

SUMMARY
Vipp1 is highly conserved and essential for photosynthesis, but its function is unclear as it does not participate directly in light-dependent reactions. We analyzed Vipp1 localization in live cyanobacterial cells and show that Vipp1 is highly dynamic, continuously exchanging between a diffuse fraction that is uniformly distributed throughout the cell and a punctate fraction that is concentrated at high curvature regions of the thylakoid located at the cell periphery. Experimentally perturbing the spatial distribution of Vipp1 by relocalizing it to the nucleoid causes a severe growth defect during the transition from non-photosynthetic (dark) to photosynthetic (light) growth. However, the same perturbation of Vipp1 in dark alone or light alone growth conditions causes no growth or thylakoid morphology defects. We propose that the punctuated dynamics of Vipp1 at the cell periphery in regions of high thylakoid curvature enable acquisition of photosynthetic competency, perhaps by facilitating biogenesis of photosynthetic complexes involved in light-dependent reactions of photosynthesis.
Keywords: thylakoid biogenesis, photosynthesis, cyanobacteria, live-cell imaging, membrane curvature
Abbreviated Summary
In live and growing cyanobacterial cells, cytosolic Vipp1 continuously concentrates on and off at regions of high thylakoid curvature. These punctuated dynamics of Vipp1 represent events of Vipp1 oligomerization on the thylakoid membrane which are essential for turning on photosynthetic metabolism, perhaps by mediating functional assembly of photosynthetic machinery.
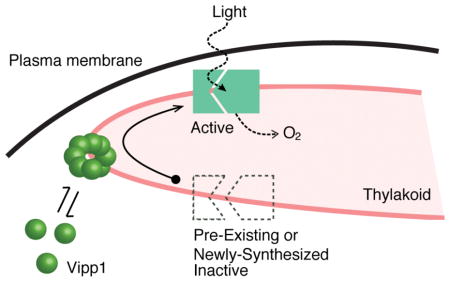
INTRODUCTION
Oxygenic photosynthesis is a metabolic process that must be dynamically modulated to accommodate varying light conditions and cellular needs (Eberhard et al., 2008). If extraneous light energy is not productively engaged in a timely manner, phototoxic damage can arise and lead to cellular death (Apel and Hirt, 2004). These light-dependent reactions are catalyzed by photosynthetic protein complexes that are assembled in a specialized membrane system which forms the thylakoid compartment in both chloroplasts and cyanobacterial cells (Rast et al., 2015). Although much is known about the structure and function of individual photosynthetic complexes, it is still unclear how the thylakoid membrane system is formed or maintained, and how the photosynthetic complexes are assembled in the thylakoid membrane (Pribil et al., 2014; Rast et al., 2015). These two issues are interrelated – thylakoid structure depends on the presence of photosynthetic complexes (Pribil et al., 2014; Zhang et al., 2014), and assembly of photosynthetic complexes depends on the presence of thylakoid membrane (Nickelsen and Rengstl, 2013; Yang et al., 2015). Thus, experimentally uncoupling the factors required for thylakoid membrane formation from those required for photosynthesis is difficult.
Vipp1 (Vesicle-inducing protein in plastids 1), a conserved protein in plants, algae and cyanobacteria, has been proposed to play a role in thylakoid formation (Kroll et al., 2001; Vothknecht et al., 2012; Heidrich et al., 2017). Vipp1 is not known to participate in photosynthetic reactions and its deletion in plants causes loss of photosynthesis and thylakoid organization, as well as loss of viability (Kroll et al., 2001; Westphal et al., 2001). The biophysical properties of Vipp1 are consistent with a membrane-related function – in vitro studies of Vipp1 revealed that it is a soluble protein with high affinity for lipid components (Otters et al., 2013; McDonald et al., 2015). On membrane surfaces prepared in vitro, recombinant Vipp1 forms higher order oligomers and, under certain conditions, mediates vesicle fusion (Hennig et al., 2015). Thus, Vipp1 may facilitate thylakoid membrane formation, with one recent model proposing that oligomeric Vipp1 is an intermembrane lipid transporter, possibly transporting lipids from the plasma (or chloroplast inner) membrane to the thylakoid membrane (Heidrich et al., 2017).
However, not all experiments are consistent with the above hypothesis. When expression of Vipp1 is reduced to a low level in cyanobacteria (Gao and Xu, 2009), photosynthetic output was severely diminished but thylakoid morphology remained intact, suggesting that Vipp1 plays a role in photosynthesis in addition to, or instead of, being required for thylakoid formation. This observation, combined with the fact that deletion of a core photosynthetic complex induces a thylakoid morphology defect (Zhang et al., 2014), raises the possibility that Vipp1 may not directly facilitate thylakoid formation but instead its function may be related to photosynthesis. If this is true, knockdown or deletions of Vipp1 could generate a thylakoid morphology defect as a secondary consequence. Given the pleiotropy of vipp1 phenotypes and the contrasting conclusions reached by other studies (Aseeva et al., 2007; Fuhrmann, Gathmann, et al., 2009), the role of Vipp1 in thylakoid membrane formation and in photosynthetic complexes formation remains unclear.
Previous microscopy work demonstrated that Vipp1 manifests as both diffuse and rare concentrated signals (Nordhues et al., 2012; Zhang et al., 2012; Bryan et al., 2014). In cyanobacteria exposed to damaging high-light induced stress, the diffuse form of Vipp1 concentrates into long-lived immobile foci at the cell periphery, which were suggested to be important for light-induced stress protection (Bryan et al., 2014). However, the native localization and dynamics of Vipp1 in normal (non-stressful) growth conditions has not been well characterized. Furthermore, without a perturbation that targets Vipp1 localization, the functional importance of this localization for photosynthesis or thylakoid membrane formation is not clear.
Here we show that in normal growth conditions, fluorescently labeled Vipp1 is highly dynamic, continuously exchanging between two fractions – a punctate fraction at the cell periphery that is concentrated at high curvature regions of the thylakoid, and a diffuse fraction that is uniformly distributed in the cytoplasm. By rapidly perturbing the spatial distribution of Vipp1 in living cells, we show that native Vipp1 localization is not required for thylakoid membrane formation, but is essential during the transition from non-photosynthetic to photosynthetic metabolism. We propose that the punctate fraction of Vipp1 is the cytological manifestation of the oligomeric and membrane-bound forms of Vipp1, whose role is to enable acquisition of photosynthetic competence. We hypothesize that Vipp1 facilitates the functional assembly of photosynthetic complexes.
RESULTS
Vipp1 forms transient puncta at regions of high thylakoid curvature at the cell periphery
To gain insight into the cellular role of Vipp1 we studied its localization and dynamics in live cells of Synechocystis sp. PCC 6803. To fluorescently label endogenous Vipp1 we integrated a Vipp1-mGFPmut3 fusion construct (hereafter Vipp1-GFP) at the native vipp1 locus via homologous recombination. Both the expression level of Vipp1-GFP protein and the bulk growth rate of the strain harboring the construct were similar to that of the wild type parental strain (Fig. S1A–B), as was reported previously (Bryan et al., 2014). In actively growing cells (doubling time ~7 h) maintained at moderate light intensity (Fig. S1B), Vipp1-GFP is present in two fractions: as fluorescent diffraction-limited spots (hereafter puncta), and as a diffuse signal in the cytoplasm (Fig. 1A and Fig. S1C). Since Vipp1 puncta form when Vipp1 is fused to a known monomeric GFP variant that is least prone to induce multimerization of various bacterial targets (Landgraf et al., 2012) and also when fused to the SNAP-tag (Keppler et al., 2003) (Fig. S1D), which is not known to induce artificial clustering, we conclude that the observed Vipp1 puncta formation and diffuse signal reflect native spatial localization.
Fig. 1.

