

Abstract
The development of a gold(III) catalyzed direct enantioconvergent 1,5-enyne cycloisomerization and kinetic resolution reaction is described. The transformation results in highly enantioenriched bicyclo[3.1.0]-hexenes at all levels of conversion, with no racemization or symmetrization taking place during the course of the reaction, and simultaneously affords optically enriched 1,5-enynes. This report marks the first highly enantioselective transformation catalyzed by a well-defined cationic gold-(III) catalyst and demonstrates the unique potential of gold(III) complexes in enantioselective catalysis.
Cycloisomerization reactions have been studied extensively because they rapidly and efficiently build structural complexity from much simpler acyclic compounds.1 The field of homogeneous gold catalysis2 has been the subject of intense investigation during the past decade, elevating gold(I) complexes to among the most useful catalysts for a diverse range of cycloisomerization reactions.3 Despite significant progress in the development of enantioselective gold(I) catalysis,4,5 challenges still remain largely as a result of the linear geometry that places the active site distant from the chiral information. This challenge can potentially be addressed by employing square-planar geometry gold(III) complexes as catalysts; however, the majority of gold(III) catalyzed reactions employ simple gold halide salts as catalysts or ligands that produce unstable or unreactive complexes (Scheme 1).6 We recently reported on an oxidative addition strategy to prepare well-defined and stable gold(III) catalysts that, as a result of the square-planar geometry, place an ancillary ligand much closer to the active site (Scheme 1).7 This new class of organometallic gold(III) complexes takes advantage of a cyclometalated C–C ligand framework to stabilize the catalytically active cationic species, and offers the possibility to leverage an L-type ligand to introduce chiral information.
Scheme 1.

Ligand Environment of AuI and AuIII Complexes
The gold(I)-catalyzed cycloisomerization of 1,5-enynes was first reported over a decade ago,8 but a highly enantioselective variant has yet to be developed.9 A challenge inherent to this reaction is that the (R) and (S) enantiomer of the 1,5-enyne can transform to either the (R,R) or the (S,S) enantiomer of the bicyclo[3.1.0]hexene, and transfer of chirality has been observed for certain classes of substrates with achiral catalysts (Scheme 2).10 To develop an enantioselective variant of this cycloisomerization, the chiral catalyst ideally will selectively produce one enantiomer of the bicyclo[3.1.0]hexane adduct, overriding the transfer of chirality.
Scheme 2.

Proposed Mechanism for Au(I) Catalyzed 1,5-Enyne Cycloisomerization
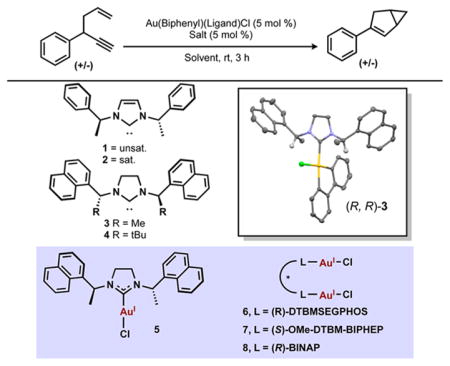
Initial investigations began by testing a number of gold(III) catalysts bearing chiral N-heterocyclic carbene (NHC) ligands (Table 1).11 The gold(III) complex bearing a saturated NHC ligand catalyzed the formation of the bicylic adduct in higher enantioselectivity when compared to the unsaturated analog (entries 1–2). Among the saturated NHC ligands, the more sterically encumbered naphthyl-substituted ligand resulted in higher enantioselectivity than the phenyl-substituted ligand (entry 3). Increasing the steric bulk of the NHC ligand by changing the methyl substitution to t-butyl resulted in a large drop in enantioselectivity as well as catalytic activity (entry 4). To confirm that in situ reductive elimination12 of biphenylene from the gold(III) species to form an active gold(I) species was not the operative mechanism, the gold(I) analog was tested and resulted in no enantioselectivity as well as decreased catalytic activity (entry 5). Additionally, the gold(III) catalyst was reisolated from the reaction mixture with no observable reductive elimination to gold(I), and showed the same catalytic activity when resubjected to the reaction conditions (see SI). A range of gold(I) catalysts bearing chiral phosphine ligands were also tested and showed complete conversion to product; however, the enantioselectivity was significantly lower than that obtained with the gold(III) complex as catalyst (entries 6–8).
