.1096/fj.201600539. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi S., Sato M., Kasakoshi T., Tsutsui T., Sugimoto M., Osaki M., Okada F., Igarashi K., Hiratake J., Homma T. Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. J. Biol. Chem. 2015;290:8778–8788. doi: 10.1074/jbc.M114.625053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kožich V., Krijt J., Sokolová J., Melenovská P., Ješina P., Vozdek R., Majtán T., Kraus J.P. Thioethers as markers of hydrogen sulfide production in homocystinurias. Biochimie. 2016;126:14–20. doi: 10.1016/j.biochi.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Majtan T., Krijt J., Sokolová J., Křížková M., Ralat M.A., Kent J., Gregory J.F., 3rd, Kožich V., Kraus J.P. Biogenesis of hydrogen sulfide and thioethers by cystathionine beta-synthase. Antioxid. Redox Signal. 2018;28:311–323. doi: 10.1089/ars.2017.7009. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y., Tu Y., De Maria A., Mecham R.P., Bassnett S. Development, composition, and structural arrangements of the ciliary zonule of the mouse. Invest. Ophthalmol. Vis. Sci. 2013;54:2504–2515. doi: 10.1167/iovs.13-11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S., Kühnisch J., Mustafa A., Lhotak S., Schlachterman A., Slifker M.J., Klein-Szanto A., High K.A., Austin R.C., Kruger W.D. Mouse models of cystathionine beta-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 2009;23:883–893. doi: 10.1096/fj.08-120584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maclean K.N., Sikora J., Kožich V., Jiang H., Greiner L.S., Kraus E., Krijt J., Overdier K.H., Collard R., Brodsky G.L. A novel transgenic mouse model of CBS-deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol. Genet. Metab. 2010;101:153–162. doi: 10.1016/j.ymgme.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009;284:11601–11612. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabil O., Vitvitsky V., Xie P., Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 2011;15:363–372. doi: 10.1089/ars.2010.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L., Yang G., Untereiner A., Ju Y., Wu L., Wang R. Hydrogen sulfide impairs glucose utilization and increases gluconeogenesis in hepatocytes. Endocrinology. 2013;154:114–126. doi: 10.1210/en.2012-1658. [DOI] [PubMed] [Google Scholar]
- 29.Golbahar J., Aminzadeh M.A., Kassab S.E., Omrani G.R. Hyperhomocysteinemia induces insulin resistance in male Sprague-Dawley rats. Diabetes Res. Clin. Pract. 2007;76:1–5. doi: 10.1016/j.diabres.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 30.Orendác M., Zeman J., Stabler S.P., Allen R.H., Kraus J.P., Bodamer O., Stöckler-Ipsiroglu S., Kvasnicka J., Kozich V. Homocystinuria due to cystathionine β-synthase deficiency: novel biochemical findings and treatment efficacy. J. Inherit. Metab. Dis. 2003;26:761–773. doi: 10.1023/B:BOLI.0000009963.88420.c2. [DOI] [PubMed] [Google Scholar]
- 31.Elshorbagy A.K., Nurk E., Gjesdal C.G., Tell G.S., Ueland P.M., Nygård O., Tverdal A., Vollset S.E., Refsum H. Homocysteine, cysteine, and body composition in the Hordaland Homocysteine Study: does cysteine link amino acid and lipid metabolism? Am. J. Clin. Nutr. 2008;88:738–746. doi: 10.1093/ajcn/88.3.738. [DOI] [PubMed] [Google Scholar]
- 32.Elshorbagy A.K., Kozich V., Smith A.D., Refsum H. Cysteine and obesity: consistency of the evidence across epidemiologic, animal and cellular studies. Curr. Opin. Clin. Nutr. Metab. Care. 2012;15:49–57. doi: 10.1097/MCO.0b013e32834d199f. [DOI] [PubMed] [Google Scholar]
- 33.Namekata K., Enokido Y., Ishii I., Nagai Y., Harada T., Kimura H. Abnormal lipid metabolism in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. J. Biol. Chem. 2004;279:52961–52969. doi: 10.1074/jbc.M406820200. [DOI] [PubMed] [Google Scholar]
- 34.Majtan T., Park I., Bublil E.M., Kraus J.P. Enzyme replacement therapy prevents loss of bone and fat mass in murine homocystinuria. Hum. Mutat. 2017 doi: 10.1002/humu.23360. Published online October 16, 2017. [DOI] [PubMed] [Google Scholar]
- 35.Flowers M.T., Ntambi J.M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 2008;19:248–256. doi: 10.1097/MOL.0b013e3282f9b54d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parrot F., Redonnet-Vernhet I., Lacombe D., Gin H. Osteoporosis in late-diagnosed adult homocystinuric patients. J. Inherit. Metab. Dis. 2000;23:338–340. doi: 10.1023/a:1005618927729. [DOI] [PubMed] [Google Scholar]
- 37.Gupta S., Wang L., Kruger W.D. Betaine supplementation is less effective than methionine restriction in correcting phenotypes of CBS deficient mice. J. Inherit. Metab. Dis. 2016;39:39–46. doi: 10.1007/s10545-015-9883-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mudd S.H., Skovby F., Levy H.L., Pettigrew K.D., Wilcken B., Pyeritz R.E., Andria G., Boers G.H., Bromberg I.L., Cerone R. The natural history of homocystinuria due to cystathionine β-synthase deficiency. Am. J. Hum. Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- 39.De Maria A., Wilmarth P.A., David L.L., Bassnett S. Proteomic analysis of the bovine and human ciliary zonule. Invest. Ophthalmol. Vis. Sci. 2017;58:573–585. doi: 10.1167/iovs.16-20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramsey M.S., Yanoff M., Fine B.S. The ocular histopathology of homocystinuria. A light and electron microscopic study. Am. J. Ophthalmol. 1972;74:377–385. doi: 10.1016/0002-9394(72)90895-1. [DOI] [PubMed] [Google Scholar]
- 41.Kraus J.P., Hasek J., Kozich V., Collard R., Venezia S., Janosíková B., Wang J., Stabler S.P., Allen R.H., Jakobs C. Cystathionine gamma-lyase: clinical, metabolic, genetic, and structural studies. Mol. Genet. Metab. 2009;97:250–259. doi: 10.1016/j.ymgme.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allen R.H., Stabler S.P., Lindenbaum J. Serum betaine, N,N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism. 1993;42:1448–1460. doi: 10.1016/0026-0495(93)90198-w. [DOI] [PubMed] [Google Scholar]
- 43.Krijt J., Vacková M., Kozich V. Measurement of homocysteine and other aminothiols in plasma: advantages of using tris(2-carboxyethyl)phosphine as reductant compared with tri-n-butylphosphine. Clin. Chem. 2001;47:1821–1828. [PubMed] [Google Scholar]
- 44.Krijt J., Dutá A., Kozich V. Determination of S-Adenosylmethionine and S-Adenosylhomocysteine by LC-MS/MS and evaluation of their stability in mice tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009;877:2061–2066. doi: 10.1016/j.jchromb.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serkova N., Litt L., James T.L., Sadée W., Leibfritz D., Benet L.Z., Christians U. Evaluation of individual and combined neurotoxicity of the immunosuppressants cyclosporine and sirolimus by in vitro multinuclear NMR spectroscopy. J. Pharmacol. Exp. Ther. 1999;289:800–806. [PubMed] [Google Scholar]