

Abstract
The gain/amplification of the CKS1B gene on chromosome 1q21 region is associated with a poor outcome in patients with multiple myeloma (MM). However, there are limited data on the outcome of patients with CKS1B amplification after a single high-dose chemotherapy and autologous hematopoietic stem cell transplantation (auto-HCT). We retrospectively evaluated the outcome of patients with CKS1B amplification who received an auto-HCT between June 2012 and July 2014 at our institution. We identified 58 patients with MM and CKS1B gene amplification detected by fluorescent in situ hybridization (FISH). We compared their outcomes with a propensity score matched control group of 58 patients without CKS1B amplification that were treated at approximately the same time. The primary objective was to compare the progression-free (PFS) and overall survival (OS) between the CKS1B and the control group. Stratified log-rank test with the matched pairs as strata and double robust estimation under the Cox model were used to assess the effect of CKS1B gene amplification on PFS or OS in the matched cohort. Patients in the CKS1B and control groups were well matched for age, gender, disease status, year of auto-HCT, response to pre-transplant therapy and baseline hemoglobin level. In both groups, 57% patients were in first remission and 43% had relapsed disease at auto-HCT. Twenty-seven (47%) patients with CKS1B amplification had concurrent monosomy 13 or 13q deletion [del(13q)]; 6 (10%) by conventional cytogenetics only, 16 (28%) by FISH only, and 5 (9%) by both. Median follow up after auto-HCT was 25.4 months. The median PFS of the CKS1B and the control group were 15.0 months and 33.0 months (p= 0.002), respectively. The median OS have not been reached yet. The 2-year OS rates in the CKS1B and the control group were 62% and 91% (p=0.02), respectively. In conclusion, Patients with CKS1B amplification are more likely to have additional high-risk cytogenetic abnormalities, and a shorter PFS and OS after an auto-HCT.
Keywords: CKS1B, 1q21, multiple myeloma, stem cell transplantation
INTRODUCTION
There has been significant recent progress in the understanding of the molecular pathogenesis of multiple myeloma (MM) [1], which has led to the recognition of several chromosomal and molecular abnormalities that may play a role in its development and progression. Conventional cytogenetic (CC) and fluorescence in-situ hybridization (FISH) studies have traditionally been used to identify these high-risk chromosomal abnormalities [2]. Among these, t(4;14), t(14;16), deletion 17p and 1q gain have been associated with shorter survival, and have emerged as independent predictors of outcome [3]. A majority of MM patients with these high-risk chromosomal or molecular abnormalities continue to have a poor outcome even with the availability of immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs) and high-dose chemotherapy autologous hematopoietic stem cell transplantation (auto-HCT) [4, 5]. Recent data, however, have shown that the adverse impact of t(4;14) can be significantly improved by the use of bortezomib in induction and maintenance therapy [6–8]. This highlights the importance of detecting these abnormalities upfront, as they not only predict the outcome but also help in selecting appropriate therapy according to risk stratification.
Gain of chromosome band 1q21, which results in the overexpression of the CKS1B gene, is one of the high-risk chromosomal abnormalities [9–11]. CKS1B is a cofactor for ubiquitination and degradation of the cell cycle inhibitor p27Kip1 [12, 13], which may contribute to a worse outcome. CKS1B amplification is associated with other cytogenetic abnormalities, such as deletion of 13q (del(13q)) [11, 13, 14]. Although CKS1B gene amplification, detected by fluorescence in situ hybridization (FISH) studies, has been identified as a prognostic marker, more needs to be learned about the clinical characteristics, response to therapy and overall outcome in patients with this abnormality. Several recent reports highlighted the adverse outcome in patients with CKS1B gene amplification detected by FISH [14, 15]. Of note, in contrast to other cytogenetic abnormalities such as deletion 17p or deletion 13q, gain of 1q21(CKS1B) still indicates a worse prognosis in patients treated with PI-based induction [16]. However, its role as an independent risk factor needs to be further elucidated [14, 17–20]. To evaluate its role as an independent predictor of outcome, we performed a retrospective study on 58 patients with MM and CKS1B amplification, identified by FISH, who underwent an auto-HCT at our institution.
METHODS
Patients
A total of 475 patients received auto-HCT for MM at MD Anderson Cancer Center between June 2012, and July, 2014. We identified 58 patients (12%) with CKS1B amplification on FISH prior to auto-HCT. Using a propensity score matched analysis [21, 22] we identified a matched control for each of the 58 patients with CKS1B amplification, who did not have CKS1B amplification on FISH and also received auto-HCT between June 2012 and July 2014. Clinical characteristics, treatment type and responses, outcome and patient demographic data were obtained from a retrospective chart review under an Institutional Review Board (IRB)-approved protocol with a waiver of informed consent.
