

Abstract
Human embryonic stem cell-derived mesenchymal stem cells (hE-MSCs) have greater proliferative capacity than other human mesenchymal stem cells (hMSCs), suggesting that they may have wider applications in regenerative cellular therapy. In this study, to uncover the anti-senescence mechanism in hE-MSCs, we compared hE-MSCs with adult bone marrow (hBM-MSCs) and found that hepatocyte growth factor (HGF) was more abundantly expressed in hE-MSCs than in hBM-MSCs and that it induced the transcription of RAD51 and facilitated its SUMOylation at K70. RAD51 induction/modification by HGF not only increased telomere length but also increased mtDNA replication, leading to increased ATP generation. Moreover, HGF-treated hBM-MSCs showed significantly better therapeutic efficacy than naive hBM-MSCs. Together, the data suggest that the RAD51-mediated effects of HGF prevent hMSC senescence by promoting telomere lengthening and inducing mtDNA replication and function, which opens the prospect of developing novel therapies for liver disease.
Keywords: hepatocyte growth factor, human MSC, liver fibrosis, RAD51
Graphical Abstract
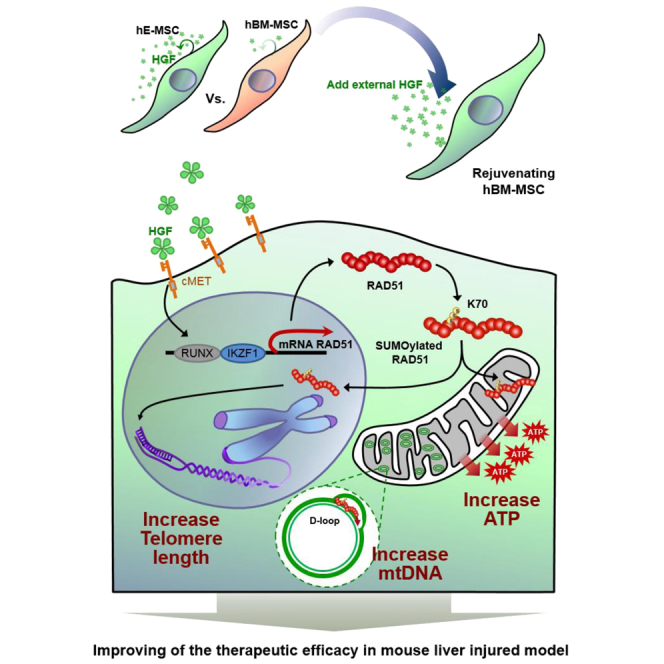
RAD51 is the actual effector by HGF to accomplish anti-senescence. HGF induces transcription of RAD51 and activates RAD51 via sumoylation. Action mechanisms of RAD51 by HGF are telomere lengthening at nuclei and induction of mtDNA replication at mitochondria.
Introduction
Preclinical and clinical studies of human bone-marrow-derived mesenchymal stem cells (hBM-MSCs) have yielded their promising results and suggested a powerful paradigm to treat human diseases.1, 2, 3 However, clinical applications using human mesenchymal stem cells (hMSCs), including hBM-MSCs, are hampered by their finite self-renewal ability.
Senescence caused by repetitive proliferation in vitro is one of several factors that determine the self-renewal ability of stem cells. Although the underlying molecular mechanism is unclear, Bertolo et al.4 characterized the senescent state of hMSCs by estimating their differentiation ability, population doubling time (PDT), senescence-associated β-galactosidase activity, and telomere length. Normal cells reportedly have restricted telomerase activity, meaning that their telomeres shorten as they proliferate in vitro, eventually leading to proliferation arrest.5, 6 Replicative senescence of hMSCs has been associated with gradual loss of telomeric DNA.7, 8, 9, 10 Therefore, senescence phenotypes according to repetitive proliferation in vitro are related to telomere length.
We previously obtained hMSCs from human embryonic stem cells (hE-MSCs) and demonstrated that they could be consistently produced, maintained, and expanded more effectively than other hMSCs, such as hBM-MSCs.11, 12 To build upon this previous research, we sought to decipher the mechanism underlying this difference in senescence and regeneration capability between hE-MSCs and hBM-MSCs and to identify a factor that can rejuvenate senescence-prone hBM-MSCs.
In this study, hE-MSCs were found to have a shorter PDT and longer telomeres than hBM-MSCs. Systemic comparison of hE-MSCs and hBM-MSCs using a human growth factor array revealed that hepatocyte growth factor (HGF) was the only growth factor whose expression was significantly higher in the former. Treatment with HGF increased telomere length in hBM-MSCs, whereas inhibition of HGF decreased telomere length in hE-MSCs. RAD51, rather than telomerase reverse transcriptase (TERT), controlled senescence downstream of HGF. RAD51 expression was higher in hE-MSCs than in hBM-MSCs, and HGF treatment induced RAD51 expression in hBM-MSCs. HGF enhanced transcription of RAD51 through two transcription factors: IKZF1 and RUNX1. HGF also facilitated a post-translational (PTM) modification of RAD51, SUMOylation at K70. Induction of RAD51 by HGF not only increased telomere length but also increased mtDNA replication, leading to enhanced ATP generation in hBM-MSCs. Finally, we confirmed that hBM-MSCs rejuvenated by HGF treatment had a better capability than untreated hBM-MSCs to regenerate the damaged liver in a mouse model after cell transplantation.
HGF is a well-known pleiotropic growth factor,13, 14 a critical factor in the development and regeneration of the liver, and an anti-apoptotic factor in hepatocytes.15, 16 One study reported that mouse MSCs pretreated with HGF and FGF4 have better therapeutic potential than naive MSCs in the repair of injured liver.17 Another study reported that rat BM-MSCs cultured with HGF had an improved therapeutic effect for the repair of damaged liver.15 However, to our knowledge, no study has reported RAD51-mediated HGF effects on telomere length and mtDNA replication. This study provides mechanistic insights into why stem/progenitor cells differ in terms of senescence and how senescence is prevented by HGF. These findings could help improve the efficacy of hBM-MSCs and lead to the development of clinical applications for the treatment of liver disease.
Results
HGF Regulates Telomere Length in hMSCs: Comparative Analysis of hE-MSCs and hBM-MSCs
To identify a factor that can rejuvenate the senescence-prone hBM-MSCs, we compared hBM-MSCs at passage (P) 8 with hE-MSCs at P15. In our previous report, hE-MSCs expanded over p30.12 Thus, we selected p15 as a founder to identify the mechanism to maintain the higher stemness in hE-MSCs than in hBM-MSCs. We purchased hBM-MSCs from Lonza, which recommended hBM-MSCs to be used by P5, and we chose P8 as senescence-prone hBM-MSCs (http://bio.lonza.com/uploads/tx_mwaxmarketingmaterial/Lonza_ManualsProductInstructions_Poietics_Human_Mesenchymal_Stem_Cells.pdf).
PDT was shorter in hE-MSCs (∼40 hr) than in hBM-MSCs (60 hr) (Figure 1A). To confirm this difference, we performed fluorescence-activated cell sorting (FACS) to check the proliferative activity of these hMSCs. A larger percentage of hE-MSCs than hBM-MSCs expressed proliferating cell nuclear antigen (PCNA), an S-phase-specific marker (28.85% ± 3.77% of hE-MSCs versus 6.42% ± 0.43% of hBM-MSCs) (Figure 1B). As expected, relative telomere length (RTL) was 2.5-fold longer in hE-MSCs than in hBM-MSCs (Figure 1C).
Figure 1.

Differences in Senescence between hE-MSCs, P15, and hBM-MSCs, P8, and Comparison of Secreted Factors Using a Human Growth Factor Array
(A) Comparison of PDT between hBM-MSCs (P8) and hE-MSCs (P15). hE-MSCs had a shorter PDT than hBM-MSCs. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (B) FACS analysis demonstrated that expression of the S phase marker PCNA was higher in hE-MSCs than in hBM-MSCs. (C) Analysis of RTL by real-time genomic DNA PCR demonstrated that telomeres were longer in hE-MSCs than in hBM-MSCs. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (D) In a human cytokine array, the levels of only two cytokines, HGF and IGFBP1, were higher in the culture supernatant of hE-MSCs than in that of hBM-MSCs. (E) RTL in hBM-MSCs treated with various cytokines. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (F) PDT was reduced upon treatment with human rHGF. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (G) Interphase FISH analysis. PNA was used to detect telomeres and was visualized with Alexa Fluor 488. Twenty cells were quantified using the TeloFISH program, as described in the Materials and Methods. Means with error bars for counted 20 cells each group. The bars show SD and present plus and minus values.
