

Abstract
During viral replication in the nucleus, the DNA genomes of adenoviruses are accessible to cellular DNA-binding proteins. Human adenovirus type 5 (Ad5) targets the cellular Mre11-Rad50-Nbs1 complex (MRN) to evade detection by the DNA damage response (DDR). Ad5 mutants that cannot target MRN have reduced viral propagation. Previous studies showed that diverse adenovirus serotypes interact differently with MRN. While these studies revealed diverse MRN interactions among serotypes, it remains unclear how these differences influence viral replication. Here, we examined effects of the DDR on several adenovirus serotypes. We demonstrate that wild-type Ad9 and Ad12 do not overcome MRN impairment. We also examined viral proteins involved in targeting MRN and found that unlike Ad5-E4orf3, expression of Ad9-E4orf3 is not sufficient for MRN mislocalization observed during infection. We conclude that adenovirus serotypes target MRN in distinct ways, and the MRN complex can impair DNA replication of wild-type viruses across the adenovirus family.
Keywords: Adenovirus, DNA damage response, serotypes, MRN, ATM, E4orf3
Introduction
Adenoviruses are nuclear-replicating viruses with linear double-stranded DNA genomes. Adenovirus genomes produce early viral proteins from viral genomic regions E1, E2, E3, and E4, each of which expresses multiple proteins through alternatively spliced transcripts [1]. Human adenovirus serotypes are classified into seven subgroups (A–G) [2]. Different serotypes infect distinct tissues and cause a variety of illnesses, including respiratory infections, conjunctivitis, and gastrointestinal disorders [1]. While there are differences in tissue tropism and clinical manifestations, all adenoviruses have analogous genome structures, replicate in the cell nucleus, and follow a similar replication cycle [1, 2]. Replication within the host cell nucleus provides adenoviruses access to host proteins that can be harnessed for viral DNA replication but also renders viral genomes susceptible to detection by host DNA-binding proteins that can impair viral replication [3]. Thus, adenoviruses have evolved several strategies to evade detection and overcome detrimental downstream effects.
A network of proteins comprises the cellular DNA damage response (DDR). These proteins recognize various forms of damage in cellular DNA and activate cascades of kinase signaling to arrest the cell cycle and repair damage [4]. Double-stranded DNA breaks (DSBs) are recognized by the Mre11-Rad50-Nbs1 complex (MRN) [5–7], which activates downstream signaling by promoting autophosphorylation of the phosphatidylinositol-like kinase ATM [5, 8, 9]. ATM signaling results in cell cycle arrest and eventually leads to repair of DSBs by either non-homologous end-joining or homologous recombination [10]. Resection of DNA ends during double-strand break repair can activate the ATR kinase, which responds to accumulation of single-stranded DNA [11]. Studies with adenovirus demonstrated that the DDR can also be activated by viral infection [8, 12], and subsequent work has shown that several other viruses also manipulate DDR signaling to promote viral replication [13–18]. The vast majority of our understanding of how adenovirus interacts with the DDR has come from studies using wild-type and mutant strains of adenovirus type 5 (Ad5, subgroup C). The degree of conservation of these interactions across the adenovirus family is unclear. Differences between adenovirus serotypes and their interactions with the DDR could contribute to the evolution and diverse tissue tropism of these viruses. Additionally, common cellular targets of viral manipulation are likely to be important host anti-viral defenses. Therefore, understanding the interactions with the DDR across the adenovirus family could contribute to our understanding of both viral evolution and cellular intrinsic defenses.
An early indication that the DDR could respond to Ad5 infection came from the observation that infection with E4-deleted Ad5 mutants resulted in fusion of viral genomes into concatemers [19]. This was later shown to be dependent on MRN, DNA-PK, and DNA ligase IV [12, 20, 21]. Subsequent studies with Ad5 mutants demonstrated that cellular proteins involved in the DDR can impair viral DNA accumulation, viral protein production, and virion formation [22–27]. To circumvent these obstacles, wild-type Ad5 inactivates DDR pathways through multiple mechanisms. The viral early proteins E1b55K (expressed from the E1 region) and E4orf6 (expressed from the E4 region) associate with cellular proteins to form an E3 ubiquitin ligase that is required for the degradation of several host proteins [12, 20, 28]. The E4orf6 protein recruits cellular E3 ubiquitin ligase components [28, 29], and the E1b55K protein is thought to provide substrate specificity to the ligase complex [29–31]. Another early viral protein from the E4 region, E4orf3, polymerizes to form long tracks in the nucleus and disrupts PML bodies [32–35]. The MRN complex is sequestered into these tracks, keeping it away from viral replication centers in a process that has been referred to as “mislocalization” [12, 23–25]. The Ad5-E4orf3 protein is necessary and sufficient to disrupt PML bodies and to induce MRN mislocalization [12, 23–25, 32–35]. In addition, Ad5-E4orf3 forms cytoplasmic foci and perinuclear aggresomes in which cellular proteins such as MRN are sequestered [36]. Manipulation of MRN during Ad5 infection also affects downstream signaling through the DDR. Since MRN is required for full activation of ATM [37], degradation and mislocalization of MRN during wild-type Ad5 infection prevents ATM activation at viral replication centers [8, 25]. However, there is evidence of pan-nuclear MRN-independent ATM activation at late stages of infection [25, 38]. ATR activation is also suppressed by MRN degradation and mislocalization during wild-type Ad5 infection [8, 39, 40]. Thus, wild-type Ad5 utilizes several mechanisms to manipulate MRN and disable downstream DDR signaling.
Targeting MRN through degradation or mislocalization is critical for optimal replication of Ad5 [22, 24]. Infection with Ad5 mutants that cannot target MRN due to the absence of E4orf3 and the E1b55K/E4orf6 complex results in MRN association with viral replication centers, activation of downstream ATM signaling, concatemerization of viral genomes, and dramatic inhibition of viral replication [8, 12, 22–26]. Ad5 mutants that retain the ability to target MRN through either degradation or mislocalization are not impaired for DNA replication by MRN [22], demonstrating that each mechanism is sufficient to overcome MRN’s inhibitory effects on Ad5 replication. ATM signaling has been suggested to impair Ad5 mutants [25, 27], and DNA repair mechanisms downstream of MRN result in concatemer formation [12, 21]. In addition, MRN directly impairs viral replication independently of downstream signaling [22]. Thus, targeting MRN allows wild-type Ad5 to evade direct inhibition of viral DNA replication by MRN as well as detrimental downstream effects.
It has been well established that the DDR is an obstacle for wild-type Ad5 replication and that Ad5 employs redundant mechanisms to evade its negative effects. However, there has been relatively little research into the interactions between other adenovirus serotypes and the DDR. Analysis of known Ad5 degradation substrates during infection with other Ad serotypes revealed that all serotypes examined to date lead to degradation of DNA ligase IV and thereby prevent genome concatemerization [40]. In contrast, some serotypes appear not to degrade MRN, p53, or integrin α3, and only Ad12 has been found to degrade the DDR regulatory protein TOPBP1 [40–43]. Interestingly, substrate degradation by non-Ad5 serotypes does not always correlate with interaction with E1b55K [44], suggesting that degradation of host proteins by adenoviruses may be regulated in additional unknown ways. Furthermore, infection with some serotypes does not result in MRN mislocalization to tracks or to aggresomes [40, 45, 46]. These findings demonstrate that although the ability to evade recognition by MRN is critical for optimal wild-type Ad5 replication, this may not necessarily be representative across the whole adenovirus family.