Vipp1 forms peripheral puncta that are located at regions of high thylakoid curvature. A) Representative live-cell epifluorescence images in the Vipp1 (GFP) and the thylakoid (far-red) channels. Images represent Maximum Z-Intensity Projections of 3D Z-stacks. Scale bar = 2 μm. B) Distribution of the number Vipp1 puncta per cell in a population of cells obtained from an exponentially growing culture. The mean and the variance of the distribution is 1.36. Puncta were identified by thresholding and segmenting filtered 3D Z-stacks. C) Analysis of the positioning of Vipp1 puncta relative to the cell radius. For each Vipp1 puncta identified at a given mid-cell plane (see inset diagram for an illustration), the corresponding distances to the cell center (labeled as “C”) and to the closest cell edge (labeled as “E”), as defined by brightfield segmentation masks, were obtained. The sum of the two distances defined the radius (blue dashed line) that was scaled from 0 to 1 on the x-axis. On the left y-axis, the mean of the thylakoid signal intensity along the line profiles (i.e. radii) connecting the cell centers, the Vipp1 puncta and the closest cell edge points is shown. The right y-axis shows the range of intensities (sizes) of all Vipp1 puncta analyzed. To limit the variability in size coming from dividing cells only cells whose area shape met an eccentricity value of 0.6 or less were analyzed. D) 3D rendering of a super-resolution image of a live cell of Synechocystis PCC 6803 expressing Vipp1-GFP (green) and CurT-mTurquoise2 (blue). Thylakoids (red) are distributed at the cell periphery which by fluorescence microscopy show up as peripheral sheets. Vipp1 puncta are localized at the edge of thylakoid enrichments, at the same regions where the thylakoid membrane protein CurT is concentrated. Images were obtained with a laser scanning confocal system equipped with an Airyscan detector (Zeiss LSM880) which affords increased spatial resolution. Bar = 1 μm. For a full rendering of the same cell see Video S1. E) Representative live-cell confocal fluorescence image of a cell expressing both Vipp1-GFP and CurT-CFP obtained by Airyscan imaging. As diagramed on the left, the rows show Vipp1, thylakoid and CurT channels at the top, middle and the bottom of a cell. Two Vipp1 puncta are shown at mid-cell slice (orange arrowheads) which co-localize with CurT enrichments and with gaps in the thylakoid signal. A profile line (dashed in light grey) running circumferentially through the peripheral thylakoids was used to extract the intensities of Vipp1, CurT and thylakoid fluorescence and plotted in panel F. Bar = 1 μm. F) Intensities of the Vipp1, CurT and thylakoid signals along the mid-cell circumferential curved line profile traced in panel E. Raw signal intensities were normalized from 0 (minimum) to 1 (maximum) on the y-axis. Orange arrowheads indicate colocalization of Vipp1 puncta with CurT, which is enriched at regions of high thylakoid signal changes (edges).
In live cells growing in light on the microscope stage (see Experimental Procedures), we find that the number of Vipp1 puncta per cell is well-described by a Poisson distribution with a mean and variance of 1.36 (Fig. 1B), suggesting that the formation of each punctum is an independent event. By measuring the positioning of each punctum relative to the cell boundary we find that most Vipp1 puncta localize near the cell periphery (Fig. 1C and Fig. S1E). At this region, the thylakoids are highly abundant (Fig. 1C), as estimated by the fluorescence emitted by the endogenous photosynthetic proteins in the far-red portion of the visible spectrum (Vermaas et al., 2008). Moreover, within the thylakoids, we observed that Vipp1 puncta tended to localize to regions where the thylakoid signal is low (Fig. 1A and Fig. S1C). These low thylakoid signal regions correspond to the edges of the thylakoid stacks and are known as zones of high thylakoid membrane curvature (Heinz et al., 2016). To confirm localization at sites of high thylakoid curvature, we asked whether Vipp1 puncta co-localize with CurT, a membrane protein enriched at these regions (Heinz et al., 2016). For this, we used super-resolution imaging to determine the localization of Vipp1, CurT, and the thylakoid signal in living cells (see Experimental Procedures). We find that in any given cell, Vipp1 puncta localize at the edge of thylakoid stacks where CurT is concentrated (Fig. 1D–F, Video S1). To quantify the relationship between Vipp1 puncta and CurT, we extracted and compared the intensity profiles of 76 arc lines that are each centered on a Vipp1 punctum found at the mid cell plane (Fig. S2A–B). We found that in general, Vipp1 signal positively correlated with the CurT signal and negatively with the thylakoid signal (Fig. S2A–B). We also used automatic 3D object-based colocalization analysis to evaluate the spatial relationship between Vipp1 puncta with CurT enrichments and find that the majority of puncta overlap with the volume of CurT objects (Fig. S2C–D). Based on these observations we conclude that in live cells the punctate fraction of Vipp1 localizes at cell periphery at or near regions of high thylakoid membrane curvature.
To better localize Vipp1 at the cell periphery and determine its relation to the thylakoid membranes we used immunoelectron microscopy. For immunodetection we used anti-GFP primary antibodies and gold-conjugated secondary antibodies to stain ultrasections (60 nm thickness) of freeze-substituted cells obtained from vipp1-gfp and wild type strains. We find that immunogold signals specific for Vipp1-GFP were enriched near the edges of thylakoids (high curvature regions which appear as tips in 2D representations) which typically converge near the plasma membrane (Fig. 2 and Fig. S3). Even though the antibodies can only bind the antigens from the surface of ultrasections, and therefore probe only a sparse subset of Vipp1-GFP, occasional clusters of 2–3 nanogold signals were observed near the thylakoid edges (Fig. 2C, Fig. S3A, Fig. S3F). This distribution is consistent with the idea that a fluorescent Vipp1 punctum consists of multiple Vipp1-GFP molecules closely juxtaposed in space. Combining the fluorescent and electron microscopy observations we conclude that the regions of high thylakoid membrane curvature are the sites of Vipp1 puncta localization.
Fig. 2.

Detection of Vipp1-GFP by immuno-electron microscopy. Representative immunoelectron microscopy image of vipp1-gfp (A and C) or wild type cells (B) stained with anti-GFP primary antibodies detected by gold-conjugated secondary antibodies (black arrowheads). Arrows show the stacked thylakoids which run parallel to the plasma membrane. Scale bar = 100 nm (inset scale bar = 50 nm). D) Count of gold particles categorized by the proximity to the nearest cellular feature (plasma membrane, thylakoid edges, thylakoid (excluding edges), cell outside and the cytoplasm) in 35 independent whole cell sections obtained from either vipp1-gfp or wild type cells, respectively.
At a given time, many cells appear to contain no Vipp1 puncta (Fig. 1B), raising the question of whether only a fraction of cells are capable of forming Vipp1 puncta or if all cells can form puncta that are relatively short lived. We used time-lapse fluorescent microscopy to investigate Vipp1 dynamics in cells growing photosynthetically on the microscope stage and find that sparse Vipp1 puncta dynamically appear and disappear in all cells over time (Video S2). Furthermore, we also observed that in a small minority of cells (under 5%), Vipp1 is present exclusively as very bright and stable puncta, but these are observed only in non-growing cells (Fig. S4A). Thus, transient Vipp1 puncta likely exist in all growing cells over time, explaining the zero puncta bin seen in the distribution of Vipp1 counts per cell (Fig. 1B).
To determine if Vipp1 puncta form directly on the membrane or if they form in the cytoplasm and diffuse to the membrane, we imaged photosynthetically growing cells at high temporal resolution to capture the appearance, disappearance, and the movement of Vipp1 puncta. We find that the majority of Vipp1 puncta rise and fall in intensity on a time scale of 1–2 min (Fig. 3A–B, Fig. S4B, Video S3) and their mobility at the cell periphery is greatly limited (Fig. 3C and Video S4). This constrained mobility and the rise and fall in fluorescence within a diffraction limited volume is consistent with a Vipp1 assembly process occurring on the membrane. Given that Vipp1 binds membranes as a homo-oligomeric complex in vitro and exists as a distribution of oligomeric states in cell lysates (Fuhrmann, Bultema, et al., 2009; Hennig et al., 2015; McDonald et al., 2015; Heidrich et al., 2016), we infer that the dynamically forming Vipp1 puncta, occurring at or near the highly curved regions of the thylakoid compartment, represent events of punctuated oligomerization and de-oligomerization between its cytosolic and its membrane-bound forms.
Fig. 3.