Next, we investigated the cause of the difference in RTL between these two types of hMSCs. Cytokines and growth factors affect cells through autocrine and paracrine signaling and trigger outside-in stimulation to activate downstream cellular signals, which regulate various events. Therefore, we compared hE-MSCs and hBM-MSCs using a human cytokine array containing 41 representative cytokines. The levels of two factors, HGF and insulin-like growth factor-binding protein 1 (IGFBP1), differed between the two types of hMSCs (Figure 1D).
To validate these findings, ELISA was performed using the culture supernatants of hE-MSCs and hBM-MSCs (Figure S1A). Consistent with the findings of the cytokine array, levels of HGF and IGFBP1 secretion were significantly higher in hE-MSCs than in hBM-MSCs. The concentration of HGF (10 ng/mL) was significantly higher than that of other cytokines (100–200 pg/mL) in the culture supernatant of hE-MSCs. A loss-of-function experiment in hE-MSCs was performed to determine whether HGF controls telomere length. RTL was analyzed after treating hE-MSCs with a neutralizing HGF antibody. Telomere length was decreased to ∼50% (Figure S1B) and PDT was delayed to ∼80 hr (Figure S1C) upon loss of HGF function in hE-MSCs.
Next, we investigated whether HGF affected telomere length in hBM-MSCs via outside-in stimulation. Telomeres were noticeably longer in hBM-MSCs treated with recombinant HGF (rHGF) than in those treated with other cytokines (Figure 1E), including IGFBP1, epidermal growth factor (EGF), FGF2, and vascular endothelial growth factor (VEGF), which are known cytokines expressed by hMSCs.18 Additionally, PDT was shorter in hBM-MSCs treated with rHGF (Figure 1F).
Recently, quantitative real-time PCR-based techniques have emerged as an alternative method for quantifying telomere length.19 Thus, we used quantitative real-time PCR to measure RTL, which is less time-consuming than other conventional methods. Additionally, we visualized telomeres using peptide-conjugated nucleic acid (PNA), which specifically binds to telomeres. Interphase telomere fluorescence in situ hybridization (FISH) showed that treatment of hBM-MSCs with rHGF increased the number and intensity of PNA foci in the nucleus (Figure 1G).
RAD51 Is Key for HGF-Induced Telomere Lengthening in MSCs
To investigate whether HGF-induced telomere lengthening in MSCs is dependent on TERT, we first compared the protein level of TERT between hE-MSCs and hBM-MSCs. However, neither the TERT protein level (Figure 2A) nor TERT activity determined by the TRAP assay (Figure 2B) differed between these two types of hMSCs. Therefore, we considered a TERT-independent pathway. RAD51 is a critical mediator of alternative mechanisms in the maintenance of telomere length (ALT).20, 21, 22 The RAD51 protein level (Figure 2C) and RAD51 activity determined by the HR assay (Figure 2D) were higher in hE-MSCs than in hBM-MSCs. Moreover, when we treated hBM-MSCs with rHGF, the activity and protein level of RAD51 increased (Figures 2D and 2E), whereas the TERT protein level did not change (Figure 2E). Conversely, when we blocked HGF in hE-MSCs using a neutralizing antibody, the protein level of RAD51 decreased, whereas that of TERT did not change (Figure S2A). ZCAN4, a recently reported regulator of telomeres in embryonic stem cells23 and stemness in induced pluripotent stem cells,24 was not expressed in either hE-MSCs or hBM-MSCs (Figure S2B). These findings suggest that RAD51 is a key factor for telomere lengthening in hE-MSCs and rHGF-treated hBM-MSCs. Specifically, HGF increases telomere length in hBM-MSCs via RAD51, similar to the phenomenon in hE-MSCs.
Figure 2.

Mechanism Underlying HGF-Dependent Telomere Lengthening in hMSCs: A Role for RAD51
(A and B) TERT protein expression (A) and TERT activity (B) determined by the TRAP assay did not significantly differ between hE-MSCs and hBM-MSCs. (C) Western blot analysis to compare the protein level of RAD51 between hE-MSCs and hBM-MSCs. (D) In the HR assay, rHGF treatment upregulated RAD51 activity in hBM-MSCs, similar to the high level in hE-MSCs. A high-density band generated using the “HR product primer” represents high RAD51 activity, whereas a high-density band generated using the “incomplete plasmid primer” represents low RAD51 activity. (E) Protein expression of RAD51 in hBM-MSCs but not TERT expression was increased upon rHGF treatment. Means with error bars for measured intensity of band for n = 3 experiments independently. The bars show SD and present plus values only. (F–H) RAD51 mRNA (F) and RAD51 protein levels (G), and RTL (H) upon siRAD51 treatment in rHGF-treated hBM-MSCs. (F) Means with error bars for triple replication; (G) means with error bars for measured intensity of band for n = 3 experiments independently; (H) means with error bars for n = 3 independent experiments. All bars show SD and present plus values only. (I) Overexpression of RAD51 increased RTL. Means with error bars for n = 3 independent experiments. The bars show SD and present plus values only. (J) Exogenous RAD51 was detected by western blotting.
To confirm the function of RAD51 in telomere lengthening, we performed a loss-of-function experiment in rHGF-treated hBM-MSCs using RAD51-targeting small interfering RNA (siRNA, called siRAD51). The mRNA and protein levels of RAD51 were increased by rHGF treatment and decreased by siRAD51 treatment (Figures 2F and 2G). These results correlated with telomere length (Figure 2H). In the gain-of-function experiment using the lentiviral CMV-RAD51 vector, telomere length increased (Figure 2I) when the RAD51 protein level was increased (Figure 2J). In hE-MSCs, when the RAD51 mRNA level was decreased by siRAD51 treatment (Figure S2C), its protein level was reduced (Figure S2D) and RTL dramatically declined (Figure S2E). Thus, RAD51 is involved in a mechanism regulating telomere lengthening in hE-MSCs.
Transcription of RAD51 Is Activated by RUNX1 and IKZF1 in the Absence of DNA Damage
RAD51 is activated in response to DSBs, although there is debate about the level of its expression.25, 26, 27 Therefore, we investigated whether the increase in RAD51 upon rHGF treatment is associated with DSBs. To this end, we labeled γ-H2AX, a DSB marker. γ-H2AX and RAD51 were both detected in the nucleus of hBM-MSCs upon induction of DSBs by UV irradiation (Figure 3A). On the other hand, in the absence of irradiation, γ-H2AX was not detected in rHGF-treated hBM-MSCs, in which the level of RAD51 was increased (Figure 3A). To investigate whether chromosomes were normal, karyotyping was performed in rHGF-treated hBM-MSCs. No aberrations were detected (Figure 3B). These findings indicate that RAD51 is induced by HGF per se in hBM-MSCs, but not by incidental DNA damage.
Figure 3.

IKZF1 and RUNX1 Activate Transcription of RAD51 in Response to HGF
(A) Immunofluorescence staining of RAD51 and γ-H2AX, a marker of DSBs. UV irradiation induced DNA damage and expression of γ-H2AX, leading to expression of RAD51. HGF increased RAD51 expression in hBM-MSCs in the absence of DNA damage and γ-H2AX expression. (B) In G-banding to assess chromosome stability, hBM-MSCs demonstrated a normal karyotype and telomeres regardless of rHGF treatment. (C–E) RAD51 mRNA expression (C), RAD51 protein expression (D), and telomere length (E) were increased in the rHGF-treated hBM-MSCs. These effects of HGF were blocked by treatment with siRNA targeting IKZF1 or RUNX1. (C) Means with error bars for triple replication; (E) means with error bars for n = 3 independent experiments. The bars show SD and present plus values only.