Table 1.
Development of the Enantioselective 1,5-Enyne Cycloisomerization*
| |||||
|---|---|---|---|---|---|
| entry | catalyst | salt | solvent | conversion (%) | ee (%) |
| 1 | 1 | AgSbF6 | CH2Cl2 | 100 | −24 |
| 2 | 2 | AgSbF6 | CH2Cl2 | 100 | −34 |
| 3 | 3 | AgSbF6 | CH2Cl2 | 100 | 37 |
| 4 | 4 | AgSbF6 | CH2Cl2 | 4 | −2 |
| 5 | 5 | AgSbF6 | CH2Cl2 | 23 | 0 |
| 6 | 6 | AgSbF6 | CH2Cl2 | 100 | 9 |
| 7 | 7 | AgSbF6 | CH2Cl2 | 100 | 6 |
| 8 | 8 | AgSbF6 | CH2Cl2 | 100 | 2 |
| 9 | 2 | AgOMs | CH2Cl2 | 0 | |
| 10 | 2 | AgTRIP | CH2Cl2 | 0 | |
| 11 | 2 | AgOTf | CH2Cl2 | 31 | 48 |
| 12 | 2 | AgNTf2 | CH2Cl2 | 100 | 21 |
| 13 | 2 | AgPF6 | CH2Cl2 | 68 | 44 |
| 14 | 2 | AgBF4 | CH2Cl2 | 91 | 53 |
| 15 | 2 | NaBArF24 | CH2Cl2 | 72 | 19 |
| 16 | 2 | AgBF4 | DMF | 0 | |
| 17 | 2 | AgBF4 | MeCN | 8 | 58 |
| 18 | 2 | AgBF4 | THF | 68 | 29 |
| 19 | 2 | AgBF4 | Toluene | 41 | 73 |
| 20 | 2 | AgBF4 | Toluene | 61 | 54 |
General reaction conditions: Enyne (1.00 equiv), gold(III) catalyst (0.05 equiv), salt (0.05 equiv), solvent (0.3 M), rt, 3 h. Conversion determined by 1H NMR spectroscopy, enantiomeric excess (% ee) determined by chiral phase HPLC.
It has been frequently reported that the identity of the counterion in homogeneous gold catalysis can have a large effect on the outcome of a reaction.13 Screening of possible counterions in the gold(III) system revealed that several counterions were incompatible (entries 9–10). Presumably, the decrease in catalyst activity results from tight binding of the counterion to the cationic gold(III) center, which has been shown to have hard Lewis acidity and high oxophilicity.6 Ultimately, tetrafluoroborate proved to be the optimal counterion retaining catalytic activity and increasing enantioselectivity (entries 11–15).
We next looked to optimize the solvent conditions, and found low reactivity in solvents that provided poor solubility or were capable of coordination to the cationic gold(III) (entry 16). On the other hand, a significant increase in enantioselectivity was observed when dichloromethane was replaced with toluene as solvent, albeit with a decrease in reactivity (entry 19). When the reaction was run for 16 h, conversion increased but a significant decrease in enantioselectivity was also observed (entry 20).
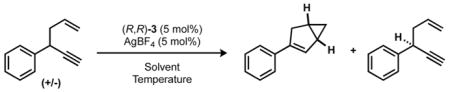
This decrease in product enantioselectivity with increased conversion suggested a potential kinetic resolution; however, at complete conversion the product remained enantioenriched (vide inf ra). Isolation of the starting 1,5-enyne and analysis by chiral HPLC showed that the enyne was highly enantioenriched in addition to the bicyclo[3.1.0]hexene. Complete optimization of the kinetic resolution resulted in the highest s-factor when the reaction was run in CHCl3 at −40 °C (Table 2).
Table 2.
