

Abstract
Epithelial-to-mesenchymal transition (EMT) has been closely linked with therapy resistance and cancer stem cells (CSCs). However, EMT pathways have proven challenging to therapeutically target. MicroRNA 145 (miR-145) targets multiple stem cell transcription factors and its expression is inversely correlated with EMT. Therefore, we hypothesized that miR-145 represents a therapeutic target to reverse snail family transcriptional repressor 1 (SNAI1)-mediated stemness and radiation resistance (RT). Stable expression of SNAI1 in DLD1 and HCT116 cells (DLD1-SNAI1; HCT116-SNAI1) increased expression of Nanog and decreased miR-145 expression compared to control cells. Using a miR-145 luciferase reporter assay, we determined that ectopic SNAI1 expression significantly repressed the miR-145 promoter. DLD1-SNAI1 and HCT116-SNAI1 cells demonstrated decreased RT sensitivity and, conversely, miR-145 replacement significantly enhanced RT sensitivity. Of the five parental colon cancer cell lines, SW620 cells demonstrated relatively high endogenous SNAI1 and low miR-145 levels. In the SW620 cells, miR-145 replacement decreased CSC-related transcription factor expression, spheroid formation, and radiation resistance. In rectal cancer patient-derived xenografts, CSC identified by EpCAM+/aldehyde dehydrogenase (ALDH)+ demonstrated high expression of SNAI1, c-Myc, and Nanog compared with non-CSCs (EpCAM+/ALDH−). Conversely, patient-derived CSCs demonstrated low miR-145 expression levels relative to non-CSCs. These results suggest that the SNAI1:miR-145 pathway represents a novel therapeutic target in colorectal cancer to overcome RT resistance.
Keywords: SNAI1, microRNA-145, colorectal cancer, radiation, stem cell, resistance
Cancer stem cells are recognized as a critical mediator of radiation resistance, although our ability to therapeutically target this cell population is limited. Camp and colleagues show that transcriptional factor SNAI1-driven stemness and therapy resistance can be effectively targeted by delivery of microRNA 145.
Introduction
Colorectal cancer is the third most prevalent cancer and the second highest cause of cancer-related deaths in the United States.1 Rectal cancer, a subset of colorectal cancer, is located in the pelvis within 12 cm from the anus. Patients with rectal cancer with either a muscle-invasive primary tumor (American Joint Committee on Cancer [AJCC] stage 2) or involved lymph nodes (AJCC stage 3) are particularly challenging, with high rates of disease recurrence. Nearly 13 years ago, the landmark randomized German Rectal Cancer Trial (CAO/ARO/AIO-94) showed that rectal cancer patients treated with neoadjuvant 5-fluorouracil (5FU) chemoradiation (5FU/RT) had fewer treatment-associated complications and less local disease recurrence compared with patients receiving surgery followed by 5-FU/RT.2 This regimen remains the current standard of care today. Although additional agents have shown promise in colorectal cancer cell lines in vitro and in vivo, therapies combining these agents with 5FU/RT have significantly increased toxicity in patients, without improvement in clinical response.3, 4, 5, 6 The lack of recent advances in rectal cancer therapy highlights the critical need for precision medicine strategies and novel therapy targets to improve outcomes.
Studies in a variety of cancer types have shown that cancer stem cells (CSCs) are enriched following both chemotherapy and radiation, supporting the concept that CSCs are resistant to conventional therapies, including radiotherapy.7, 8, 9 The existence of CSCs offers a potential explanation for cancer treatment failure and suggests that CSCs should be targeted to enhance conventional therapies. In epithelial tumors such as colorectal cancer, CSC phenotype and epithelial-mesenchymal transition (EMT) program cooperate to impact tumor progression, metastasis, and therapeutic resistance. Growing evidence supports that cancer cells enter the EMT process and gain CSC pluripotency simultaneously. The enriched CSC population induced by EMT events leads to a therapeutic resistant phenotype.10, 11, 12 The zinc finger molecule snail family transcriptional repressor 1 (SNAI1, alternatively referred as Snail) is a transcriptional factor that plays a critical role in driving the EMT program.11 However, the impact of EMT and CSC-related molecular alteration on radiotherapy sensitivity in colorectal cancer is largely unexplored.
MicroRNAs (miRNAs) are small non-coding RNAs, with a distinct capacity to regulate multiple gene expressions simultaneously. For example, microRNA 145 (miR-145) has demonstrated tumor suppressor properties in various cancers, including colorectal cancer.13, 14, 15 miR-145 represses mediators of EMT, such as insulin-like growth factor 1 receptor (IGF1R), fascin-1, and paxillin, to suppress cell proliferation and metastasis in colorectal cancer.16, 17, 18 Importantly, miR-145 is also an essential component of the p53 regulatory network.19 p53 is a critical repressor of c-myc mediated through miR-145 induction. Besides the role in cancer, miR-145 is a master regulator of differentiation in human embryonic stem cells as a central repressor of transcription factors OCT4, SOX2, and KLF4, which critically maintain the stemness.20 Therefore, we hypothesize that a reciprocal relationship exists between miR-145 and EMT that influences the CSC phenotype and radiation response in colorectal cancer. We further postulate that a SNAI1:miR-145 signaling axis facilitates the CSC phenotype mediated by stem cell self-renewal mediators, such as Nanog and c-Myc.21, 22, 23
Results
SNAI1 Level Is Consistently Elevated in Rectal Cancer Tissue Samples
Oncomine databases were reviewed to determine SNAI1, SNAI2, ZEB1, and ZEB2 mRNA expression levels in human rectal cancers. Compared with SNAI2, ZEB1, and ZEB2 levels, SNAI1 was consistently elevated in all cohorts evaluated, ranging from 1.3- to 4.5-fold greater than normal rectal tissue samples (Table 1). Similarly, data mined from the TCGA using bioportal demonstrated SNAI1 has the highest frequency of amplification and/or overexpression in colorectal adenocarcinoma compared with SNAI2, ZEB1, and ZEB2 (Figure 1). Considering the clinical relevance of SNAI1 and the association with a CSC phenotype, we established SNAI1-overexpressing DLD1 and HCT116 stable cell lines (DLD1-SNAI1; HCT116-SNAI1) to further explore the therapeutic significance of SNAI1 as a mediator of radiation resistance. Expression of SNAI1 mRNA and protein was confirmed in the two SNAI1-overexpressing cell lines (Figures S1A and S1B).
Table 1.
EMT Transcription Factor Expression in Rectal Cancer Specimens
Figure 1.