Response and outcome measures
Clinical response, relapse and progression were defined by International Myeloma Working Group (IMWG) criteria [23]. Toxicity grading was assessed using the Common Terminology Criteria for Adverse Events version 4.0. Neutrophil engraftment was defined as the first of 3 consecutive days with an absolute neutrophil count of more than 0.5 × 109/L. Platelet engraftment was defined as the first of 7 consecutive days with a platelet count of more than 20,000 /uL without receiving a platelet transfusion.
Routine cytogenetics and FISH analyses for assessment of CKS1B gene amplification
Chromosomal abnormalities involving 1q21 were detected by CC analyses, which were performed either at initial diagnosis or when patients first presented to our institution. A minimum of 20 metaphases were analyzed, and a clonal abnormality was defined as the presence of at least two abnormal metaphases with the same structural abnormality. FISH analyses were performed on plasma cells enriched in BM aspirates by using a magnetic cell-sorting procedure to select CD138+ cells [24] for the presence of CKS1B gene amplification, IgH gene rearrangement, monosomy 13/del(13q), as well as for monosomy 17/del(17q)-TP53 deletion using probes from Abbott Molecular, Inc. (Abbott Park, IL). A total of 200 interphases were analyzed for each FISH abnormality. CKS1B amplification testing was performed using a Cytocell LSI CKS1B dual-color probe with a total of 200 interphases analyzed. The 95% (P<0.05) confidence limit of the CKS1B probe was used on twenty normal samples using the Beta Inverse Method of calculation at the MDACC Cytogenetics Laboratory to establish the cutoff value for CSK1B gain/amplification. The cutoff for CKS1B gain/amplification with three signals is 6% of interphase nuclei evaluated, whereas 4.4% for the interphase nuclei with 4 or more signals.
Statistical Analysis
Patient demographic and clinical characteristics were summarized by using descriptive statistics when appropriate. The Student t-test/ANOVA and Wilcoxon/Kruskal-Wallis tests were used to compare continuous variables between different groups.
Differences between categorical variables were determined using Fisher’s exact test or Pearson χ2 test. To reduce the impact of selection bias in control patients on the estimation of PFS and OS, we conducted a propensity score matched analysis with the following covariates in the multivariate logistic model to create the propensity scores: age at auto-HCT, gender, disease status at auto-HCT, response to pre-transplant therapy, baseline hemoglobin level, and time from diagnosis to auto-HCT. In addition, the interaction between age at transplant and time from diagnosis to transplant was also included in the logistic regression model to improve balance between the two groups (CKS1B vs control). We identified 1:1 matched doublets, one case for each of the two groups, using a 5 to 1 digit greedy match algorithm. We used absolute standardized differences to assess balance in the covariates between the case cohort and control cohort. The distributions of PFS and OS were estimated by the Kaplan-Meier method. PFS was defined as the time from transplant to the time of progression or death, whichever occurred first, or to the time of last contact, and OS defined as the time from transplant to the time of death, or to the time of last contact. For the propensity-score matched cohort, the stratified log-rank test with the matched pairs as strata was fitted to evaluate the difference of PFS or OS between the two groups. Stratified proportional hazards regression model of Cox with the matched pairs as strata was used to estimate the hazard ratio of progression or death for the matched case cohort compared to the matched control cohort. P-values less than 0.05 were considered statistically significant. All analyses were conducted using SAS (version 9.2, Cary, NC) and S-plus (version 8.04, TIBCO Software Inc., Palo Alto, CA) statistical software.
RESULTS
Patient Characteristics
We identified 58 patients with a positive FISH study for CKS1B gene amplification that received an auto-HCT at our institution between June 2012 and July 2014. Using a propensity score matched analysis, we identified a control group of 58 MM patients without CKS1B amplification on FISH, who also received auto-HCT between June 2012 and July 2014. Patient characteristics are summarized in Table 1. Patients in both groups were well matched for age, gender, disease status at auto-HCT, year of auto-HCT, response to pre-transplant therapy and hemoglobin level as measured by the absolute standardized differences (Table 1). In both CKS1B and Control groups, 33 patients (57%) were in first remission and 25 patients (43%) had relapsed disease (p=1.00) at auto-HCT. In terms of pre-auto-HCT response, 4 (7%), 14 (24%) and 28 patients (48%) patients had CR, VGPR or PR, respectively, in the CKS1B group with an overall response rate of 79%. In the control group, 10 (17%), 10 (17%) and 27 patients (47%) patients had CR, VGPR or PR respectively, with an overall response rate of 81% (p=0.88).