Vipp1 forms transient puncta of limited mobility at the cell periphery. A) Time-series montage of a live cell growing photosynthetically on the microscope stage. The images were acquired every 30 seconds in the Vipp1 and thylakoid channels for a duration of 10 minutes. The montage shows only the mid-cell (obtained from a filtered epifluorescence Z-stack) for the first 2.5 minutes. The blue arrows highlight the appearance and disappearance of a Vipp1 puncta in consecutive time points. The dashed circumferential line was used to extract the intensity profiles of the two channels and build the kymogram displayed below the montage. The Vipp1 puncta appear and disappear at the edge the thylakoids which manifest as horizontal streaks. Two examples of Vipp1 intensity profiles in time (i and ii) are graphed on the right to illustrate how the full-width at half-maximum (FWHM) durations and peak intensities were obtained for a given punctum. The contrast in the Vipp1 channel was linearly adjusted to enhance puncta visualization. B) Distribution of Full-Width at Half Maximum (FWHM) durations relative to their peak intensities for a representative subset of Vipp1 puncta. The values were extracted from the individual kymograms of 15 growing cells that were imaged every 30 seconds for a duration of 20 min (see examples in Video S3). All images were acquired in epifluorescence as Z-stacks at the indicated times and processed as described in Experimental Procedures. C) Example of Vipp1 puncta mobility recorded by continuous imaging (frame rate 81 milliseconds) in epifluorescence mode. On the left, the Laplacian-of-Gaussian filtered image of a cell containing two peripheral Vipp1 puncta is shown. A circumferential line profile traversing the cell periphery (dashed line) was used to extract the kymogram on the right to illustrate the constrained mobility of Vipp1 puncta over time. The kymogram also captures the birth of a new Vipp1 punctum as evidenced by the appearance of a middle horizontal streak over time. See Video S4 for examples of other cells imaged at similar time scale resolution. Scale bar =1 μm.
Spatio-temporal distribution of Vipp1 enables photosynthetic competency
To test the functional importance of the spatial distribution and dynamics of Vipp1 in the cell, we designed a perturbation in which Vipp1 was rapidly and reversibly localized to the cell center, away from the cell periphery, without changing its overall concentration. To achieve this perturbation we used the anchor-away technique (Liberles et al., 1997; Haruki et al., 2008) in which a protein of interest can be rapidly moved away from its native location by inducing a drug-dependent dimerization to another protein, known as the anchor, that is in a different sub-cellular location (Fig. 4A). For the anchor we used the histone-like HU protein that binds to DNA in the nucleoid and is localized at the center of the cell (Wang et al., 2011). In the presence of the drug rapamycin, cells expressing the components of the anchor-away system rapidly alter the native localization of Vipp1 by coalescing the entire pool of Vipp1 to the nucleoid region of the cell, away from the cell periphery (Fig. 4B, Fig. S5). We note that this perturbation of Vipp1 localization inherently alters the dynamics of puncta formation occurring at the cell periphery.
Fig. 4.

Perturbation of Vipp1 localization and characterization of the growth conditions in which the perturbation was applied. A) Diagram illustrating how Vipp1 can be relocalized to the nucleoid, away from the cell periphery using the anchor-away approach. A schematic cell is shown, with red disks representing thylakoids, the black circle in the center representing the nucleoid, and green dots representing the diffuse and punctate fractions of Vipp1. Human FKBP12 (12 kDa FK506-binding protein) was fused to Vipp1-GFP and the FRB (FKBP-rapamycin binding) domain was fused to the histone-like HU protein. In the presence of the cell permeable drug rapamycin, the Vipp1-GFP-FKBP12 chimera tethers to HU-FRB and forms a ternary complex enriched at the nucleoid due to the high binding affinity of HU to DNA. B) Rapamycin-dependent relocalization of Vipp1 to the nucleoid region at the cell center is effective and rapid. Raw images of Maximum Z-Intensity Projections in the brightfield and Vipp1 channels of exponentially growing cells expressing the Vipp1-GFP anchor-away system in the presence or absence (solvent only) of rapamycin after 20 min incubation. Nucleoid-localized Vipp1-GFP manifests as bright concentrations of Vipp1-GFP signal at the cell centers. The grayscale contrast is the same for both conditions. Cell outlines based on the brightfield channel are overlaid in red. C) The three growth conditions used for testing the functional importance of native Vipp1 localization via anchor-away: Dark Growth, based on glucose, during which thylakoids are multiplied while photosynthetic competency is highly reduced; Dark-to-Light Transition, during which dark-grown cells are shifted to light in glucose-free media so that cells can regain their photosynthetic competency over time (“delta” symbols in front represent changes: glucose drop out and light on); and Light Growth, during which the cells grow in constant photosynthetic conditions. In each panel, a cell is shown growing from a smaller size to a larger size, and thylakoids are shown in orange if not competent for photosynthesis and in green if they do support photosynthesis. Photosynthetic competency is illustrated as the ability to produce O2 when light is applied (dotted arrows). Approximate growth rates shown were estimated by monitoring the optical density of liquid cultures and are consistent with previously published measurements (Anderson and McIntosh, 1991). D) Thylakoids are present in both dark- and light-grown cells. Top - representative electron microscopy images of dark- and light grown cells showing a similar thylakoids arrangement consisting of several cell peripheral stacks of 2–4 thylakoid sheets (arrows). Scale bar = 0.5 μm. Bottom - thylakoid fluorescence images of two dark-grown cells before (left) and after 24 hours of growth in light (right) directly on the microscope stage in glucose-free media. In both conditions, the thylakoid signal is enriched at the cell periphery with occasional gaps in fluorescence intensity that correspond to the thylakoid stacks’ edges. The same greyscale contrast was applied to both images. Only the mid-Z planes of filtered image stacks are shown. Scale bar = 2 μm. The dark-grown cells were obtained by growing a culture in darkness on glucose for 144 h (~ 8 generations), as described in Experimental Procedures. E) Both dark- and light-grown cells contain photosynthetic complexes and pigments. Whole-cell absorption (dashed lines) and low-temperature (77K) fluorescence emission (solid lines) spectra of dark-grown (144 h in darkness) and light-grown (24 h post-dark) cultures. The chlorophyll (445 and 680 nm) and the phycocyanin (625 nm) peaks in the absorption spectra are present in both growth conditions, albeit at slightly lower level in dark-grown culture. The fluorescence emission spectra (excitation at 445 nm) reveal the signature peak of the photosystem I at 725 nm present in both cultures, and the peaks associated with active photosystem II (685 nm and 690 nm) which appear conspicuous in the light-grown culture only, as was reported previously (Barthel et al., 2013). F) Dark-grown cells are photosynthetically incompetent. Rates of whole-cell oxygen production of dark- and light-grown cultures. Light-grown cells were obtained by shifting the dark-grown cells to light for 24 h in glucose-free medium. Linear rates of oxygen production were obtained from cells maintained in darkness (respiration, i.e. oxygen consumption) or in saturating light (photosynthesis, i.e. oxygen evolution) for the duration of the measurement. Respiratory activity which originates from the protein complexes that also localize in thylakoid membranes is similar between the dark- and light-grown samples. Error bars are standard error of the mean obtained from three biological replicates.
To reveal the functional role of native Vipp1 localization, we chose to apply the anchor-away perturbation in three different physiological conditions that enable us to disentangle interactions between thylakoid formation and photosynthesis – Dark Growth, Dark-to-Light Transition, and Light Growth (Fig. 4C). During Dark Growth (Anderson and McIntosh, 1991), cells rely on glucose from the media to grow and divide in the absence of photosynthetic light, while still multiplying their thylakoid membranes and associated pigments. However, dark-grown cells are not photosynthetically competent (Barthel et al., 2013), meaning they are incapable of immediate oxygen production upon application of light. In the Dark-to-Light Transition, photosynthetic competency is induced when dark-grown cells are shifted to light conditions for at least 6 - 8 hours (Barthel et al., 2013). During Light Growth, cells rely on photosynthesis to grow and divide in the absence of glucose, and also continuously multiply their thylakoids and photosynthetic components. We confirmed in our own experiments that dark-grown cells, even after multiple cell divisions, still maintain and produce thylakoids and photosynthetic pigments as estimated by microscopy, and spectrophotometry respectively (Fig. 4D–E). We note that in electron microscopy images, the thylakoid sheets in dark-grown cells appeared less tightly stacked than in light-grown cells, however they still maintained a peripheral arrangement that was also observed in fluorescence images of live cells, which later grew and divided when shifted to Light Growth (Fig. 4D). We also confirmed that dark-grown cells are photosynthetically incompetent as evidenced by their inability to immediately evolve oxygen in the presence of saturating light (Fig. 4F). When these dark-grown cells are shifted to Light Growth in glucose free media, the photosynthetic competency is restored within 24 h (Fig. 4F) as shown previously (Barthel et al., 2013).