Since cellular restriction factors can influence tissue tropism and virulence, we reasoned that there may be differences among serotypes in their ability to overcome MRN inhibition. While previous studies have demonstrated that some serotypes do not degrade or mislocalize MRN, it remains unknown how the different interactions with MRN impact virus replication. Given the importance of inactivating MRN and downstream responses during wild-type Ad5 replication, it is possible that virulence and/or tissue tropism of adenoviruses are partially influenced by their potential to evade inhibition by MRN. Furthermore, it is unclear whether MRN targeting by other serotypes is accomplished by the analogous viral proteins as Ad5. Here, we examined more closely the fate of MRN during infection with multiple adenovirus serotypes representing several subgroups, and we determined the impact on wild-type viral DNA replication. Consistent with previous reports [40, 41], we identified serotypes that target MRN through both degradation and mislocalization, and other serotypes incapable of one or both of these mechanisms. We found that serotypes Ad9 and Ad12 can target MRN by mislocalization or degradation but are still impaired for DNA replication, demonstrating differences between these serotypes and Ad5. By examining the viral proteins that target MRN, we found that Ad9-E4orf3 alone is not sufficient to induce MRN mislocalization even though it is observed during Ad9 infection, suggesting that MRN mislocalization by Ad9 may be regulated through additional viral mechanisms. This work adds to our growing understanding of adenoviruses and the DDR, and suggests that diverse strategies have evolved across the adenovirus family to overcome MRN during wild-type virus infections.
Materials and Methods
Cell lines
U2OS were purchased from the American Tissue Culture Collection. Immortalized NBS cells (ILB1) transduced to express Nbs1 or empty vector were previously described [47, 48]. Immortalized A-T cells (AT22IJE-T) and matched cells complemented with ATM as previously described were gifts from Y. Shiloh [10, 49]. All cells were maintained in Dulbecco modified Eagle medium (Corning MT10-013-CV) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen 15140122) at 37°C in a humidified incubator with 5% CO2. Acceptor cells for the generation of doxycycline-inducible cell lines were provided by E. Makeyev and were used as previously described [50]. Briefly, FLAG-Ad9-E4orf3 was PCR amplified from the pL2-FLAG-Ad9-E4orf3 plasmid described below and inserted into the inducible plasmid backbone. The inducible plasmid containing FLAG-Ad9-E4orf3 was transfected into U2OS acceptor cells together with a plasmid expressing the Cre recombinase. Recombined clones were selected with 1 μg/mL Puromycin. Cells were induced with 0.2 μg/mL doxycycline for 24 hours to express FLAG-Ad9-E4orf3. Expression was confirmed by immunoblot and immunofluorescence. Inducible cells were maintained in medium supplemented with tetracycline-free fetal bovine serum.
Plasmids and transfections
The Ad9-E4orf3 cDNA was obtained from cells infected with Ad9, PCR amplified, and cloned into the pL2-FLAG plasmid backbone (described in [45]). Transfections were performed using the standard protocol for Lipofectamine 2000 (Invitrogen).
Viruses and infections
Wild-type Ad5, Ad2, Ad4, Ad9, Ad12, and Ad35 were purchased from American Tissue Culture Collection. Mutant Ad5 viruses dl1004, dl110, and dl1006 were previously described [51, 52] and were gifts from G. Ketner and D. Ornelles. Wild-type Ad5, Ad2, Ad4, Ad9, Ad12, Ad35, dl110, and dl1006 were propagated on 293 cells. The E4-deleted virus dl1004 was propagated on W162 cells. All viruses were purified by two sequential rounds of ultracentrifugation of cesium chloride gradients and stored in 40% glycerol at −20°C. Viral titers were determined by plaque assay on 293 cells. Infections were carried out by standard protocols using a multiplicity of infection of 20 (Ad5 wild-type and mutants, Ad2, Ad4, Ad12, Ad35) or 50 (Ad9). Viruses were diluted in Dulbecco modified Eagle medium supplemented with 2% fetal bovine serum and 1% penicillin-streptomycin and added to cell monolayers. Cells were incubated with the virus for 2 hours at 37°C before supplementing infection medium with medium containing 10% fetal bovine serum.
Antibodies and inhibitors
Primary antibodies to cellular proteins were purchased from commercial sources: Mre11 (Novus NB100-142), Rad50 (GeneTex [13B3] GTX70228), Nbs1 (Novus NB100-143), ATM pS1981 (Epitomics 2152-1 and Abcam [EP1890Y] ab81292), ATM (Abcam [Y170] ab32420 and Epitomics 1549-1), Actin (Sigma a5441), RPA32 (Abcam ab2175 and Bethyl A300-244A), PML (Santa Cruz [PG-M3] sc-966), and FLAG (Sigma F3165 and F7425). Primary antibodies to adenoviral proteins DBP and E4orf3 were gifts from A. Levine and T. Dobner, respectively. Horseradish peroxidase-conjugated secondary antibodies for immunoblotting were purchased from Jackson Laboratories. Fluorophore-conjugated secondary antibodies for immunofluorescence were purchased from Life Technologies. The ATM kinase inhibitor KU55933 was purchased from Abcam. The proteasome inhibitor MG132 was purchased from Sigma-Aldrich.
Immunoblotting
Immunoblot analysis was carried out using standard methods. Briefly, protein samples were prepared in lithium dodecyl sulfate loading buffer (NuPage) with 10% dithiothreitol and boiled. Equal amounts of protein were separated by electrophoresis. Proteins were transferred to nitrocellulose membranes (GE Healthcare Amersham) and blocked in 5% milk in tris buffered saline with Tween (TBST). Proteins were detected by enhanced chemiluminescence (Thermo Scientific) on film (HyBlot CL) or on a Syngene G-Box.
Immunofluorescence
Cells were plated on glass coverslips. Cells were washed with phosphate buffered saline (PBS) and fixed with cold 4% paraformaldehyde for 15 minutes. Cells were permeabilized for ten minutes with 0.5% Triton X-100 and coverslips were blocked for 1 hour with 3% bovine serum albumin (BSA) in PBS, incubated with each primary antibody diluted in 3% BSA for one hour, and incubated with a mixture of secondary antibodies and 4,6-diamidino-2-phenylindole (DAPI) in 3% BSA for one hour. Coverslips were mounted onto glass slides using ProLong Gold AntiFade Reagent (Life Technologies) and fluorescence was visualized using a Zeiss LSM 710 confocal microscope. Images were processed using ImageJ and Adobe Creative Suite 6.
Virus genome accumulation by quantitative PCR
Cells were infected and harvested by Trypsin at 4 hours post-infection (hpi) and at the times indicated. Total DNA was isolated using the PureLink Genomic DNA kit (Invitrogen). Quantitative PCR was performed using primers specific for a conserved sequence in the viral genome (5′ atcaccaccgtcagtgaa and 5′ gtgttattgctgggcga) or cellular tubulin (5′ ccagatgccaagtgacaagac and 5′ gagtgagtgacaagagaagcc). Values for viral DNA were normalized internally to tubulin and externally to the 4 hour time point to control for any variation in virus input. Quantitative PCR was performed using Sybr Green (Thermo) and data were collected using the ViiA 7 Real-Time PCR System (Thermo). At least three biological replicates were included, and statistical analyses were performed with the Prism v7 software (GraphPad).