To elucidate the transcriptional regulatory mechanism underlying HGF-dependent RAD51 induction, we screened the promoter region of RAD51. Four alternative transcripts are expressed from chromosome 15, all of which have a common putative promoter region located 2.3 kb upstream of the transcriptional start site. We utilized the TFSEARCH V1.3 database to explore putative transcriptional regulatory factors and found binding sites of IKZF1 and RUNX1 (Figure S3A).
To confirm that IKZF1 and RUNX1 are transcriptional regulators of RAD51 in response to HGF, we knocked down these genes in rHGF-treated hBM-MSCs. RAD51 mRNA and protein levels were increased by rHGF treatment (Figures 3C and 3D), which was prevented by knockdown of IKZF1 or RUNX1 in rHGF-treated hBM-MSCs (Figures 3C and 3D). RTL also correlated with the RAD51 level and was significantly reduced by knockdown of IKZF1 or RUNX1 (1 in control hBM-MSCs versus 3.48 ± 0.68 in rHGF-treated hBM-MSCs, p = 0.0002; 3.28 ± 0.46 in siScramble- and rHGF-co-treated hBM-MSCs versus 0.15 ± 0.04 in siIKZF1- and rHGF-co-treated hBM-MSCs versus 0.21 ± 0.12 in siRUNX- and rHGF-co-treated hBM-MSCs) (Figure 3E). We also knocked down IKZF1 and RUNX1 in hE-MSCs to validate their function. The knockdown efficiencies were confirmed by real-time PCR (Figure S3B). RUNX1 or IKZF1 knockdown decreased RAD51 protein expression (Figure S3C) and RTL (Figure S3D). In summary, HGF induces transcription of RAD51 in hMSCs through two factors: IKZF1 and RUNX1.
SUMOylation Is Critical for RAD51-Dependent Telomere Lengthening
In order to perform its functions, RAD51 must undergo PTMs for efficient binding to its different protein partners. Western blot analysis of whole cell lysates showed that a portion of RAD51 migrated to a position above the predicted one (∼37 kD), corresponding to a molecular weight of ∼100 kD after electrophoresis on SDS-polyacrylamide gels, suggesting that it was ubiquitinated or SUMOylated. The intensity of the band was higher when the cells were treated with rHGF (Figure 4A). Considering that ubiquitination induces degradation, we excluded the possibility of ubiquitination and other PTMs. The ∼32 kD presumed isoform of RAD51 was not detected in the nucleus (Figure S4A) and therefore was not considered to be relevant to the study.
Figure 4.

SUMOylation of RAD51 Is Important for Its Function
(A) Multiple bands of endogenous RAD51 in hBM-MSCs. Cells were treated with 10 ng/mL human rHGF for 3 days. (B) hBM-MSCs were treated with 100 μM GA and 10 ng/mL human rHGF. (C) RTL was determined using the aforementioned conditions. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (D) Diagram showing the RAD51 constructs, in which the predicted SUMOylation sites were mutated. The constructs harbored Myc and His tags to facilitate immunoprecipitation. (E and F) Western blotting (E) demonstrated that precipitated Myc-RAD51 was SUMOylated. GAPDH was used as an endogenous control for the input samples. hBM-MSCs were transfected with RAD51 mutant constructs and then RTL (F) was determined by real-time PCR. All qPCR samples were normalized using a CMV promoter-specific primer to account for variation in the transfection efficiency. Means with error bars for n = 3 independent experiments. The bars show SD and present plus values only.
Next, we tested whether RAD51 was SUMOylated using ginkgolic acid (GA), a known inhibitor of SUMOylation.28 The RAD51 protein band migrating to ∼100 kD after rHGF treatment disappeared upon co-treatment with GA (Figure 4B). We also checked the indices of RAD51 function. RTL was increased by rHGF treatment and this was blocked by GA (Figure 4C). However, GA is a pan-inhibitor of SUMOylation with cytotoxic potential.29 Therefore, we confirmed SUMOylation of RAD51 by performing immunoblotting with SUMO antibodies on immunoprecipitated RAD51, with and without mutant SUMOylation sites. To identify specific SUMOylation sites in RAD51, we performed an in silico analysis based on an established database to predict SUMOylation sites. Two common sites were identified by two different bioinformatics programs (GPS SUMO2.0 [http://sumosp.biocuckoo.org] and SUMOplot [http://www.abgent.com/sumoplot]), which identify amino acid sequences that can be SUMOylated (Table 1). K70 is a non-consensus type II sequence and K57 is a type I consensus sequence for SUMOylation in RAD51.
Table 1.
Prediction of RAD51 SUMOylation Sites
| Position | Peptide | Score | Cutoff | Type |
|---|---|---|---|---|
| 57 | YAPKKEL | 0.341 | 0.13 | type I: Ψ-K-X-E |
| 70 | SEAKADK | 3.588 | 2.64 | type II: non-consensus |
We used pcDNA3.1 Myc/His(-) to construct CMV RAD51 vectors harboring a lysine-to-arginine mutation of each of the SUMOylation sites (K70R and K57R) and a Myc-His tag (Figure 4D). Upon immunoprecipitation with an anti-Myc antibody, less SUMO1 was detected with the K70R mutant than with the K57R mutant (Figure 4E). To validate the effect of RAD51 SUMOylation, we checked RTL. RTL was increased in cells transfected with the wild-type RAD51 and K57R vectors, but not in cells transfected with the K70R vector (Figure 4F).
HGF Induces Binding of Cytoplasmic RAD51 to mtDNA and Increases Its Replication
While observing the immunofluorescence staining pattern of RAD51 (Figure 5A), we noticed cytoplasmic RAD51. This pool of RAD51 was different from nuclear RAD51, which induced telomere lengthening. Recent reports show that RAD51 functions in mitochondria to increase the mtDNA copy number, which is not related to telomeres.30, 31 mtDNA encodes genes that are involved in production of ATP, an energy source of all cells in living organisms. Hence, it can indirectly reflect cell viability. Along with these reports, many studies demonstrate that the mtDNA copy number and telomere length are indices of senescence in healthy human cells.32 However, a single factor linking mtDNA and telomeres has not been reported. We hypothesized that RAD51 could be present in both nuclear and mitochondria foci upon stimulation by HGF. We first stained hBM-MSCs expressing RAD51-GFP for TOM20, a mitochondrial marker, to determine whether they co-localized. TOM20 completely co-localized with RAD51-GFP in the cytoplasmic region (Figure 5A). Next, to compare the mtDNA copy number between rHGF-treated and non-treated cells, we designed a primer set to amplify a mtDNA-specific region33 that is absent from genomic DNA (Figure S5A). After normalization to AIB1, a reference gene with only a single copy on the chromosome, the relative mtDNA copy number was 1.5-fold higher in rHGF-treated cells than in control cells (Figure 5B). Furthermore, RAD51 knockdown decreased the mtDNA copy number in rHGF-treated cells (Figure 5B), similar to the effect on telomere length. ATP synthesis was measured to validate the functionality of mitochondria. The ATP concentration was increased by rHGF treatment (1 in control cells versus 2 ± 0.46 in rHGF-treated cells) and this was dependent on RAD51 (1 in control cells versus 2.4 ± 0.26 in RAD51-overexpressing cells versus 0.59 ± 0.04 in siRAD51-treated cells) (Figure 5C).
Figure 5.