We asked whether the rapid drug-induced relocalization of Vipp1 to the nucleoid in the three conditions (Dark, Dark-to-Light and Light) would elicit either a growth or thylakoid morphology defect. For each of the conditions, we added rapamycin to induce Vipp1 relocalization and used time-lapse fluorescent microscopy to monitor thylakoid content, Vipp1 localization and cell growth for at least half of the cell cycle (Fig. 5A–C). For longer timescales, we measured growth by monitoring turbidity in bulk culture (Fig. 6A–B) and examined thylakoid morphology by both fluorescence and electron microscopy (Fig. 6C). We find that Vipp1 relocalization causes no significant growth or thylakoid morphology defects in Dark or Light conditions (Fig. 5A–B, Fig. 6A–C, Fig. S6), but causes a severe growth defect when Vipp1 is relocalized during the Dark-to-Light Transition (Fig. 5C, Fig. 6A, Video S5). The growth defect observed during the Dark-to-Light Transition is not due to nucleoid-related changes or to drug addition, since a control strain in which heterologously-expressed GFP is relocalized to the nucleoid exhibits no growth phenotype (Fig. 6B). The severity of the growth defect correlates with the timing of Vipp1 relocalization during the Dark-to-Light Transition, with less of a growth defect observed when the perturbation occurs later in the transition (Fig. S7A). Intriguingly, the most severe growth defect is observed when relocalization is induced at the start of the Dark-to-Light Transition, which coincides with the peak burst in Vipp1 puncta formation in normal conditions (Fig. S7B, Video S6).
Fig. 5.

Relocalization of Vipp1 to the nucleoid has no significant effect on Dark or Light Growth but elicits a severe growth defect during Dark-to-Light Transition. A) Relocalization of Vipp1 induces no growth defect during Dark Growth. Left: Example images (in raw epifluorescence) of dark-grown cells (Maximum Z-Intensity Projections, 12 Z-steps) in brightfield, thylakoid and Vipp1 channels before addition of rapamycin (0 h) and after 23 hours of growth in darkness. A mix of two strains are shown, one (yellow outlines) in which the Vipp1-GFP-FKBP12 construct is expressed alone, and another (red outlines) in which the HU-FRB anchor construct is also expressed in trans (i.e. responsive to rapamycin-based relocalization). 20 μM rapamycin was added to the agarose pad holding the cells on the microscope stage immediately after time 0 h. Right: quantification of cell growth as areas (extracted from automatic segmentations in the brightfield channel - open circles) over time of ten random cells obtained from each strain (with or without HU-FRB anchor) in the same field of view. On the bottom x-axis, normalized time is shown as the time it took for “-anchor” cells to increase the average cell area by 50 %. On the top x-axis (dashed grey line), time is shown in hours. Arrows indicate the addition of rapamycin to the agarose pad. Average growth trace is shown as a black bold curve. The mean and standard error of the mean of area expansions rates of the 10 cells are shown in the lower right corners. We found no significant difference between the means of “+anchor” and “- anchor” cells as calculated with the two-sample two-tailed t-test at 5% significance level. B) Relocalization of Vipp1 induces no significant growth defect during Light Growth. Similar to panel A - on the left: selected images of light-growing cells obtained from a timelapse movie (8 Z-steps, every 30 min for 7.5 hours total). Right: quantifications of cell growth as areas over time of ten random cells obtained from each strain from the same field of view. No significant differences between the means of “+anchor” and “- anchor” cells rates (shown at the lower right corner) as calculated with the two sample two-tailed t-test at 5% significance level were found. C) Relocalization of Vipp1 during Dark-to-Light Transition induces a severe growth defect. Similar to panel A - on the left: selected images of dark-grown cells shifted to light growth obtained from a timelapse movie (10 Z-steps, every 20 min for 12 hours). To remove the glucose-containing media, the cells were briefly washed in glucose-free media prior to imaging. Photosynthetic light and rapamycin were applied immediately after the first timepoint. On the right: quantification of cell growth as areas over time of ten random cells obtained from each strain from the same field of view. The difference between the means of “+anchor” and “- anchor” cells rates (shown in the lower right corner) was found to be significant (** P < 0.001) based on the two-sample two-tailed t-test at 5% significance level.
Fig. 6.

Vipp1 relocalization produces no growth or thylakoid morphology defect in Dark or Light Growth cultures but elicits a severe growth defect in the Dark-To-Light Transition. A) Effect of Vipp1 relocalization on growth in bulk culture. Dark- and light growing vipp1-gfp-fkbp12 cells of the two strains - with or without the HU-FRB anchor construct expressed in trans - were resuspended in fresh media in pairs of flasks at the same starting inoculum and incubated in dark or in light for 48 h. 20 μM rapamycin (arrows) or solvent only (dimethyl sulfoxide, denoted as “-”) was added at time 0 h. The effect on growth was similar regardless if the glucose from the Dark growth media was washed out or still present before shifting the cells to light. B) Control cultures showing that relocalization of GFP alone at the nucleoid elicits no growth defects. Dark- and light growing gfp-fkbp12 cells of the two strains - with or without the HU-FRB anchor construct expressed in trans - were inoculated in fresh media in pairs of flasks at the same starting inoculum and incubated in dark or in light for 48 h. 20 μM rapamycin (arrows) or solvent only (dimethyl sulfoxide, denoted as “-”) was added at time 0 h. C) Relocalization of Vipp1 during Dark or Light Growth induces no changes in overall thylakoid content or morphology. Top - filtered images in the thylakoid channel (mid-cell plane) obtained from cells expressing all the components of Vipp1 anchor-away in the presence or absence of rapamycin after 48 h of growth in darkness (left) or in light (right). The images are displayed at the same grayscale contrast for rapamycin and no rapamycin treated cells. The overall thylakoid morphology (i.e. peripherally localized signal with occasional gaps in fluorescence intensity that correspond to regions of high curvature of thylakoid membranes) in the rapamycin-treated cells is similar to that of the control cells. Scale bar = 2 μm. Bottom - corresponding electron microscopy images of representative cells from each of the conditions tested. Stacked thylakoids (arrows) are present in both dark- and light-grown cells and their morphology and arrangement remain unaffected by Vipp1 perturbation. Scale bar = 0.5 μm. See Fig. S6 for more electron microscopy examples of cells from these conditions.
Based on the following observations, we conclude that the native localization of Vipp1 is not necessary for thylakoid membrane growth, but is necessary for enabling acquisition of photosynthetic competency. First, since perturbing Vipp1 in the Dark condition (in which there is active thylakoid growth) triggers no defect in thylakoid growth or morphology (Fig. 6C, Fig. S6A–B), we conclude that Vipp1 localization is not required for thylakoid membrane growth. However, since perturbing Vipp1 in the Dark-to-Light condition (in which cells must enable photosynthesis in order to grow) elicits a severe growth defect (Fig. 5C, Fig. 6A), we infer that native Vipp1 localization is necessary to enable acquisition of photosynthetic competency. As photosynthetic competency is already established during Light Growth and Vipp1 perturbation does not trigger a growth or thylakoid morphology defect (Fig. 5B, Fig. 6C, Fig. S6C–D), we infer that Vipp1 localization is not critical for maintaining thylakoid membranes or supporting active and pre-existing photosynthetic capacity.