Results
Effect of adenovirus infection on MRN protein levels and localization
We selected five serotypes to investigate MRN during adenovirus infection, each serotype representing a different adenovirus subgroup. We included Ad2, a subgroup C virus that is closely related to Ad5 (92.6% genome identity), as well as viruses that are less closely related to Ad5: Ad12 (subgroup A, 56.8% genome identity), Ad35 (subgroup B, 63.9% genome identity), Ad9 (subgroup D, 61.0% genome identity), and Ad4 (subgroup E, 61.4% genome identity). We first defined their impact on MRN by examining MRN protein levels by western blot over a time course of infection. Infections for Ad2, Ad4, Ad5, Ad9, and Ad35 were confirmed by western blot for the viral DNA-binding protein (DBP) (Figure 1A). The antibody generated against Ad5 DBP does not recognize DBP produced from Ad12, and therefore Ad12 infection was instead confirmed by the presence of cytopathic effect (data not shown). We observed that Ad2, Ad4, Ad5, and Ad12 degrade MRN, as indicated by decreased protein levels of Mre11, Rad50, and Nbs1 during infection (Figure 1A). Ad9 does not degrade MRN, and the protein levels for Mre11, Rad50, and Nbs1 remained steady throughout infection (Figure 1A). Interestingly, Mre11 and Nbs1 protein levels remained steady throughout infection with Ad35, but Rad50 protein levels were dramatically reduced (Figure 1A). To confirm that the decrease in Rad50 levels during Ad35 infection was due to degradation, we treated infected cells with the proteasome inhibitor MG132 and compared to results obtained during Ad5 infection (Figure 1B). MG132 treatment rescued Rad50 levels, suggesting that Ad35 somehow leads to degradation of Rad50 but not Mre11 or Nbs1 (Figure 1B).
Figure 1. Effect of adenovirus infection on MRN protein levels.
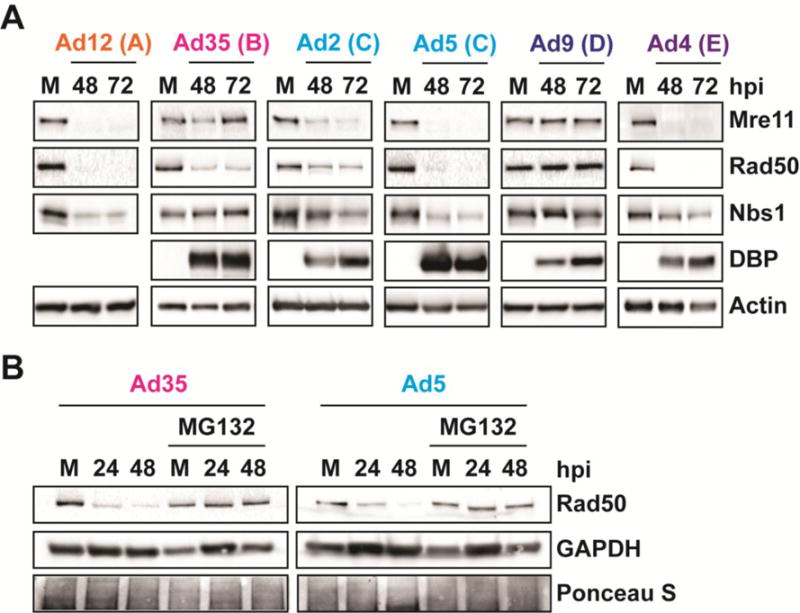
(A) Western blot analysis of Mre11, Rad50, and Nbs1 using infected cell lysates. U2OS cells were infected with serotypes from subgroups A-E and harvested at 48 and 72 hours post-infection (hpi). Subgroups are indicated in parentheses. Viral DBP confirms infection for all serotypes except Ad12. (B) Western blot analysis of Rad50 during Ad35 and Ad5 infection in the presence of the proteasome inhibitor MG132. Cells were treated with 20 uM MG132 or equal volume DMSO 8 hpi and harvested at the indicated time points. MG132 and DMSO were refreshed every 24 hours.
We also examined subcellular localization of Mre11 in relation to viral replication centers (VRCs) by immunofluorescence (Figure 2). VRCs were visualized using antibodies for viral DBP or cellular RPA32, which are both known to localize to sites of single-stranded adenovirus DNA [45, 53]. VRCs begin as small foci, which transition to large, donut-shaped structures as viral DNA replication progresses [53]. In an asynchronous infection, there is a mixture of cells with small and large VRCs, depending on the stage of viral replication. We examined Mre11 localization in cells with small and large VRCs to determine how Mre11 localization is affected at different stages of infection. Representative images from early and late stages of infection are shown in Figure 2. During Ad2, Ad4, Ad9, and Ad5 infections, Mre11 was redistributed to sites distinct from VRCs early during infection (Figure 2). We previously demonstrated that Nbs1 can colocalize with VRCs during late stages of Ad4 infection, although much of the Nbs1 was reorganized in structures separate from VRCs earlier during Ad4 infection [45]. The results presented here suggest that the effect of infection on Nbs1 localization can differ from that of Mre11. This is consistent with other reports, where Nbs1 was found colocalized with VRCs during late stages of Ad5 infection even though Mre11 was mislocalized to nuclear tracks or degraded [24]. During late stages, Mre11 was undetectable in Ad2, Ad4, and Ad5 infections, consistent with MRN degradation by these serotypes (Figure 2). Mre11 was detected during late stages of Ad9 infection but remained sequestered from VRCs. In contrast, during infection with Ad12 and Ad35, Mre11 colocalized with VRCs at early stages of infection, demonstrating that these serotypes do not mislocalize Mre11 (Figure 2). Mre11 was undetectable at late stages of Ad12 infection, consistent with degradation (Figure 2), but remained colocalized with Ad35 VRCs late during infection since Mre11 is not degraded by this serotype (Figure 2). In line with previous reports [40, 41, 45], we conclude that these representative adenovirus serotypes interact differently with MRN: some serotypes degrade and mislocalize MRN (Ad2, Ad4, and Ad5), and some only degrade (Ad12) or only mislocalize (Ad9) MRN complex members (Table 1). In the case of Ad35, it appears that this serotype can selectively degrade a single component of the MRN complex without degrading the entire complex (Table 1). This could be through direct interaction and targeting of Rad50 or indirectly by removal of an additional protein required for its stability within the complex.
Figure 2. Effect of adenovirus infection on Mre11 localization.
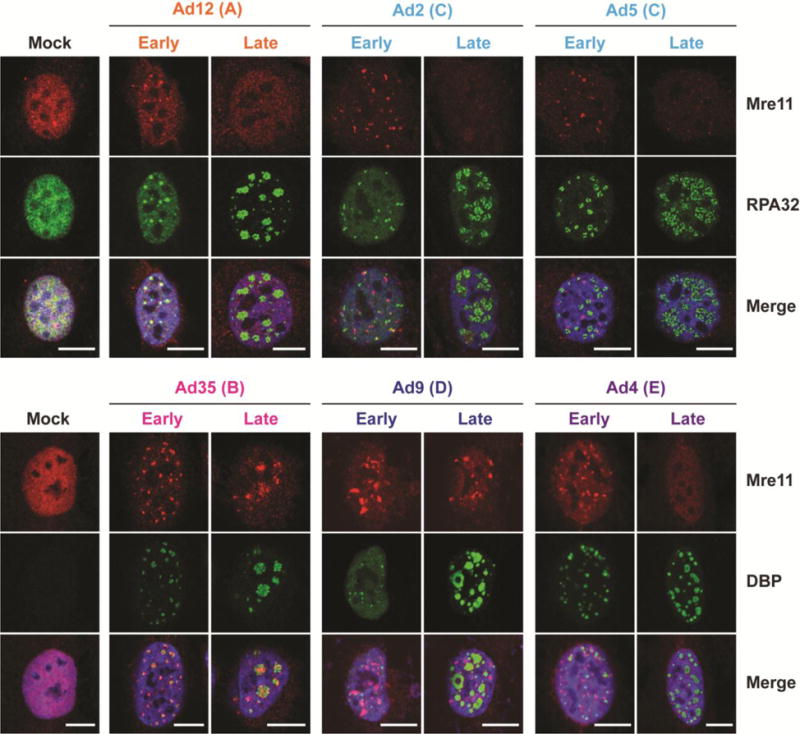
Immunofluorescence results of Mre11 (red) during infection of U2OS cells with each serotype at 18–24 hpi. Cellular RPA32 or viral DBP (green) mark viral DNA replication centers (VRC), which enlarge over the course of infection. Representative early and late infection images based on VRC size are shown. Merged images include DAPI stain in blue. Scale bar = 10 μm.