RAD51 Regulates mtDNA Replication in the Cytoplasm in Response to HGF
(A) The mitochondrial marker TOM20 (red) and RAD51 (green) were co-stained and visualized in hBM-MSCs by immunofluorescence microscopy. Nuclei were counterstained with DAPI. The bottom right image is a merge of TOM20 and RAD51. (B) mtDNA copies were quantified by real-time PCR. The mtDNA copy number was increased by 1.5-fold in rHGF-treated hBM-MSCs and decreased in siRAD51-treated cells. Means with error bars for triple replication. The bars show SD and present plus values only. (C) ATP production was determined by calculating luciferase activity and was converted to the fold change. hBM-MSCs were treated with rHGF and transfected with a RAD51 overexpression construct or siRAD51. Means with error bars for n = 3 independent experiments. The bars show SD and present plus value only. (D and E) The mtDNA copy number (D) and ATP concentration (E) were determined using the aforementioned conditions. These indices were increased by HGF and decreased by GA. (D) Means with error bars for triple replication; (E) means with error bars for n = 3 independent experiments. The bars show SD and present plus values only. (F) hBM-MSCs were transfected with RAD51 mutant constructs and then the mtDNA copy number was assessed by real-time PCR. All qPCR samples were normalized using a CMV promoter-specific primer to account for variation in the transfection efficiency. Means with error bars for triple replication. The bars show SD and present plus values only. (G) CytoChIP analysis using an anti-RAD51 antibody to precipitate mtDNA from the cytoplasmic fraction. The presence of the D-loop and ND-2 regions of mtDNA was confirmed using specific primers. An anti-polymerase γ antibody was used as a positive control to confirm direct mtDNA binding. Immunoglobulin G (IgG) was used as a negative control. (H) qPCR analysis to validate the semiquantitative PCR data. Means with error bars for triple replication. The bars show SD and present plus values only. (I) Real-time PCR analysis of the D-loop region after CytoChIP. Relative binding of RAD51 to the D-loop detected by CytoChIP was decreased in cells co-treated with GA and rHGF. Means with error bars for triple replication. The bars show SD and present plus values only.
Then, we tested whether RAD51 SUMOylation affects mitochondria using GA. Similar to RTL, the mtDNA copy number and ATP concentration were higher after rHGF treatment and lower after inhibition of SUMOylation (Figures 5D and 5E). The mtDNA copy number was increased in cells transfected with the wild-type RAD51 and K57R vectors, but not in cells transfected with the K70R vector (Figure 5F).
Next, we investigated whether RAD51 binds to mtDNA to facilitate its replication. There have not been previous reports of specific mtDNA-binding sites for RAD51 to bind on mtDNA, although one study reported that RAD51 physically associates with whole mtDNA using ChIP assay when cells are exposed to ionizing radiation.30 To identify the sites of mtDNA for RAD51 to bind on, we performed CytoChIP assay, which employs the sheared cytoplasmic fraction to obtain mtDNA fragments (Figure S5B). The D-loop and ND2 reference regions are reported to be poly (ADP-ribose) polymerase 1-binding sites in mtDNA,34 and the D-loop region was reported to be a start site for mtDNA replication35 (Figure S5C). Polymerase γ binds to the D-loop region as a main factor of the replication initiation complex and was therefore used as a D-loop-binding control. The nuclear and cytoplasmic fractions were well separated (Figure S5D), and DNA was sheared into suitably sized fragments of 100–500 bp (Figure S5E). PCR analysis of the AIB1 gene and D-loop region demonstrated clear separation of the nuclear and cytoplasmic fractions (Figure S5F).
Using CytoChIP, we demonstrated that RAD51 bound to mtDNA directly via the D-loop region (Figure 5G). Binding of polymerase γ to the D-loop was increased upon rHGF treatment, similar to that of RAD51 (Figure 5G). Real-time PCR analysis confirmed that rHGF treatment increased binding of RAD51 by more than 3-fold (3.17 ± 0.2) (Figure 5H) as well as binding of polymerase γ (Figure 5H).
Finally, we detected binding of SUMOylated RAD51 to the D-loop using CytoChIP with GA (Figure 5I). Binding of RAD51 to the D-loop was induced by HGF, which was inhibited by co-treatment with GA. Taken together, we conclude that HGF-induced RAD51 is SUMOylated at K70 and that this PTM is critical for telomere lengthening and mtDNA replication.
rHGF Treatment Enhances the Therapeutic Efficacy of hBM-MSCs
Given that HGF treatment induced RAD51 in hBM-MSCs and thereby prevented senescence and improved mitochondrial energy production, we investigated whether HGF promotes the therapeutic function of hBM-MSCs in vivo using a thioacetamide (TAA)-induced liver fibrosis mouse model. DiI-labeled hBM-MSCs were systemically administered via intra-cardiac injection 1 day after the first injection of TAA. TAA was injected every 3 days to maintain its effect (Figure 6A). Histological analysis using Picro Sirius red, which detects collagen deposits, demonstrated liver portal-to-portal septa formation indicative of fibrosis upon TAA treatment. The staining showed that transplantation of hBM-MSCs significantly prevented liver fibrosis. The fibrotic percentage area at day 14 was significantly smaller in mice administered rHGF-treated hBM-MSCs than in mice administered non-treated hBM-MSCs (0.83% ± 0.06% in normal liver versus 9.9% ± 0.19% in TAA-treated liver versus 6.6% ± 0.11% in hBM-MSC-injected liver versus 4.8% ± 0.06% in rHGF-primed hBM-MSC-injected liver, p < 0.0001, n = 5) (Figure 6B). Additionally, there was less undulation, indicative of a lower degree of fibrosis, in liver tissue of mice administered rHGF-treated hBM-MSCs than in that of mice administered non-treated hBM-MSCs (Figure 6B).
Figure 6.

rHGF Treatment Enhances the Therapeutic Efficacy of hBM-MSCs
(A) Experimental scheme of the systemic administration of rHGF-treated hBM-MSCs via intracardiac injection into the livers of TAA-treated mice. (B) Picrosirius red staining of fibrotic areas at 14 days after cell transplantation. TAA-induced liver fibrosis was significantly prevented by transplantation of hBM-MSCs and even more so by transplantation of rHGF-treated hBM-MSCs. Means with error bars for n = 5, measured fibrotic area in 3 pictures taken at random from each group. The bars show SD and present plus value only. (C) Confocal microscopy images of liver tissues after 14 days showed the persistence of systemically administered hBM-MSCs in the liver. DiI-labeled hBM-MSCs were counted in liver tissues at 14 days after systemic administration. rHGF treatment improved the homing, engraftment, and survival of hBM-MSCs (p = 0.0022). Means with error bars for n = 5, measured cells in 3 pictures taken at random from each group and calculated cell number per 0.125 mm2. The bars show SD, and present plus value only. (D) Immunostaining of transplanted hBM-MSCs to determine their fate. CRBP1 was used as a marker of quiescent HSCs.
After 14 days, the number of DiI-labeled hBM-MSCs per 0.125 mm2 of liver tissue was higher in mice administered rHGF-treated hBM-MSCs than in mice administered non-treated hBM-MSCs, demonstrating that rHGF treatment enhanced the engraftment efficacy of these cells (5.25 ± 2.08 in non-treated hBM-MSC-injected mice versus 8.125 ± 1.49 in rHGF-primed hBM-MSC-injected mice) (Figure 6C). To investigate the fate of the transplanted hBM-MSCs, we stained liver tissue for cellular retinol-binding protein 1 (CRBP1) as a marker of quiescent hepatic stellate cells (HSCs), which can be converted into myofibroblasts under fibrotic stimulation (Figure 6D). DiI-positive hBM-MSCs expressed CRBP1, suggesting that transplanted hBM-MSCs differentiated into quiescent HSCs rather than myofibroblasts, leading to prevention of liver fibrosis.
Discussion
HGF Rejuvenates hMSCs by Elongating Telomeres and Enhancing Mitochondrial Function
In our study, we confirmed that hE-MSCs were younger or less senescent and had longer telomeres than hBM-MSCs. Examination of two mechanisms underlying telomere lengthening in hE-MSCs revealed that the effector molecule was RAD51 rather than TERT. In a human 41 growth factor array, HGF was the only growth factor that was highly produced by hE-MSCs but rarely produced by hBM-MSCs. When hBM-MSCs were treated with HGF, RAD51 was induced, and this led to telomere lengthening. Induction of RAD51 was mediated at the transcriptional level via IKZF1 and RUNX1 as well as at the post-translational level via SUMOylation. Induction of RAD51 not only lengthened telomeres in the nucleus, it also increased mtDNA replication in the cytoplasm, leading to efficient energy production in hBM-MSCs. The in vitro finding of hBM-MSC rejuvenation by HGF treatment was confirmed using an in vivo liver injury mouse model, in which liver fibrosis was more effectively prevented by rHGF-primed hBM-MSCs than by naive hBM-MSCs.