DISCUSSION
The presence of Vipp1 as diffuse and punctate signals has been previously reported in cyanobacteria exposed to two extreme physiological conditions: low light (8 μE m−2 s−1 intensity) and high light (600 μE m−2 s−1 intensity) (Bryan et al., 2014). In each of these conditions, both suboptimal for growth (Kopecná et al., 2012), Vipp1 was predominantly distributed either as a diffuse signal in low light or as stable, long-lived puncta in high light (Bryan et al., 2014). Here we characterized in detail the localization and dynamics of these two Vipp1 fractions in exponentially growing cells, maintained at moderate light intensity (100 μE m−2 s−1 intensity), and show that Vipp1 continuously exchanges between the diffuse and punctate fractions on a timescale of minutes (Fig. 1C, Fig. 3A–B, Video S3). This punctate fraction is short-lived, displays limited mobility (Fig. 3) and localizes to regions of high thylakoid membrane curvature (Fig. 1D–F, Fig. S2). The localization of Vipp1 is consistent with the known biochemical properties of Vipp1 oligomerization and membrane binding (Fuhrmann, Bultema, et al., 2009; Otters et al., 2013; Hennig et al., 2015), particularly the high affinity to membrane substrates manifesting high stored curvature elastic stress (McDonald et al., 2015). Combining these observations with the spatiotemporal dynamics of Vipp1, we propose that transient formation of Vipp1 puncta at regions of high thylakoid curvature represent events of assembly and disassembly of Vipp1 oligomers that accompanies normal cellular growth. The diffuse and cytosolic Vipp1 fraction may consist of a range of lower order oligomerization states (ex. dimers, tetramers etc.) of Vipp1 that are not readily distinguishable in situ as puncta but observed previously in soluble fractions (Fuhrmann, Bultema, et al., 2009).
The ability of Synechocystis PCC6803 cells to grow non-photosynthetically, while still maintaining and multiplying their thylakoids (Fig. 4D) – which are also the sites of respiratory activity (Mullineaux, 2014) – allowed us ask whether the localization and dynamics of Vipp1 is important for thylakoid membrane growth alone. By experimentally perturbing the native spatial distribution of Vipp1 in this Dark condition, we show that thylakoid membrane growth does not require native Vipp1 localization (Fig. 6C). This suggests that thylakoids (at least the ones generated in Dark growth) can be made even when Vipp1 is not available at the thylakoids or cell periphery, casting doubt on the hypothesis that Vipp1 is involved in lipid (or other components) transport from the plasma membrane to thylakoids (Heidrich et al., 2017).
During the Dark-To-Light Transition the cells must either turn on pre-existing inactive photosynthetic protein components or synthesize new active ones. As native Vipp1 spatial distribution is critical in this condition (Fig. 5C, Fig. 6A, Video 5), and given the previously described requirement of Vipp1 to support photosynthesis (Gao and Xu, 2009), we infer that in Dark-to-Light cells Vipp1 must be involved in the light-dependent synthesis or maturation of photosynthetic machinery. In other words, native Vipp1 localization enables the acquisition of photosynthetic competency when light is available. Therefore, we speculate that the native function of Vipp1 is to facilitate activation of photosynthetic components at the molecular level. If this is true, Vipp1 function should also be important during Light Growth which is based on photosynthesis. However, when we relocalized Vipp1 in Light Growth conditions, this perturbation had no significant effect on growth rate (Fig. 5B, Fig. 6A). We suspect that the lack of a significant effect on growth is due to the fact that these cells already contain a reservoir of active photosynthetic components, and that our short-term perturbation of Vipp1 was not limiting for photosynthetic output. Taken together, we conclude that native localization of Vipp1 is not required for thylakoid growth, but for enabling acquisition of photosynthetic competency.
Our interpretation of Vipp1’s native function rests on the key assumptions that localization and function of Vipp1 are linked to each other and that this function remains the same in the three conditions tested (Dark, Dark-to-Light and Light). The simplest explanation is that Vipp1’s function does not change, and the need for that function is revealed in a sensitized context such as the Dark-to-Light Transition.
Although relocalization of Vipp1 to the nucleoid alters both the diffuse and the punctate fractions, we postulate that the punctuated dynamics of Vipp1 (which are abolished during the relocalization experiment) at high curvature regions of the thylakoid are important for enabling the acquisition of photosynthetic competency. We base this on our observations that only in actively growing cells Vipp1 continuously exchanges between the diffuse and punctate fractions, in contrast to non-growing cells in which cellular Vipp1 remains “locked” as stable puncta (Fig. S4). In addition, at the beginning of the Dark-to-Light condition when the need to initiate photosynthesis is high, the rate of Vipp1 puncta formation is also greatly increased as is the sensitivity of cells to Vipp1 relocalization (Fig. S7). Finally, Vipp1 puncta form at regions of thylakoids marked by CurT enrichments (Fig. 1D–F), a specialized zone previously proposed to act as biogenesis centers for new photosynthetic complexes (Heinz et al., 2016). We note that the association between Vipp1 and CurT that we measured cytologically (Fig. S2) has not been confirmed in previous biochemical interaction studies (Bryan et al., 2014; Heinz et al., 2016), which suggests only an indirect connection between Vipp1 and CurT at these regions of the thylakoid.
An apparent implication of our results is that Vipp1 should be genetically dispensable during Dark Growth in Synechocystis PCC 6803. We note however that previous attempts to obtain a fully segregated vipp1 knockout in such conditions were not successful (Gao and Xu, 2009). This may mean that either a small amount of Vipp1 not relocalized in our experiments and is sufficient to carry out its (essential) function or relocalization of Vipp1 does not abrogate that function. The ultimate test of the latter premise would be to deplete Vipp1 levels in Dark Growth and determine if growth or viability is affected.
What could be the molecular mechanisms by which Vipp1 enables acquisition of photosynthetic competency? Combining our analysis of Vipp1 localization and dynamics with its known biochemical properties, and the phenotypes observed, we speculate that punctuated oligomerization of Vipp1 at regions of high thylakoid curvature mediates functional light-dependent assembly of photosystem(s) complexes. Perhaps due to its membrane-binding and/or lipid rearrangement properties (Hennig et al., 2015), the oligomerized form of Vipp1 could facilitate efficient translation and/or assembly of newly made photosynthetic complexes in the membrane – this may be accomplished by alleviating the membrane curvature elastic stress potentially emerging at thylakoid edges (McDonald et al., 2015), assisting complex assembly by providing lipid cofactors, as previously proposed (Nordhues et al., 2012; McDonald et al., 2017), or mediating putative transient intermembrane contacts (Hennig et al., 2015; Heidrich et al., 2017). Our conclusions are consistent with the previous analysis of Vipp1 depletion in Synechocystis, which showed that Vipp1 function is related to photosynthesis and only indirectly to thylakoid membrane biogenesis (Gao and Xu, 2009). We further clarify that Vipp1 is necessary for establishing new photosynthetic capacity and not for maintaining the existing capacity, thus narrowing its potential mechanism of action to synthesis and/or assembly of functional photosynthetic machinery. This proposal is consistent with the phenotype of Vipp1 deletion in Synechococcus PCC 7002 (obtained in a special genetic background) which implicates Vipp1 primarily in the translation or assembly of photosystem I, not in thylakoid membrane biogenesis (Zhang et al., 2014). Additionally, Vipp1 has been shown to interact with specific chaperones, translation factors and core photosynthetic proteins, again supporting a role for Vipp1 in the translation or assembly of photosystems (Bryan et al., 2014).
In future work it will be important to determine the molecular mechanisms that allows Vipp1 to enable photosynthetic activity. Our three growth conditions and the conditional perturbation of Vipp1 localization offer a promising avenue to investigate these details. In particular, the examination of the composition, activity and turnover of photosynthetic complexes during the Dark and Dark-to-Light Growth and identifying the changes caused by Vipp1 relocalization may help illuminate which aspects of general photosynthetic establishment require Vipp1.
Our results highlight the importance of Vipp1 dynamic behavior at high curvature areas of the thylakoid, which could act as sites of activation or synthesis of new photosynthetic complexes. Additionally, we present a new rapid perturbation of Vipp1 as a tool to biochemically probe which photosynthetic component(s) requires Vipp1 in its functional assembly. Finally, this work highlights how single-cell time lapse imaging and rapid perturbations can complement biochemical and genetic observations in understanding complex processes such photosynthesis that change in space and time.