Table 1. Summary of MRN degradation and mislocalization during adenovirus infection.
| Serotype | Subgroup | MRN degradation | Mre11 mislocalization |
|---|---|---|---|
| Ad12 | A | ✓ | – |
| Ad35 | B | Rad50 only | – |
| Ad2 | C | ✓ | ✓ |
| Ad5 | C | ✓ | ✓ |
| Ad9 | D | – | ✓ |
| Ad4 | E | ✓ | ✓ |
ATM is activated during infection with multiple serotypes
Since the MRN complex is required for full activation of ATM in response to DNA breaks [5, 8], we examined how differences in MRN manipulation by diverse adenovirus serotypes affect ATM activity. Previous research has shown that ATM substrate KAP1 is phosphorylated during infection with several serotypes [40], but no studies have examined ATM activation directly or ATM localization during infection with serotypes other than Ad5. We assessed ATM activation by western blot and immunofluorescence using an antibody specific to phosphorylation at serine 1981, the ATM autophosphorylation site [9]. The E4-deleted Ad5 (dl1004 [51]) served as a positive control for ATM activation [8]. We found that ATM autophosphorylation increased during infection with all serotypes except Ad5 (Figure 3A and 3B) and that phosphorylated ATM colocalized with DBP or RPA32-stained VRCs (Figure 3A). These data suggest that ATM is activated in response to viral DNA during infection with these serotypes. Most cells infected with Ad5 did not show ATM activation (representative image, Figure 3A), although in some cells a small amount of phosphorylated ATM colocalized with VRCs (data not shown). The phosphorylated ATM signal with Ad5 was much less intense than in cells infected with the E4-deleted Ad5 (Figure 3B). Together, these data suggest that wild-type Ad5 suppresses ATM activation at VRCs, but that ATM signaling is activated during infection with wild-type forms of other serotypes.
Figure 3. ATM is activated during infection with multiple serotypes.
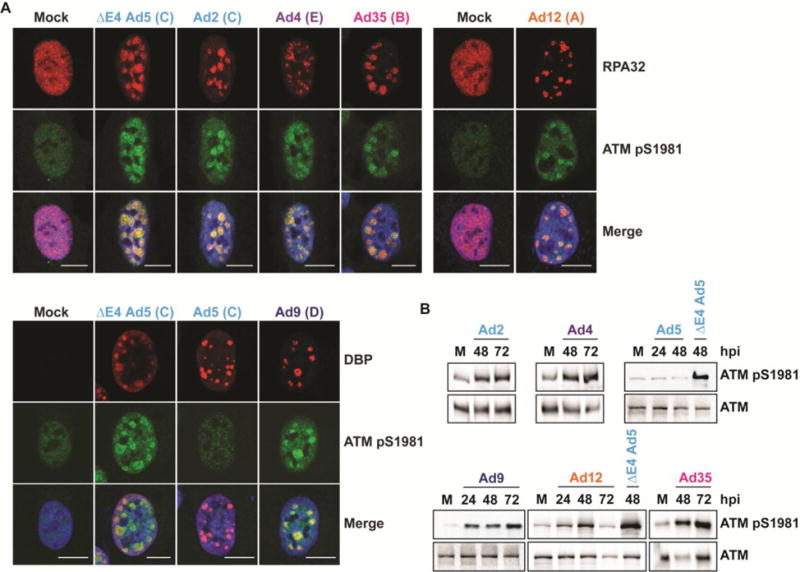
(A) Immunofluorescence of phosphorylated ATM (pS1981) (green) during infection of U2OS cells with each serotype at 24 hpi. The E4-deleted Ad5 mutant dl1004 serves as a positive control for ATM phosphorylation. Cellular RPA32 or viral DBP (red) mark sites of viral replication. Merged images include DAPI stain in blue. Scale bar = 10 μm. Representative images are shown. (B) Western blots of phosphorylated ATM (pS1981) and total ATM with infected cell lysates. U2OS cells were infected with each serotype and harvested at the indicated time points.
MRN impairs DNA replication for Ad9 and Ad12 serotypes
Based on observed differences for MRN components during infection with different wild-type Ad serotypes, we asked to what extent MRN inhibits replication of the different serotypes. To determine whether the observed differences between serotypes affect viral DNA replication, we measured viral DNA accumulation by quantitative PCR in the presence and absence of a functional MRN complex (Figure 4). NBS-ILB1 cells harbor a hypomorphic Nbs1 mutation that prevents formation of the MRN complex [48], and complementation of these cells with wild-type Nbs1 restores MRN complex formation [47]. We infected NBS-ILB1 cells (NBS+Vector) and matched cells expressing wild-type Nbs1 (NBS+Nbs1) with each serotype, as well as with Ad5 mutants. As expected, replication of wild-type Ad5 was similar in the presence or absence of the MRN complex (Figure 4A). We also observed that the presence of Nbs1 did not impact replication of Ad5 mutants that were E1b55K-deleted (dl110 [52], retains mislocalization of MRN), or E4orf1-3-deleted (dl1006 [51], retains degradation of MRN). In contrast, DNA replication of complete E4-deleted virus (dl1004 [51]) was inhibited in cells complemented with Nbs1 to generate the functional MRN complex, but was rescued in cells that lack functional Nbs1. This demonstrates that in wild-type Ad5 infection, either mislocalization or degradation of MRN is sufficient to overcome the inhibitory effects of the MRN complex, as previously reported [22]. Similar to Ad5, neither Ad2 nor Ad4 was affected by MRN, since replication of each virus was similar in mutant and complemented cells (Figure 4A). This is consistent with our observation of MRN degradation and mislocalization by both viruses (Figures 1 and 2). Interestingly, Ad35, which does not mislocalize or degrade Mre11, was not impaired in the presence of functional Nbs1. In fact, Ad35 replication significantly decreased in the absence of Nbs1. It was also interesting to observe that replication of Ad9, which mislocalizes but does not degrade MRN, was significantly increased in the absence of functional Nbs1 at multiple stages of infection (Figure 4A–B). Similarly, replication of Ad12, which degrades but does not mislocalize MRN, was significantly increased in the absence of the functional MRN complex (Figure 4A–B). We verified that Ad9 and Ad12 retained the ability to manipulate MRN in these cells by examining Mre11 by immunofluorescence (Figure 4C). Together, these data suggest that serotypes differ in their susceptibility to inhibition by the MRN complex. Ad5, Ad2, Ad4, are not affected by MRN, and Ad35 replication is enhanced in the presence of MRN. In contrast, MRN impairs replication of Ad9 and Ad12, despite being targeted by each of these viruses. These data suggest that in contrast to Ad5, neither MRN mislocalization by Ad9 nor MRN degradation by Ad12 is sufficient to overcome inhibition of viral DNA replication by the MRN complex during wild-type virus infection.
Figure 4. MRN impairs Ad9 and Ad12 replication.