The mechanism underlying telomere lengthening, which we confirmed to be a determinant of cellular senescence, has received attention in cancer therapy research. Telomere lengthening in normal cells has also been studied in stem cell research.36 ALT is another mechanism of telomere maintenance distinct from that of the telomerase-dependent pathway. Lafferty-Whyte et al.37 analyzed 297 gene signatures and identified the cell lines in which ALT signaling pathways are activated. These are human mesenchymal cell lines with repressed TERT. In this context, we explored whether MSCs use RAD51 for telomere maintenance and found that telomere length was increased by RAD51 after HGF treatment. Although HGF is known to increase telomerase activity in hepatocyte primary cultures,38 we propose that HGF lengthens telomeres in hMSCs via activation of RAD51 signaling. However, further study is required to clarify whether RAD51 exercised its role in classical ALT or via another telomerase-independent mechanism for the rejuvenation by HGF.
RAD51 is an important factor for the regulation of telomere lengthening and genome stability. It also repairs DNA double-strand breaks through homologous recombination (HR).20, 21, 22, 39 Recent reports demonstrated that RAD51 contributes to the homology search process and captures sister chromatin in HR-based telomere maintenance as a component of the HR machinery.40 Additionally, RAD51 has been studied in mitochondria.30 However, it has not been investigated how RAD51 is activated or how its expression and stabilization are regulated.
In this study, we found that RAD51 bound to the D-loop of mtDNA using a method that precipitated cytoplasmic DNA without precipitating nuclear DNA. We developed this alternative method to detect the precise mechanism of interaction between RAD51 and mtDNA. The mtDNA copy number was increased by binding of RAD51 to mtDNA. Considering that mitochondrial function is directly related with the mtDNA copy number, cell viability, and aging,32, 41 RAD51 activation in hBM-MSCs upon HGF treatment lengthens telomeres in nuclear DNA, increases ATP production by increasing the cytoplasmic mtDNA copy number, and could augment the capability to repair damaged organs.
HGF Triggers Transcription of RAD51 via IKZF1 and RUNX1 in the Absence of DNA Damage
DSBs can be triggered by external sources, including exposure to infrared (IR), UV, and environmental toxins or endogenous sources.42 Several DNA repair pathways deal with these distinct types of DNA damage. Among these, HR is an important error-free mechanism to maintain genome stability/variability.39 RAD51 is a RecA recombinase that plays a central role in HR as the loader at sites of DSBs.25, 43
Here, we demonstrated a novel mechanism that triggers RAD51 activation to elongate telomeres in hMSCs, which is induced by HGF but not by DNA damage. The formation of γ-H2AX foci is critical for an efficient response to DNA damage. We observed γ-H2AX foci and RAD51 activation after irradiation of hBM-MSCs. However, we also observed RAD51 activation upon rHGF treatment without irradiation (DNA damage), which suggests that induction of RAD51 by HGF is not triggered by DSBs.
The mechanism by which RAD51 transcription is regulated has not been clarified. Thus, we sought to identify the mediator of the HGF-RAD51 axis. We first examined the exons and introns of all known RAD51 transcripts and predicted the promoter region. Four alternative transcripts of RAD51 are encoded by chromosome 15. Three transcripts share the same transcription start site, including exon 1 and exon 3. The other transcript has a different transcription start site, including exon 2, which starts +51 bp from exon 1 (Figure S3A). We identified binding sites for IKZF1 and RUNX1 by screening for transcription-factor-binding sites in the ∼2.3 kb RAD51 putative promoter region (Figure S3A). IKZF1 has been reported to play a role in lymphocyte development and homeostasis,44 whereas RUNX1 was reported to be essential for proliferation of blood cells and MSCs as well as fate commitment in myofibroblast differentiation.43, 45 However, there have been no previous studies linking IKZF1 or RUNX1 to RAD51. In the current study, we demonstrated that IKZF1 and RUNX1 are transcriptional activators of RAD51 in hMSCs. Knockdown of IKZF1 and RUNX1 downregulated RAD51 expression, which was accompanied by telomere shortening and a reduced mtDNA copy number in rHGF-treated hBM-MSCs. The same results were observed in hE-MSCs, in which HGF is highly expressed. These results suggest IKZF1 and RUNX1 are novel transcriptional activators of RAD51 in response to HGF in hMSCs.
SUMOylation of RAD51
This study revealed that RAD51 has distinct functions in nuclei and mitochondria of hMSCs. In gel electrophoresis, a portion of RAD51 protein migrated as a more slowly migrating band than the naive form on SDS-PAGE gels and its intensity increased after rHGF treatment. Changes in protein mobility during PAGE are usually due to PTMs, such as phosphorylation, ubiquitination, and SUMOylation, and in the case of RAD51, these have been shown to be associated with changes in its function, localization during DNA repair, and binding partners.46 SUMOylation was the only PTM that could explain the >60 kD increase in molecular weight. Furthermore, a pan-inhibitor of SUMOylation, GA, resulted in the loss of the slow migrating band and decreased the telomere lengthening and higher mtDNA copy number induced by HGF.
SUMOylated RAD51 accumulates at sites of DNA damage to repair DNA, and a SUMO-interacting domain has been identified in RAD51.47 We demonstrated that the K70 residue of RAD51 was SUMOylated upon HGF stimulation by showing that the increases in telomere length and mtDNA copy number were abolished in the K70R mutant. Hence, we suggested that K70 is a direct SUMOylation site for the activation of RAD51 in mitochondria and on telomeres. Although in-depth research is needed to determine how SUMOylated RAD51 can perform both functions, we predict that it undergoes conformational changes and binds to different partner proteins, which underlies its distinct localizations and functions.
Safety and Therapeutic Efficacy of the hBM-MSC Rejuvenation Protocol Based on HGF Priming
Spontaneous telomere elongation/maintenance is common in cancer cells and is a target of cancer therapy.48 Telomere elongation/maintenance is required in stem cells and is used for regenerative medicine.8, 9 A previous study attempted to elongate telomeres by introducing human TERT, but this causes c-Myc activation in human mammary epithelial cells and thus its use in therapeutic applications has been criticized.49 In the current study, we found that HGF secretion was much higher in hE-MSCs having longer telomeres than in hBM-MSCs. Treatment of hBM-MSCs with rHGF elongated short telomeres via RAD51 induction. This result points to a safer way to rejuvenate hMSCs via treatment with rHGF without the need for genetic manipulation. DNA damage may trigger induction of RAD51. Here, we confirmed that induction of RAD51 by HGF in hBM-MSCs was not associated with DNA damage, as demonstrated by the normal chromosome status detected by karyotyping using G-banding and telomere FISH.
We investigated the therapeutic potency of rHGF-treated hMSCs in a TAA-induced liver injury mouse model. Histological analysis showed that the level of fibrosis was significantly lower in mice administered rHGF-treated hBM-MSCs than in mice administered naive hBM-MSCs. Safety was confirmed by the absence of ectopic mass formation in other organs. We injected 5 × 104 cells per mouse, a high dose, because our previous study showed that the minimum effective dose was 1 × 105 cells per rat.50 On average, rats are 10-fold heavier than mice. Although we have yet to determine the optimal cell dose in mice, we evaluated the therapeutic efficacy, engraftment, and safety of hBM-MSCs subjected to the HGF-based rejuvenation protocol. Taken together, we suggest that the activation of RAD51 by HGF could be used to improve the therapeutic efficacy of hBM-MSCs by increasing telomere length and mitochondrial ATP generation.
Materials and Methods
hMSC Culture, rHGF Treatment, and HGF Neutralization
hE-MSCs were cultured in EGM-2MV medium (Lonza, Basel, Switzerland) and hBM-MSCs (purchased from Lonza) were cultured in MSCGM medium (Lonza) at 37°C with 5% CO2. hE-MSCs were used at P15 and hBM-MSCs were used at P8. hE-MSCs were treated with an HGF-neutralizing antibody (2.5 μg/mL; Abcam, Cambridge, UK) every 8 hr to block HGF function. hBM-MSCs were treated with rHGF (10 ng/mL; R&D Systems, Minneapolis, MN) every day for 5 days. Medium was replaced daily with each addition of rHGF.