EXPERIMENTAL PROCEDURES
Strains and growth conditions
Synechocystis sp. PCC 6803 GT strain (kind gift of Dr. Wim Vermaas) (Trautmann et al., 2012) was used for all the work presented. Strains were propagated on BG11-1.5 % GelRite (PlantMedia) containing 10 mM HEPES-KOH pH 8.0 in the presence of the appropriate antibiotic. Photosynthetically-grown cultures were grown at 30 °C either in Nalgene plastic flasks tilted at low angle on an orbital shaker (70 rpm) or in 40 ml glass tubes bubbled with 1% CO2-air mix. Philips cool fluorescent tube light bulbs (F40T12/841 Alto) provided the light whose intensity was set to 30 (when flasks were used) or 100 μE m2 s−1 (when air was bubbled) as measured with a LI-COR 190R sensor. Depending on the conditions, the doubling time in the log-phase ranged from 10 to 5 hours (see example of a typical growth curve in Fig. S1B) For all growth or imaging experiments, the cells were prepared and maintained in media with no antibiotics. Prior to imaging, cells were acclimated for ~30 min on the microscope stage under a thin agarose pad made with BG-11 medium at 30 °C while illuminated by an external LED white light source (set to ~100 μE m2 s−1 intensity light measured at the objective lens) (See Light Microscopy and Image Analysis Section below). To sustain non-photosynthetic growth (Dark Growth), the cultures were supplemented with 27 mM glucose and kept in darkness and stimulated with a daily 5 min exposure of 5 μE m2 s−1 light as was previously established to promote their viability (Anderson and McIntosh, 1991). The cultures were grown in dark up to 144 hours on an orbital shaker (70 rpm) and diluted to an OD750 of 0.1 when their OD750 reached 1 or above (doubling time ~16 h). To limit any light effect during Dark Growth perturbation of Vipp1, the cultures were kept in continuous darkness for the duration of the experiment.
Genetic manipulations
To obtain the GFP-tagged allele of Vipp1, we cloned into pGem-3Zf(+) (Promega) between BamHI and SphI using Gibson Assembly (Gibson, 2011) a chimeric construct containing the upstream region and the open reading frame of sll0617 translationally fused (SGGGG linker) at the C-terminus to the codon-optimized variant of mGFPmut3 (Landgraf et al., 2012), the putative native 3′ untranslated region of sll0617 (68 bp), the spectinomycin resistance cassette (aadA) and a ~ 1 kb homology region downstream of sll0617. The resulting plasmid (pAGH42) was used to transform the naturally competent Synechocystis according to published procedures (Zang et al., 2007). As for the vipp1-gfp construct, the SNAP-tagged version of Vipp1 - also codon optimized - was cloned and transformed into Synechocystis cells.
To make the Vipp1-GFP-FKBP12 construct for the anchor-away relocalization system (Liberles et al., 1997) we fused the codon optimized fragment of the human FKBP12 to the C-terminus of GFP (GSGG linker) in the Vipp1-GFP construct described above using the Gibson assembly procedure. The resulting plasmid (pAGH50) was transformed into the appropriate Synechocystis strain to replace the native vipp1 locus. No differences in growth or Vipp1 localization and dynamics were observed between the vipp1-gfp-fkbp12 and vipp1-gfp strains.
To make the HU-anchor fusion, we designed a construct synthesized by Integrated DNA Technologies (Coralville, IA) in which the codon optimized FRB fragment- was fused to the C-terminus (GSG linker) of HU (encoded by sll1712). The DNA construct included the putative native promoter and 5′ untranslated region (265 nt) of the sll1712 gene. At the 3′end, the 68 bp 3′ untranslated region of vipp1 was used. The HU-anchor construct was cloned by Gibson assembly downstream of the kanamycin resistance cassette in a pBR322-based plasmid designed to recombine into the slr0168 neutral site of the Synechocystis chromosome (Gao and Xu, 2009).
A GFP-FKBP12 control construct (pAGH93) was made similarly to vipp1-gfp-fkbp12, except that the L03 synthetic promoter was used to drive its expression (Huang and Lindblad, 2013).
In all cases, the colonies obtained after transformation were re-streaked onto fresh selection media and the genetic modification was confirmed by PCR amplification with primers flanking the locus of interest. For long-term storage, all Synechocystis strains were kept at −80 °C in BG11 with 8% dimethyl sulfoxide. The main DNA constructs used in this study have been deposited in the Addgene plasmid bank.
Light microscopy and image analysis
All epifluorescence imaging was performed on a Zeiss AxioObserver.Z1 inverted microscope equipped with an environmental chamber, a Definite Focus module, a hardware-triggered Hamamatsu ORCA Flash4.0 V2+ camera and a Plan-Apochromat 63x/1.40 Oil DIC M27 (NA 1.4) objective. Unless noted, all images were acquired at Nyquist sampling rate as Z-stacks in Zen 2.1 software (Zeiss). Sola SE (Lumencor, Beaverton, OR) white light source was used to image Vipp1 and the thylakoid in the GFP and the far-red fluorescence channels (Zeiss Filter Sets 38HE and 50). All brightfield imaging was done in Köhler illumination with light passing through the same filter cube as the one used for Vipp1 channel.
For superresolution imaging we used Zeiss LSM 880 laser confocal system equipped with an Airyscan unit, environmental control chamber, and 1.4 NA Plan-Apochromat 63x DIC M27 oil objective. Similar to epifluorescence imaging procedures, cells were acclimated on the microscope stage for ~30 min, while being illuminated by photosynthetic light. To limit any potential laser-induced stress on cells (and also changes to Vipp1 localization) we identified fields of view with the use 594 nm laser set to the lowest power at maximal detector gain. Image stacks were acquired for 16 Z-planes (512 x 512) with a voxel size 40 x 40 x 180 nm centered at mid-cell where the focus was the sharpest. Laser lines 458 nm, 515 nm and 594 nm were passed through the same beamsplitter (MBS 458/514/594) for excitation of CurT-mTurquoise2, Vipp1-GFP (mGFPmut3 variant: excitation maximum - 501 nm, emission maximum - 511 nm) and thylakoid respectively. Emissions from the CurT and Vipp1 channels passed through a dual bandpass filter BP 420–480, BP 495–550). Pixel dwell time (set at the fastest) and laser power were adjusted to avoid saturation and bleaching effects while the detector gain was set to 800. For Airscan processing, Zen Black 2.1 was used to process the image stacks by performing the filtering, deconvolution and pixel reassignments in 3D mode at default settings. This processing confers the increased spatial resolution (by a factor of 1.5 to 1.7x both laterally and axially) and an enhanced signal to noise ratio (Korobchevskaya et al., 2017).
For live-cell imaging, an aliquot of cells (obtained from an OD750 of 0.2 – 0.3 culture) was placed onto the glass of a 35 mm MatTek dish (P35G-0.170-14-C) (MatTek, Ashland, MA) under a thin BG11 - 1% agarose pad (2 mm thickness) prepared in advance and covered with a coverslip to reduce evaporation. The dish was placed onto the objective in the stage insert and around the agarose pad a basin of water was poured to create a humidified atmosphere. The temperature was maintained at 30 °C while the photosynthetic light was provided by a white light LED ring: RL1360 or DF198 (Advanced Illumination, Rochester, VT) that was positioned on the stage insert and controlled by an analog signal through Zen 2.1. The LED white light was programmed to switch off only during the fluorescence image acquisition. The light intensity reaching the cells at the objective lens was 100 μE m2 s−1 which permitted a growth rate comparable to the rate measured in bulk culture. To limit photobleaching and phototoxicity, the imaging light power was limited to 20% by the FL attenuator and the light exposure for image acquisition in the GFP channel (20 to 30 ms) was adjusted to generate a signal-to-background ratio of less than 1.5. Also, the Z-series were limited to 12 steps or fewer. For thylakoid channel, all exposures used were 3.2 ms. Cell doubling time under these conditions was approximately 8 h.
Multispectral datasets were aligned using the affine transformation plugin in Zeiss Zen 2.1 using a standard prepared from TetraSpeck (Invitrogen, Carlsbad, CA) multicolor beads immobilized on a 1.5 coverslip.