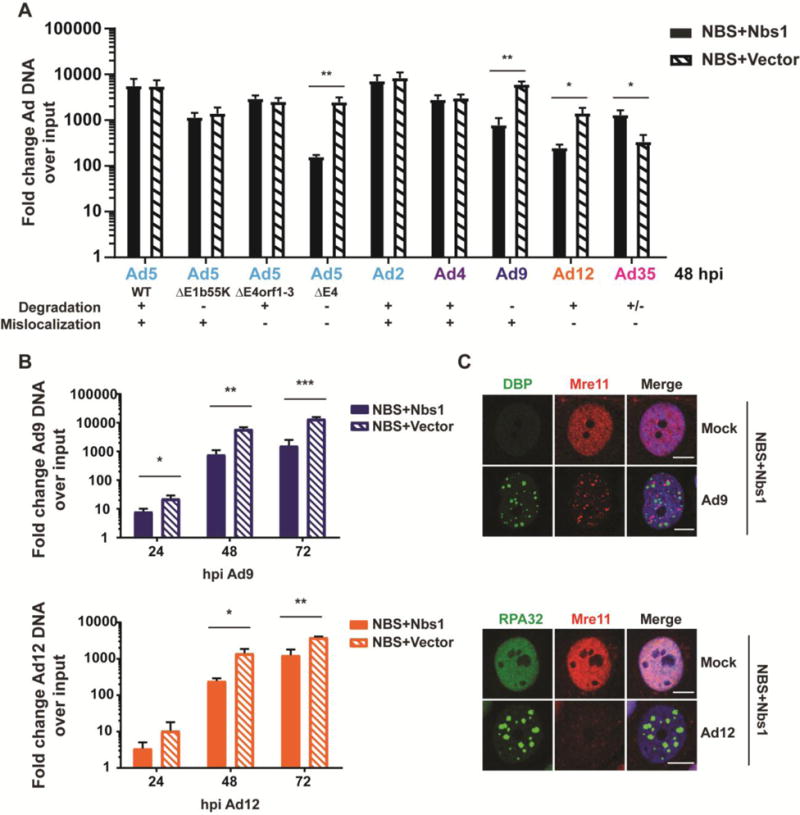
(A) Hypomorphic Nbs1 cells complemented with wild-type Nbs1 (NBS+Nbs1) or empty vector (NBS+Vector) were infected to determine the effect of MRN on viral replication. Cells were harvested 48 hpi, and viral DNA accumulation was measured by quantitative PCR using primers specific for a conserved region of the viral genome. Values were normalized internally to tubulin and also to a 4-hour time point to control for input virus. Fold increase over input is shown, and error bars represent standard deviation from at least three biological replicates. Statistical significance was determined by a student’s T test (* = p < 0.05, ** = p < 0.01). (B) Viral DNA accumulation was measured in NBS+Vector and NBS+Nbs1 cells as in panel A over a time course of infection with Ad9 and Ad12. MRN impairs DNA accumulation at multiple time points of infection. Error bars represent standard deviation from at least three biological replicates. Statistical significance was determined by a student’s T test (* = p < 0.05, ** = p < 0.01, *** = p < 0.001). (C) Immunofluorescence of complemented NBS cells (NBS+Nbs1) 48 hpi confirms that Ad9 mislocalizes MRN and that Ad12 decreases MRN levels in these cells. Mre11 is shown in red. Viral DBP and cellular RPA32 (green) mark sites of viral DNA replication, and merged images include DAPI in blue. Scale bar = 10μm.
ATM does not impair Ad9 or Ad12
Since neither MRN targeting by Ad9 nor Ad12 was sufficient to overcome inhibition by MRN, we investigated these serotypes further to identify potential reasons for their inability to overcome MRN. ATM signaling has been suggested to impair infection of certain Ad5 mutants [25, 27]. Since ATM signaling is activated during infection by wild-type Ad9 and Ad12 (Figure 3), we examined whether inhibition of Ad9 and Ad12 by MRN could be due to the downstream effects of ATM activation. To determine the effect of ATM activity on viral replication, we measured viral genome accumulation by quantitative PCR in cells treated with the ATM inhibitor KU55933 [54]. ATM inhibition was demonstrated by decreased signals for the autophosphorylation mark at S1981 (Figure 5A–B). We found that ATM inhibition did not affect accumulation of viral DNA genomes for Ad9 but led to decreased DNA accumulation for Ad12 (Figure 5A–B). We also assessed the impact of ATM by infecting A-T cells, which are ATM deficient, and matched cells complemented with ATM [10, 49]. Neither Ad9 nor Ad12 DNA replication was impaired by ATM in these cells, but Ad12 replication was greater in cells with ATM (Figure 5C–D). We conclude that ATM does not impair replication of these serotypes, and therefore inhibition of viral DNA replication by MRN is unlikely to be through ATM.
Figure 5. ATM does not impair Ad9 or Ad12.
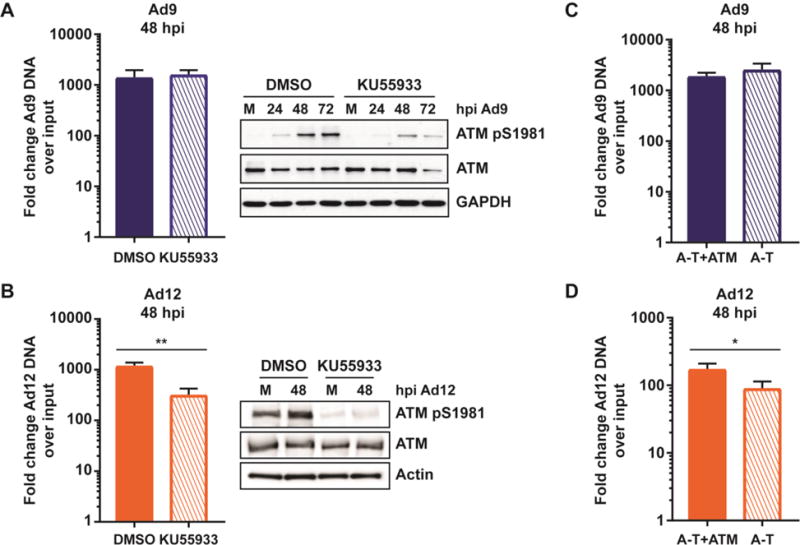
(A–B) U2OS cells were treated with the ATM inhibitor KU55933 or DMSO at 1 hour prior to infection with Ad9 (A) or Ad12 (B). Cells were harvested 48 hpi and viral genome accumulation measured by quantitative PCR as in Figure 3. Averages from at least three biological replicates are shown. Statistical analyses were performed using a student’s T test. Western blots demonstrate reduced ATM phosphorylation in cells treated with KU55933. The phospho-ATM blot in panel A was performed with an antibody from Epitomics, and the phospho-ATM blot in panel B was performed with an antibody from Abcam. This difference may explain the different intensity of phospho-ATM in mock samples between panels A and B. (C–D) ATM-deficient A-T cells or matched cells complemented with ATM were infected with Ad9 (C) or Ad12 (D). Cells were harvested 48 hpi and viral genome accumulation was measured by quantitative PCR as described in Figure 3. Averages from at least three biological replicates are shown. Statistical significance was determined using a student’s T test (* = p < 0.05).