Elevated SNAI1 Expression in Human Colorectal Cancer Datasets
Whole exome and RNA Seq data of colorectal adenocarcinoma from TCGA was mined for the frequency of SNAI1, SNAI2, ZEB1, and ZEB2 using bioportal.53
Overexpression of SNAI1 Sustains Stemness Maintenance in Colorectal Cancer Cell Lines
Initially, we examined the expression of stem cell transcription factors in five established colorectal cancer cell lines and observed that Nanog and SNAI1 were highly expressed in the metastatic SW620 cell line when compared to the other four cell lines (Figure 2A). The SW620 cell line was derived from a metastatic lymph node from a Duke’s C colon cancer patient with widely metastatic disease.24 To further explore the role of SNAI1 in sustaining cellular stemness, we selected the DLD1 and HCT116 colon cancer cell lines that demonstrated relatively low expression of established CSC transcription factors, such as c-Myc, Nanog, and Sox-2 (Figure 2A). Ectopic expression of SNAI1 in DLD1-SNAI1 and HCT116-SNAI1 cells induced the expression of Nanog compared with vector control cells (Figure 2B). In addition, we observed overexpression of SNAI1 slightly decreased E-cadherin in both the DLD1-SNAI1 and HCT116-SNAI1 cells compared to the vector control cells (Figure S1B). Recent evidence suggests that although human SNAI1 and SNAI2 share a 48% identical amino acid sequence,11 ectopic expression of SNAI1 and SNAI2 exhibited opposing functions, especially on Nanog-driven induced pluripotent stem cell (iPSC) programming.25 It is possible that alternative transcriptional repressors play a dominant role in regulating E-cadherin expression in our cell lines.
Figure 2.

Ectopic Expression of SNAI1 Induces CSC Phenotype
(A) Western blot analysis for expression of critical EMT mediators and CSC transcription factors in parental colon cancer cell lines. (B) Western blot analysis and relative density quantitation of EMT biomarker and stem-cell-related transcription factors in ectopic-expressing SNAI1 cell lines compared with empty vector control cells. (C and D) Limited dilution spheroid assay comparing DLD1-SNAI1 (C) and HCT116-SNAI1 (D) cells with empty vector (Vec) control cells (data are shown as mean ± SD; n = 12; * indicated p < 0.05).
To evaluate the impact of SNAI1 on the cancer cell self-renewal ability, we performed an in vitro spheroid assay, with limited dilution of DLD1-SNAI1 and HCT116-SNAI1 cells. Compared with the vector control cells (DLD1-Vec; HCT116-Vec), both DLD1-SNAI1 and HCT116-SNAI1 cells were able to generate significantly more spheroids than the empty vector controls (Figures 2C and 2D). Our data indicated that colorectal cancer cells with high SNAI1 expression acquired a CSC phenotype associated with high expression of critical cancer stem cell transcription factors.
Overexpression of SNAI1 Confers a Radiation-Resistant Phenotype in Colorectal Cancer Cells
Based on the association of EMT with increased cellular survival, we decided to investigate whether overexpression of SNAI1 resulted in radiation resistance. At 10 days following radiation, the DLD1-SNAI1 cells demonstrated increased colony formation compared with DLD1-Vec cells at 2, 4, and 6 Gy radiation (p < 0.05 at all doses) (Figure 3A). The maximal difference was observed at 4 Gy, with DLD1-SNAI1 cells demonstrating a 3-fold greater colony formation than DLD1-Vec cells. Short-term cell viability following radiation demonstrated similar findings (Figure 3B). DLD1-SNAI1 cells displayed radiation resistance at 96 hr, with a 1.5-fold increased cell viability observed in DLD1-SNAI1 cells compared to DLD1-Vec cells at the 4-Gy dose. The differences in viability were consistent across all doses tested (p < 0.05 at all doses). Similarly, SNAI1 overexpression also induced radiation resistance in HCT116 cells comparing to vector control cells (Figure S2). Oxaliplatin is a platinum-based chemotherapy drug for advanced colorectal cancer treatment and is being tested as an agent to enhance current neoadjuvant chemoradiation strategies for rectal cancer.26, 27 Therefore, we also assessed the oxaliplatin therapeutic sensitivity on DLD1-SNAI1 and HCT116-SNAI1 cells based on cell viability at 72 hr after treatment. In addition to radiation resistance, SNAI1 overexpression also decreased oxaliplatin sensitivity in both DLD1 and HCT116 cells compared to vector control cells (p < 0.05 at all doses) (Figures S6A and S6B).
Figure 3.

Ectopic Expression of SNAI1 Increases Resistance to Radiation Therapy
(A) Clonogenic assay on DLD1-SNAI1 cells and vector control cells treated with increasing doses of radiation. (B) Ectopic SNAI1 expression enhanced cancer cell viability after radiation treatment (data are shown as mean ± SD; n = 3; * indicated as p < 0.05).
Expression of miR-145 Inversely Correlates with Stem-Cell-Associated Biomarkers
Previous reports have demonstrated that miR-145 directly targets critical stem cell transcription factors and EMT mediators.20, 28, 29 Considering the ability of SNAI1 to mediate stem-like properties, we decided to explore the relationship between SNAI1 and miR-145. We determined the expression of miR145 in five colorectal cancer cell lines and observed SW620 with high endogenous SNAI1 had the lowest miR-145 expression (Figure 4A). In DLD1-SNAI1 cells, a significant decrease (36%) in miR-145 expression was observed compared with the control cells (Figure 4B). Similarly, miR-145 expression was even further reduced in the HCT116-SNAI1 cells when compared with the vector control cells (Figure 4C).
Figure 4.

SNAI1 Represses miR-145 Promoter Activity and Expression in Colorectal Cancer Cells
(A) Relative miR-145 expression level assessed by real-time PCR in parental colorectal cancer cell lines: HCT116 cells versus other four colorectal cancer cell lines (data are shown as mean ± SD; n = 3; * indicated p < 0.05). (B and C) Relative miR-145 expression level of (B) DLD1-SNAI1 cells and (C) HCT116-SNAI1 cells compared to vector control cells (data are shown as mean ± SD; n = 3; * indicated p < 0.05). (D and E) Luciferase reporter activity of (D) DLD1-SNAI1 and (E) HCT116-SNAI1 cells compared to vector control cells (data are shown as mean ± SD; n = 3; *p < 0.05).
SNAI1 Represses miR-145 Promoter Activity in Colorectal Cancer Cells
Our data indicated that there is an inverse correlation between the expression of miR-145 and SNAI1 in the parental colorectal cancer panel of cell lines and SNAI1-overexpressing stable cell lines. These results prompted us to investigate SNAI1-mediated negative regulation of miR-145 as a potential mechanism associated with radiation resistance. In the miR-145 promoter region, we identified the SNAI1 consensus binding site core sequence CA(C/G)(C/G)TG with the upstream CT rich region.30, 31 To determine whether SNAI1 represses the miR-145 promoter activity, we transfected the ectopic SNAI1-overexpressing colorectal cancer cell lines with the luciferase reporter plasmid containing the putative miR-145 promoter sequence.32 In both the DLD1-SNAI1 and HCT116-SNAI1 cell lines, miR-145 promoter activity was suppressed by 95% and 97%, respectively, when compared with vector-transfected controls (Figures 4D and 4E). Findings were similar to previous reports that demonstrated ZEB2 binds to the E-box region of the miR-145 promoter.33
Introduction of miR-145 Can Sensitize SNAI1-Overexpressing Cells to Radiation Therapy
In order to assess miR-145 as an effective therapeutic target, we introduced miR-145 to DLD1-SNAI1 and SW620 cells to test if miR-145 delivery can overcome SNAI1-mediated radiation resistance. We employed the clonogenic assay to evaluate cancer cell survival post-transfection of miR-145, followed by radiation therapy. miR-145 delivery increased the radiation sensitivity in DLD1-SNAI1 cells compared with the scramble (scr) control (Figures 5A and 5B). Interestingly, miR-145 did not increase radiation sensitivity in the control cells, suggesting the effect was limited to cancer cells with high SNAI1 levels and a CSC phenotype. In SW620 parental cells that have high endogenous levels of SNAI1, we observed a significant increase in radiation sensitivity when miR-145 was delivered compared with scr control (Figures 5C and S4B). miR-145 expression after transfection was confirmed by real-time PCR (Figures S3A, S3B, and S4A). In addition, the delivery of miR-145 also sensitized SW620 cells to oxaliplatin therapy (Figure S6C).
