

Abstract
Canavan disease, a leukodystrophy caused by loss-of-function ASPA mutations, is characterized by brain dysmyelination, vacuolation, and astrogliosis (“spongiform leukodystrophy”). ASPA encodes aspartoacylase, an oligodendroglial enzyme that cleaves the abundant brain amino acid N-acetyl-L-aspartate (NAA) to L-aspartate and acetate. Aspartoacylase deficiency results in a 50% or greater elevation in brain NAA concentration ([NAAB]). Prior studies showed that homozygous constitutive knockout of Nat8l, the gene encoding the neuronal NAA synthesizing enzyme N-acetyltransferase 8-like, prevents aspartoacylase-deficient mice from developing spongiform leukodystrophy. We now report that brain Nat8l knockdown elicited by intracerebroventricular/intracisternal administration of an adeno-associated viral vector carrying a short hairpin Nat8l inhibitory RNA to neonatal aspartoacylase-deficient AspaNur7/Nur7 mice lowers [NAAB] and suppresses development of spongiform leukodystrophy.
Keywords: Canavan disease, Nat8l knockdown, leukodystrophy
Graphical Abstract
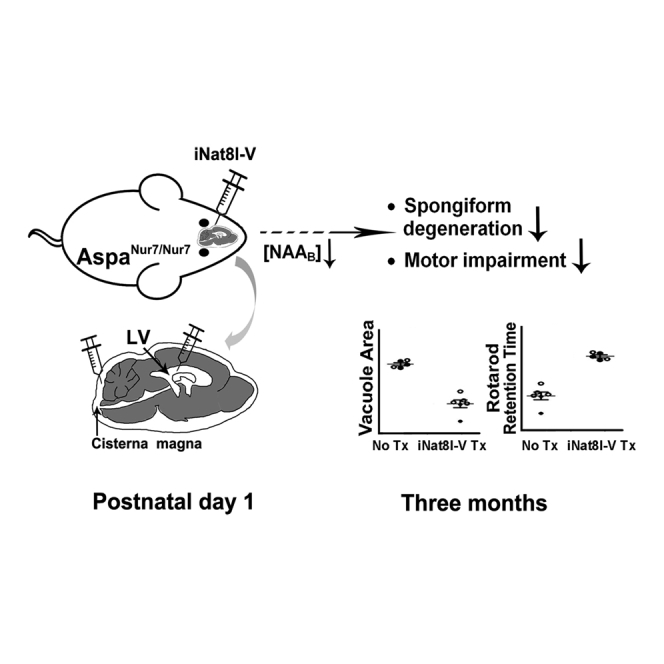
Prior studies demonstrated that constitutive Nat8l knockout prevents aspartoacylase-deficient mice from developing spongiform leukodystrophy. Bannerman et al. now report that neonatal intracerebroventricular/intracisternal administration of an adeno-associated viral vector carrying an Nat8l inhibitory short hairpin RNA preserves motor function and suppresses spongiform leukodystrophy in 3-month-old aspartoacylase-deficient mice.
Introduction
Canavan leukodystrophy, a recessively inherited CNS disease of infancy and childhood caused by ASPA (EC 3.5.1.15) loss-of-function mutations,1, 2 is characterized clinically by progressive impairment of cognitive and motor function and pathologically by forebrain, cerebellar, and brainstem myelin intraperiod line splitting and astroglial vacuolation (“spongiform leukodystrophy”).3, 4, 5, 6 ASPA encodes aspartoacylase, an oligodendrocyte-enriched enzyme that cleaves N-acetyl-L-aspartate (NAA) to L-aspartate and acetate.7 The lack of aspartoacylase in Canavan disease results in a substantial elevation in the brain concentration of NAA ([NAAB]).8
How might aspartoacylase deficiency cause spongiform leukodystrophy? One hypothesis is that brain dysmyelination in Canavan disease is attributable to inability of aspartoacylase-deficient oligodendroglia to derive acetate essential for synthesis of acetyl-CoA and myelin lipids from NAA.9, 10, 11, 12 This “oligodendroglial starvation” hypothesis has been weakened by the demonstration that brains of mice devoid of detectable NAA owing to homozygous constitutive knockout of Nat8l (EC 2.3.1.17), which encodes the neuronal NAA-synthesizing enzyme N-acetyltransferase 8-like (also known as N-acetylaspartate synthase),12, 13 become fully myelinated,14
An alternative hypothesis is that brain [NAAB] elevation is the proximate cause of spongiform leukodystrophy in the aspartoacylase-deficient CNS.14, 15, 16 Astroglia, which are linked to oligodendroglia via gap junctions that are essential for myelin maintenance,17 and which express an Na+-coupled dicarboxylate transporter (Nadc3) with high affinity for NAA,18 may accumulate excess NAA when [NAAB] is high, and this could elicit astroglial vacuolation and perturb brain myelin stability. In order to test this “NAA toxicity” hypothesis, we and others crossed mice deficient in aspartoacylase owing to the nonsense mutation nur7, which develop spongiform leukodystrophy by postnatal day 21,19 with mice in which Nat8l, the neuronal NAA-synthetic enzyme,12, 13 had been constitutively ablated. [NAAB] was below detection limits and spongiform leukodystrophy and brain neuron loss were prevented in AspaNur7/Nur7 mice that lacked both Nat8l alleles (AspaNur7/Nur7/Nat8l−/− mice), and, in AspaNur7/Nur7 mice with only one functional Nat8l allele (AspaNur7/Nur7/Nat8l+/− mice), the severities of spongiform leukodystrophy and brain neuron loss were substantially less than in AspaNur7/Nur7 mice with two intact Nat8l alleles (AspaNur7/Nur7/Nat8l+/+ mice).14, 20, 21 These observations suggested that elevated [NAAB] causes leukodystrophy in the aspartoacylase-deficient brain, and nominated Nat8l as a therapeutic target. To further evaluate this treatment option, we administered an adeno-associated virus (AAV) carrying a short hairpin sequence designed to knockdown Nat8l expression into the cerebral ventricles and cisterna magna of neonatal AspaNur7/Nur7 mice, and determined the effects on brain Nat8l mRNA abundance, [NAAB], motor function, and extents of spongiform leukodystrophy and cerebral cortical neuron survival 3 months later.