MRN colocalizes with E4orf3 and PML during Ad9 infection
We reasoned that mechanistic differences between Ad5, Ad9, and Ad12 targeting of MRN may explain the inability of Ad9 and Ad12 to overcome the inhibitory effects of MRN. We therefore more closely examined MRN mislocalization and degradation by each of these serotypes. During wild-type Ad5 infection, MRN is colocalized with E4orf3 and PML into nuclear tracks [12]. We used an antibody raised against the Ad5-E4orf3 to detect E4orf3 expressed during Ad9 infection by immunofluorescence (Figure 6A). We found that Ad9-E4orf3 formed nuclear structures similar to those characterized for Ad5-E4orf3 [32, 33]. We also found that Mre11 colocalized with E4orf3 during Ad9 infection (Figure 6A). Immunofluorescence of Ad9 infected cells showed that PML was also disrupted from PML bodies into track-like structures that partially colocalized with Mre11 (Figure 6B). Staining for Mre11 and Nbs1 showed colocalization into these structures, suggesting that MRN components are redistributed as a complex during Ad9 infection (Figure 6C). These results suggest that Ad9 disrupts PML and mislocalizes MRN to nuclear structures containing E4orf3 and PML, similar to Ad5.
Figure 6. MRN colocalizes with E4orf3 and PML during Ad9 infection.
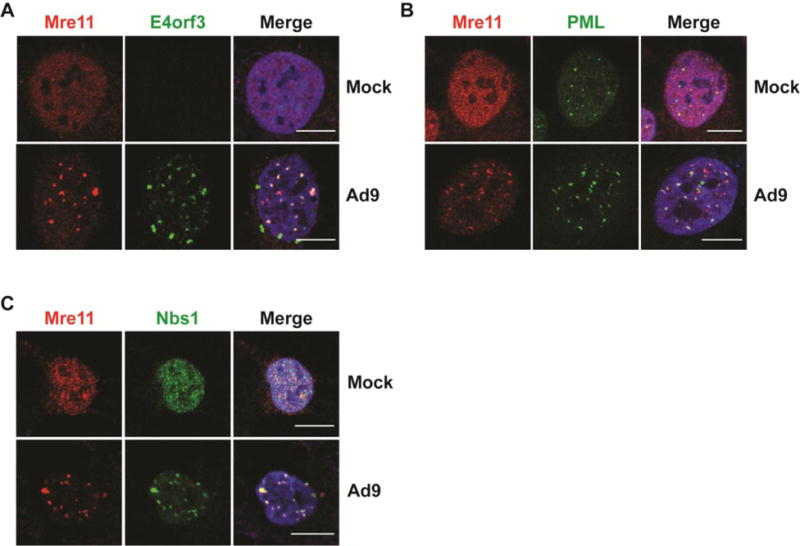
(A) Representative immunofluorescence results from Ad9-infected U2OS cells (24 hpi) showing Mre11 (red) and Ad9-E4orf3 (A), PML (B), or Nbs1 (C) in green. Merged images include DAPI in blue. Scale bar = 10 μm.
Ad9-E4orf3 is not sufficient to alter MRN localization
Since Ad5-E4orf3 is sufficient for MRN mislocalization and disruption of PML bodies by transfection [12, 33], we investigated the role of Ad9-E4orf3 in MRN mislocalization to PML tracks. We transfected an expression vector for FLAG-tagged Ad9-E4orf3 and found that it formed characteristic track-like structures, although the E4orf3 tracks formed in the absence of infection are notably longer than those formed during infection (Figure 7A, compare to Figures 6A and 7B). Ectopically expressed Ad9-E4orf3 was sufficient to reorganize PML into tracks (Figure 7A) similar to Ad5-E4orf3. However, we found that Ad9-E4orf3 was not able to alter the localization of MRN, since Mre11 retained a diffuse nuclear pattern when Ad9-E4orf3 was expressed (Figure 7B). Additional immunofluorescence showed that Mre11 results are representative of all three MRN components (data not shown). FLAG-Ad9-E4orf3 expressed from a doxycycline-inducible cell line was also insufficient to alter Mre11 localization (Figure 7C). However, the MRN complex colocalized with Ad9-E4orf3 when transfected cells were subsequently infected with Ad9 (Figure 7B). Together with data from Figure 5, these observations show that although Ad9 mislocalizes MRN to E4orf3-PML tracks during infection, Ad9-E4orf3 is not sufficient to mislocalize MRN. This suggests that expression of additional viral proteins or viral-induced changes are required for MRN mislocalization by Ad9 during infection.
Figure 7. Ad9-E4orf3 is not sufficient to alter MRN localization.
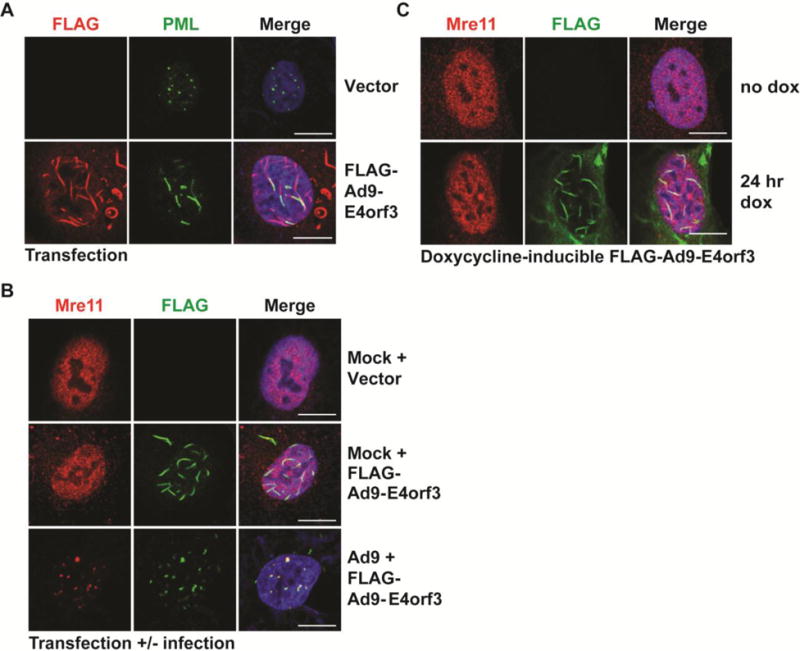
(A) Immunofluorescence results from U2OS cells transfected with FLAG-tagged Ad9-E4orf3 showing the effect of Ad9-E4orf3 expression on PML (green). Ad9-E4orf3 was visualized using an antibody for FLAG (red). Merged images include DAPI in blue. Scale bar = 10 μm. (B) Immunofluorescence of U2OS cells transfected with FLAG-tagged Ad9-E4orf3 with or without Ad9 infection. Cells were transfected 2 hpi and harvested 24 hours post-infection. FLAG-Ad9-E4orf3 is shown in green, Mre11 in red, and merged images include DAPI in blue. Scale bar = 10 μm. (C) Immunofluorescence of U2OS cells with doxycycline-inducible FLAG-Ad9-E4orf3. Cells were treated with doxycycline (+dox) for 24 hours. Mre11 is shown in red, FLAG-Ad9-E4orf3 in green, and merged images include DAPI in blue. Scale bar = 10 μm.