Figure 5.

miR-145 Sensitizes SNAI1-Overexpressing Colorectal Cancer Cells to Radiation Therapy
(A and B) Clonogenic assay (A) and quantitation (B) following radiation of DLD1-SNAI1 cells after transfection of miR-145 compared with scr vector (data are shown as mean ± SD; n = 3; * indicated p < 0.05, DLD1-SNAI1/scr versus DLD1-Vec/scr; ** indicated p < 0.05, DLD1-SNAI1/miR-145 versus DLD1-SNAI1/scr). (C) Clonogenic assay following radiation of SW620 cells after transfection of miR-145 compared with scr control for 48 hr (data are shown as mean ± SD; n = 3; * indicated p < 0.05). (D) Limited dilution in vitro spheroid assay of SW620 cells after transfection of miR-145 compared with scr control for 48 hr (data are shown as mean ± SD; n = 6; * indicated p < 0.05). (E) Western blot analysis of EMT mediators and miR-145 targets in SW620 cells after transfection with miR-145 and scr control for 48 hr.
To further explore the potential mechanism of the miR-145 therapeutic effect, we evaluated the ability of miR-145 to inhibit the stem cell phenotype, including cancer cell self-renewal capacity and expression of critical stem cell transcription factors. Using an in vitro spheroid formation assay, we evaluated SW620 cells after miR-145 transfection in a limiting dilution fashion. Our results showed that miR-145 significantly reduced spheroid formation in SW620 cells compared to scr control after transfection (Figure 5D). We also determined the expression of stem-cell-related transcription factors and EMT biomarkers after miR-145 transfection in SW620 cells. Prior reports have demonstrated that miR-145 directly targets c-Myc, OCT4, and KLF4,20, 32 but not SNAI1 and Nanog. In the SW620 cells, miR-145 significantly downregulated expression of multiple transcription factors, including c-Myc, KLF4, SNAI1, Oct4, and Nanog (Figure 5E). We further confirmed both SNAI1 and Nanog do not contain the 7-mer miR-145 seed site in their 3′ UTRs and transcript coding region by the TargetScan-microRNA target prediction program.34 Therefore, miR-145 may indirectly downregulate expression of SNAI1 and Nanog, possibly through c-Myc. In sum, miR-145 represents a key molecular regulator of both the CSC phenotype and SNAI1-mediated radiation resistance. miR-145 delivery may represent a future molecular agent to specifically target the CSC in order to overcome radiation resistance.
Detection of Functional SNAI1:miR145 Axis in EpCAM+/ALDH+ Enriched CSC Population in Colorectal Cancer PDX Tumors
Prior studies have confirmed aldehyde dehydrogenase (ALDH) as an effective CSC marker in fresh human colorectal cancer samples.1, 35 Therefore, to evaluate the clinical relevance of the SNAI1:miR-145 axis, we established multiple rectal cancer patient-derived xenografts (PDXs) and isolated CSCs using EpCAM and ALDH biomarkers. In three PDX models evaluated (referred to as MRC02, MRC07, and MRC13), we found the percentage of the EpCAM+/ALDH+ double positive population in tumor tissue ranges from 0.26% to 3.47% (Table 2). Representative flow cytometry analysis data are provided in Figure S5. We used in vitro and in vivo approaches to assess the self-renewal ability of EpCAM+/ALDH+ cells as a functional assessment of the CSC. We observed that the EpCAM+/ALDH+ cell subpopulation could generate tumor spheroids, but the EpCAM+/ALDH− subpopulation did not (Table 3). The in vivo tumorigenicity assay, in which EpCAM+/ALDH+ and EpCAM+/ALDH− cells were injected subcutaneously in a limited dilution fashion into NSG mice, confirmed that EpCAM+/ALDH+ cells displayed greater tumorigenicity than the EpCAM+/ALDH− population (Table 4). These results indicate that EpCAM+/ALDH+ cancer cells isolated from our rectal cancer PDXs have an enhanced self-renewal ability consistent with CSCs. To examine whether the SNAI:miR-145 axis is active in the patient-derived CSCs, we employed a digital droplet PCR analysis in the EPCAM+/ALDH+ and EpCAM+/ALDH− populations. Interestingly, we observed that the EpCAM+/ALDH+ enriched CSC population showed increased expression of SNAI1, SNAI2, Nanog, and c-Myc (Figure 6A). Conversely, the expression levels of miR-145 were significantly decreased (Figure 6B). Of interest, IGF1R, an instrumental mediator for Nanog to maintain the CSC self-renewal ability,36 also showed increased expression in the EpCAM+/ALDH+ CSC population. Thus, colorectal cancer CSCs isolated by ALDH activity demonstrate a phenotype with an active SNAI1:miR-145 axis.
Table 2.
CSC Population in PDX Tumor
| PDX Model Gen2 | EpCAM+/ALDH+ (%) |
|---|---|
| MRC02 | 0.26 |
| MRC07 | 3.47 |
| MRC13 | 1.47 |
Table 3.
In Vitro Spheroid Formation from EpCAM/ALDH-Sorted Patient-Derived Cancer Cells
| Spheroid Formation |
Significance |
||
|---|---|---|---|
| EpCAM+/ALDH− | EpCAM+/ALDH+ | ||
| MRC02 | 0 | 26 ± 6 | p < 0.05 |
| MRC07 | 0 | 34 ± 9 | p < 0.05 |
| MRC13 | 0 | 45 ± 4 | p < 0.05 |
Table 4.
Limited Dilution In Vivo Tumorigenesis from EpCAM/ALDH-Sorted Patient-Derived Cancer Cells
| 500 Cells |
5,000 Cells |
|||
|---|---|---|---|---|
| EpCAM+/ALDH− | EpCAM+/ALDH+ | EpCAM+/ALDH− | EpCAM+/ALDH+ | |
| MRC07 | 0/6 | 3/6 | 0/6 | 5/6* |
| MRC13 | 0/6 | 1/6 | 0/6 | 6/6* |
* indicated p < 0.05, EpCAM+/ALDH+ versus EpCAM+/ALDH−.
Figure 6.