Results
Brain Vector Administration and Transduction
An AAV-Nat8l-shRNA vector (referred to henceforth as “iNat8l-V”), which included an Nat8l inhibitory short-hairpin RNA (shRNA) sequence driven by the U6 promoter and an EGFP coding sequence driven by the eSyn promoter, was administered into the cerebral ventricles and cisterna magna of AspaNur7/Nur7 and Aspa+/+ mice on postnatal day 1 (P1). Three months later, immunoreactive EGFP was detected in 15.5% ± 1.2% of NeuN+ cortical neurons in the AspaNur7/Nur7 mice and 14.0% ± 1.2% of NeuN+ cortical neurons of Aspa+/+ mice (mean ± SEM, n = 4, p = 0.84). Fewer than 1% of cerebral cortical Gfap+ astroglia, Olig2+ oligodendroglial lineage cells, and Iba1+ microglia were EGFP+ in cerebral cortex of these 3-month-old iNat8l-V-treated AspaNur7/Nur7 and Aspa+/+ mice (Figure 1). Immunoreactive EGFP was also detected at age 3 months in Calbindin+ cerebellar Purkinje cells (67.0% ± 3.1% in Aspa+/+ mice and 64.0% ± 3.1% in AspaNur7/Nur7 mice (mean ± SEM, n = 4 mice/group, p = 0.68) that had been treated with intracerebroventricular/intracisternal iNat8l-V on P1.
Figure 1.

Cerebral Cortical EGFP Transduction by iNat8l-V
Cerebral cortical expression of immunoreactive EGFP (shown in green) 3 months after intracerebroventricular/intracisternal iNat8l-V administration to postnatal day 1 (P1) Aspa+/+ and AspaNur7/Nur7 mice. (A) Colabeling (yellow) of NeuN+ neurons colabeled with EGFP; immunoreactive EGFP was detected in 15.5% ± 1.2% of NeuN+ cortical neurons in the AspaNur7/Nur7 mice and 14.0% ± 1.2% of NeuN+ cortical neurons of Aspa+/+ mice (mean ± SEM, n = 4, p = 0.84). (B) Colabeling of cerebral cortical Gfap+ astroglia with EGFP was rarely detected. (C) Colabeling of cerebral cortical Olig+ oligodendroglial lineage cells with EGFP was rarely detected. (D) Colabeling of cerebral cortical Iba1+ microglia/macrophages with EGFP was rarely detected.
P1 iNat8l-V Treatment Diminishes Brain Nat8l mRNA Abundance in AspaNur7/Nur7 and Aspa+/+ Mice
Brain Nat8l mRNA abundance, assayed by qRT-PCR, was similar in untreated 3-month-old Aspa+/+ and AspaNur7/Nur7 mice, thus suggesting that elevated [NAAB] in the aspartoacylase-deficient brain does not exert feedback inhibition on brain Nat8l gene transcription. P1 iNat8l-V administration lowered brain Nat8l mRNA abundance to a similar extent in 3-month-old Aspa+/+ and AspaNur7/Nur7 mice, whereas intracerebroventricular/intracisternal administration on P1 of a control AAV vector carrying an shRNA sequence designed not to inhibit any known mammalian gene (referred to henceforth as “Cont-V”) did not significantly alter brain Nat8l mRNA abundance in 3-month-old Aspa+/+ or AspaNur7/Nur7 mice (Figure 2).
Figure 2.

Brain Nat8l mRNA Knockdown by iNat8l-V
qRT-PCR for brain Nat8l mRNA abundance in 3-month-old mice, normalized to brain Hsp90 mRNA abundance. Each circle represents an individual mouse; open circles males, filled circles females. Mouse treatment group sizes were: Aspa+/+ no treatment (no Tx) 7; Aspa+/+ + Cont-V 13; Aspa+/+ + iNat8l 13; AspaNur7/Nur7 no Tx 8; AspaNur7/Nur7 + Cont-V 6; AspaNur7/Nur7 + iNat8l-V 7. Means ± SEMs are shown. Brain Nat8l mRNA abundance was diminished in both Aspa+/+ and AspaNur7/Nur7 mice by P1 iNat8l-V administration, but not by P1 Cont-V administration. Brain Nat8l mRNA abundance in no Tx or Cont-V treated Aspa+/+ mice did not differ significantly from those in untreated or Cont-V treated Aspanur7/nur7 mice. Two-way ANOVA indicated no significant effects of gender. **p < 0.01; n.s., not significant.