Table 1.
Clinical characteristics of patients with Multiple Myeloma and auto-HCT with and without CKS1B gene amplification.
| Group statistics
|
P-value
|
||
|---|---|---|---|
| Patients without CKS1B gene amplfification (n=58) | Patients with CKS1B gene amplfification (n=58) | ||
|
|
|
||
| Median age at TP (range) | 61 (33–75) | 60 (34–78) | 0.737 |
| Sex (F/M) | 29/29 | 26/32 | 0.577 |
| Hb, g/dl, median (range)* | 10.0 (5.4–16.3) | 9.9 (5.5–16.7) | 0.688 |
| Calcium, mg/dl, median (range)* | 9.6 (8.3–16.1) | 9.5 (8.0–14.2) | 0.812 |
| Creatinine, mg/dl, median (range)* | 1.0 (0.6–10.6) | 1.0 (0.5–11.8) | 0.911 |
| LDH, U/l, median (range)*, (n=34 and 44) | 428 (3–953) | 433 (115–2720) | 0.467 |
| % Bone marrow plasma cells, (range)* | 50 (1–100) | 60 (5–96) | 0.295 |
| ISS Stage III, n (%) (n= 42 and 50) | 15 (37) | 18 (36) | 0.954 |
| Induction therapy bortezomib-based | 53 (91) | 39 (70) | 0.004 |
| Time from diagnosis to auto-HCT, years, median (range) | 0.86 (0.30–12.35) | 0.61 (0.3–11.77) | 0.142 |
| Disease status at auto-HCT, n (%) | |||
| First remission | 33 (57) | 33 (57) | 1.000 |
| Relapse | 25 (43) | 25 (43) | |
| CR/VGPR/PR before auto-HCT, n (%) | 47 (81) | 46 (79) | 0.887 |
| CR, n (%) | 4 (7) | 10 (17) | |
| VGPR, n (%) | 14 (24) | 10 (17) | |
| PR, n (%) | 28 (48) | 27 (47) | |
| Prep regimen: Melphalan alone, n (%) | 43 (74) | 45 (78) | 0.664 |
at diagnosis, Hb indicates Hemoglobin, CR indicates complete response; NR, no response; PD, progressive disease; PR, partial response; SD, stable disease; VGPR, very good partial response; TP, Transplantation; auto-HCT, autologous hematopoietic stem cell transplantation; ISS, International Staging-System.
Induction Therapy
Novel agents, including immunomodulatory drugs (IMiDs) and/or proteasome inhibitors (PI) were used for induction in 52 patients (90%) in the CKS1B group and in 54 patients (95%) in the control group. All patients, except one in the control group who received carfilzomib, received bortezomib as the only PI. In the CKS1B group, 8 patients (14%) received an IMiD-based induction without bortezomib, 18 patients (31%) received bortezomib-based induction without IMiDs, while 25 (43%) received both an IMiD and a PI. In the control group 4 patients (7%) received an IMiD-based induction, 25 patients (43%) received bortezomib-based induction, and 26 (45%) received both an IMiD and a PI. Forty-seven (81%) and 48 (82%) patients received only one induction regimen in CKS1B and control group, respectively, while 11 (19%) and 10 (17%) patients in CKS1B and control group received >1 induction regimen (p=1.00). Interestingly, patients with CKS1B amplification were less likely to receive induction with bortezomib (± IMiDs) prior to transplant (70% in the CKS1B group vs. 91% in the control group, p=0.004).
Preparative Regimen
Melphalan alone was used as preparative regimen in 45 patients (78%) in the CKS1B group, and in 43 (74%) in the control group (p=0.66). Five patients (9%) in the CKS1B group received melphalan with lenalidomide, and 8 patients (14%) received melphalan with busulfan. In the control group, 8 patients (14%) received melphalan with lenalidomide and 7 patients (12%) received melphalan with busulfan.
Maintenance
Maintenance therapy with IMiDs was used in 44 (76%) patients in the CKS1B group and 47 (81%) control patients (p=0.65). Of the 44 CKS1B patients receiving maintenance, 25 patients (43%) received lenalidomide alone, and 1 (2%) pomalidomide alone and 18 (30%) received bortezomib alone or in combination. In the control group, 38 (65%) received maintenance with lenalidomide alone, and 1 (2%) with pomalidomide alone, and 8 (13%) received a PI alone or in combination. Overall, a significantly higher number of patients in the CKS1B vs. control group (30% vs. 13%, p=0.04) received a bortezomib-based maintenance.