SNAI1 and miR-145 Expression in CSC and Non-CSC Population in Patient-Derived Rectal Cancer Xenografts
(A) Droplet digital PCR analysis of genes involved in the SNAI1:miR-145 axis. (B) miR-145 in FACS-sorted non-CSC (EpCAM+/ALDH−; black bars) and CSC (EpCAM+/ALDH+; gray bars) cells from two patient-derived rectal cancer xenografts (* indicated p < 0.05).
Discussion
In 2004, the German Rectal Cancer Trial (CAO/ARO/AIO-94) established neoadjuvant 5FU/RT as the current gold standard option for the majority of rectal cancer patients.2 Unfortunately, mechanisms of radiation resistance in colorectal cancer are still poorly understood and we continue to lack therapeutic strategies to enhance clinical outcomes. A growing body of work implicates the closely aligned EMT pathways and CSCs in chemotherapy and RT resistance, although therapeutically targeting these pathways has remained challenging.10, 11, 12 The existence of CSCs offers a potential explanation for cancer treatment failure and suggests that CSCs should be targeted to enhance conventional therapy, such as radiation. Thus, the primary objectives of this investigation were to define molecular mechanisms by which CSC promotes radiation resistance and to use this information to explore specific CSC-directed therapeutic strategies.
In this study, our group is the first to report that SNAI1 represses miR-145 promoter activity and subsequently its expression. We also demonstrated that highly expressing SNAI1 cells are able to induce the expression of the CSC-related transcription factor Nanog, and further confer colorectal cancer cells a radiation-resistant phenotype. In addition, our findings suggest that the SNAI1:miR-145 axis is clinically relevant because patient-derived rectal cancer stem cells demonstrate a similar genotype. These novel findings implicate the SNAI1:miR-145 axis as a mediator of radiation resistance and highlights a promising molecular pathway to target the CSC for clinical therapy. We theorize that delivery of miR-145 to target the SNAI1:miR-145 axis will enhance treatment efficacy because miRNAs have a distinct capacity to regulate multiple gene expressions simultaneously.
After delivering miR-145 to radiation-resistant SNAI1-expressing colon cancer cells, we observed downregulation of established miR-145 targets, including c-Myc and KLF4, along with repression of the non-miR-145 targets SNAI1 and Nanog.20 There are two canonical Myc-binding sites located in the human SNAI1 promoter; therefore, one possible mechanism for miR-145 repression of SNAI1 is via c-Myc.37, 38 It has been reported that SNAI1-induced Nanog expression in non-small-cell lung cancer (NSCLC) and SNAI1-activated Nanog driving induced pluripotent stem cells.25, 39 Similarly, in our SNAI1-overexpressing DLD1 and HCT116 cells, SNAI1 consistently induced Nanog expression compared to the vector control cells. Further investigation is needed to demonstrate if the SNAI1:miR-145-axis-mediated radiation resistance is Nanog dependent and verify Nanog as an indispensable partner of SNAI1 to activate the SNAI:miR-145 axis or just one of the components among SNAI1 effectors.
Therapy-resistant CSCs have gained interest as a novel therapeutic target to overcome resistance to traditional cancer therapy, such as radiation.7, 8, 9 Dissecting molecular pathways driving the CSC phenotype holds potential to identify future novel therapy targets. Our findings confirm the strong relationship among EMT pathways, the CSC phenotype, and cellular survival.25, 40, 41, 42 Prior reports implicate that EMT mediators, such as the SNAIL family of transcriptional repressors, can drive the CSC phenotype and mediate chemotherapy resistance, highlighting novel candidate pathways to target in order to improve chemotherapy sensitivity.40 However, less is understood regarding molecular mechanisms leading to resistance to radiation in colorectal cancer. Thus, our work implicated CSC in mediating colorectal cancer radiation resistance and identified a critical molecular pathway underlying the mechanisms of resistance. Importantly, the SNAI1:miR-145 axis was identified in patient-derived rectal cancer stem cells that were highly tumorigenic, supporting the significance in the context of rectal cancer neoadjuvant chemoradiation.
Our work identified SNAI1-mediated stemness as an attractive molecular pathway to improve radiation therapy. However, transcriptional factors are notoriously difficult to therapeutically target. Strong evidence exists that miR-145 is a potent tumor suppressor and is inversely related to EMT.15, 43, 44, 45, 46 Further, miR-145 delivery represents a viable therapeutic strategy and has been complexed with polyethylenimine as a vehicle to target cancer xenografts.47, 48, 49, 50 In this report, we identified an inverse relationship between miR-145 and SNAI1. We also demonstrated that miR-145 delivery inhibits both SNAI1-mediated stemness and expression of SNAI1-driven expression of critical CSC transcription factors. miR-145 effectively inhibited multiple mediators downstream of SNAI1 to disrupt the CSC phenotype. In addition, miR-145 enhanced radiation sensitivity specifically in the stem-like SNAI1-overexpressing cells, suggesting that miR-145 may represent an effective CSC-targeted therapeutic agent.
In summary, the SNAI1:miR-145 axis (Figure 7) significantly influences colorectal cancer radiation sensitivity. The crosstalk between SNAI1 and miR-145 regulates expression of central CSC transcriptional factors to modulate the response to radiation. Therapeutic targeting of SNAI1-mediated stemness by miR-145 delivery represents a promising strategy to improve neoadjuvant therapy and overcome radiation resistance in rectal cancer.
Figure 7.
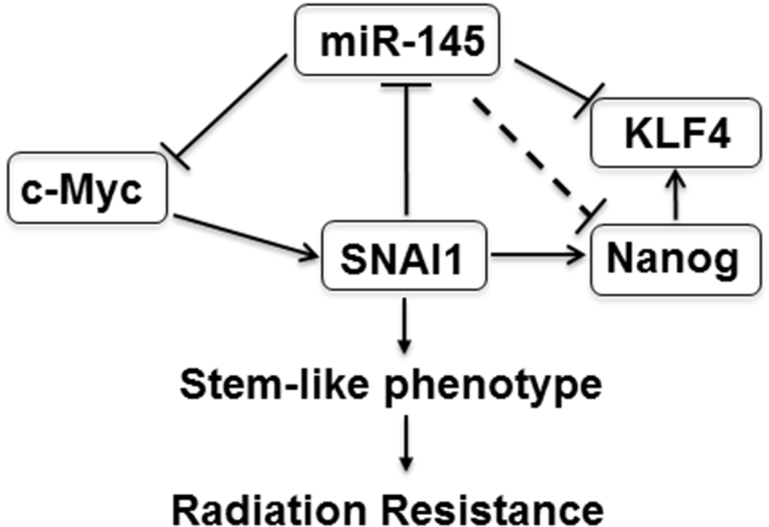
Therapeutic Mechanism of miR-145 Delivery to Overcome SNAI1-Mediated Colorectal Cancer Radiation Resistance
SNAI1 can activate Nanog and repress miR-145. Thus, SNAI1 mediates the CSC program, resulting in a radiation-resistant colorectal cancer cell population. Delivery of miR-145 to target multiple stemness genes, including Nanog, KLF4, and c-Myc, can impair CSC self-renewal capacity and increase sensitivity of radiation therapy.