Enyne Cycloisomerization s-Factor Optimization*
| ||||
|---|---|---|---|---|
| entry | solvent | temperature (°C) | conversion (%) | s-factor |
| 1 | CH2Cl2 | rt | 30 | 6 |
| 2 | CH2Cl2 | 0 | 31 | 9 |
| 3 | CH2Cl2 | −20 | 46 | 12 |
| 4 | CH2Cl2 | −30 | 55 | 14 |
| 5 | CH2Cl2 | −40 | 51 | 21 |
| 6 | CH2Cl2 | −50 | 0 | |
| 7 | CH2Cl2 | −40 | 51 | 21 |
| 8 | PhCl | −40 | 14 | 18 |
| 9 | DCE | −40 | 44 | 16 |
| 10 | CHCl3 | −40 | 44 | 23 |
General reaction conditions: enyne (1.00 equiv), gold(III) catalyst (0.05 equiv), AgBF4 (0.05 equiv), solvent (0.3 M), 3 h. Conversion determined by 1H NMR spectroscopy, enantiomeric excess (ee) determined by chiral phase HPLC. Selectivity (s-factor) calculated as s = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)].
With optimized reaction conditions in hand, the scope of the transformation was explored (Table 3). Substrates with more sterically hindered ortho substitution required higher temperatures but maintained similarly high levels of enantioselectivity and s-factor (enynes 11 and 12). Despite the potential for nonproductive coordination to the hard Lewis-acidic cationic gold(III), we found that heteroarene substrates were tolerated and in one case greatly exceeded the selectivity of the model substrate (enynes 16–18). Substrates in which the propargyl substitution was changed from aryl to alkyl were compatible with the reaction but with a decreased selectivity (enyne 19). On the other hand, the use of a silyl-substituted alkyne dramatically slowed the rate of reaction and resulted in no kinetic resolution or product enantioselectivity (enyne 20).
Table 3.
Scope of the Enantioconvergent Kinetic Resolution*

|
General reaction conditions: enyne (1.00 equiv), catalyst (0.05 equiv), AgBF4 (0.05 equiv), solvent (0.3 M), 3 h. Conversion determined by 1H NMR spectroscopy, ee determined by chiral phase HPLC. Selectivity (s-factor) calculated as s = ln[(1 − C)(1 − eeSM)]/ln [(1 − C)(1 + eeSM)].
−20 °C for 3 h.
−30 °C for 3 h.
60 °C for 24 h.
To further explore the interplay between the kinetic resolution and the enantioselective product formation, we first isolated enantioenriched starting material and subjected it to the reaction conditions with an achiral gold(I) and gold(III) catalyst. The bicyclo[3.1.0]hexene product was isolated with 44% and 43% enantiomeric excess respectively, indicating that the reaction proceeded with some, but not complete, chirality transfer (Scheme 3). Additionally, when the racemic starting material was reacted under our conditions to complete conversion, the product was enantioenriched. This observation stands in contrast to a standard kinetic resolution where there would be no enantioenrichment at complete conversion. Moreover, the inherent chirality transfer of the cycloisomerization must be overridden when using the chiral gold(III) catalyst for one enantiomer of starting material, and enforced in the case of the other enantiomer.
Scheme 3.

Contribution of Chirality Transfer to Enantiomeric Excess
The decrease in the enantiomeric excess of the bicyclo[3.1.0]-hexene over the course of the reaction implies that each enantiomer of starting material must also be transforming to product with different enantiomeric ratios. This phenomenon can be modeled and used as a predictive system (Scheme 3b, see SI). Given the enantiomeric excess data from at least two different values of conversion, it is possible to solve for the enantiomeric ratios that each starting material is converting to product. Using this method, the (R)-enyne starting material was found to transform to the bicyclo[3.1.0]hexene product with 89% ee catalyzed by (R,R)-3, whereas the (S)-enyne converted to the same product with 42% ee under our optimized reaction conditions (Scheme 3c). These values were then compared to experimental values and found to be in close agreement (see SI). Using this data, it was possible to create a model to determine product enantioenrichment at any conversion, which was compared to a similar plot for a standard kinetic resolution (Figure 1). From the comparison, it is clear that the enantioconvergent kinetic resolution results in more highly enantioenriched product at synthetically useful conversions (above 40%) when s factors are identical, while simultaneously maintaining the high levels of starting material enantioenrichment seen in a standard kinetic resolution.14
Figure 1.