Additional Cytogenetic abnormalities with CKS1B amplification
Monosomy 13/del(13q) was seen in 27 (47%) patients in the CKS1B group; 6 (10%) by conventional cytogenetics only, 16 (27%) by FISH only, and 5 (9%) by both. Nineteen (33%) of these had monosomy 13 and 8 (14%) had deletion 13q. In order to account for the high frequency of chromosome 13 abnormalities we performed a subgroup analysis of CKS1B patients with and without monosomy/del(13q). The median PFS in the CKS1B Group with chromosome 13 abnormalities (monosomy 13 or deletion 13p) and without chromosome 13 abnormalities was 11 months (95%CI: 9–14) and 16 months (95%CI: 6–26), respectively, which was not significantly different (log rank test, p=0.26). The Median OS was not reached for both groups. In the control group neither the median PFS nor the median OS was reached for patients with or patients without chromosome 13 abnormalities. No significant differences were detected in any of the disease related characteristics (e.g. hemoglobin, calcium, creatinine, lactate dehydrogenase and ISS-Stage at diagnosis) between the two groups. Seven patients (12%) in the CKS1B group also had monosomy 17/del(17p), and 3 (5%) had t(11;14)(q13;q32). In comparison, only ten patients (17%) in the control group had monosomy13/del(13q) and ten patients (17%) had t(11:14). None of the control patients had deletion 17/17p. Frequencies of cytogenetic abnormalities in both groups are summarized in Table 3.
Table 3. Cytogenetic abnormalities.
Cytogenetic abnormalities in patients with Multiple Myeloma and auto-HCT with and without CKS1B gene amplification.
| Cytogenetic Abnormality
|
Group (N, %)
|
|
|---|---|---|
| Control (58, 100) | CKS1B (58, 100) | |
| Complex (≥3 abnormalities) | 12 (21) | 25 (43) |
| monosomy 13 / deletion 13q | 10 (17) | 27 (47) |
| IGH gene rearrangement, | 8 (14) | 8 (14) |
| t(11;14) | 10 (17) | 3 (5) |
| Hyperdiploidy | 2(3) | 7 (12) |
| Pseudodiploidy | 4 (7) | 5 (9) |
| Del etion17p | 0 (0) | 7 (12) |
| Trisomy 11 | 3 (5) | 3 (5) |
| t(14;16) | 0 (0) | 3 (5) |
| t(4;14) | 0 (0) | 3 (5) |
| Hypodiploidy | 3 (5) | 1 (2) |
| Trisomy 9 | 1 (2) | 1 (2) |
| add(X)(p22.1) | 1 (2) | 0 (0) |
| Trisomy 13 | 1 (2) | 0 (0) |
| Deletion 14 | 1 (2) | 0 (0) |
| del(2)(q31) | 0 (0) | 1 (2) |
| inv(9)(p12q13) | 0 (0) | 1 (2) |
| Trisomy 5 | 1 (2) | 1 (2) |
| Tetrasomy 11 | 1 (2) | 0 (0) |
| del(15)(q21) | 0 (0) | 1 (2) |
| t(5;10)(q31;p15) | 0 (0) | 1 (2) |
| Hypertriploidy | 0 (0) | 1 (2) |
| Hypotriploidy | 0 (0) | 1 (2) |
| Hypotetraploidy | 1 (2) | 1 (2) |
| ins(5;2) | 1 (2) | 0 (0) |
| Deletion 16q | 1 (2) | 0 (0) |
| del(X)(q22) | 1 (2) | 0 (0) |
| del(11)(q23) | 1 (2) | 0 (0) |
| Trisomy 12 | 0 (0) | 1 (2) |
| del(9)(q12) | 1 (2) | 0 (0) |
| Trisomy 3 | 1 (2) | 1 (2) |
Engraftment and Toxicity
The median time to neutrophil (absolute neutrophil count of 500 cells/μl) and platelet engraftment (platelet count of 20.000/μl) in the CKS1B group was 11 (range: 9–13, n=56) and 12 days (range: 8–18, n=56), respectively. Similarly, in the control group the median time to neutrophil and platelet engraftment was 11 (range: 9–20, n= 57) and 12 days (range: 0–63), respectively. Grade 3 infectious adverse events requiring antibiotics were reported in 32 (55%) patients in the CKS1B group, and in 22 (38%) patients in the control group (p=0.09). Otherwise, there was no significant difference in adverse events between the two groups.