Materials and Methods
Data Mining
To determine the expression levels of EMT transcription factors in human rectal cancer, whole exome and RNA Seq data from the cancer genome atlas database of colorectal adenocarcinoma was mined for the frequency of SNAI1, SNAI2, ZEB1, and ZEB2 genetic alteration using bioportal.51, 52, 53 SNAI1, SNAI2, ZEB1, and ZEB2 expression values were also extracted from the public datasets specifically for rectal cancer from Oncomine 4.4 Research Edition (Compendia Bioscience, Ann Arbor, MI). To find the resulting dataset, the following filters were used: Gene: SNAI1, SNAI2, ZEB1, and ZEB2; Analysis Type: Cancer versus Normal; and Cancer Type: Rectal cancer. The datasets were ordered by overexpression; p values and the datasets selected for analysis and visualization were the largest available datasets with complete data.
Cell Lines and Culture Conditions
Human colorectal cancer cell lines (DLD1, HCT116, HT29, SW480, and SW620) were obtained from ATCC (Manassas, VA) and cultured according to ATCC recommendations. DLD1 and HCT116 were transfected with empty vector pCMV-3Tag-1 and pCMV-SNAI1 construct using Lipofectamine 3000 according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA) to make overexpressing SNAI1 stable cell lines, i.e., DLD1-SNAI1 and HCT116-SNAI1. Transfected cells were selected in medium containing G418 at a final concentration of 400 μg/mL (Invitrogen, Carlsbad, CA). The expression of SNAI1 was confirmed by western blot analysis.
Clonogenic Assay
DLD1-SNAI1 and HCT116-SNAI1 cells were plated in a 6-well plate and compared with empty vector control cells. Plates were radiated at 0, 2, 4, and 6 Gy X-ray delivered by Precision X-Ray (Model X-RAD 320, North Branford, CT). In a second experiment, DLD1-SNAI1 and HCT116-SNAI1 cells were transfected with either 2.5 μg miR-145 expression construct or pEZX vector with scr (Genecopoeia, Rockville, MD) by Lipofectamine 3000 (Invitrogen, Carlsbad, CA). Following radiation, cells were re-plated at a cell density of 400, 800, 1,000, and 1,200 per well in 6-well plates in triplicates to determine the survival fraction. After 10 days of incubation, the colonies were washed with 1x PBS three times, fixed with 70% ethanol, and stained with 0.5% Crystal Violet. After staining, the number of colonies was counted and recorded.
SRB Cytotoxicity Assay
Cells (3 × 103) were seeded in 96-well plates for next-day radiation. 96 hr post treatment, cells were fixed at 4°C with 1% trichloroacetic acid (TCA), followed by staining at room temperature for 30 min in 0.4% (w/v) sulforhodamine B (SRB) dissolved in 1% acetic acid. After 4 washes with 1% acetic acid, the protein-bound dye was extracted with 10 mM unbuffered Tris base.54 The plates were read at 560 nm.
Cell Viability Assay
Cell viability was evaluated by CellTiter-Glo Assay (Promega, Madison, WI) according to the manufacturer’s protocol. 5,000 cells per well were plated in 96-well plates. After 24-hr incubation, cells were treated with DMSO and 0.1 μM, 1 μM, and 2 μM oxaliplatin in triplicates for 72 hr. After 72-hr treatment, 20 μL CellTiter-Blue reagent was added to each well for 4-hr incubation at 37°C. The plates were read at 570 nm, and cell viability was analyzed.
Reverse Transcription PCR
Total RNA from cultured cells was extracted using the RNeasy Plus Mini kit (QIAGEN, Valencia, CA). Total RNA (0.8 μg) was reverse transcribed in a 20-μL reaction using iScript (Bio-Rad, Hercules, CA). Real-time PCR was performed with 5 μL of a 1/16 dilution cDNA for cell line samples using the Universal Probe Library (UPL) mono-color probes in the Roche LightCycler 480 machine (Roche Diagnostics, Basel, Switzerland). The conditions for all genes were pre-incubated at 95°C for 10 min, followed by 55 cycles of denaturation at 95°C for 15 s and amplification/extension at 60°C for 30 s; after cycle completion, cooling was held for 30 s at 40°C. Triplicate reactions were run for each cDNA sample. Data were normalized to GAPDH and confirmed with replicate samples. Sequences for gene-specific primers and probe numbers can be found elsewhere.43
Droplet Digital PCR
Complementary DNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The ddPCR reaction mixture consisted of 10 μL 2x ddPCR master mix (Bio-Rad, Hercules, CA), 2 μL primer mix, and 5 μL sample nucleic acid solution in a final volume of 20 μL. The entire reaction mixture was loaded into a disposable droplet generator cartridge, together with droplet generation oil, and placed in the droplet generator (Bio-Rad, Hercules, CA). The droplets generated from each sample were transferred to a 96-well PCR plate. After PCR amplification, the plate was loaded on the droplet reader (Bio-Rad) and the droplets from each well of the plate were read automatically. ddPCR data were analyzed with QuantaSoft analysis software (Bio-Rad, Hercules, CA), and the quantification of the target molecule was presented as the number of copies per μL of PCR mixture.
MicroRNA Analysis
Total RNA was extracted using the RNeasyPlus Mini Kit (QIAGEN, Valencia, CA) following the manufacturer’s instructions. cDNA was synthesized using miR-145- or RNU6B-specific primers and the Applied Biosystems reverse transcription kit (Thermo Fisher Scientific, Waltham, MA). Real-time PCR (qPCR) was performed using Taqman Master Mix and 1 μL cDNA as a template on the Roche LightCycler 480 machine (Roche, Nutley, NJ). miR-145 and RNU6B Taqman primers were purchased from Applied Biosystems. The relative expression of miR-145 was quantified on the basis of Ct value measured against an internal standard curve for each specific set of primers using the software provided by the instrument manufacturer (Roche, Nutley, NJ). RNU6B was used as the reference gene.
Luciferase Promoter Assay
The luciferase reporter plasmid containing the putative 1.4-kb miR-145 promoter in pGL3 basic vector (Promega, Madison, WI, USA) was generously provided by Dr. Yin-Yuan Mo (University of Mississippi).19 Luciferase assays were carried out in DLD1-SNAI1, HCT116-SNAI1, and empty vector stably transfected cells. Cells were transfected with miR-145 promoter and Renilla luciferase plasmids in 12-well plates. The cells were lysed for the luciferase assay 48 hr after transfection. Luciferase assays were performed using the dual luciferase assay kit (Promega, Madison, WI) according to the manufacturer’s protocol.
Western Blot Analysis
Cells were suspended in RIPA protein lysis buffer (Thermo Fisher Scientific, Waltham, MA) with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Protein concentration was quantified using BCA protein assay (Thermo Fisher Scientific, Waltham, MA). 50 μg total protein was resolved with SDS-PAGE (10% polyacrylamide gel) and transferred to a nitrocellulose membrane (GE Healthcare, Marlborough, MA). Blots were probed with primary antibodies E-Cadherin (BD Biosciences, San Diego, CA), SNAI1, SNAI2, c-Myc, Nanog, Vimentin, KLF4, Sox2, Oct4, and Actin (Cell Signaling Technology, Danvers, MA) and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Immunoblots were visualized with enhanced chemiluminescence (GE Healthcare, Marlborough, MA) by the LI-COR Odyssey Imaging System (LI-COR BioSciences, Lincoln, NE).