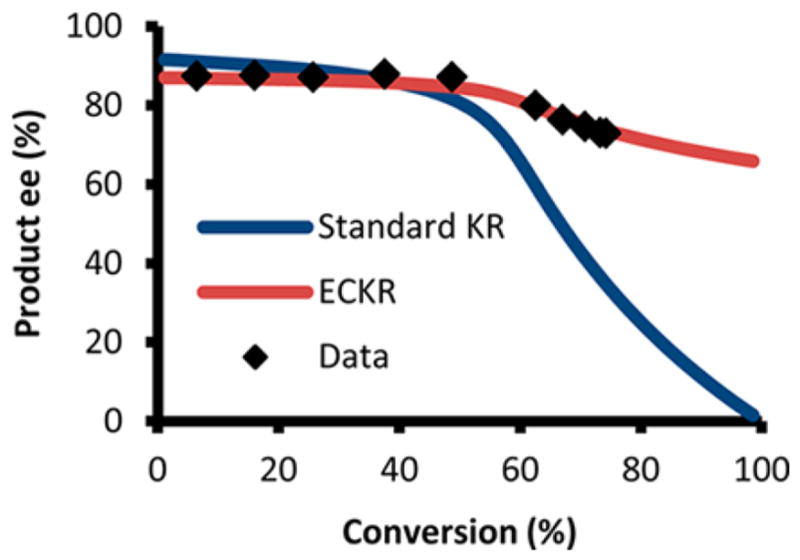
Enantioselectivity of a kinetic resolution with s = 23 (blue) and enantioselectivity of the enantioconvergent kinetic resolution with s = 23 (data = black, model = red).
The previously proposed mechanism for this reaction accounts for the chirality transfer of the reaction by considering the half-chair transition states (Scheme 4). In the matched case the (R)-enyne preferentially forms the (S,S)-bicyclo[3.1.0]-hexene, and this preference is reinforced when the transformation is performed with (R,R)-3 as the catalyst (Scheme 4). In the mismatched case the (S)-enyne preferentially forms the (R,R)-bicyclo[3.1.0]hexene. However, the chiral gold(III) catalyst overrides this preference and the major product becomes the (S,S)-bicyclo[3.1.0]hexene. These observations can be best explained by examining the selectivity determining transition state. The chiral catalyst either induces the large phenyl substituent to be in the axial position of the half-chair or it induces the cyclopropane ring to fold in to a half-boat conformation. Because both enantiomers of starting material convert to the same enantiomer of product without any racemization or symmetrization processes, this gold(III)-catalyzed cycloisomerization reaction falls under the rare class of direct enantioconvergent transformations.15
Scheme 4.

Direct Enantioconvergence of the 1,5-Enyne Cycloisomerization Reaction
In summary, we have developed an enantioconvergent kinetic resolution that allows access to enantioenriched 1,5-enynes and bicyclo[3.1.0]hexenes that has not been shown to be possible with gold(I). This transformation represents a rare mode of direct enantioconvergent kinetic resolution wherein product enantioenrichment is increased at synthetically useful conversions and starting material enantioenrichment is maintained when compared to a standard kinetic resolution. More importantly, these transformations mark the first highly enantioselective reactions catalyzed by a well-defined gold(III) complex and set the stage for further exploration and development.
Supplementary Material
Acknowledgments
We gratefully acknowledge NIGMS (R35 GM118190) for financial support. We thank Dr. Mark Levin, Andrew Samant, Dr. Johnathan Brantley, and Dr. Steven Jacob for helpful discussions, Xingyu Jiang for SFC assistance, and Richard Thornbury and Nicholas Settineri for X-ray crystallography assistance.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06025.
Experimental details and characterization data (PDF)
Crystallographic data for 3 (CIF)
References
- 1.For reviews of transition metal-catalyzed cycloisomerization reactions, see: Stathakis CI, Gkizis PL, Zografos AL. Nat Prod Rep. 2016;33:1093–1117. doi: 10.1039/c6np00026f.Watson IDG, Toste FD. Chem Sci. 2012;3:2899–2919.Marinetti A, Jullien H, Voituriez A. Chem Soc Rev. 2012;41:4884–4908. doi: 10.1039/c2cs35020c.Aubert C, Fensterbank L, Garcia P, Malacria M, Simonneau A. Chem Rev. 2011;111:1954–1993. doi: 10.1021/cr100376w.Michelet V, Toullec Genêt JP. Angew Chem, Int Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589.