P1 iNat8l-V Treatment Diminishes [NAAB] in AspaNur7/Nur7 and Aspa+/+ Mice
[NAAB] was more than 2-fold higher in untreated 3-month-old AspaNur7/Nur7 mice than in age-matched untreated Aspa+/+ mice. P1 iNat8l-V treatment reduced [NAAB] by approximately one third in both 3-month-old AspaNur7/Nur7 and Aspa+/+ mice, whereas P1 Cont-V did not significantly alter [NAAB] in either 3-month-old AspaNur7/Nur7 or Aspa+/+ mice (Figure 3).
Figure 3.

iNat8l-V Lowers [NAAB] in Aspa+/+ and AspaNur7/Nur7 Mice
HPLC for [NAAB] in 3-month-old mice, expressed as μmol/gram wet weight of brain. Each symbol represents an individual mouse; open circles males, filled circles females. Mouse treatment group sizes were: Aspa+/+ no Tx 8; Aspa+/+ + Cont-V 6; Aspa+/+ + iNat8l-V 6; AspaNur7/Nur7 no Tx 6; AspaNur7/Nur7 + Cont-V 4; AspaNur7/Nur7 + iNat8l-V 8. Means ± SEMs are shown. P1 Nat8l-V administration diminished [NAAB] in both Aspa+/+ and AspaNur7/Nur7 mice, whereas P1 Cont-V administration did not. [NAAB] in P1 iNat8l-V treated AspaNur7/Nur7 mice, though lower than in untreated or Cont-V treated AspaNur7/Nur7 mice, remained significantly above levels in untreated 3-month-old Aspa+/+ mice. Results in male and female mice within each group did not significantly differ. Two-way ANOVA indicated no significant effects of gender. ***p < 0.001; **p < 0.01; n.s., not significant.
P1 iNat8l-V Treatment Improves Motor Function in AspaNur7/Nur7 Mice
Accerating rotarod retention times of untreated 3-month-old AspaNur7/Nur7 mice were approximately half as long as those in untreated or P1 Cont-V treated Aspa+/+ mice. P1 iNat8l-V, but not P1 Cont-V, lengthened rotarod retention times in AspaNur7/Nur7 mice tested at age 3 months, but did not alter accelerating rotarod retention times in 3-month-old Aspa+/+ mice (Figure 4).
Figure 4.

Enhanced Motor Performance in iNat8l-V-Treated AspaNur7/Nur7 Mice
Accelerating rotarod retention times in 3-month-old AspaNur7/Nur7 mice are enhanced by P1 iNat8l-V treatment. Each circle represents an individual mouse; open circles males, filled circles females. Treatment group sizes were: Aspa+/+ no Tx 8; Aspa+/+ + Cont-V 8; Aspa+/+ + iNat8l-V 6; AspaNur7/Nur7 No Tx 8; AspaNur7/Nur7 + Cont-V 6; AspaNur7/Nur7 + iNat8lV 5. Means ± SEMs are shown. Retention times in P1 iNat8l-V treated AspaNur7/Nur7 mice were higher than in untreated or Cont-V treated AspaNur7/Nur7 mice, and not significantly different from retention times in the 3 Aspa+/+ mouse groups. Two-way ANOVA indicated no significant effects of gender. ***p < 0.001; n.s., not significant.
P1 iNat8l-V Diminishes Brain Vacuolation and Astrogliosis in AspaNur7/Nur7 Mice
There was prominent cerebellar white matter vacuolation in untreated 3-month-old AspaNur7/Nur7 mice, but much less so in P1 iNat8l-V treated 3-month-old AspaNur7/Nur7 mice (Figure 5). P1 iNat8l-V also diminished reactive astrogliosis, visualized by GFAP indirect immunofluorescence, in 3-month-old AspaNur7/Nur7 mouse cerebral cortex (Figure 6), and lowered whole brain Gfap mRNA abundance, assayed by qRT-PCR, in 3-month-old AspaNur7/Nur7 mice (Figure 7).
Figure 5.

Diminished Cerebellar Vacuolation in iNat8l-V-Treated AspaNur7/Nur7 Mice
Top: cerebellar fields (low magnification top, and high magnification of boxed area below) from (left) an untreated 3-month-old Aspa+/+ mouse, (center) an untreated 3-month-old AspaNur7/Nur7 mouse, and (right) a P1 iNat8l-V treated 3-month-old AspaNur7/Nur7 mouse. The sections were immunostained for MBP (green) and Gfap (red), with DAPI nuclear counterstaining. P1 iNat8l-V treatment diminished cerebellar vacuolation in 3-month-old AspaNur7/Nur7 mice. MBP immunostaining suggested that P1 iNat8l-V treatment also diminished the extent of myelin disruption in the 3-month-old AspaNur7/Nur7 mice. Scale bars, 100 μm (low magnification images) and 25 μm (high magnification images). Bottom (graphs): NIH ImageJ was used to quantify the proportion of cerebellar white matter that was vacuolated (left) and average cerebellar white matter vacuole area (in μm2) in groups of four Aspa+/+ no Tx, AspaNur7/Nur7 no Tx, and AspaNur7/Nur7 + iNat8l-V. Means ± SEMs are shown. ***p < 0.001, one-way ANOVA.