Discussion
Cellular proteins can serve as obstacles to virus infection, and viruses have therefore evolved strategies to overcome these intrinsic defenses. Extensive work from our lab and others has demonstrated that proteins within the DDR can inhibit adenovirus DNA replication, late protein production, and viral propagation. In particular, the MRN complex has been suggested to impair viral replication both directly and indirectly through downstream responses [12, 22, 24–26]. The multiple ways that wild-type Ad5 targets the MRN complex have presumably evolved to overcome this inhibition. Previous work has demonstrated that adenovirus serotypes differ in their interactions with MRN and other proteins in the DDR network [40, 41, 44–46]. In this study, we further examined the relationship between MRN and serotypes across the adenovirus family, with representatives from different adenovirus subgroups (A-E). We found that adenovirus serotypes in different subgroups could target MRN complex proteins, suggesting that MRN is a ubiquitous obstacle to viral DNA replication across the adenovirus family. We specifically asked whether adenovirus serotypes differed in their susceptibility to MRN inhibition and found that unlike Ad5, some serotypes are unable to overcome impairment by MRN. Previous work demonstrated that MRN can impair mutants of Ad5 (subgroup C) and Ad4 (subgroup E) that cannot target MRN [22, 24, 26]. Here, we demonstrate that MRN can also restrict replication of wild-type serotypes from subgroup A (Ad12) and subgroup D (Ad9) (Figure 4). We were surprised to find that even though Ad9 can redistribute MRN away from viral replication centers, wild-type Ad9 genome levels were significantly reduced in the presence of functional MRN complex (Figure 4). This suggests that mislocalization by Ad9 is not sufficient to overcome inhibition by the MRN complex. Results with Ad12 were also unexpected, since MRN significantly impaired wild-type Ad12, despite being degraded during infection. The subgroup B serotype Ad35 did not degrade or mislocalize Mre11, similar to prior findings with other subgroup B serotypes, Ad7 and Ad11 [40]. However, another study demonstrated that transfection with E1b55K and E4orf6 from subgroup B serotypes Ad16 and Ad34 leads to a decrease in Mre11 levels [41], raising the possibility that interactions with MRN could vary even within a subgroup. While Ad35 did not degrade or mislocalize Mre11, it did result in Rad50 degradation, demonstrating that this serotype can target a single component of the complex. Surprisingly, degradation of Rad50 did not affect Mre11 or Nbs1 levels, nor did it affect Mre11 localization to VRCs. Interestingly, wild-type Ad35 DNA replication appeared to be enhanced in the presence of MRN formation (Figure 4). It is possible that Ad35 prevents inhibition of DNA replication by MRN through its degradation of Rad50 or through an alternative, undefined mechanism. Results with Ad35 raise the possibility that Ad35 could even exploit Mre11 or Nbs1 to benefit viral replication, and these observations merit further investigation.
While previous studies have demonstrated that MRN can inhibit replication of mutants of Ad5 that do not manipulate MRN [22, 24, 26], we demonstrate for the first time that MRN can inhibit replication of wild-type viruses Ad9 and Ad12 despite the fact that Ad9 and Ad12 alter MRN localization or protein levels. Previous studies showed that concatemers do not form during infection with either of these serotypes, likely due to ligase IV degradation [40]. We explored the role of ATM to determine if inhibition could be through downstream signaling, since ATM can inhibit certain Ad5 mutants [25, 27]. We first investigated how infection with each of these serotypes affects ATM signaling. As previously reported [8], wild-type Ad5 limited ATM activation at VRCs, but infection with the E4-deleted Ad5 mutant dl1004 resulted in robust ATM activation at VRCs (Figure 3). ATM was activated and colocalized with VRCs during infection with all other serotypes examined (Figure 3). These data indicate that the ATM activation observed during infection with these serotypes is in response to viral DNA or replication, rather than the global ATM activation sometimes observed during Ad5 infection [25]. Since all but one of the serotypes we studied can target MRN through either degradation or mislocalization, ATM activation at VRCs indicates that either (1) ATM is activated independently of MRN during these infections, or (2) there is sufficient residual MRN at VRCs to activate ATM. Pan-nuclear MRN-independent ATM activation has been observed during late stages of wild-type Ad5 infection [25], but MRN is required for full ATM activation at Ad5 VRCs [8, 25]. Therefore, we expect that the ATM activation at VRCs is due to residual MRN at VRCs. Furthermore, since MRN inhibited wild-type Ad9 and Ad12, it is likely that there is some MRN at VRCs of these two serotypes. We found that ATM did not impair replication of Ad9 or Ad12 (Figure 5), excluding the possibility that MRN inhibition of these serotypes is through downstream ATM signaling. Ad12 replication may benefit from ATM (Figures 5B and D). Other DNA viruses, such as herpes simplex virus type 1 and human papillomavirus, also benefit from ATM [18, 55–57], but the mechanism by which ATM influences these viruses remains unclear. It is possible that viruses exploit the kinase activity of ATM to regulate proteins involved in viral replication.
Since wild-type Ad9 and Ad12 viruses did not overcome MRN inhibition of viral DNA replication, we investigated whether mislocalization or degradation by these serotypes occurred through mechanisms different than Ad5. We reasoned that different mechanisms could render these serotypes less effective at evading MRN recognition and overcoming inhibition of viral DNA replication. We found that MRN colocalizes with E4orf3 and PML in nuclear tracks during Ad9 infection (Figure 6), similar to MRN localization during Ad5 infection [12]. However, unlike Ad5, we found that Ad9-E4orf3 alone was not able to alter MRN localization, even though it was sufficient to disrupt PML bodies (Figure 7). It is possible that during Ad9 infection another viral protein is responsible for MRN mislocalization, either in conjunction with E4orf3 or by itself. The responsible Ad9 protein may only partially sequester MRN from VRCs, allowing sufficient MRN to accumulate at viral DNA and impair virus replication. Another possibility is that Ad9 infection promotes changes to the cellular environment, to MRN, or to E4orf3 that facilitate mislocalization. For example, there could be post-translational modifications to Ad9-E4orf3 or to MRN that occur during infection and promote E4orf3 interaction with MRN. Such a requirement could delay mislocalization until after some MRN had already associated with Ad9 VRCs and inhibited replication. Since Ad12 was also inhibited by MRN during infection despite degradation of MRN proteins, we further examined MRN degradation but were unable to identify any differences between Ad5 and Ad12 degradation in this study (data not shown). Previous work suggested that the ubiquitin ligase formed by Ad12-E1b55K and Ad12-E4orf6 utilizes Cullin 2, in contrast to the Cullin 5 used by Ad5 [41]. It is possible that this difference renders Ad12 degradation less effective at overcoming MRN, or that differences in degradation substrates between Ad12 and Ad5 [40–42] create distinct cellular environments that influence MRN function.
An alternative explanation for the observed inhibition of wild-type Ad9 and Ad12 viruses could be that each serotype is uniquely adapted to its natural cell type. In this study, we used a single cell type in each experiment to examine serotypes with diverse tissue tropisms. This experimental design allowed us to uncover differences in interactions with MRN between these serotypes that may not be observed in their natural cell types. It is possible that Ad9 and Ad12, which respectively cause conjunctivitis and gastrointestinal disorder, are able to escape MRN inhibition in conjunctival or gastrointestinal cells but not in the fibroblasts or osteosarcoma epithelial cells used in our experiments [47, 48]. This could be due to unidentified differences in MRN levels, regulation, or activity between cell types. Ad9 and Ad12 could potentially be used to uncover differences between MRN from different cell types. It is possible that MRN may have provided selective pressure for adenovirus evolution. Previously published work suggests that the species-specific interaction between Nbs1 and another nuclear-replicating DNA virus, herpes simplex virus type 1, impacts species tropism [58]. Given the negative impact of MRN on adenovirus replication, differences in tissue tropism between human adenovirus serotypes could be partially due to an inability to evade MRN-mediated restriction in certain cell types. Together, the findings from this study suggest that MRN is an obstacle for serotypes across the human adenovirus family and raise the possibility that MRN has provided selective pressure and influenced adenovirus evolution and tissue tropism.
Highlights.
Adenoviruses have diverse effects on MRN and ATM.
Wild-type adenoviruses Ad9 and Ad12 are impaired by MRN but not by ATM.