To obtain filtered images, a custom MATLAB script (available upon request) was written to computationally enhance both diffraction-limited spots and contours of thylakoids by applying a local maxima filter that fit a 3D Gaussian to every position in the dataset and returned the Maximum Likelihood Estimate (MLE) of the amplitude, background and square-error. The amplitude component of the MLEs reports a quantitative visualization of the diffraction limited signal since it is separated from its background component. See Fig. S1C for an example of the raw and MLE amplitude filtered image. Unless noted, all filtered images presented are MLE visualizations of the signal amplitudes. There are no parameters to adjust for this computational filter.
To automatically segment cells in the brightfield channel a custom MATLAB script was written that implemented correlation imaging as described previously (Julou et al., 2013). In summary, the edge of a cell is uniquely defined by its Z intensity profile given a 3D brightfield Z-stack. Therefore, to computationally find all cell edges, a brightfield Z-stack was filtered in Z by cross-correlating this unique Z-intensity profile using the MATLAB function convn. This cross-correlated dataset was then thresholded and all enclosed objects were then segmented by MATLAB’s function regionprops.
Fiji (Schindelin et al., 2012) was used to prepare the kymograms, extract the intensity profiles, and convert the final images. Unless noted, in all images displayed, the grayscale contrast was auto-adjusted linearly before converting to bitmaps.
Electron microscopy
Synechocystis cultures were grown to an OD750 of 0.2, incubated briefly in media supplemented with 100 mM mannitol and centrifuged. Aliquots of the pellet were high pressure frozen in a Wohlwend Compact 02 High Pressure Freezer at the University of Colorado-Boulder Electron Microscopy Facility and stored under liquid nitrogen. Frozen samples were then freeze-substituted at −80 C in 0.05% uranyl acetate in acetone and embedded in Lowicryl HM20 in a Leica AFS. For immunodetection, thin sections (50–60 nm) were labeled with rabbit polyclonal anti-GFP (generous gift of Pam Silver, Harvard University) diluted 1:500 in 1% non-fat dry milk in PBST, followed by secondary labeling with 5 nm gold-goat anti-rabbit Ig diluted 1:20 in PBST. Labeled grids were further contrasted with 2% aqueous uranyl acetate and lead citrate. Electron micrographs were obtained with an FEI Tecnai Spirit BioTwin TEM operating at 100 kV equipped with an AMT 2Kx2K side entry CCD camera.
To obtain transmission electron microscopy images of cells from dark- and light grown cultures presented in Fig. 6 and Fig. 6S, the cells were collected by centrifugation, resuspended in a 100 μl of 20% BSA in BG-11 medium, loaded onto 100 μm-deep carriers and frozen in a Wohlwend Compact 01 High Pressure Freezer at the Electron Microscopy Facility of HHMI/Janelia Research Campus. Samples were freeze-substituted in 2% osmium tetroxide/0.1 % uranyl acetate/3% water in acetone using a fast freeze-substitution method (McDONALD and Webb, 2011) and embedded in Eponate 12 resin. Ultrathin sections (60–65 nm) were post-stained with uranyl acetate/lead citrate and imaged in a Tecnai 12 electron microscope (FEI, Hillsboro, OR) operating at 80kV equipped with an Ultrascan 4000 digital camera (Gatan Inc, CA).
Spectrophotometry
Whole-cell absorption and fluorescence emission spectra and were obtained on Spectramax i3 multimode reader (Molecular Devices, Sunnyvale, CA). For low-temperature measurements the cells were suspended in 50% glycerol and flash frozen in liquid nitrogen.
Oxygen production measurements
Initial rates of oxygen production were obtained from dark- (144 hours) and light-growing (24 hours post-dark) cultures. Cells were diluted in BG11 medium supplemented with 10 mM NaHCO3 to an OD750 of 0.3, which corresponds to a cell density of ~2.5*108 cells per ml, as estimated by a Multisizer 3 Coulter Counter (Beckman Coulter). Measurements were performed with an InLab® OptiOx sensor connected to the S9 Seven2Go Pro dissolved oxygen meter (Mettler Toledo) at 23° C, with moderate stirring in a closed vessel. Linear rates of oxygen production were obtained in complete darkness (respiration) or in light (photosynthesis) at 300 μE m2 s−1 provided by a white light LED ring (DF198, Advanced Illumination, Rochester, VT).
Supplementary Material
Acknowledgments
We thank Courtney Ozzello and Thomas Giddings of University of Colorado-Boulder Electron Microscopy Facility for their immunoelectron microscopy work and discussions on the project. We also thank Amalia Pasolli of HHMI/Janelia for her electron microscopy imaging of dark- and light grown cells and Eric Wait of Advanced Imaging Center at HHMI/Janelia for his help in generating the 3D rendering of Vipp1-GFP and CurT-mTurquoise2 expressing cells. We are grateful to Jeff Moffitt, Joe Markson and members of the O’Shea laboratory for their comments on the manuscript. This work was funded by the Howard Hughes Medical Institute. Research by FC and FC salary were supported by grants to Nancy Kleckner from the NIH (R01-GM025326; R01-GM044794) and a Harvard University Office of Technology Development Physical Sciences Accelerator Award to Nancy Kleckner (368056). Salary to FC was also provided by an NIH pre-doctoral training grant to Harvard University, NIH T32 GM007598, and an NSF SAVI Student Research Network in the Physics of Living Systems (PoLS) grant from the NSF to Harvard University (PHY-1219334).
Footnotes
AUTHOR CONTRIBUTIONS:
AG, FC and EKO conceived and designed the research. AG performed the experiments. AG and FC analyzed the data, prepared the figures and wrote the initial manuscript draft. EKO supervised research and revised manuscript draft.
References
- Anderson SL, McIntosh L. Light-activated heterotrophic growth of the cyanobacterium Synechocystis sp. strain PCC 6803: a blue-light-requiring process. J Bacteriol. 1991;173:2761–2767. doi: 10.1128/jb.173.9.2761-2767.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- Aseeva E, Ossenbühl F, Sippel C, Cho WK, Stein B, Eichacker LA, et al. Vipp1 is required for basic thylakoid membrane formation but not for the assembly of thylakoid protein complexes. Plant Physiol Biochem. 2007;45:119–128. doi: 10.1016/j.plaphy.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Barthel S, Bernat G, Seidel T, Rupprecht E, Kahmann U, Schneider D. Thylakoid Membrane Maturation and PSII Activation Are Linked in Greening Synechocystis sp. PCC 6803 Cells. Plant Physiol. 2013;163:1037–1046. doi: 10.1104/pp.113.224428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan SJ, Burroughs NJ, Shevela D, Yu J, Rupprecht E, Liu L-N, et al. Localisation and interactions of the Vipp1 protein in cyanobacteria. Mol Microbiol. 2014 doi: 10.1111/mmi.12826. http://dx.doi.org/10.1111/mmi.12826. [DOI] [PMC free article] [PubMed]
- Eberhard S, Finazzi G, Wollman FA. The dynamics of photosynthesis. Annu Rev Genet. 2008;42:463–515. doi: 10.1146/annurev.genet.42.110807.091452. [DOI] [PubMed] [Google Scholar]
- Fuhrmann E, Bultema JB, Kahmann U, Rupprecht E, Boekema EJ, Schneider D. The vesicle-inducing protein 1 from Synechocystis sp. PCC 6803 organizes into diverse higher-ordered ring structures. Mol Biol Cell. 2009;20:4620–4628. doi: 10.1091/mbc.E09-04-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann E, Gathman S, Rupprecht E, Golecki J, Schneider D. Thylakoid membrane reduction affects the photosystem stoichiometry in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol. 2009;149:735–744. doi: 10.1104/pp.108.132373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Xu X. Depletion of Vipp1 in Synechocystis sp. PCC 6803 affects photosynthetic activity before the loss of thylakoid membranes. FEMS Microbiol Lett. 2009;292:63–70. doi: 10.1111/j.1574-6968.2008.01470.x. [DOI] [PubMed] [Google Scholar]