Patient-Derived Rectal Cancer Xenograft Model
Based on the approved protocol by the Institutional Review Board (IRB protocol number 30678) at Medical University of South Carolina, human tumor tissue was obtained from consenting stage 2 and 3 rectal cancer patients by biopsy. Tissue fragments were dissociated in DMEM containing 0.26 U/mL Liberase DH (Roche, Nutley, NJ) and cell suspension (5 × 105 cells) in 100 μL Matrigel (Corning) was implanted subcutaneously in female NSG mice (Jackson Laboratories, Bar Harbor, ME). Initial tumor xenografts were dissociated in a similar fashion and re-implanted into multiple mice (up to 10) to obtain the first passaged cohort that was used to obtain the CSC fraction. PDX tumors were inspected twice per week and measured for tumor volume (V), as determined by the formula V = LW2/2 (L is the length and W is the width of the tumor).
Isolation of Cancer Stem Cells
Identification of aldehyde dehydrogenase positive (ALDH+) cells was performed using the ALDEFLUOR kit (STEMCELL Technologies, Cambridge, MA) according to the manufacturer’s instruction. Dissociated cells from the patient-derived xenografts were resuspended in ALDEFLUOR assay buffer (4 × 105 cells per mL) and incubated with 1.5 μM ALDH substrate BODIPY-aminoacetaldehyde. One aliquot of this cell mixture was immediately transferred to the tube containing the ALDH inhibitor diethylaminobenzaldehyde (DEAB) to serve as the negative control. These samples were incubated for 45 min at 37°C to allow the generation of fluorescent product. Cells were then incubated with anti-human EpCAM-PE (epithelial marker, Miltenyi Biotec, Auburn, CA) for 30 min on ice to label EpCAM cells. ALDH+ and EpCAM+ cells were sorted using FACS (BD Aria IIu Cell Sorter) by comparing the fluorescence of test samples against that of control samples that were treated with the ALDH inhibitor DEAB.
Limited Dilution Spheroid Assay
The ability to form spheres in a 96-well ultra-low-attachment plate was evaluated as described previously.40 The culture medium consisted of DMEM/F12 supplemented with 1X B27, 20 ng/mL epidermal growth factor, 20 ng/mL fibroblast growth factor basic, 0.5 mg/mL BSA, 4 μg/mL heparin, 55 μM 2-mercaptoethanol, and 1x penicillin/streptomycin (Invitrogen, Carlsbad, CA). After 7 days of incubation, the total number of spheres greater than 50 μm in diameter was quantified under light microscopy.
In Vivo Tumorigenicity
Tumorigenicity of EpCAM+/ALDH+ and EpCAM+/ALDH− sorted cells was determined by subcutaneous injection in 6- to 8-week-old female NSG mice. Before injection, cells were suspended at a 1:1 ratio of DMEM:Matrigel in a total volume of 100 μL. EpCAM+/ALDH+ and EpCAM+/ALDH− sorted cells (500 and 5,000 cells) were injected in the flanks of NSG mice. The tumors (>100 mm3) were quantified at 50 days after injection.
Statistical Analysis
Statistical analyses were performed using the Student’s t test for paired data. p < 0.05 was considered significant. The patient data were analyzed using GraphPad Prism Software.
Author Contributions
Conceptualization, E.R.C. and V.J.F.; Methodology, E.R.C. and V.J.F.; Investigation, Y.Z., S.A.B., C.W., K.H., and L.M.N.; Writing – Original Draft, Y.Z. and E.R.C.; Writing – Review and Editing, Y.Z., S.A.B., C.W., K.H., L.M.N., V.J.F., and E.R.C.; Funding Acquisition, E.R.C.
Acknowledgments
This work was supported in part by pilot research funding, Hollings Cancer Center’s Cancer Center Support Grant P30 CA138313 at the Medical University of South Carolina.
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.12.023.
Contributor Information
Yun Zhu, Email: zhuyun@musc.edu.
E. Ramsay Camp, Email: campe@musc.edu.
Supplemental Information
References
- 1.Wu J., Hirata I., Zhao X., Gao B., Okazaki M., Kato K. Influence of alkyl chain length on calcium phosphate deposition onto titanium surfaces modified with alkylphosphonic acid monolayers. J. Biomed. Mater. Res. A. 2013;101:2267–2272. doi: 10.1002/jbm.a.34545. [DOI] [PubMed] [Google Scholar]
- 2.Sauer R., Becker H., Hohenberger W., Rödel C., Wittekind C., Fietkau R., Martus P., Tschmelitsch J., Hager E., Hess C.F., German Rectal Cancer Study Group Preoperative versus postoperative chemoradiotherapy for rectal cancer. N. Engl. J. Med. 2004;351:1731–1740. doi: 10.1056/NEJMoa040694. [DOI] [PubMed] [Google Scholar]
- 3.Willett C.G., Duda D.G., di Tomaso E., Boucher Y., Ancukiewicz M., Sahani D.V., Lahdenranta J., Chung D.C., Fischman A.J., Lauwers G.Y. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J. Clin. Oncol. 2009;27:3020–3026. doi: 10.1200/JCO.2008.21.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velenik V., Ocvirk J., Music M., Bracko M., Anderluh F., Oblak I., Edhemovic I., Brecelj E., Kropivnik M., Omejc M. Neoadjuvant capecitabine, radiotherapy, and bevacizumab (CRAB) in locally advanced rectal cancer: results of an open-label phase II study. Radiat. Oncol. 2011;6:105. doi: 10.1186/1748-717X-6-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewdney A., Cunningham D., Tabernero J., Capdevila J., Glimelius B., Cervantes A., Tait D., Brown G., Wotherspoon A., Gonzalez de Castro D. Multicenter randomized phase II clinical trial comparing neoadjuvant oxaliplatin, capecitabine, and preoperative radiotherapy with or without cetuximab followed by total mesorectal excision in patients with high-risk rectal cancer (EXPERT-C) J. Clin. Oncol. 2012;30:1620–1627. doi: 10.1200/JCO.2011.39.6036. [DOI] [PubMed] [Google Scholar]
- 6.Czito B.G., Willett C.G., Bendell J.C., Morse M.A., Tyler D.S., Fernando N.H., Mantyh C.R., Blobe G.C., Honeycutt W., Yu D. Increased toxicity with gefitinib, capecitabine, and radiation therapy in pancreatic and rectal cancer: phase I trial results. J. Clin. Oncol. 2006;24:656–662. doi: 10.1200/JCO.2005.04.1749. [DOI] [PubMed] [Google Scholar]