- 2.For recent reviews on gold catalysis, see: Joost M, Amgoune A, Bourissou D. Angew Chem, Int Ed. 2015;54:15022–15045. doi: 10.1002/anie.201506271.Miro J, del Pozo C. Chem Rev. 2016;116:11924–11966. doi: 10.1021/acs.chemrev.6b00203.Wei Y, Shi M. ACS Catal. 2016;6:2515–2524.
- 3.For select reviews on gold(I)-catalyzed cycloisomerization reactions, see: Harris RJ, Widenhoefer RA. Chem Soc Rev. 2016;45:4533–4551. doi: 10.1039/c6cs00171h.Dorel R, Echavarren AM. Chem Rev. 2015;115:9028–9072. doi: 10.1021/cr500691k.Fensterbank L, Malacria M. Acc Chem Res. 2014;47:953–965. doi: 10.1021/ar4002334.Obradors C, Echavarren AM. Acc Chem Res. 2014;47:902–912. doi: 10.1021/ar400174p.
- 4.For examples of gold(I)-catalyzed enantioselective cycloisomerization reactions of 1,6-enynes, see: Guitet M, Zhang P, Marcelo F, Tugny C, Jiménez-Barbero J, Buriez O, Amatore C, Mouriés-Mansuy V, Goddard JP, Fensterbank L, Zhang Y, Roland S, Ménand M, Sollogoub M. Angew Chem, Int Ed. 2013;52:7213–7218. doi: 10.1002/anie.201301225.Yavari K, Aillard P, Zhang Y, Nuter F, Retailleau P, Voituriez A, Marinetti A. Angew Chem, Int Ed. 2014;53:861–865. doi: 10.1002/anie.201308377.Teller H, Corbet T, Mantilli L, Gopakumar G, Goddard R, Thiel W, Fürstner A. J Am Chem Soc. 2012;134:15331–15342. doi: 10.1021/ja303641p.Deschamps NM, Elitzin VI, Liu B, Mitchell MB, Sharp MJ, Tabet EA. J Org Chem. 2011;76:712–715. doi: 10.1021/jo102098y.Sethofer S, Mayer T, Toste FD. J Am Chem Soc. 2010;132:8276–8277. doi: 10.1021/ja103544p.Chao CM, Beltrami D, Toullec PY, Michelet V. Chem Commun. 2009:6988–6990. doi: 10.1039/b913554e.Muñoz MP, Adrio J, Carretero JC, Echavarren AM. Organometallics. 2005;24:1293–1300.
- 5.For recent reviews on enantioselective gold(I) catalysis see: Li Y, Li W, Zhang J. Chem - Eur J. 2017;23:467–512. doi: 10.1002/chem.201602822.Zi W, Toste FD. Chem Soc Rev. 2016;45:4567–4589. doi: 10.1039/c5cs00929d.Wang YM, Lackner AD, Toste FD. Acc Chem Res. 2014;47:889–901. doi: 10.1021/ar400188g.Cera G, Bandini M. Isr J Chem. 2013;53:848–855.
- 6.Hashmi ASK, Weyrauch JP, Rudolph M, Kurpejovicm E. Angew Chem, Int Ed. 2004;43:6545–6547. doi: 10.1002/anie.200460232.Johnson MW, DiPasquale AG, Bergman RG, Toste FD. Organometallics. 2014;33:4169–4172. doi: 10.1021/om500663m.Lo VKY, Liu Y, Wong M-K, Che CM. Org Lett. 2006;8:1529–1532. doi: 10.1021/ol0528641.For a discussion on the stability of gold(III) in catalysis, see: Hashmi ASK, Blanco MC, Fischer D, Bats JW. Eur J Org Chem. 2006;2006:1387–1389.During the course of our investigation, Wong and coworkers reported a single example of enantioselective (41% ee) catalysis C,O-chelated BINOL/Au(III) complexes Cui J, Ko H, Shing K, Deng J, Lai NC, Wong M. Angew Chem, Int Ed. 2017;56:3074–3079. doi: 10.1002/anie.201612243.