Figure 6.

Diminished Cerebral Cortical Astrogliosis in iNat8l-V-Treated AspaNur7/Nur7 Mice
Low magnification cerebral cortical images, immunostained for GFAP and Iba1 with higher magnifications of the boxed areas, are shown from a 3-month-old untreated Aspa+/+ mouse, a 3-month-old untreated AspaNur7/Nur7 mouse, and a P1 iNat8l-V treated 3-month-old AspaNur7/Nur7 mouse. Note prominent astrogliosis in the untreated 3-month-old AspaNur7/Nur7 mouse, with less prominent astrogliosis in the P1 iNat8l-V treated 3-month-old AspaNur7/Nur7 mouse. Scale bars, 100 μm (upper panels) and 25 μm (lower panels).
Figure 7.

Diminished Brain Gfap mRNA Expression in iNat8l-V-Treated AspaNur7/Nur7 Mice
Each circle represents an individual 3-month-old mouse; open circles represent males, filled circles represent females. Treatment group sizes were Aspa+/+ no Tx 11; Aspa+/+ + Cont-V 13; Aspa+/+ + iNat8l-V 13; AspaNur7/Nur7 no Tx 8; AspaNur7/Nur7 + Cont-V 6; and AspaNur7/Nur7 + iNat8l-V 7. Means ± SEMs are shown. Two-way ANOVA indicated no significant effects of gender. ***p < 0.001.
P1 iNat8l-V Prevents Cortical Neuron Depletion in AspaNur7/Nur7 Mice
NeuN+ neurons per unit area in frontal motor cortex (“motor cortex neuron density”) was significantly lower in 3-month-old untreated AspaNur7/Nur7 mice than in 3-month-old untreated Aspa+/+ mice. P1 iNat8l-V significantly increased motor cortex neuron density in 3-month-old AspaNur7/Nur7 mice, but not in 3-month-old Aspa+/+ mice. P1 Cont-V treatment did not alter motor cortex neuron density in either 3-month-old Aspa+/+ or 3-month-old AspaNur7/Nur7 mice (Figure 8).
Figure 8.

Diminished Loss of Cortical Neurons in iNat8l-V-Treated AspaNur7/Nur7 Mice
Cerebral cortical NeuN+ neurons per unit area were diminished in untreated 3-month-old AspaNur7/Nur7 mice, but not in P1 iNat8l-V treated 3-month-old AspaNur7/Nur7 mice. Each circle represents an individual mouse, and there were four mice in each treatment group. Non-biased stereology was employed to determine the numbers of NeuN+ neurons per unit area of cerebral cortex. Means ± SEMs are shown. One-way ANOVA, ***p < 0.001; **p < 0.01; n.s., not significant.
Discussion
Administration of an AAV-ASPA vector directly into the brain parenchyma of ASPA-deficient children with pre-existing spongiform leukodystrophy was only minimally effective in ameliorating their neurological deficits8, perhaps because brain ASPA transduction efficiency was low. However, both direct intracranial AAV-mediated brain Aspa gene transduction and brain Aspa transduction via systemic administration of an Aspa AAV vector of a serotype that can cross the blood-brain barrier have been reported to prevent spongiform leukodystrophy in aspartoacylase-deficient mice.22, 23, 24, 25, 26 In one of those mouse studies, substantial protection against development of spongiform leukodystrophy was achieved by selective neonatal astroglial Aspa transduction,25 thus arguing against an obligatory oligodendroglial cell-autonomous requirement for aspartoacylase. Noteably in that study, the engineered astroglial aspartoacylase expression substantially lowered [NAAB] in aspartoacylase-deficient mice.
Prior studies established that homozygous constitutive Nat8l knockout, profoundly lowers [NAAB] and prevents AspaNur7/Nur7 mice from developing leukodystrophy.14, 20 Constitutive knockout of a single Nat8l allele, which reduced [NAAB] in these mice toward but not below the normal range, reduced leukodystrophy severity.20, 21
Though initial progress has been made in developing specific Nat8l antagonists,27 no druggable Nat8l inhibitor is yet available. We therefore tested the therapeutic efficacy in AspaNur7/Nur7 mice of in vivo brain Nat8l knockdown via neonatal combined intracerebroventricular/intracisternal administration of an AAV vector driving transduction of an Nat8l inhibitory shRNA construct. Evaluation of these P1 iNat8l-V treated AspaNur7/Nur7 mice at age 3 months showed that [NAAB] was lower than that in untreated AspaNur7/Nur7 mice, accelerating rotarod performance was normal, brain vacuolation and astrogliosis were markedly attenuated, and cerebral cortical neurons had been “rescued.” These results are consistent with the NAA toxicity hypothesis.