Response after Auto-HCT
Eight (13%) and 19 (32%) patients in the CKS1B and control group, respectively, achieved a CR (p=0.02) (Table 2). Twenty-eight (48%) and 27 (46%) patients in the CKS1B and control group, respectively, achieved a VGPR (p=1.00). The overall response (CR+VGPR+PR) post auto-HCT was seen in 51 (88%) and 53 (91%) patients in the CKS1B and control group, respectively (p= 0.76). Thirty-five (60%) patients in the CKS1B group and 40 (69%) in the control group had an upgrade in their response post-auto-HCT. Two (3%) patients in the CKS1B group and 1 (2%) patients in the control group had evidence of progressive disease post auto-HCT.
Table 2. Response after auto-HCT.
CR indicates complete response; sCR, stringent complete response; PD, progressive disease; PR, partial response; SD, stable disease; VGPR, very good partial response; auto-HCT, autologous hematopoietic stem cell transplantation
|
Patients without CKS1B gene amplfification (n=58) |
|
|---|---|
| ORR | 53 (91) |
| ≥CR | 19 (32) |
| VGPR | 27 (46) |
| PR | 7 (12) |
| SD | 4 (7) |
| PD | 1 (2) |
Survival and outcomes
The median follow-up was 25.4 months. The median PFS of the CKS1B and control group was 15.0 and 33.0 months, respectively (p=0.0018, stratified log-rank test) (Figure 1a). The median OS in both CKS1B and the control cohort has not been reached yet. The 2-year OS rates in the CKS1B and the control groups were 62% and 91%, respectively (p=0.018, stratified log-rank test) (Figure 1b).
Figure 1.
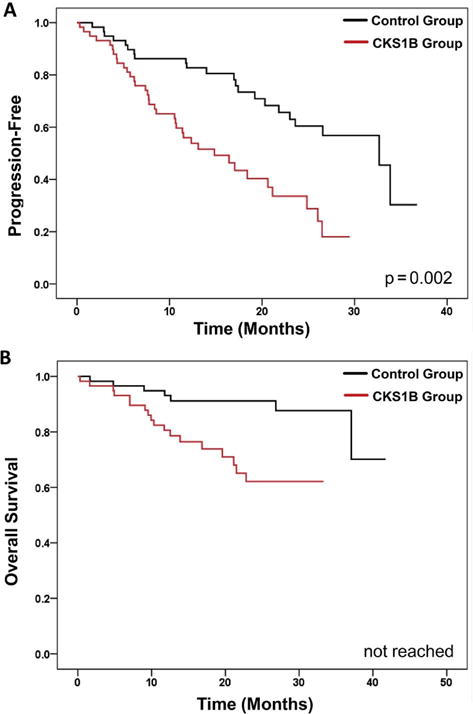
Probabilities of Progression-Free Survival (A) and Overall Survival (B).
On multivariate analysis for PFS, CKS1B amplification, indicated by three signals on 6% or four or more signals on 4.4% of interphase nuclei, and relapsed disease status at transplant were associated with worse outcome. Specifically, the patients with CKS1B amplification had larger hazard of having progression than the controls (HR (95% CI) = 3.0 (1.7, 5.3), p=0.0001, Table 4). Similarly, on multivariate analysis for OS, CKS1B amplification and relapsed disease at transplant were associated with worse outcome. The patients with CKS1B amplification had inferior OS than the controls (HR (95% CI) = 3.9 (1.5, 9.8), p=0.0043, Table 5).
Table 4.
CKS1B amp indicates CKS1B amplification; REF: Reference; TP: Transplantation; HR: Hazard Ratio
| Multivariate Analysis for PFS
|
|||
|---|---|---|---|
| HR | 95 % CI | P-value | |
| Presence of CKS1B amp (REF: absence of CKS1B amp) | 3.0 | 1.7–5.3 | 0.0001 |
| Relapsed disease status at TP (REF: in first remission at TP) | 2.7 | 1.6–4.6 | 0.0002 |
Table 5.
CKS1B amp indicates CKS1B amplification; REF: Reference; TP: Transplantation; HR: Hazard Ratio
| Multivariate Analysis for OS
|
|||
|---|---|---|---|
| HR | 95 % CI | P-value | |
| Presence of CKS1B amp (REF: absence of CKS1B amp) | 3.9 | 1.5–9.8 | 0.0043 |
| Relapsed disease status at TP (REF: in first remission at TP) | 4.8 | 1.9–12.2 | 0.0008 |
DISCUSSION
In this study we report that CKS1B amplification is an independent adverse prognostic factor for patients with multiple myeloma who underwent an auto-HCT at our institution. We used a propensity score matching analysis to identify a control group without CKS1B amplification that was well-matched with the CKS1B group in almost all the demographic characteristics. Compared to this evenly matched control group, more patients with CKS1B gene amplification had concurrent high-risk chromosomal abnormalities like monosomy 13/del(13q), a lower CR rate (13% vs. 32%), a significantly shorter PFS (15 vs. 33 months) and a shorter OS. These findings are mostly consistent with two prior reports [14, 15] with a few differences.