- 7.Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 8.Li X., Lewis M.T., Huang J., Gutierrez C., Osborne C.K., Wu M.F., Hilsenbeck S.G., Pavlick A., Zhang X., Chamness G.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 9.Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N., Qian D., Lam J.S., Ailles L.E., Wong M. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rolland D., Ribrag V., Haioun C., Ghesquieres H., Jardin F., Bouabdallah R., Franchi P., Briere J., De Kerviler E., Chassagne-Clement C. Phase II trial and prediction of response of single agent tipifarnib in patients with relapsed/refractory mantle cell lymphoma: a Groupe d’Etude des Lymphomes de l’Adulte trial. Cancer Chemother. Pharmacol. 2010;65:781–790. doi: 10.1007/s00280-009-1185-4. [DOI] [PubMed] [Google Scholar]
- 11.Arndt G.M., Dossey L., Cullen L.M., Lai A., Druker R., Eisbacher M., Zhang C., Tran N., Fan H., Retzlaff K. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer. 2009;9:374. doi: 10.1186/1471-2407-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raponi M., Dossey L., Jatkoe T., Wu X., Chen G., Fan H., Beer D.G. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69:5776–5783. doi: 10.1158/0008-5472.CAN-09-0587. [DOI] [PubMed] [Google Scholar]
- 13.Yin Y., Yan Z.P., Lu N.N., Xu Q., He J., Qian X., Yu J., Guan X., Jiang B.H., Liu L.Z. Downregulation of miR-145 associated with cancer progression and VEGF transcriptional activation by targeting N-RAS and IRS1. Biochim. Biophys. Acta. 2013;1829:239–247. doi: 10.1016/j.bbagrm.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Yamada N., Noguchi S., Mori T., Naoe T., Maruo K., Akao Y. Tumor-suppressive microRNA-145 targets catenin δ-1 to regulate Wnt/β-catenin signaling in human colon cancer cells. Cancer Lett. 2013;335:332–342. doi: 10.1016/j.canlet.2013.02.060. [DOI] [PubMed] [Google Scholar]
- 15.Gao P., Xing A.Y., Zhou G.Y., Zhang T.G., Zhang J.P., Gao C., Li H., Shi D.B. The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer. Oncogene. 2013;32:491–501. doi: 10.1038/onc.2012.61. [DOI] [PubMed] [Google Scholar]
- 16.Mata M., Raponi M. Circulating tumor cells: utility for predicting response to anti-EGFR therapies? Expert Rev. Mol. Diagn. 2009;9:115–119. doi: 10.1586/14737159.9.2.115. [DOI] [PubMed] [Google Scholar]
- 17.Raponi M., Buratti E., Dassie E., Upadhyaya M., Baralle D. Low U1 snRNP dependence at the NF1 exon 29 donor splice site. FEBS J. 2009;276:2060–2073. doi: 10.1111/j.1742-4658.2009.06941.x. [DOI] [PubMed] [Google Scholar]
- 18.Shibata T., Ichikawa Y., Okubo M., Tahara T., Ishizuka T., Hirata I. Gastric pyogenic granuloma detected due to abdominal symptoms and treated with endoscopic resection. Intern. Med. 2013;52:2749–2752. doi: 10.2169/internalmedicine.52.1133. [DOI] [PubMed] [Google Scholar]
- 19.Sachdeva M., Liu Q., Cao J., Lu Z., Mo Y.Y. Negative regulation of miR-145 by C/EBP-β through the Akt pathway in cancer cells. Nucleic Acids Res. 2012;40:6683–6692. doi: 10.1093/nar/gks324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu N., Papagiannakopoulos T., Pan G., Thomson J.A., Kosik K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 21.Fraga T.R., Courrol Ddos S., Castiblanco-Valencia M.M., Hirata I.Y., Vasconcellos S.A., Juliano L., Barbosa A.S., Isaac L. Immune evasion by pathogenic Leptospira strains: the secretion of proteases that directly cleave complement proteins. J. Infect. Dis. 2014;209:876–886. doi: 10.1093/infdis/jit569. [DOI] [PubMed] [Google Scholar]
- 22.Cruz-Silva I., Neuhof C., Gozzo A.J., Nunes V.A., Hirata I.Y., Sampaio M.U., Figueiredo-Ribeiro Rde C., Neuhof H., Araújo Mda S. Using a Caesalpinia echinata Lam. protease inhibitor as a tool for studying the roles of neutrophil elastase, cathepsin G and proteinase 3 in pulmonary edema. Phytochemistry. 2013;96:235–243. doi: 10.1016/j.phytochem.2013.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Costa H.M., Freitas Júnior A.C., Amaral I.P., Hirata I.Y., Paiva P.M., Carvalho L.B., Jr., Oliveira V., Bezerra R.S. Metal-sensitive and thermostable trypsin from the crevalle jack (Caranx hippos) pyloric caeca: purification and characterization. Chem. Cent. J. 2013;7:166. doi: 10.1186/1752-153X-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirata I., Kimizu T., Ikeda T., Kimura S., Mogami Y., Yanagihara K., Mano T., Toribe Y., Suzuki Y. [A fatal case of lamotrigine associated drug-induced hypersensitivity syndrome with fulminant myocarditis] No To Hattatsu. 2013;45:243–244. [PubMed] [Google Scholar]
- 25.Akao Y., Iio A., Itoh T., Noguchi S., Itoh Y., Ohtsuki Y., Naoe T. Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol. Ther. 2011;19:395–399. doi: 10.1038/mt.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuta S., Matsuo K., Yagasaki F., Yujiri T., Hatta Y., Kimura Y., Ueda Y., Kanamori H., Usui N., Akiyama H. Pre-transplant imatinib-based therapy improves the outcome of allogeneic hematopoietic stem cell transplantation for BCR-ABL-positive acute lymphoblastic leukemia. Leukemia. 2011;25:41–47. doi: 10.1038/leu.2010.228. [DOI] [PubMed] [Google Scholar]
- 27.Tsujimura A., Kiyoi H., Shiotsu Y., Ishikawa Y., Mori Y., Ishida H., Toki T., Ito E., Naoe T. Selective KIT inhibitor KI-328 and HSP90 inhibitor show different potency against the type of KIT mutations recurrently identified in acute myeloid leukemia. Int. J. Hematol. 2010;92:624–633. doi: 10.1007/s12185-010-0692-8. [DOI] [PubMed] [Google Scholar]
- 28.Huang S., Guo W., Tang Y., Ren D., Zou X., Peng X. miR-143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells. Oncol. Rep. 2012;28:1831–1837. doi: 10.3892/or.2012.2015. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z., Zeng H., Guo Y., Liu P., Pan H., Deng A., Hu J. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J. Exp. Clin. Cancer Res. 2010;29:151. doi: 10.1186/1756-9966-29-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 31.Cano A., Pérez-Moreno M.A., Rodrigo I., Locascio A., Blanco M.J., del Barrio M.G., Portillo F., Nieto M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 32.Sachdeva M., Zhu S., Wu F., Wu H., Walia V., Kumar S., Elble R., Watabe K., Mo Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ono T., Takeshita A., Iwanaga M., Asou N., Naoe T., Ohno R., Japan Adult Leukemia Study Group Impact of additional chromosomal abnormalities in patients with acute promyelocytic leukemia: 10-year results of the Japan Adult Leukemia Study Group APL97 study. Haematologica. 2011;96:174–176. doi: 10.3324/haematol.2010.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamatani A., Nakagawa Y., Akao Y., Maruyama N., Nagasaka M., Shibata T., Tahara T., Hirata I. Downregulation of anti-oncomirs miR-143/145 cluster occurs before APC gene aberration in the development of colorectal tumors. Med. Mol. Morphol. 2013;46:166–171. doi: 10.1007/s00795-013-0020-5. [DOI] [PubMed] [Google Scholar]