- Gibson DG. Enzymatic assembly of overlapping DNA fragments. Experimental Procedures Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutu A, O’Shea EK. Two antagonistic clock-regulated histidine kinases time the activation of circadian gene expression. Mol Cell. 2013;50:288–294. doi: 10.1016/j.molcel.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Heidrich J, Thurotte A, Schneider D. Specific interaction of IM30/Vipp1 with cyanobacterial and chloroplast membranes results in membrane remodeling and eventually in membrane fusion. Biochim Biophys Acta. 2017;1859:537–549. doi: 10.1016/j.bbamem.2016.09.025. [DOI] [PubMed] [Google Scholar]
- Heidrich J, Wulf V, Hennig R, Saur M, Markl J, Sönnichsen C, Schneider D. Organization into Higher Ordered Ring Structures Counteracts Membrane Binding of IM30, a Protein Associated with Inner Membranes in Chloroplasts and Cyanobacteria. J Biol Chem. 2016;291:14954–14962. doi: 10.1074/jbc.M116.722686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Rast A, Shao L, Gutu A, Gügel IL, Heyno E, et al. Thylakoid Membrane Architecture in Synechocystis Depends on CurT, a Homolog of the Granal CURVATURE THYLAKOID1 Proteins. Plant Cell. 2016 doi: 10.1105/tpc.16.00491. http://dx.doi.org/10.1105/tpc.16.00491. [DOI] [PMC free article] [PubMed]
- Hennig R, Heidrich J, Saur M, Schmüser L, Roeters SJ, Hellmann N, et al. IM30 triggers membrane fusion in cyanobacteria and chloroplasts. Nat Commun. 2015;6:7018. doi: 10.1038/ncomms8018. [DOI] [PubMed] [Google Scholar]
- Huang HH, Lindblad P. Wide-dynamic-range promoters engineered for cyanobacteria. J Biol Eng. 2013;7:10. doi: 10.1186/1754-1611-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julou T, Mora T, Guillon L, Croquette V, Schalk IJ, Bensimon D, Desprat N. Cell-cell contacts confine public goods diffusion inside Pseudomonas aeruginosa clonal microcolonies. Proc Natl Acad Sci U S A. 2013;110:12577–12582. doi: 10.1073/pnas.1301428110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Kopecná J, Komenda J, Bucinska L, Sobotka R. Long-Term Acclimation of the Cyanobacterium Synechocystis sp. PCC 6803 to High Light Is Accompanied by an Enhanced Production of Chlorophyll That Is Preferentially Channeled to Trimeric Photosystem I. Plant Physiol. 2012;160:2239–2250. doi: 10.1104/pp.112.207274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobchevskaya K, Lagerholm B, Colin-York H, Fritzsche M. Exploring the Potential of Airyscan Microscopy for Live Cell Imaging. Photonics. 2017;4:41. [Google Scholar]
- Kroll D, Meierhoff K, Bechtold N, Kinoshita M, Westphal S, Vothknecht UC, et al. VIPP1, a nuclear gene of Arabidopsis thaliana essential for thylakoid membrane formation. Proc Natl Acad Sci U S A. 2001;98:4238–4242. doi: 10.1073/pnas.061500998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nat Experimental Procedures. 2012;9:480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberles SD, Diver ST, Austin DJ, Schreiber SL. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc Natl Acad Sci U S A. 1997;94:7825–7830. doi: 10.1073/pnas.94.15.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Jovanovic G, Wallace BA, Ces O, Buck M. Structure and function of PspA and Vipp1 N-terminal peptides: Insights into the membrane stress sensing and mitigation. Biochim Biophys Acta. 2017;1859:28–39. doi: 10.1016/j.bbamem.2016.10.018. [DOI] [PubMed] [Google Scholar]
- McDonald KL, Webb RI. Freeze substitution in 3 hours or less. J Microsc. 2011;243:227–233. doi: 10.1111/j.1365-2818.2011.03526.x. [DOI] [PubMed] [Google Scholar]
- van de Meene AML, Hohmann-Marriott MF, Vermaas WFJ, Roberson RW. The three-dimensional structure of the cyanobacterium Synechocystis sp. PCC 6803. Arch Microbiol. 2006;184:259–270. doi: 10.1007/s00203-005-0027-y. [DOI] [PubMed] [Google Scholar]
- Mullineaux CW. Co-existence of photosynthetic and respiratory activities in cyanobacterial thylakoid membranes. Biochim Biophys Acta. 2014;1837:503–511. doi: 10.1016/j.bbabio.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Nickelsen J, Rengstl B. Photosystem II assembly: from cyanobacteria to plants. Annu Rev Plant Biol. 2013;64:609–635. doi: 10.1146/annurev-arplant-050312-120124. [DOI] [PubMed] [Google Scholar]
- Nordhues A, Schöttler MA, Unger AK, Geimer S, Schönfelder S, Schmollinger S, et al. Evidence for a role of VIPP1 in the structural organization of the photosynthetic apparatus in Chlamydomonas. Plant Cell. 2012;24:637–659. doi: 10.1105/tpc.111.092692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escudé C, Boudier T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29:1840–1841. doi: 10.1093/bioinformatics/btt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otters S, Braun P, Hubner J, Wanner G, Vothknecht UC, Chigri F. The first α-helical domain of the vesicle-inducing protein in plastids 1 promotes oligomerization and lipid binding. Planta. 2013;237:529–540. doi: 10.1007/s00425-012-1772-1. [DOI] [PubMed] [Google Scholar]
- Pribil M, Labs M, Leister D. Structure and dynamics of thylakoids in land plants. J Exp Bot. 2014;65:1955–1972. doi: 10.1093/jxb/eru090. [DOI] [PubMed] [Google Scholar]
- Rast A, Heinz S, Nickelsen J. Biogenesis of thylakoid membranes. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2015;1847:821–830. doi: 10.1016/j.bbabio.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Experimental Procedures. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann D, Voss B, Wilde A, Al-Babili S, Hess WR. Microevolution in cyanobacteria: re-sequencing a motile substrain of Synechocystis sp. PCC 6803. DNA Res. 2012;19:435–448. doi: 10.1093/dnares/dss024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas WFJ, Timlin JA, Jones HDT, Sinclair MB, Nieman LT, Hamad SW, et al. In vivo hyperspectral confocal fluorescence imaging to determine pigment localization and distribution in cyanobacterial cells. Proc Natl Acad Sci U S A. 2008;105:4050–4055. doi: 10.1073/pnas.0708090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vothknecht UC, Otters S, Hennig R, Schneider D. Vipp1: a very important protein in plastids? ! J Exp Bot. 2012;63:1699–1712. doi: 10.1093/jxb/err357. [DOI] [PubMed] [Google Scholar]
- Wait E, Winter M, Bjornsson C, Kokovay E, Wang Y, Goderie S, et al. Visualization and correction of automated segmentation, tracking and lineaging from 5-D stem cell image sequences. BMC Bioinformatics. 2014;15:328. doi: 10.1186/1471-2105-15-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li GW, Chen C, Xie XS, Zhuang X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science. 2011;333:1445–1449. doi: 10.1126/science.1204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal S, Heins L, Soll J, Vothknecht UC. Vipp1 deletion mutant of Synechocystis: a connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 2001;98:4243–4248. doi: 10.1073/pnas.061501198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Liu J, Wen X, Lu C. Molecular mechanism of photosystem I assembly in oxygenic organisms. Biochim Biophys Acta. 2015;1847:838–848. doi: 10.1016/j.bbabio.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Zang X, Liu B, Liu S, Arunakumara KKIU, Zhang X. Optimum conditions for transformation of Synechocystis sp. PCC 6803. J Microbiol. 2007;45:241–245. [PubMed] [Google Scholar]
- Zhang L, Kato Y, Otters S, Vothknecht UC, Sakamoto W. Essential role of VIPP1 in chloroplast envelope maintenance in Arabidopsis. Plant Cell. 2012;24:3695–3707. doi: 10.1105/tpc.112.103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Shen G, Li Z, Golbeck JH, Bryant DA. Vipp1 is essential for the biogenesis of Photosystem I but not thylakoid membranes in Synechococcus sp. PCC 7002. J Biol Chem. 2014;289:15904–15914. doi: 10.1074/jbc.M114.555631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.