Two recent studies found that elevating [NAAB] in Aspa+/+ mice, either by oral administration of NAA-methyl ester28 or by engineering neuronal transgenic overexpression of Nat8l,26 was not sufficient to cause brain vacuolation. These results suggest that both aspartoacylase deficiency and elevated [NAAB] are necessary to elicit spongiform leukodystrophy in Canavan disease.
One consideration in evaluating the desirability of brain Nat8l knockdown as a potential therapy for Canavan disease is that small interfering RNAs can exert off-target effects.29 Of particular concern, some shRNAs have proven to be neurotoxic after direct administration into brain parenchyma.30, 31 In the present study, however, in which the vectors were administered into the cerebrospinal fluid on P1, cerebral cortical neuron density was not diminished in Aspa+/+ mice at age 3 months, and P1 iNat8l-V treatment prevented the decrease in neuron density in motor cortex that was seen in untreated AspaNur7/Nur7 mice. This iNat8l-V mediated “rescue” of cerebral cortical neurons in AspaNur7/Nur7 mice resembled that previously reported in adult AspaNur7/Nur7 mice to result from constitutive Nat8l knockout.21 Another possible deleterious effect of brain Nat8l knockdown is suppression of synthesis of the neurotransmitter/neuromodulator N-acetyl-L-aspartyl-L-glutamate (NAAG), for which NAA is an obligatory precursor.32 However, whereas a diminution in brain NAAG concentration has been documented in homozygous constitutive Nat8l knockout mice, in which [NAAB] was undetectably low, brain NAAG concentration in heterozygous constitutive Nat8l knockout mice, in which [NAAB] was similar to that obtained by Nat8l knockdown, was maintained within the normal range.20
Though the present study has shown that P1 intracerebroventricular/intracisternal iNAT8l-V administration lowers [NAAB] and suppresses development of spongiform leukodystrophy in aspartoacylase-deficient mice, questions remain. First, because different promoters were incorporated into iNat8l-V to drive transduction of EGFP and of Nat8l shRNA, the selective neuronal distribution of brain EGFP immunoreactivity in the iNat8l-V-treated mice cannot be assumed to accurately reflect the cellular specificity of Nat8l shRNA transduction, thus opening the possibility of yet undetected off-target shRNA effects on brain non-neuronal cells as well as on neurons. Second, because the treated AspaNur7/Nur7 mice were all sacrificed 3 months after iNat8l-V administration, the longer-term effects of Nat8l knockdown on [NAAB] and brain pathology in this murine Canavan disease model have not yet been determined.
Materials and Methods
AAV Vector Design
Four predicted mouse Nat8l (RefSeq: NM_001001985) inhibitory shRNA sequences, based on the Broad TRC algorithm, http://www.broadinstitute.org/rnai/public/seq/search, were screened in an Nat8l cDNA-transduced HEK293 cell line, with silencing testing performed by qRT-PCR 48 hr after cell transfections. Nat8l mRNA knockdown efficiencies by these sequences ranged between 44% and 84%. The full length of the most potent sequence, GCTGACATTGAGCAGTACTAC, was used to construct the Nat8l shRNA CACCGCTGACATTGAGCAGTACTACCTCGAGGTAGTACTGCTCAATGTCAGCTTTTTG, which included the hairpin loop sequence CTCGAG.
An AAV2/AAV8 hybrid backbone was chosen for AAV-Nat8l-shRNA vector construction, based on reports that intracerebroventricular administration of AAV vectors of these serotypes yield widespread brain neuronal transduction in neonatal mice.33, 34 The Nat8l shRNA sequence, driven by a U6 promoter, was incorporated in this vector.35 An EGFP transcript driven by the neuron-specific eSyn promoter35 was also included to facilitate visualization of the distribution of brain transduction by the AAV vector. We refer to this Nat8l knockdown vector, which was manufactured by Vector Biolabs (Malvern, PA, USA) as “iNat8l-V.” The iNat9l-V titer administered to mice in this study was 4.0 × 1013 GC/mL.
To evaluate possible non-specific effects on the brain of P1 intracranial AAV administration and shRNA transduction, a control vector was also designed. This control vector, referred to as “Cont-V,” obtained from Vector Biolabs, was also constructed on an AAV2/AAV8 hybrid backbone, and also employed the U6 promoter to drive an shRNA sequence. Cont-V differed from iNat-V in two important respects: (1) the Cont-V shRNA sequence was selected not to inhibit any known mammalian gene and (2) the EGFP sequence in this vector was driven by a hybrid CMV immediate early enhancer/chicken β-actin promoter,36 rather than by eSyn. The Cont-V titer administered to mice in this study was 4.1 × 1013 GC/mL.
Mice
AspaNur7/+ mice, obtained originally from the Jackson Laboratory (RRID: MGI:5771788), were maintained on a C57BL/6J background. Mouse pups were derived from crosses between AspaNur7/+ males and females, and were genotyped by Transnetyx (Cordova, TN, USA) by a probe-based method. AspaNur7/Nur7 pups were born in the expected 1:4 Mendelian ratio. All mouse procedures were approved by the UC Davis Institutional Animal Care and Use Committee.