Although there was no significant difference in the use of IMiD or PI for induction therapy between the two groups, where >90% patients in each group received an IMiD or PI-based regimen, a higher proportion of patients in the control group (91% vs. 70 %) received a PI for induction. The beneficial effects of a bortezomib-based induction have been demonstrated for various myeloma subgroups, including treatment-refractory and high-dose therapy ineligible patients [18, 25]. Moreover, additional studies indicate that a bortezomib-based induction can potentially overcome the adverse prognosis of cytogenetic abnormalities such as del13q [26]. Hence, one may speculate that lower use of bortezomib-based induction in patients with CKS1B patients may have contributed to their inferior outcome.
Overall, there was no significant difference in proportion of patients receiving post-transplant maintenance therapy between the CKS1B and the control group (76% vs. 81%). In contrast to induction, a higher proportion of patients in the CKS1B group received a PI-based maintenance (30% vs. 13%) than the control, and still had a shorter PFS and OS.
Historically, the PFS after auto-HCT for multiple myeloma patients receiving an auto-HCT in first remission without maintenance is 24–30 months [27, 28] and 41–46 months with maintenance therapy with lenalidomide [29, 30]. Similarly, PFS after an auto-HCT for patients transplanted for relapsed disease is 12–16 months [8, 31, 32]. The PFS of 15 months in our report is slightly lower than what had been previously reported by Chang et al. for patients with CKS1B amplification after auto-HCT, which was 18.5 months [14]. The relatively shorter PFS in our study could be due to a higher proportion of patients with relapsed disease at auto-HCT, a slightly older population and a higher proportion of patients with ISS-Stage III at diagnosis (36% vs. 12% in Chang et al.) [14]. In another study, by Fonseca et al., 1q21 gain by FISH was associated with an OS of 21.9 months, which was significantly shorter than an OS of 38 months for patients without 1q21/CKS1B gain [15]. Similarly, patients with CKS1B overexpression by gene expression profiling (GEP) had a significantly shorter OS of 12.8 months vs. not reached for those without CKS1B overexpression [15]. Consistent with these prior two studies, a significant proportion of patients in our study had coexisting high-risk chromosomal abnormalities, like del(13) and del(17p).
The PFS of only 15 months for patients with CKS1B amplification in our study, where approximately 60% patients were in first remission, highlights the importance of CKS1B gene amplification as a marker of poor outcome. Unlike the two previous reports, CKS1B amplification emerged as an independent predictor of shorter PFS and OS. Furthermore, our findings suggest a poor outcome for patients with CKS1B amplification even with a PI or an IMiD-based induction in >90% of patients, and the use of post-transplant maintenance therapy in approximately 80% of patients. Similar outcomes were reported by Nahi et al., who found that patients with 1q21 gain had a shorter overall survival, which was not overcome by treatment with PI, IMiD or auto-HCT [33]. In contrast to induction, a higher proportion of patients in the CKS1B group received a PI-based maintenance (30% vs. 13%) than the control, and still had a shorter PFS and OS. Despite the availablity of therapies that improve outcomes for patients with high-risk cytogenetic abnormalities, CKS1B was still associated with a significantly worse prognosis.
Myeloid cell-leukemia 1 (Mcl-1) is another factor that maps to the 1q21 region [34]. It has been demonstrated that this is a pro-survival factor crucial for myeloma progression and treatment resistance. However, therapeutic strategies are being tested, such as novel proteasome inhibitors like carfilzomib or ixazomib that specifically cause degradation of Mcl-1 [35]. Myeloma patients with cytogenetic abnormalities in the 1q21 region may specifically benefit from these strategies but this remains to be seen. Furthermore, the availability of several new agents like elotuzumab, daratumumab, and panabinostat may also lead to improved outcome in this and other high-risk patient populations [36–39].
More than 40% of patients in this study had relapsed at least once before the auto-HCT. These patients generally have inferior outcomes compared to patients transplanted upfront [31, 32, 40]. Here we show that relapsed disease at transplant is an independent predictor of shorter PFS and OS in a multivariate analysis, thereby reinforcing the point that high-dose therapy and auto-HCT should be utilized early in the course of disease, especially in patients with high-risk disease [41, 42].