Human embryonic stem cell (hESC)-MSCs were obtained using a previously reported protocol.12 In brief, mechanically fragmented SNUhES3 hESCs (Institute of Reproductive Medicine and Population, Medical Research Center, Seoul National University Hospital, Seoul, Korea) were allowed to form embryonic bodies (EBs) in Petri dishes without fibroblast growth factor-2 (FGF2) for 14 days. Then, round EBs were attached to gelatin-coated dishes and cultivated for 16 days in low-glucose DMEM (Invitrogen) with 10% FBS (Invitrogen). Finally, the cells were expanded in EGM-2MV media (Lonza, Basel, Switzerland) and characterized by their differentiation into mesodermal lineages using FACS with MSC-specific markers. The absence of hESC contamination was confirmed by FACS with hESC markers and by testing for teratoma formation.12 After characterization, the cells were named hESC-MSCs and used in experiments.
Real-Time PCR Analysis
QIAshredder and an RNeasy mini kit (QIAGEN, Hilden, Germany) were used to prepare total RNA according to the manufacturer’s instructions. Subsequently, 1 μg RNA was transcribed into cDNA using a PrimeScript 1st strand cDNA Synthesis kit (Takara, Tokyo, Japan). PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, USA). Real-time PCR samples were run on an ABI PRISM 7500 sequence detection system (Applied Biosystems). GAPDH was used as the internal control and for normalization.
The real-time PCR primers were as follows: Oct4, forward: 5′-GAGGCAACCTGGAGAATTTG-3′, reverse: 5′-TAGCCTGGGGTACCAAAATG-3′; Nanog, forward: 5′-TTCCTTCCTCCATGGATCTG-3′, reverse: 5′-TGCTGGAGGCTGAGGTATTT-3′; RAD51, forward: 5′-GCATAAATGCCAACGATGTG-3′, reverse: 5′-GTGGTGAAACCCATTGGAAC-3′; IKZF1, forward: 5′-GGATATTGTGGCCGAAGCTA-3′, reverse: 5′-GTTTGGCGACGTTACTTGCT-3′; RUNX1, forward: 5′-CGAAGACATCGGCAGAAACT-3′, reverse: 5′-TGCCTTGTATCCTGCATCTG-3′; and GAPDH, forward: 5′-TGTGAGGAGGGGAGATTCA-3′, reverse: 5′-CAACGAATTTGGCTACAGCA-3′.
Western Blot Analysis
All hMSCs were lysed with protein lysis buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate, and 0.5% deoxycholate) containing a protease inhibitor cocktail (Roche, Indianapolis, IN). Total lysates (25 μg) were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore, Billerica, USA) by electro-blotting. Membranes were incubated with the following primary antibodies overnight at 4°C: anti-Oct4 (1:2,000; Santa Cruz Biotechnology, Santa Cruz, USA), anti-Nanog (1:1,000; Abcam), anti-RAD51 (1:500; Merck, New York, USA), and anti-TERT (1:1,000; Abcam). An anti-α-tubulin antibody (1:5,000; Sigma-Aldrich, St. Louis, MO) was used as internal housekeeping control. The immunoblotted membranes were incubated with HRP-conjugated secondary antibodies (1:2,000; Sigma-Aldrich) for 2 hr at room temperature. Quantitative analysis of immunoreactive bands was performed using ImageJ (NIH, Bethesda, USA).
TRAP and HR Assays
The TRAPEZE Telomerase Detection kit (EMD Millipore) was used to detect telomerase activity. DNA samples were separated by 12% DNA-polyacrylamide gel electrophoresis. The polyacrylamide gels consisted of 40% acrylamide, TBE electrophoresis buffer, 10% ammonium persulfate, and TEMED.
The Homologous Recombination Assay Kit (Norgen Biotek, Ontario, Canada) was used to evaluate the efficiency of HR according to the manufacturer’s instructions. PCR products were quantified by analyzing the gel images using ImageJ.
Real-Time qPCR Analysis of Telomere Length
Real-time qPCR was used to evaluate telomere length as described previously.19 Reactions were performed on an ABI PRISM 7500 with 40 cycles of 95°C for 15 s and 54°C for 2 min. For PCR of 36B4, a single copy gene, the program included 40 cycles of 95°C for 15 s and 58°C for 1 min. The primer sequences used to analyze telomere length and 36B4 were as follows: Tel1: 5′-GGTTTTTGAGGGTGAGGGTGAGGGTGAGGGTGAGGGT-3′, Tel2: 5′-TCCCGACTATCCCTATCCCTATCCCTATCCCTATCCCTA-3′, 36B4u: 5′-CAGCAAGTGGGAAGGTGTAATCC-3′, and 36B4d: 5′-CCCATTCTATCATCAACGGGTACAA-3′.
FACS Analysis
hMSCs were washed with FACS Dulbecco’s PBS (DPBS) buffer (Gibco, NY, USA) containing 2.5% fetal bovine serum (Gibco) and incubated for 30 min with an anti-PCNA antibody conjugated to fluorescein isothiocyanate (1:100; AbD Serotec, CA, USA) for FACS. At least 104 events were analyzed on a FACSCalibur system (BD Biosciences, CA, USA) with CellQuest software.
Loss- and Gain-of-Function Experiments
Specific gene knockdown was achieved by mixing siRNA with Metafectene PRO transfection reagent (Biontex, München, Germany). Before transfection, siRNAs targeting RAD51, IKZF1, and RUNX1 (all from Dharmacon, Lafayette, CO) and Metafectene PRO were diluted in culture medium lacking fetal bovine serum at room temperature. The two solutions were then combined and incubated for 15 min at room temperature. After incubation, the RNA-transfection reagent complexes were added dropwise to the culture dish. After incubation for 6 hr at 37°C in a CO2 incubator, the medium was replaced with fresh culture medium.
A CMV-lentivirus for RAD51 overexpression was purchased from Sirion Biotech (Am Klopferspitz, Germany). The lentivirus supernatant was diluted with complete culture medium containing 1 μg/mL polybrene and added to hMSCs. More than 1 × 107 lentiviral particles were added to each culture plate (50%–60% cell confluency). After 24 hr, the medium was replaced with fresh culture medium and cultures were incubated at 37°C to allow the virus-infected cells to grow.
HGF Array and ELISA
HGF array C1 (Raybiotech, Norcross, USA) was used to quantitatively compare differences in secreted growth factors between hE-MSCs and hBM-MSCs. Culture supernatants of hMSCs were processed according to the manufacturer’s protocols. Imaging was performed with a chemiluminescence bioimage analyzer (Alpha Innotech, San Jose, USA).
To measure secreted HGF and IGFBP1, culture supernatants of hMSCs were analyzed using a human HGF Quantikine ELISA kit or a human free IGFBP1 Quantikine ELISA kit (R&D Systems). Results were read on an ELISA reader (Multiskan GO Microplate Spectrophotometer, Thermo Scientific, Waltham, USA) at 450 nm.
G-banding and Telomere FISH
Cell division was blocked at mitotic metaphase by treatment with 0.1 g/mL colcemid (Gibco-Invitrogen) for 2 hr. Then, hBM-MSCs were trypsinized and resuspended in 0.075 M KCl for 20 min at 37°C. After incubation, the cells were fixed in cold methanol:acetic acid (3:1). G-band standard staining was used to visualize chromosomes. When at least 20 cells were detected, karyotypes were analyzed and reported according to the International System for Human Cytogenetic Nomenclature.
For telomere FISH, mitotic cells were collected by mitotic shake-off and swollen in hypotonic solution (0.075 M KCl) at 37°C for 20 min. Cells were then fixed in freshly prepared methanol:glacial acetic acid (3:1). Fixed cells were dropped onto pre-cleaned slides and left to dry overnight. FISH was performed according to the manufacturer’s instructions (Cellay, MA, USA). In brief, the slides were hybridized with Pan-Telomere OligoFISH probes (Cellay), incubated for 10 min at 37°C, washed in 2 × SSC under agitation to float off the coverslips, and then incubated in IsoThermal Wash Solution (Cellay) for 5 min at room temperature. Finally, the slides were rinsed in 2 × SSC, mounted with antifade mounting medium containing DAPI, and analyzed by fluorescence microscopy.