AAV Vector Administration
P1 male and female AspaNur7/Nur7 and Aspa+/+ pups were anesthetized on ice, and received 2.5 μL of either iNat8l-V or Cont-V by slow injection into each lateral ventricle and into the cisterna magna (a total of 7.5 μL per mouse) using a 5 μL Hamilton syringe and a 33-gauge needle. The pups were then warmed and returned to their mothers. Additional “no treatment” (no Tx) AspaNur7/Nur7 and Aspa+/+ pups were not anesthetized, and did not receive intracerebroventricular/intracisternal injections.
Accelerating Rotarod Retention Times
An accelerating rotarod apparatus with a starting speed of 4 rotations per min (rpm) and a speed step of 1.2 rpm every 10 s was used. The mice received a 5-min training session on the apparatus each day for 5 days, by which point each had achieved the reported plateau performance.
Brain Harvest
Three-month-old mice were deeply anesthetized with ketamine/xylazine, and their brains (including forebrain, cerebellum, and upper brainstem) were harvested following perfusion with 20 ml of cold PBS. To permit multiple assays from each mouse, the brain was cut sagitally, and the left hemisphere was cut coronally at the bregma with the anterior portion flash-frozen for RNA assay, while the posterior portion (predominantly cerebellum and upper brainstem) was flash-frozen for NAA assay. The right hemisphere and attached upper brainstem and cerebellum were used for immunohistology. These were fixed in 4% paraformaldehyde in PBS for 48 hr at 4°C, followed by washing in PBS, cryoprotection in 30% sucrose, and embedding in OCT prior to cryostat sectioning.
[NAAB]
NAA was measured by high-performance liquid chromatography (HPLC),21, 37 and expressed in μmol/g brain wet weight.
RNA Extraction and qRT-PCR for Nat8l, Gfap, and Hsp90 mRNAs
Brain mRNA was isolated by RNeasy Lipid Tissue Mini Kit (QIAGEN, catalog #74804) and subjected to on-column DNase digestion to eliminate genomic DNA contamination. RNA concentration was measured by Nanodrop 2000, and the quality of RNA was evaluated by Agilent Bioanalyzer, demonstrating RNA integrity numbers (RINs) greater than 6.8. cDNA was synthesized by Omniscript RT Kit (QIAGEN, catalog #205111). One μL of cDNA (equivalent to 5 ng of RNA) was used as template for subsequent real-time quantitative RT-PCR. RT-PCR was performed by using the SYBR green-based method. The following primers were used:
Nat8l-forward 5′-ATCTTCTACGACGGCATCTTGG-3′
Nat8l-reverse 5′-GCGGGTCACAGCAAAACAG-3′
Gfap-forward 5′-CGGAGACGCATCACCTCTG-3′
Gfap-reverse 5′-AGGGAGTGGAGGAGTCATTCG-3′
Hsp90-foreward 5′-GTCCGCCGTGTGTTCATCAT-3′
Hsp90-reverse 5′-GCACTTCTTGACGATGTTCTTGC-3′.
The mRNA levels of Nat8l and Gfap (y axis) were normalized to that of the internal control gene Hsp90 (Hsp90ab1), using the equation: 2(Ct value for Hsp90 − Ct value for Nat8l or Gfap), where Ct (cycle threshold) was defined as the number of cycles required for the SYBR green fluorescent signal to cross the threshold.
Immunohistology
Ten micron thick cryostat sections prepared from forebrain and cerebellum were air-dried at room temperature, then incubated in 10% normal donkey serum (v/v) and 0.1% Triton X-100 (v/v) in PBS for 1 hr at room temperature. Primary antibodies included goat anti-EGFP (1:400, Rockland Immunochemicals #600-102-215; RRID: AB_218187); mouse anti-Gfap (1:100, Millipore Sigma #MAB360; RRID: AB_2109815); rat anti-MBP, aa 36-50 (1:5, Millipore Sigma #MAB395; RRID: AB-240845); rabbit anti-Olig2 (1:500, Millipore AB9610; RRID: AB_570666); rabbit anti-Calbindin (1:200, Spring Bioscience #E10340; RRID: AB_1660695); and rabbit anti-IBA1 (1:500, WAKO Chemicals #019-19471; RRID: AB_2665520). All species-specific secondary antibodies were obtained from Jackson ImmunoResearch, and were used at 1:500. Images were created with a Nikon laser-scanning confocal microscope and processed using NIS-Elements software. Low magnification images were constructed from 8 consecutive stacked 1 μm confocal optical slices, and high magnification images were constructed from three consecutive stacked 1μm confocal optical slices. Laser settings across treatment groups remained consistent. The images shown in Figures 1, 5, and 6 were captured initially in RGB then converted to CMYK using Adobe Photoshop. Vacuole area and size in cerebellar white matter were measured using the NIS-Elements Annotate and Measure tool. Quantification of EGFP+ and EGFP− cerebral cortical NeuN+ neurons and cerebellar Calbindin+ Purkinje neurons was done using NIH ImageJ software.
Stereological Quantification of Cortical Neurons
Coronal forebrain sections at bregma −1.1 were used for NeuN+ neuron counts, which were performed using an Olympus BX61-DSU microscope with motorized stage and Stereo Investigator software. The optical dissector and guard zone were set at 8 and 1 μm, respectively, and the NeuN+ counts were performed at 40× magnification.21
Data Management and Statistics
Assays were performed by blinded observers. Means ± SEs are indicated in the figures. Statistical analysis was by two-way ANOVA (when effects of both treatment and sex were analyzed) or one-way ANOVA (when only effects of treatment were analyzed), in either case followed by the Tukey post hoc test to conduct pairwise comparisons between groups.