MRN is mislocalized to E4orf3 and PML-containing tracks during Ad9 infection.
Ad9-E4orf3 is not sufficient to mislocalize MRN.
Acknowledgments
We thank A. Levine, Y. Shiloh, E. Makeyev, G. Ketner, T. Dobner, and D. Ornelles for generously providing reagents. We thank current and former members of the Weitzman lab for generation of reagents, helpful discussions, and critical reading of the manuscript.
Funding statement
This work was supported by a grant from the National Institutes of Health (CA097093 to M.D.W.) and funds from the Children’s Hospital of Philadelphia (M.D.W.). N.J.P. was supported in part by a National Institutes of Health T32 training grant (NS007180).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
We have no competing interests.
References
- 1.Berk AJ. Adenoviridae. In: Knipe DMH PM, editor. Fields Virology. Lippincott Williams & Wilkins; Philadelphia, PA: 2013. pp. 1704–1731. [Google Scholar]
- 2.Davison AJ, Benko M, Harrach B. Genetic content and evolution of adenoviruses. J Gen Virol. 2003;84(Pt 11):2895–908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 3.Reyes ED, et al. Identifying host factors associated with DNA replicated during virus infection. Mol Cell Proteomics. 2017 doi: 10.1074/mcp.M117.067116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle. 2005;4(6):737–40. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 6.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13(9):458–62. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 7.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12(2):90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carson CT, et al. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22(24):6610–20. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 10.Ziv Y, et al. Cellular and molecular characteristics of an immortalized ataxia-telangiectasia (group AB) cell line. Cancer Res. 1989;49(9):2495–501. [PubMed] [Google Scholar]
- 11.Jazayeri A, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8(1):37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 12.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418(6895):348–52. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 13.Turnell AS, Grand RJ. DNA viruses and the cellular DNA-damage response. J Gen Virol. 2012;93(Pt 10):2076–97. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 14.Luftig MA. Viruses and the DNA Damage Response: Activation and Antagonism. Annu Rev Virol. 2014;1(1):605–25. doi: 10.1146/annurev-virology-031413-085548. [DOI] [PubMed] [Google Scholar]
- 15.Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15(3):119–26. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses. 2015;7(5):2542–91. doi: 10.3390/v7052542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan EL, Hollingworth R, Grand RJ. Activation of the DNA Damage Response by RNA Viruses. Biomolecules. 2016;6(1):2. doi: 10.3390/biom6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pancholi NJ, Price AM, Weitzman MD. Take your PIKK: tumour viruses and DNA damage response pathways. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732) doi: 10.1098/rstb.2016.0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiden MD, Ginsberg HS. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc Natl Acad Sci U S A. 1994;91(1):153–7. doi: 10.1073/pnas.91.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker A, et al. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol. 2007;81(13):7034–40. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263(2):307–12. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- 22.Lakdawala SS, et al. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J Virol. 2008;82(17):8362–72. doi: 10.1128/JVI.00900-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans JD, Hearing P. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J Virol. 2003;77(9):5295–304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol. 2005;79(10):6207–15. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah GA, O’Shea CC. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell. 2015;162(5):987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathew SS, Bridge E. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology. 2007;365(2):346–55. doi: 10.1016/j.virol.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 27.Gautam D, Bridge E. The kinase activity of ataxia-telangiectasia mutated interferes with adenovirus E4 mutant DNA replication. J Virol. 2013;87(15):8687–96. doi: 10.1128/JVI.00376-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Querido E, et al. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15(23):3104–17. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harada JN, et al. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol. 2002;76(18):9194–206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Querido E, et al. Identification of three functions of the adenovirus e4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J Virol. 2001;75(2):699–709. doi: 10.1128/JVI.75.2.699-709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz RA, et al. Distinct requirements of adenovirus E1b55K protein for degradation of cellular substrates. J Virol. 2008;82(18):9043–55. doi: 10.1128/JVI.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho T, et al. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol. 1995;131(1):45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doucas V, et al. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996;10(2):196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 34.Leppard KN, Everett RD. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J Gen Virol. 1999;80(Pt 4):997–1008. doi: 10.1099/0022-1317-80-4-997. [DOI] [PubMed] [Google Scholar]
- 35.Ou HD, et al. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell. 2012;151(2):304–19. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araujo FD, et al. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol. 2005;79(17):11382–91. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 38.Nichols GJ, Schaack J, Ornelles DA. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J Virol. 2009;83(12):5987–98. doi: 10.1128/JVI.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carson CT, et al. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009;28(6):652–62. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forrester NA, et al. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J Virol. 2011;85(5):2201–11. doi: 10.1128/JVI.01748-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng CY, et al. The E4orf6/E1B55K E3 ubiquitin ligase complexes of human adenoviruses exhibit heterogeneity in composition and substrate specificity. J Virol. 2011;85(2):765–75. doi: 10.1128/JVI.01890-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackford AN, et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc Natl Acad Sci U S A. 2010;107(27):12251–6. doi: 10.1073/pnas.0914605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bridges RG, et al. The Adenovirus E4-ORF3 Protein Stimulates SUMOylation of General Transcription Factor TFII-I to Direct Proteasomal Degradation. MBio. 2016;7(1):e02184–15. doi: 10.1128/mBio.02184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng CY, et al. Role of E1B55K in E4orf6/E1B55K E3 ligase complexes formed by different human adenovirus serotypes. J Virol. 2013;87(11):6232–45. doi: 10.1128/JVI.00384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stracker TH, et al. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J Virol. 2005;79(11):6664–73. doi: 10.1128/JVI.79.11.6664-6673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanchette P, et al. Aggresome formation by the adenoviral protein E1B55K is not conserved among adenovirus species and is not required for efficient degradation of nuclear substrates. J Virol. 2013;87(9):4872–81. doi: 10.1128/JVI.03272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerosaletti KM, et al. Retroviral expression of the NBS1 gene in cultured Nijmegen breakage syndrome cells restores normal radiation sensitivity and nuclear focus formation. Mutagenesis. 2000;15(3):281–6. doi: 10.1093/mutage/15.3.281. [DOI] [PubMed] [Google Scholar]
- 48.Kraakman-van der Zwet M, et al. Immortalization and characterization of Nijmegen Breakage syndrome fibroblasts. Mutat Res. 1999;434(1):17–27. doi: 10.1016/s0921-8777(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 49.Ziv Y, et al. Recombinant ATM protein complements the cellular A–T phenotype. Oncogene. 1997;15(2):159–67. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 50.Khandelia P, Yap K, Makeyev EV. Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci U S A. 2011;108(31):12799–804. doi: 10.1073/pnas.1103532108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bridge E, Ketner G. Redundant control of adenovirus late gene expression by early region 4. J Virol. 1989;63(2):631–8. doi: 10.1128/jvi.63.2.631-638.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babiss LE, Ginsberg HS. Adenovirus type 5 early region 1b gene product is required for efficient shutoff of host protein synthesis. J Virol. 1984;50(1):202–12. doi: 10.1128/jvi.50.1.202-212.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pombo A, et al. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 1994;13(21):5075–85. doi: 10.1002/j.1460-2075.1994.tb06837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hickson I, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 55.Lilley CE, et al. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102(16):5844–9. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillespie KA, et al. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 2012;86(17):9520–6. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85(17):8981–95. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lou DI, et al. An Intrinsically Disordered Region of the DNA Repair Protein Nbs1 Is a Species-Specific Barrier to Herpes Simplex Virus 1 in Primates. Cell Host Microbe. 2016;20(2):178–88. doi: 10.1016/j.chom.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]