Immunocytochemistry
Cultured hMSCs were fixed in acetone for 3 min at −20°C and washed three times with 1 × PBS. Non-specific binding was blocked with DPBS (Gibco) containing 0.1% bovine serum albumin and 0.05% Triton X-100 for 1 hr. hMSCs were subsequently incubated overnight at 4°C with specific antibodies. After three washes for 5 min, the cells were incubated with secondary antibodies conjugated to Alexa Fluor fluorescent dyes (1:200; Invitrogen) for 2 hr at room temperature. Images were obtained by confocal microscopy (Carl Zeiss LSM710, Germany). UV irradiation was performed for 2 hr under a UV lamp on a clean bench (100–180 nm).
Mouse Liver Fibrosis Model and Cell Transplantation
BALB/c-nude mice (male, 12–13 weeks old, 20–25 g) were used for all animal experiments. Mice were administered TAA (200 mg/kg; Sigma-Aldrich) by intraperitoneal injection three times per week for 14 days. The negative control group was injected with 0.9% saline. Cells were transplanted by intracardiac injection 1 day after the first TAA injection (Figure 6A). Mice in the negative control group and the TAA-injected positive control group were administered DPBS. Mice in the cell transplantation groups were administered non-treated hBM-MSCs or hBM-MSCs treated with 10 ng/mL rHGF for 5 days. After recovery for 2 days, mice were injected with TAA three times per week until 14 days after transplantation.
Mice were injected with stained hBM-MSCs to track these cells in liver tissue. Cells were stained with 4 μg/mL CellTracker CM-DiI (Invitrogen, MA, USA) at 37°C for 24 hr. At 14 days after cell transplantation, liver tissue was harvested for histology.
CytoChIP
hBM-MSCs (P8) were fixed in 0.8% formaldehyde for 10 min and collected in a tube. The supernatant was discarded, nuclear-cytoplasmic buffer (0.1% digitonin prepared in PBS containing a phosphatase inhibitor cocktail and a protease inhibitor cocktail) was added, and the sample was carefully pipetted several times. After centrifugation, the supernatant and pellet were retained as the cytoplasmic and nuclear fractions, respectively. To shear DNA, both fractions were sonicated four times with BIORUPTOR (30 s on and 30 s off per cycle). The cytoplasmic fraction was used for immunoprecipitation, and the nuclear fraction was used to confirm DNA fragmentation. The immunoprecipitation sample was supplemented with an anti-RAD51 or anti-polymerase γ antibody (1 μg) and rotated overnight. Protein A/G agarose beads were added to pull down the antibodies and sequentially washed three times with washing buffers (low and high LiCl). Samples were incubated for 4 hr at 65°C for decrosslinking and recovered using a PCR purification kit (QIAGEN). Nuclear and cytoplasmic fractionation was confirmed by western blot analysis. mtDNA precipitation using anti-RAD51 and anti-polymerase γ antibodies was analyzed by quantitative and semiquantitative PCR.
Statistical Analysis
Statistical comparisons were conducted using GraphPad Prism 6 (GraphPad Software, La Jolla, USA). Quantitative data were reported as the mean ± SEM. The unpaired t test or an analysis of variance was used to analyze each group. p values < 0.05 were regarded as statistically significant.
Study Approval
The animal experiments were approved by the Institutional Animal Care and Use Committee of Seoul National University Hospital, Korea (17-0042-S1A0).
Author Contributions
Conceptualization, E.J.L., H.-S.K; Methodology, E.J.L., I.H., J.N.P., K.C.K., J.Y.L., G.-H.K., C.-M.K., I.K; Data Analysis, E.J.L., S.-Y.L., H.-S.K; Writing of Original Draft, E.J.L., I.H., I.K.; Review and Editing of the Manuscript, E.J.L., H.-S.K.
Conflicts of Interest
The authors have declared that no conflicts of interest exist.
Acknowledgments
This study was supported by grants from the Korea Health Technology R&D Project “Strategic Center of Cell and Bio Therapy for Heart, Diabetes Center” (HI17C2085) and “Korea Research-Driven Hospital” (HI14C1277) through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (MHW), Republic of Korea. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental Information includes fives figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.12.015.
Supplemental Information
References
- 1.Parekkadan B., Milwid J.M. Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 2010;12:87–117. doi: 10.1146/annurev-bioeng-070909-105309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Detry O., Vandermeulen M., Delbouille M.H., Somja J., Bletard N., Briquet A., Lechanteur C., Giet O., Baudoux E., Hannon M. Infusion of mesenchymal stromal cells after deceased liver transplantation: a phase I-II, open-label, clinical study. J. Hepatol. 2017;67:47–55. doi: 10.1016/j.jhep.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Suk K.T., Yoon J.H., Kim M.Y., Kim C.W., Kim J.K., Park H., Hwang S.G., Kim D.J., Lee B.S., Lee S.H. Transplantation with autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: phase 2 trial. Hepatology. 2016;64:2185–2197. doi: 10.1002/hep.28693. [DOI] [PubMed] [Google Scholar]
- 4.Bertolo A., Mehr M., Janner-Jametti T., Graumann U., Aebli N., Baur M., Ferguson S.J., Stoyanov J.V. An in vitro expansion score for tissue-engineering applications with human bone marrow-derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2016;10:149–161. doi: 10.1002/term.1734. [DOI] [PubMed] [Google Scholar]
- 5.Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 6.Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S., Wright W.E. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 7.Kang S.K., Putnam L., Dufour J., Ylostalo J., Jung J.S., Bunnell B.A. Expression of telomerase extends the lifespan and enhances osteogenic differentiation of adipose tissue-derived stromal cells. Stem Cells. 2004;22:1356–1372. doi: 10.1634/stemcells.2004-0023. [DOI] [PubMed] [Google Scholar]
- 8.Shi S., Gronthos S., Chen S., Reddi A., Counter C.M., Robey P.G., Wang C.Y. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat. Biotechnol. 2002;20:587–591. doi: 10.1038/nbt0602-587. [DOI] [PubMed] [Google Scholar]
- 9.Simonsen J.L., Rosada C., Serakinci N., Justesen J., Stenderup K., Rattan S.I., Jensen T.G., Kassem M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol. 2002;20:592–596. doi: 10.1038/nbt0602-592. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi M., Takeuchi K., Kohara A., Satoh M., Shioda S., Ozawa Y., Ohtani A., Morita K., Hirano T., Terai M. Chromosomal instability in human mesenchymal stem cells immortalized with human papilloma virus E6, E7, and hTERT genes. In Vitro Cell. Dev. Biol. Anim. 2007;43:129–138. doi: 10.1007/s11626-007-9021-9. [DOI] [PubMed] [Google Scholar]
- 11.Lee E.J., Xu L.J., Kim G.H., Kang S.K., Lee S.W., Park S.H., Kim S., Choi T.H., Kim H.S. Regeneration of peripheral nerves by transplanted sphere of human mesenchymal stem cells derived from embryonic stem cells. Biomaterials. 2012;33:7039–7046. doi: 10.1016/j.biomaterials.2012.06.047. [DOI] [PubMed] [Google Scholar]
- 12.Lee E.J., Lee H.N., Kang H.J., Kim K.H., Hur J., Cho H.J., Lee J., Chung H.M., Cho J., Cho M.Y. Novel embryoid body-based method to derive mesenchymal stem cells from human embryonic stem cells. Tissue Eng. Part A. 2010;16:705–715. doi: 10.1089/ten.tea.2008.0596. [DOI] [PubMed] [Google Scholar]
- 13.Hara S., Nakashiro K., Goda H., Hamakawa H. Role of Akt isoforms in HGF-induced invasive growth of human salivary gland cancer cells. Biochem. Biophys. Res. Commun. 2008;370:123–128. doi: 10.1016/j.bbrc.2008.03.042. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi S., Morishita R., Higaki J., Aoki M., Moriguchi A., Kida I., Yoshiki S., Matsumoto K., Nakamura T., Kaneda Y. Autocrine-paracrine effects of overexpression of hepatocyte growth factor gene on growth of endothelial cells. Biochem. Biophys. Res. Commun. 1996;220:539–545. doi: 10.1006/bbrc.1996.0440. [DOI] [PubMed] [Google Scholar]