Author Contributions
P.B. and T.B. carried out the morphological studies. F.G., X.Z., Y.W., and B.K. did the molecular studies. O.C. and P.B. did the intracerebroventricular injections. N.S. and J.M. did the NAA assays. D.P., corresponding author, was overall director of this project. P.B. and F.G. contributed equally to the manuscript. All authors reviewed and critiqued the manuscript and approved it in final form.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This study was supported by NIH R21NS096004, the Shriners Hospitals for Children, and the Dana Foundation.
References
- 1.Matalon R., Michals K., Sebesta D., Deanching M., Gashkoff P., Casanova J. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am. J. Med. Genet. 1988;29:463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- 2.Mendes M.I., Smith D.E., Pop A., Lennertz P., Fernandez Ojeda M.R., Kanhai W.A., van Dooren S.J., Anikster Y., Barić I., Boelen C. Clinically distinct phenotypes of Canavan disease correlate with residual aspartoacylase enzyme activity. Hum. Mutat. 2017;38:524–531. doi: 10.1002/humu.23181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambetti P., Mellman W.J., Gonatas N.K. Familial spongy degeneration of the central nervous system (Van Bogaert-Bertrand disease). An ultrastructural study. Acta Neuropathol. 1969;12:103–115. doi: 10.1007/BF00692500. [DOI] [PubMed] [Google Scholar]
- 4.Adachi M., Schneck L., Cara J., Volk B.W. Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan’s disease). A review. Hum. Pathol. 1973;4:331–347. doi: 10.1016/s0046-8177(73)80098-x. [DOI] [PubMed] [Google Scholar]
- 5.Mirimanoff P. La dystrophie spongieuse héréditaire des enfants (Canavan: van Bogaert-Bertrand) J. Neurol. Sci. 1976;28:159–185. doi: 10.1016/0022-510x(76)90101-5. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino H., Kubota M. Canavan disease: clinical features and recent advances in research. Pediatr. Int. 2014;56:477–483. doi: 10.1111/ped.12422. [DOI] [PubMed] [Google Scholar]
- 7.Madhavarao C.N., Moffett J.R., Moore R.A., Viola R.E., Namboodiri M.A., Jacobowitz D.M. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J. Comp. Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- 8.Leone P., Shera D., McPhee S.W., Francis J.S., Kolodny E.H., Bilaniuk L.T., Wang D.-J., Assadi M., Goldfarb O., Goldman H.W. Long-term follow-up after gene therapy for Canavan disease. Science Trans. Med. 2012;4:165ra163. doi: 10.1126/scitranslmed.3003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burri R., Steffen C., Herschkowitz N. N-acetyl-L-aspartate is a major source of acetyl groups for lipid synthesis during rat brain development. Dev. Neurosci. 1991;13:403–411. doi: 10.1159/000112191. [DOI] [PubMed] [Google Scholar]
- 10.Madhavarao C.N., Arun P., Moffett J.R., Szucs S., Surendran S., Matalon R., Garbern J., Hristova D., Johnson A., Jiang W., Namboodiri M.A. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc. Natl. Acad. Sci. USA. 2005;102:5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jalil M.A., Begum L., Contreras L., Pardo B., Iijima M., Li M.X., Ramos M., Marmol P., Horiuchi M., Shimotsu K. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 2005;280:31333–31339. doi: 10.1074/jbc.M505286200. [DOI] [PubMed] [Google Scholar]
- 12.Ariyannur P.S., Moffett J.R., Manickam P., Pattabiraman N., Arun P., Nitta A., Nabeshima T., Madhavarao C.N., Namboodiri A.M. Methamphetamine-induced neuronal protein NAT8L is the NAA biosynthetic enzyme: implications for specialized acetyl coenzyme A metabolism in the CNS. Brain Res. 2010;1335:1–13. doi: 10.1016/j.brainres.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Wiame E., Tyteca D., Pierrot N., Collard F., Amyere M., Noel G., Desmedt J., Nassogne M.C., Vikkula M., Octave J.N. Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem. J. 2009;425:127–136. doi: 10.1042/BJ20091024. [DOI] [PubMed] [Google Scholar]
- 14.Guo F., Bannerman P., Mills Ko E., Miers L., Xu J., Burns T., Li S., Freeman E., McDonough J.A., Pleasure D. Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann. Neurol. 2015;77:884–888. doi: 10.1002/ana.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baslow M.H., Guilfoyle D.N. Canavan disease, a rare early-onset human spongiform leukodystrophy: insights into its genesis and possible clinical interventions. Biochimie. 2013;95:946–956. doi: 10.1016/j.biochi.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 16.Clarner T., Wieczorek N., Krauspe B., Jansen K., Beyer C., Kipp M. Astroglial redistribution of aquaporin 4 during spongy degeneration in a Canavan disease mouse model. J. Mol. Neurosci. 2014;53:22–30. doi: 10.1007/s12031-013-0184-4. [DOI] [PubMed] [Google Scholar]