- 35.Nobili V., Alisi A., Raponi M. Pediatric non-alcoholic fatty liver disease: preventive and therapeutic value of lifestyle intervention. World J. Gastroenterol. 2009;15:6017–6022. doi: 10.3748/wjg.15.6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura K., Nakano Y., Watamoto K., Koga D., Naoe T. [Significance of monitoring WT1 mRNA levels of peripheral blood and bone marrow in acute myeloid leukemia] Rinsho Ketsueki. 2010;51:1748–1755. [PubMed] [Google Scholar]
- 37.Miyawaki S., Ohtake S., Fujisawa S., Kiyoi H., Shinagawa K., Usui N., Sakura T., Miyamura K., Nakaseko C., Miyazaki Y. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood. 2011;117:2366–2372. doi: 10.1182/blood-2010-07-295279. [DOI] [PubMed] [Google Scholar]
- 38.Murase M., Nishida T., Onizuka M., Inamoto Y., Sugimoto K., Imahashi N., Murata M., Miyamura K., Kodera Y., Inoko H. Cytotoxic T-lymphocyte antigen 4 haplotype correlates with relapse and survival after allogeneic hematopoietic SCT. Bone Marrow Transplant. 2011;46:1444–1449. doi: 10.1038/bmt.2010.319. [DOI] [PubMed] [Google Scholar]
- 39.Kako S., Morita S., Sakamaki H., Ogawa H., Fukuda T., Takahashi S., Kanamori H., Onizuka M., Iwato K., Suzuki R. A decision analysis of allogeneic hematopoietic stem cell transplantation in adult patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission who have an HLA-matched sibling donor. Leukemia. 2011;25:259–265. doi: 10.1038/leu.2010.260. [DOI] [PubMed] [Google Scholar]
- 40.Fan F., Samuel S., Evans K.W., Lu J., Xia L., Zhou Y., Sceusi E., Tozzi F., Ye X.C., Mani S.A. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012;1:5–16. doi: 10.1002/cam4.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dallas N.A., Xia L., Fan F., Gray M.J., Gaur P., van Buren G., 2nd, Samuel S., Kim M.P., Lim S.J., Ellis L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Findlay V.J., Wang C., Nogueira L.M., Hurst K., Quirk D., Ethier S.P., Staveley O’Carroll K.F., Watson D.K., Camp E.R. SNAI2 modulates colorectal cancer 5-fluorouracil sensitivity through miR145 repression. Mol. Cancer Ther. 2014;13:2713–2726. doi: 10.1158/1535-7163.MCT-14-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J., Guo H., Li H., Liu Y., Liu J., Chen L., Zhang J., Zhang N. Retraction: MiR-145 regulates epithelial to mesenchymal transition of breast cancer cells by targeting Oct4. PLoS One. 2013;8:10.. doi: 10.1371/annotation/aaef10de-ee36-416f-93eb-959dfa3dc92e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyawaki S., Hatsumi N., Tamaki T., Naoe T., Ozawa K., Kitamura K., Karasuno T., Mitani K., Kodera Y., Yamagami T., Koga D. Prognostic potential of detection of WT1 mRNA level in peripheral blood in adult acute myeloid leukemia. Leuk. Lymphoma. 2010;51:1855–1861. doi: 10.3109/10428194.2010.507829. [DOI] [PubMed] [Google Scholar]
- 46.Jinnai I., Sakura T., Tsuzuki M., Maeda Y., Usui N., Kato M., Okumura H., Kyo T., Ueda Y., Kishimoto Y. Intensified consolidation therapy with dose-escalated doxorubicin did not improve the prognosis of adults with acute lymphoblastic leukemia: the JALSG-ALL97 study. Int. J. Hematol. 2010;92:490–502. doi: 10.1007/s12185-010-0672-z. [DOI] [PubMed] [Google Scholar]
- 47.Ibrahim A.F., Weirauch U., Thomas M., Grünweller A., Hartmann R.K., Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71:5214–5224. doi: 10.1158/0008-5472.CAN-10-4645. [DOI] [PubMed] [Google Scholar]
- 48.Noh J.H., Chang Y.G., Kim M.G., Jung K.H., Kim J.K., Bae H.J., Eun J.W., Shen Q., Kim S.J., Kwon S.H. MiR-145 functions as a tumor suppressor by directly targeting histone deacetylase 2 in liver cancer. Cancer Lett. 2013;335:455–462. doi: 10.1016/j.canlet.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Ostenfeld M.S., Bramsen J.B., Lamy P., Villadsen S.B., Fristrup N., Sørensen K.D., Ulhøi B., Borre M., Kjems J., Dyrskjøt L. miR-145 induces caspase-dependent and -independent cell death in urothelial cancer cell lines with targeting of an expression signature present in Ta bladder tumors. Oncogene. 2010;29:1073–1084. doi: 10.1038/onc.2009.395. [DOI] [PubMed] [Google Scholar]
- 50.Pagliuca A., Valvo C., Fabrizi E., di Martino S., Biffoni M., Runci D., Forte S., De Maria R., Ricci-Vitiani L. Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene. 2012;32:4806–4813. doi: 10.1038/onc.2012.495. [DOI] [PubMed] [Google Scholar]
- 51.Ichikawa K., Fujimori T., Moriya T., Ochiai A., Yoshinaga S., Kushima R., Nagahama R., Ohkura Y., Tanaka S., Ajioka Y. Digestive disease management in Japan: a report on the 6th diagnostic pathology summer fest in 2012. Digestion. 2013;88:153–160. doi: 10.1159/000355330. [DOI] [PubMed] [Google Scholar]
- 52.Tahara T., Maegawa S., Chung W., Garriga J., Jelinek J., Estécio M.R., Shibata T., Hirata I., Arisawa T., Issa J.P. Examination of whole blood DNA methylation as a potential risk marker for gastric cancer. Cancer Prev. Res. (Phila.) 2013;6:1093–1100. doi: 10.1158/1940-6207.CAPR-13-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamada N., Nakagawa Y., Tsujimura N., Kumazaki M., Noguchi S., Mori T., Hirata I., Maruo K., Akao Y. Role of intracellular and extracellular microRNA-92a in colorectal cancer. Transl. Oncol. 2013;6:482–492. doi: 10.1593/tlo.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abe Y., Okazaki Y., Hiasa K., Yasuda K., Nogami K., Mizumachi W., Hirata I. Bioactive surface modification of hydroxyapatite. Biomed Res. Int. 2013;2013 doi: 10.1155/2013/626452. 626452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.