- 15.Oyagi S., Hirose M., Kojima M., Okuyama M., Kawase M., Nakamura T., Ohgushi H., Yagi K. Therapeutic effect of transplanting HGF-treated bone marrow mesenchymal cells into CCl4-injured rats. J. Hepatol. 2006;44:742–748. doi: 10.1016/j.jhep.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Yu Y., Yao A.H., Chen N., Pu L.Y., Fan Y., Lv L., Sun B.C., Li G.Q., Wang X.H. Mesenchymal stem cells over-expressing hepatocyte growth factor improve small-for-size liver grafts regeneration. Mol. Ther. 2007;15:1382–1389. doi: 10.1038/sj.mt.6300202. [DOI] [PubMed] [Google Scholar]
- 17.Shams S., Mohsin S., Nasir G.A., Khan M., Khan S.N. Mesenchymal stem cells pretreated with HGF and FGF4 can reduce liver fibrosis in mice. Stem Cells Int. 2015;2015:747245. doi: 10.1155/2015/747245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodrigues M., Griffith L.G., Wells A. Growth factor regulation of proliferation and survival of multipotential stromal cells. Stem Cell Res. Ther. 2010;1:32. doi: 10.1186/scrt32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samsonraj R.M., Raghunath M., Hui J.H., Ling L., Nurcombe V., Cool S.M. Telomere length analysis of human mesenchymal stem cells by quantitative PCR. Gene. 2013;519:348–355. doi: 10.1016/j.gene.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 20.Pickett H.A., Reddel R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015;22:875–880. doi: 10.1038/nsmb.3106. [DOI] [PubMed] [Google Scholar]
- 21.Le S., Moore J.K., Haber J.E., Greider C.W. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics. 1999;152:143–152. doi: 10.1093/genetics/152.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 23.Zalzman M., Falco G., Sharova L.V., Nishiyama A., Thomas M., Lee S.L., Stagg C.A., Hoang H.G., Yang H.T., Indig F.E. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010;464:858–863. doi: 10.1038/nature08882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon Y.W., Paek J.S., Cho H.J., Lee C.S., Lee H.J., Park I.H., Roh T.Y., Kang C.M., Yang H.M., Park Y.B. Role of Zscan4 in secondary murine iPSC derivation mediated by protein extracts of ESC or iPSC. Biomaterials. 2015;59:102–115. doi: 10.1016/j.biomaterials.2015.03.031. [DOI] [PubMed] [Google Scholar]
- 25.San Filippo J., Sung P., Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 26.Chen F., Nastasi A., Shen Z., Brenneman M., Crissman H., Chen D.J. Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes Rad51 and Rad52. Mutat. Res. 1997;384:205–211. doi: 10.1016/s0921-8777(97)00020-7. [DOI] [PubMed] [Google Scholar]
- 27.Marampon F., Gravina G.L., Ju X., Vetuschi A., Sferra R., Casimiro M.C., Pompili S., Festuccia C., Colapietro A., Gaudio E. Cyclin D1 silencing suppresses tumorigenicity, impairs DNA double strand break repair and thus radiosensitizes androgen-independent prostate cancer cells to DNA damage. Oncotarget. 2016;7:64526. doi: 10.18632/oncotarget.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., Kimura K., Sodeoka M., Yoshida M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009;16:133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Berg K., Braun C., Krug I., Schrenk D. Evaluation of the cytotoxic and mutagenic potential of three ginkgolic acids. Toxicology. 2015;327:47–52. doi: 10.1016/j.tox.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Sage J.M., Gildemeister O.S., Knight K.L. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem. 2010;285:18984–18990. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sage J.M., Knight K.L. Human Rad51 promotes mitochondrial DNA synthesis under conditions of increased replication stress. Mitochondrion. 2013;13:350–356. doi: 10.1016/j.mito.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyrka A.R., Carpenter L.L., Kao H.T., Porton B., Philip N.S., Ridout S.J., Ridout K.K., Price L.H. Association of telomere length and mitochondrial DNA copy number in a community sample of healthy adults. Exp. Gerontol. 2015;66:17–20. doi: 10.1016/j.exger.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramos A., Santos C., Alvarez L., Nogués R., Aluja M.P. Human mitochondrial DNA complete amplification and sequencing: a new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis. 2009;30:1587–1593. doi: 10.1002/elps.200800601. [DOI] [PubMed] [Google Scholar]
- 34.Rossi M.N., Carbone M., Mostocotto C., Mancone C., Tripodi M., Maione R., Amati P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicholls T.J., Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014;56:175–181. doi: 10.1016/j.exger.2014.03.027. [DOI] [PubMed] [Google Scholar]
- 36.Marión R.M., Blasco M.A. Telomeres and telomerase in adult stem cells and pluripotent embryonic stem cells. Adv. Exp. Med. Biol. 2010;695:118–131. doi: 10.1007/978-1-4419-7037-4_9. [DOI] [PubMed] [Google Scholar]
- 37.Lafferty-Whyte K., Cairney C.J., Will M.B., Serakinci N., Daidone M.G., Zaffaroni N., Bilsland A., Keith W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene. 2009;28:3765–3774. doi: 10.1038/onc.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inui T., Shinomiya N., Fukasawa M., Kobayashi M., Kuranaga N., Ohkura S., Seki S. Growth-related signaling regulates activation of telomerase in regenerating hepatocytes. Exp. Cell Res. 2002;273:147–156. doi: 10.1006/excr.2001.5446. [DOI] [PubMed] [Google Scholar]
- 39.Aguilera A., Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 40.Cho N.W., Dilley R.L., Lampson M.A., Greenberg R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014;159:108–121. doi: 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bratic A., Larsson N.G. The role of mitochondria in aging. J. Clin. Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price B.D., D’Andrea A.D. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim W., Barron D.A., San Martin R., Chan K.S., Tran L.L., Yang F., Ressler S.J., Rowley D.R. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc. Natl. Acad. Sci. USA. 2014;111:16389–16394. doi: 10.1073/pnas.1407097111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida T., Landhuis E., Dose M., Hazan I., Zhang J., Naito T., Jackson A.F., Wu J., Perotti E.A., Kaufmann C. Transcriptional regulation of the Ikzf1 locus. Blood. 2013;122:3149–3159. doi: 10.1182/blood-2013-01-474916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lacaud G., Gore L., Kennedy M., Kouskoff V., Kingsley P., Hogan C., Carlsson L., Speck N., Palis J., Keller G. Runx1 is essential for hematopoietic commitment at the hemangioblast stage of development in vitro. Blood. 2002;100:458–466. doi: 10.1182/blood-2001-12-0321. [DOI] [PubMed] [Google Scholar]
- 46.Popova M., Henry S., Fleury F. Posttranslational modifications of Rad51 protein and its direct partners: role and effect on homologous recombination – mediated DNA repair. In: Kruman I., editor. DNA Repair. InTech; 2011. pp. 143–160. [Google Scholar]
- 47.Shima H., Suzuki H., Sun J., Kono K., Shi L., Kinomura A., Horikoshi Y., Ikura T., Ikura M., Kanaar R. Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage. J. Cell Sci. 2013;126:5284–5292. doi: 10.1242/jcs.133744. [DOI] [PubMed] [Google Scholar]
- 48.Lu R., Pal J., Buon L., Nanjappa P., Shi J., Fulciniti M., Tai Y.T., Guo L., Yu M., Gryaznov S. Targeting homologous recombination and telomerase in Barrett’s adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene. 2014;33:1495–1505. doi: 10.1038/onc.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J., Hannon G.J., Beach D.H. Risky immortalization by telomerase. Nature. 2000;405:755–756. doi: 10.1038/35015674. [DOI] [PubMed] [Google Scholar]
- 50.Lee E.J., Choi E.K., Kang S.K., Kim G.H., Park J.Y., Kang H.J., Lee S.W., Kim K.H., Kwon J.S., Lee K.H. N-cadherin determines individual variations in the therapeutic efficacy of human umbilical cord blood-derived mesenchymal stem cells in a rat model of myocardial infarction. Mol. Ther. 2012;20:155–167. doi: 10.1038/mt.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.