- 17.Tress O., Maglione M., May D., Pivneva T., Richter N., Seyfarth J., Binder S., Zlomuzica A., Seifert G., Theis M. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 2012;32:7499–7518. doi: 10.1523/JNEUROSCI.0392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita T., Katsukawa H., Yodoya E., Wada M., Shimada A., Okada N., Yamamoto A., Ganapathy V. Transport characteristics of N-acetyl-L-aspartate in rat astrocytes: involvement of sodium-coupled high-affinity carboxylate transporter NaC3/NaDC3-mediated transport system. J. Neurochem. 2005;93:706–714. doi: 10.1111/j.1471-4159.2005.03067.x. [DOI] [PubMed] [Google Scholar]
- 19.Traka M., Wollmann R.L., Cerda S.R., Dugas J., Barres B.A., Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J. Neurosci. 2008;28:11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier H., Wang-Eckhardt L., Hartmann D., Gieselmann V., Eckhardt M. N-acetylaspartate synthase deficiency corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J. Neurosci. 2015;35:14501–14516. doi: 10.1523/JNEUROSCI.1056-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sohn J., Bannerman P., Guo F., Burns T., Miers L., Croteau C., Singhal N.K., McDonough J.A., Pleasure D. Suppressing N-acetyl-L-aspartate synthesis prevents loss of neurons in a murine model of Canavan leukodystrophy. J. Neurosci. 2017;37:413–421. doi: 10.1523/JNEUROSCI.2013-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed S.S., Li H., Cao C., Sikoglu E.M., Denninger A.R., Su Q., Eaton S., Liso Navarro A.A., Xie J., Szucs S. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS Gene therapy in Canavan mice. Mol. Ther. 2013;21:2136–2147. doi: 10.1038/mt.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed S.S., Schattgen S.A., Frakes A.E., Sikoglu E.M., Su Q., Li J., Hampton T.G., Denninger A.R., Kirschner D.A., Kaspar B. rAAV gene therapy in a Canavan’s disease mouse model reveals immune impairments and an extended pathology beyond the central nervous system. Mol. Ther. 2016;24:1030–1041. doi: 10.1038/mt.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis J.S., Wojtas I., Markov V., Gray S.J., McCown T.J., Samulski R.J., Bilaniuk L.T., Wang D.J., De Vivo D.C., Janson C.G., Leone P. N-acetylaspartate supports the energetic demands of developmental myelination via oligodendroglial aspartoacylase. Neurobiol. Dis. 2016;96:323–334. doi: 10.1016/j.nbd.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gessler D.J., Li D., Xu H., Su Q., Sanmiguel J., Tuncer S., Moore C., King J., Matalon R., Gao G. Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. JCI Insight. 2017;2:e90807. doi: 10.1172/jci.insight.90807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Jonquieres G., Spencer Z.H.T., Rowlands B.D., Klugmann C.B., Bongers A., Harasta A.E., Parley K.E., Cederholm J., Teahan O., Pickford R. Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol. 2018;135:95–113. doi: 10.1007/s00401-017-1784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thangavelu B., Mutthamsetty V., Wang Q., Viola R.E. Design and optimization of aspartate N-acetyltransferase inhibitors for the potential treatment of Canavan disease. Bioorg. Med. Chem. 2017;25:870–885. doi: 10.1016/j.bmc.2016.11.060. [DOI] [PubMed] [Google Scholar]
- 28.Appu A.P., Moffett J.R., Arun P., Moran S., Nambiar V., Krishnan J.K.S., Puthillathu N., Namboodiri A.M.A. Increasing N-acetylaspartate in the brain during postnatal myelination does not cause the CNS pathologies of Canavan disease. Front. Mol. Neurosci. 2017;10:161. doi: 10.3389/fnmol.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson A.L., Linsley P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 30.McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin J.N., Wolken N., Brown T., Dauer W.T., Ehrlich M.E., Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011;18:666–673. doi: 10.1038/gt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neale J.H., Olszewski R.T., Zuo D., Janczura K.J., Profaci C.P., Lavin K.M., Madore J.C., Bzdega T. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J. Neurochem. 2011;118:490–498. doi: 10.1111/j.1471-4159.2011.07338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broekman M.L., Comer L.A., Hyman B.T., Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- 34.Chakrabarty P., Rosario A., Cruz P., Siemienski Z., Ceballos-Diaz C., Crosby K., Jansen K., Borchelt D.R., Kim J.Y., Jankowsky J.L. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White M.D., Milne R.V.J., Nolan M.F. A molecular toolbox for rapid generation of viral vectors to up- or down-regulate neuronal gene expression in vivo. Front. Mol. Neurosci. 2011;4:8. doi: 10.3389/fnmol.2011.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grimm D., Pandey K., Kay M.A. Adeno-associated virus vectors for short hairpin RNA expression. Methods Enzymol. 2005;392:381–405. doi: 10.1016/S0076-6879(04)92023-X. [DOI] [PubMed] [Google Scholar]
- 37.Li S., Clements R., Sulak M., Gregory R., Freeman E., McDonough J. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem. Res. 2013;38:2385–2396. doi: 10.1007/s11064-013-1151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]