There are several limitations to this study, including its retrospective nature, a high frequency of concurrent high-risk chromosomal abnormalities and heterogeneity of the patient population. However, we did try to mitigate some of these disadvantages by using a propensity-matched control group. We are aware that in addition to the covariates (age at auto-HCT, gender, disease status at auto-HCT, response to pre-transplant therapy, baseline hemoglobin level, and time from diagnosis to auto-HCT) that were used to conduct the propensity score matching analysis, other potential confounders are conceivable. However, the small sample sizes of the final group limited the number of variables for matching.
Taken together, this paper strengthens the evidence that 1q21/CKS1B is a poor prognostic marker, and these patients may need a more aggressive therapeutic strategy to overcome the poor risk [43]. Future studies should evaluate therapeutic avenues for these patients including a combination of PIs, IMiDs and auto-HCT upfront, and perhaps early incorporation of immunotherapeutic approaches like monoclonal antibodies and cellular therapy in the treatment strategy [36, 44, 45].
Highlights.
CKS1B amplification is an independent adverse prognostic factor for patients with multiple myeloma who underwent an auto-HCT.
PFS after auto-HCT for multiple myeloma patients receiving an auto-HCT in with CKS1B amplification is 15 months.
Poor outcome for patients with CKS1B amplification even with a PI or an IMiD-based induction and the use of post-transplant maintenance therapy.
Acknowledgments
The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672 and used Cancer Center Support Grant shared resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors have no potential conflicts of interest to declare.
References
- 1.Shaughnessy JD, Jr, et al. Testing standard and genetic parameters in 220 patients with multiple myeloma with complete data sets: superiority of molecular genetics. Br J Haematol. 2007;137(6):530–6. doi: 10.1111/j.1365-2141.2007.06586.x. [DOI] [PubMed] [Google Scholar]
- 2.Stella F, et al. Cytogenetic Alterations in Multiple Myeloma: Prognostic Significance and the Choice of Frontline Therapy. Cancer Invest. 2015;33(10):496–504. doi: 10.3109/07357907.2015.1080833. [DOI] [PubMed] [Google Scholar]
- 3.Avet-Loiseau H, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30(16):1949–52. doi: 10.1200/JCO.2011.36.5726. [DOI] [PubMed] [Google Scholar]
- 4.Miguel JF, Mateos MV. Current Challenges in the Management of Patients with Relapsed/Refractory Multiple Myeloma. Oncology (Williston Park) 2011;25(12 Suppl 2) [PubMed] [Google Scholar]
- 5.Kazmi SM, et al. Outcomes Among High-Risk and Standard-Risk Multiple Myeloma Patients Treated With High-Dose Chemotherapy and Autologous Hematopoietic Stem-Cell Transplantation. Clin Lymphoma Myeloma Leuk. 2015;15(11):687–93. doi: 10.1016/j.clml.2015.07.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avet-Loiseau H, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p) J Clin Oncol. 2010;28(30):4630–4. doi: 10.1200/JCO.2010.28.3945. [DOI] [PubMed] [Google Scholar]
- 7.Bergsagel PL, et al. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood. 2013;121(6):884–92. doi: 10.1182/blood-2012-05-432203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki K, et al. Impact of t(11;14)(q13;q32) on the outcome of autologous hematopoietic cell transplantation in multiple myeloma. Biol Blood Marrow Transplant. 2013;19(8):1227–32. doi: 10.1016/j.bbmt.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanamura I, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108(5):1724–32. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neben K, et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 2013;31(34):4325–32. doi: 10.1200/JCO.2012.48.4923. [DOI] [PubMed] [Google Scholar]
- 11.Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 2005;10(Suppl 1):117–26. doi: 10.1080/10245330512331390140. [DOI] [PubMed] [Google Scholar]
- 12.Zhan F, et al. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood. 2007;109(11):4995–5001. doi: 10.1182/blood-2006-07-038703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang H, et al. CKS1B nuclear expression is inversely correlated with p27Kip1 expression and is predictive of an adverse survival in patients with multiple myeloma. Haematologica. 2010;95(9):1542–7. doi: 10.3324/haematol.2010.022210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang H, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br J Haematol. 2006;135(4):486–91. doi: 10.1111/j.1365-2141.2006.06325.x. [DOI] [PubMed] [Google Scholar]
- 15.Fonseca R, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20(11):2034–40. doi: 10.1038/sj.leu.2404403. [DOI] [PubMed] [Google Scholar]