- 7.Wu C, Horibe T, Jacobsen CB, Toste FD. Nature. 2015;517:449–454. doi: 10.1038/nature14104.See also: Joost M, Estévez L, Miqueu K, Amgoune A, Bourissou D. Angew Chem, Int Ed. 2015;54:5236–5240. doi: 10.1002/anie.201500458.For an early example of organometallic diarylgold(III) catalysts, see: Casado R, Contel M, Laguna M, Romero P, Sanz S. J Am Chem Soc. 2003;125:11925–11935. doi: 10.1021/ja036049x.
- 8.Luzung MR, Markham JP, Toste FD. J Am Chem Soc. 2004;126:10858–10859. doi: 10.1021/ja046248w.For a related gold-catalyzed reaction of hydroxylated enynes, see: Mamane V, Gress T, Krause H, Fürstner A. J Am Chem Soc. 2004;126:8654–8655. doi: 10.1021/ja048094q.For the platinum-catalyzed variant, see: Harrak Y, Blaszykowski C, Bernard M, Cariou K, Mainetti E, Mouries V, Dhimane A-L, Fensterbank L, Malacria M. J Am Chem Soc. 2004;126:8656–8657. doi: 10.1021/ja0474695.
- 9.Enantioselective gold(I) catalyzed cycloisomerization reactions of (a) hydroxylated 1,5-enynes: Wu Z, Retailleau P, Gandon V, Voituriez A, Marinetti A. Eur J Org Chem. 2016;2016:70–75.1,5-silyloxyenynes: Brazeau JF, Zhang S, Colomer I, Corkey BK, Toste FD. J Am Chem Soc. 2012;134:2742–2749. doi: 10.1021/ja210388g.
- 10.Patil NT. Chem - Asian J. 2012;7:2186–2194. doi: 10.1002/asia.201200194. [DOI] [PubMed] [Google Scholar]
- 11.For examples of enantioselective catalysis with chiral NHCgold(I) complexes, see: Strand RB, Helgerud T, Solvang T, Dolva A, Sperger CA, Fiksdahl A. Tetrahedron: Asymmetry. 2012;23:1350–1359.Banerjee D, Buzas AK, Besnard C, Kündig EP. Organometallics. 2012;31:8348–8354.Handa S, Slaughter LM. Angew Chem, Int Ed. 2012;51:2912–2915. doi: 10.1002/anie.201107789.Wang YM, Kuzniewski CN, Rauniyar V, Hoong C, Toste FD. J Am Chem Soc. 2011;133:12972–12975. doi: 10.1021/ja205068j.Wang W, Yang J, Wang F, Shi M. Organometallics. 2011;30:3859–3869.Matsumoto Y, Selim KB, Nakanishi H, Yamada K, Yamamoto Y, Tomioka K. Tetrahedron Lett. 2010;51:404–406.For a review, see: Gu P, Xu Q, Shi M. Tetrahedron Lett. 2014;55:577–584.
- 12.Wolf WJ, Winston MS, Toste FD. Nat Chem. 2014;6:159–164. doi: 10.1038/nchem.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biasiolo L, Del Zotto A, Zuccaccia D. Organometallics. 2015;34:1759–1765.For a review on the counterion effect in homogeneous gold catalysis, see: Jia M, Bandini M. ACS Catal. 2015;5:1638–1652.
- 14.Vedejs E, Jure M. Angew Chem, Int Ed. 2005;44:3974–4001. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 15.For select publications on enantioconvergent catalysis, see: Ito H, Kunii S, Sawamura M. Nat Chem. 2010;2:972–976. doi: 10.1038/nchem.801.Lundin PM, Fu GC. J Am Chem Soc. 2010;132:11027–11029. doi: 10.1021/ja105148g.Chen M, Roush WR. J Am Chem Soc. 2011;133:5744–5747. doi: 10.1021/ja2010187.Langlois JB, Emery D, Mareda J, Alexakis A. Chem Sci. 2012;3:1062–1069.Delvos LB, Oestreich M. Synthesis. 2015;47:924–933.For a review, see: Mohr JT, Moore JT, Stoltz BM. Beilstein J Org Chem. 2016;12:2038–2045. doi: 10.3762/bjoc.12.192.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.