- 16.Chang H, et al. Impact of cytogenetics in patients with relapsed or refractory multiple myeloma treated with bortezomib: Adverse effect of 1q21 gains. Leuk Res. 2011;35(1):95–8. doi: 10.1016/j.leukres.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol. 2012;87(1):78–88. doi: 10.1002/ajh.22237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.San Miguel JF, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 19.Fonseca R. Strategies for risk-adapted therapy in myeloma. Hematology Am Soc Hematol Educ Program. 2007:304–10. doi: 10.1182/asheducation-2007.1.304. [DOI] [PubMed] [Google Scholar]
- 20.Chen MH, et al. Cyclin kinase subunit 1B nuclear expression predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with bortezomib. Hum Pathol. 2012;43(6):858–64. doi: 10.1016/j.humpath.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 21.Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med. 2007;26(4):734–53. doi: 10.1002/sim.2580. [DOI] [PubMed] [Google Scholar]
- 22.Austin PC. A critical appraisal of propensity-score matching in the medical literature between 1996 and 2003. Stat Med. 2008;27(12):2037–49. doi: 10.1002/sim.3150. [DOI] [PubMed] [Google Scholar]
- 23.Durie BG, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467–73. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 24.Lu G, et al. Plasma cell enrichment enhances detection of high-risk cytogenomic abnormalities by fluorescence in situ hybridization and improves risk stratification of patients with plasma cell neoplasms. Arch Pathol Lab Med. 2013;137(5):625–31. doi: 10.5858/arpa.2012-0209-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson PG, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 26.Jagannath S, et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia. 2007;21(1):151–7. doi: 10.1038/sj.leu.2404442. [DOI] [PubMed] [Google Scholar]
- 27.Attal M, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335(2):91–7. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 28.Child JA, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med. 2003;348(19):1875–83. doi: 10.1056/NEJMoa022340. [DOI] [PubMed] [Google Scholar]
- 29.Attal M, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782–91. doi: 10.1056/NEJMoa1114138. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy PL, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–81. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah N, et al. Durable remission with salvage second autotransplants in patients with multiple myeloma. Cancer. 2012;118(14):3549–55. doi: 10.1002/cncr.26662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah N, et al. Phase I/II trial of lenalidomide and high-dose melphalan with autologous stem cell transplantation for relapsed myeloma. Leukemia. 2015;29(9):1945–8. doi: 10.1038/leu.2015.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nahi H, et al. Proteasome inhibitors and IMiDs can overcome some high-risk cytogenetics in multiple myeloma but not gain 1q21. Eur J Haematol. 2015 doi: 10.1111/ejh.12546. [DOI] [PubMed] [Google Scholar]
- 34.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584(14):2981–9. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 35.Fan F, et al. Targeting Mcl-1 for multiple myeloma (MM) therapy: drug-induced generation of Mcl-1 fragment Mcl-1(128–350) triggers MM cell death via c-Jun upregulation. Cancer Lett. 2014;343(2):286–94. doi: 10.1016/j.canlet.2013.09.042. [DOI] [PubMed] [Google Scholar]
- 36.Lokhorst HM, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med. 2015;373(13):1207–19. doi: 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 37.San-Miguel JF, et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J Clin Oncol. 2013;31(29):3696–703. doi: 10.1200/JCO.2012.46.7068. [DOI] [PubMed] [Google Scholar]
- 38.Lonial S, et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2015;373(7):621–31. doi: 10.1056/NEJMoa1505654. [DOI] [PubMed] [Google Scholar]
- 39.Richardson PG, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124(7):1038–46. doi: 10.1182/blood-2014-01-548826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook G, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(8):874–85. doi: 10.1016/S1470-2045(14)70245-1. [DOI] [PubMed] [Google Scholar]
- 41.Attal M, et al. American Society of Hematology Annual Meeting. Blood; Orlando: 2015. Autologous Transplantation for Multiple Myeloma in the Era of New Drugs: A Phase III Study of the Intergroupe Francophone Du Myelome (IFM/DFCI 2009 Trial) p. 391. [Google Scholar]
- 42.Sonneveld P, et al. Treatment of Multiple Myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016 doi: 10.1182/blood-2016-01-631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nooka AK, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib and dexamethasone (RVD) in high-risk myeloma patients. Leukemia. 2014;28(3):690–3. doi: 10.1038/leu.2013.335. [DOI] [PubMed] [Google Scholar]
- 44.Lonial S, et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2015;373(7):621–31. doi: 10.1056/NEJMoa1505654. [DOI] [PubMed] [Google Scholar]
- 45.Garfall AL, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373(11):1040–7. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]